

Abstract
The introduction of targeted treatments and more recently immune checkpoint inhibitors (ICI) to the treatment of metastatic non-small cell lung cancer (NSCLC) has dramatically changed the prognosis of selected patients. For patients with oncogene-addicted metastatic NSCLC harbouring an epidermal growth factor receptor (EGFR) or v-Raf murine sarcoma viral oncogene homologue B1 (BRAF) mutation or an anaplastic lymphoma kinase (ALK) or ROS proto-oncogene 1, receptor tyrosine kinase (ROS1) gene alteration (translocation, fusion, amplification) mutation-specific tyrosine kinase inhibitors (TKI) are already first-line standard treatment, while targeted treatment for other driver mutations affecting MET, RET, human epidermal growth factor receptor (HER) 2, tropomyosin receptor kinases (TRK) 1–3 and others are currently under investigation. The role of ICI in these patient subgroups is currently under debate. This article summarises a round-table discussion organised by ESMO Open in Vienna in July 2018. It reviews current clinical data on ICI treatment in patients with metastatic oncogene-addicted NSCLC and discusses molecular diagnostic assessment, potential biomarkers and radiological methods for response evaluation of ICI treatment. The round-table panel concluded ICI should only be considered in patients with oncogene-addicted NSCLC after exhaustion of effective targeted therapies and in some cases possibly after all other therapies including chemotherapies. More clinical trials on combination therapies and biomarkers for ICI therapy based on the specific differing characteristics of oncogene-addicted NSCLC need to be conducted.
Keywords: immune checkpoint inhibitors, NSCLC, oncogene addiction
Video abstract.
Introduction
The management of patients with metastatic non-small cell lung cancer (NSCLC) underwent significant transformation in the last 10–15 years by the development of precision medicine based on molecular characterisation. Molecular analysis revealed distinct targetable driver mutations in about 10%–20% of patients with metastatic NSCLC.1 The most frequently observed targetable mutations are aberrations in the epidermal growth factor receptor (EGFR) gene (about 10%–15% in Caucasians), followed by gene rearrangements/gene fusions in the anaplastic lymphoma kinase (ALK) gene (about 5%) and ROS proto-oncogene 1, receptor tyrosine kinase (ROS1) (about 1%–3%). In addition, mutations in v-Raf murine sarcoma viral oncogene homologue B1 (BRAF) (about 4%) can be observed and targeted therapeutically with mutation-specific tyrosine kinase inhibitors (TKI).1 Besides these targetable genetic alterations, also the expression of programmed death-ligand 1 (PD-L1), an immune suppressive molecule, needs to be considered for therapeutic decision-making. The programmed cell death protein 1 (PD-1) immune checkpoint inhibitor (ICI) pembrolizumab was shown to have higher efficacy as first-line treatment compared with platin-based chemotherapy in patients without the presence of a driver oncogene alteration but PD-L1 expression in more than 50% of tumour cells2 and to increase efficacy of platin-based chemotherapy in patients with a lower PD-L1 expression level.3
Therefore, NSCLC is a molecularly heterogenous disease and initial molecular diagnosis forms the basis for systemic treatment decisions, given the clinical superiority of TKI over chemotherapy in patients harbouring a predictive molecular alteration. However, resistance to targeted therapies and progression occurs in almost all patients and especially brain metastases can be observed frequently as an area of progression.4 Treatment options on exhaustion of targeted therapies and chemotherapy are few, underscoring the need to explore the new and promising treatment category of ICI, also in patients with oncogene-addicted metastatic NSCLC.
ICI target the inhibitory T cell co-receptors and thereby increase the capacity of the tumour-specific immune response. This new category of immune-modulating therapies has revolutionised oncology as in comparison to targeted therapies and chemotherapy long lasting and durable responses can be achieved in a subfraction of patients.5 Here, PD-L1 inhibitors, PD-1 inhibitors and cytotoxic T-lymphocyte-associated protein 4 inhibitors have been investigated in patients with metastatic NSCLC.6
In the context of NSCLC, patients with oncogene addiction were frequently excluded from registration trials, resulting in so far limited clinical knowledge on the efficacy of ICI in the subcohort of molecularly altered NSCLC.7–14 This article aims to review the available clinical data on ICI treatment in patients with metastatic oncogene-addicted NSCLC and discusses molecular diagnostic assessment, potential biomarkers and radiological methods for response evaluation of ICI treatment.
Molecular diagnostic assessment in NSCLC
Personalised cancer therapy comes with increased and complex diagnostic testing. In clinical practice and as recommended by clinical guidelines,15 selection of targeted therapies for NSCLC requires testing for EGFR and BRAF mutations, rearrangements or fusion protein expression involving the ALK and ROS1 genes and the expression of PD-L1. Therefore, molecular testing should be carried out in all patients who have a definite, probable or possible diagnosis of adenocarcinoma, for whom this diagnosis cannot be reasonably excluded, and for patients with non-small cell carcinoma or for patients with squamous cell carcinoma who have a high risk of a target mutation or rearrangement (never or light smokers, very long-term ex-smokers or young women).2 16
Given the high amount of analysis to be made on often sparse tumour material, strong recommendations on tissue preservation for biomarker studies have been outlined by several guidelines.2 It is critical that pathology laboratories develop policies for integrating biomarker testing into their routine tissue-processing workflows to minimise the number of ancillary stains performed for the diagnosis and classification. The time point of molecular testing, right after pathology diagnosis as indicated by the pathologist (reflex testing) or only after additional claim by the treating clinician (bespoke testing), is currently a topic of debate and organised differently throughout centres.17 Molecular testing initiated by the pathologist immediately after diagnosis of cancer (reflex testing) provides results in 5–10 working days, in contrast to bespoke testing requested by the oncologist or the multidisciplinary team only when the test is needed. Reflex testing has the advantages of a quicker molecular profiling for clinical decisions and a higher efficiency in the diagnostic process in the laboratory. However, it increases needed resources and potentially results in costly testing in patients without therapeutic consequence18 19 (figure 1).
Figure 1.

Molecular testing parallel algorithm without next generation sequencing (adapted from Kerr and López-Ríos17). ALK, anaplastic lymphoma kinase; EGFR, epidermal growth factor receptor; FISH, fluorescence in situ hybridisation; ICI, immune checkpoint inhibitor; MDT, multidisciplinary team; NSCLC, non-small cell lung cancer; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; TKI, tyrosine kinase inhibitor
Testing of driver mutations can be performed by targeted sequencing, a combined sequencing and immunohistochemistry/immunofluorescence approach or next generation sequencing (NGS). EGFR and BRAF testing are conducted by DNA sequencing, while in several laboratories due to cost-effectiveness, ALK and ROS1 testing are mostly performed by immunohistochemistry (IHC) and/or fluorescence in situ hybridisation (FISH). Currently, the approved method for PD-L1 testing is IHC.20 NGS is rapidly emerging as an option for the delivery of multiplexed genomic testing in lung cancer, especially in academic centres. NGS testing potentially provides more data on genetic alterations than the treating clinicians would usually include in their decision-making. Alterations for which no treatment is available or for which treatment is available only through a clinical trial could therefore also be detected. Moreover, NGS approaches are becoming available for the identification of uncommon fusion genes involving ALK and ROS1, but experience of the clinical significance of these aberrations is still limited in the absence of IHC or FISH alterations.17 NGS is still relatively costly and its use will depend on whether it is considered cost-effective compared with doing several single-gene tests (figure 2).
Figure 2.
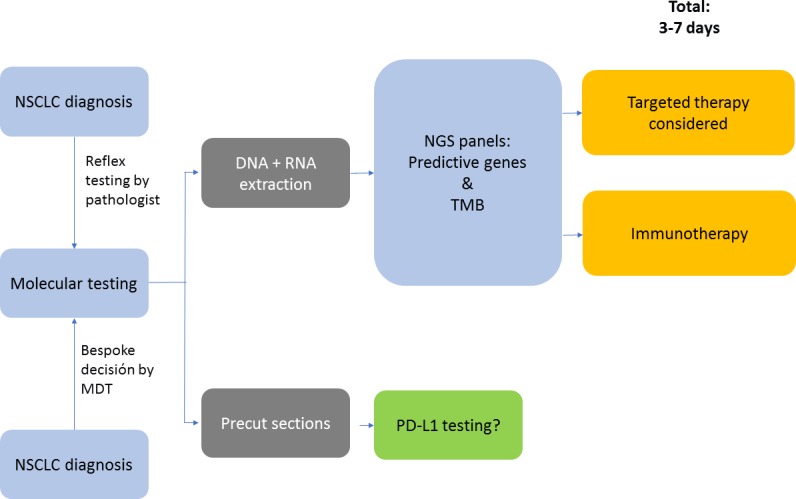
Molecular testing algorithm when NGS is commonplace (adapted from Kerr and López-Rios17). MDT, multidisciplinary team; NSCLC, non-small cell lung cancer; NGS, next generation sequencing; PD-L1, programmed death-ligand 1; TMB, tumour mutational burden.
Liquid biopsies
Liquid biopsy is a broad term that refers to the analysis of biomarkers that can be isolated from body fluid of patients with cancer. However, the analysis of cell-free DNA (cfDNA) isolated from the plasma fraction of peripheral blood is the only approach that entered clinical practice so far.21 In patients with advanced NSCLC, cfDNA testing can provide information on the presence of driver alterations at the time of therapy decision or on the mechanisms of acquired resistance to TKI in those with driver genetic alterations. Indeed, liquid biopsy is the preferred approach for the assessment of the p.T790M EGFR variant in EGFR-mutant patients who progress after treatment with first-generation or second-generation TKI.22 Additional tissue rebiopsy is usually reserved to patients whose results are negative at liquid biopsy testing. Evidence from trials with third-generation TKI in EGFR T790M-mutant patients suggests that tissue and liquid biopsy might provide complementary information.23 24 A negative liquid biopsy T790M test in patients with tumour positive for T790M is associated with a better prognosis compared with the prognosis of patients with both tissue and tumour positive. This finding most likely reflects the correlation between cfDNA levels and tumour burden and/or aggressiveness of the disease—the higher the tumour load, the higher is the amount of cfDNA. On the other hand, patients with a positive blood T790M test and negative tissue have an intermediate outcome as these patients are likely to carry a heterogeneous expression of the T790M leading to a mixed response to third-generation TKI.22 24
NGS-based analysis of liquid biopsy revealed that approximately 50% of T790M-positive resistant patients also carry additional genetic alterations.25 The presence of multiple resistance mechanisms has been associated with resistance to treatment with third-generation TKI.25–28 This highlights that the genetic background of EGFR-mutant lung cancer might significantly change over time. In fact, the molecular complexity of the disease is likely to increase after each line of treatment because of the emergence of multiple clones of resistant cells. In consequence, liquid biopsy testing with NGS-based techniques might better recapitulate the genetic landscape of the disease compared with tissue biopsy in resistant patients.29
The emergence of resistance against EGFR targeting TKI in precision treatment of NSCLC
Patients with NSCLC who harbour mutations in the EGFR gene are candidates to receive treatment with TKI. After a mean time of treatment of 10–14 months, patients usually stop responding to first-generation and second-generation TKI and in consequence show tumour progression which might be systemic, oligoprogression or restricted to the central nervous system (CNS).4 Mechanisms involved in resistance development have been extensively studied not only for first-generation or second-generation inhibitors but also for third-generation EGFR TKI.30
Resistance against first-generation and second-generation TKI
Emergence of resistance to first-generation and second-generation TKI may be due to alterations in the target gene EGFR or to the acquisition of alterations in other genes. The most frequent resistance mechanism is the acquisition of the mutation affecting the amino acid threonine located at position 790 of the EGFR protein.31 32 This mutation increases the binding of the ATP molecule, compared with the inhibitor and therefore compensates the inhibition of the EGFR. The mutation p.T790M is found in more than 50% of EGFR-mutated patients at the time of progression. It may be detected alone or simultaneously to the amplification of the EGFR gene, or to other resistance mechanisms. Other mutations affecting the EGFR gene have also been found in a limited number of patients, such as EGFR p.L747S, p.D761Y and p.T854A.33–35 Mechanisms of resistance involving genes different from EGFR have been also detected, although to a lesser extent. Among these the most recurrently found are: MET and human epidermal growth factor receptor 2 (HER2) amplification, PIK3CA and BRAF mutations and small cell histologic transformation.36 37 More recently, CDKN2A loss, MTOR mutations and FGFR3 alterations including translocations have also been implicated in mediating EGFR TKI resistance.38 39 The specific third-generation EGFR TKI osimertinib that targets the mutation p.T790M has been developed and demonstrates high efficacy in most patients.40
Resistance against third-generation TKI
Third-generation TKI show even higher effectiveness in EGFR-mutant patients with a median response rate of 80% in untreated patients, including those bearing the p.T790M mutation.41 However, resistance development also eventually occurs, although with 18.9 months significantly later than with first-generation and second-generation TKI.41 The mechanisms of resistance, as with first-generation and second-generation inhibitors, include EGFR-dependent and EGFR-independent mechanisms.40 Among the EGFR-dependent resistance mechanisms, mutations affecting the binding of the drug such as the p.C797S or the p.E709K, p.L692V and p.L798I mutations have been observed. Of note, regarding resistance to third-generation TKI, it seems that the appearance of resistance mutations, depending on the location—either in cis or trans—has different implications: if C797S and p.T790M mutations are in trans, cells will be resistant to third-generation TKI but remain sensitive to a combination of first-generation and third-generation EGFR-TKIs.42 Tumours with C797S and p.T790M mutations in cis are greatly resistant to EGFR-TKI and their combinations.42 If C797S mutation develops in T790 wild-type cells after administration of third-generation EGFR-TKI, the cells retain their sensitivity to first-generation TKI, underscoring the preclinical evidence of TKI sequencing.42 Among EGFR-independent mechanisms of resistance to third-generation TKI, MET amplification as well as amplification of genes involving receptors (such as Insulin-like growth factor 1 receptor (IGF1R)) and mutations or amplifications of genes involved in the signalling cascades (such as BRAF) have been reported.43
Molecular biomarkers for response to ICI therapy
Increasing PD-L1 levels, tumour mutational burden (TMB), CD8 T cell infiltration have been associated with increasing benefit from ICI. However, patient selection by predictive biomarkers remains controversial as no absolute predictive markers reliably differentiating between responding and not responding patients were identified yet.44 45
Expression of PD-L1
PD-L1 expressed by the tumour cells can be induced not only by the oncogenic pathways, which induce tumour development, but also by the immune response itself, especially after induction by the interferon-gamma pathway.46 Thus, in case of immune attack, the tumour cell defends itself and upregulates PD-L1. Accordingly, PD-L1 expression on tumour cells was extensively studied as a predictive marker for PD-1 axis targeting ICI. The expression by the tumour cells is heterogenous with areas of expression alternated with areas of absent expression. In line, cut-off values between 1% and 50% of PD-L1 expression tumour cells were investigated as predictive marker for PD-1 axis targeting ICI.47 48 Analysis of biopsies is challenging in this context as samples could be rated as false negative, potentially inhibiting an effective treatment option.
Furthermore, PD-L1 expression has an imperfect negative predictive value, as also a PD-L1 negative tumour can present with clinically relevant response. In addition, PD-L1 analysis might be challenging due to the expression of PD-L1 not only of tumour cells but also by cells of the microenvironment including macrophages and T cells. In addition to this variability in location, PD-L1 expression was shown to also vary over time, as treatments including chemotherapy and radiotherapy can impact the expression level.49
Importantly, oncogenes were shown to not only impact on tumour growth but also on the expression of immunosuppressive molecules including PD-L1 (figure 3). Here, the presence of ALK translocation was shown to be associated with PD-L1 expression, while the presence of EGFR mutation is inversely correlated with PD-L1 expression.50–52 In patients harbouring an EGFR mutation, those with rare EGFR mutations and not harbouring the specific T790M mutation are more likely to express PD-L1 on the tumour cells.50 53 54
Figure 3.

Relationship of programmed death-ligand 1 (PD-L1) expression with oncogene alterations.
Tumour mutational burden
TMB was recently shown to be an important predictive marker for ICI response, as cancer entities with higher TMB like melanoma or smoking-associated NSCLC were shown to have higher response rates55 56 (figure 4). The high TMB and the resulting high rate of pathological or foreign folded proteins is associated with a higher fraction of neo-epitopes potentially triggering an immune response. It is important that most responding patients have mainly clonal mutations, while non-responders have mutations present in tumour subclones, allowing immune evasion and lower response to ICI.57 Currently, the most suitable cut-off value to define ‘high’ and ‘low’ TMB as well as a uniformed method for detection still need to be defined. Recently, patients with NSCLC with a high TMB, defined as ≥10 mutations per megabase using the FoundationOne CDx assay, treated with a combination of the ICI nivolumab and ipilimumab, showed improved progression-free survival (PFS) compared with those treated with conventional chemotherapy in the first-line setting.58
Figure 4.

Relationship between mutational load and response to immunotherapies targeting PD-1/PD-L1.56 Reprinted by permission from Springer Nature. Yarchoan M, Johnson III BA, Lutz ER et al. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer 2017. NSCLC, non-small cell lung carcinoma; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; RCC, renal cell carcinoma; SCLC, small cell lung carcinoma.
Infiltration by CD8 lymphocytes and transcriptomic signature
Another potential biomarker is CD8+ T cell infiltration, which defines the notion of ‘hot’ tumour and ‘cold’ tumour based on the density of tumour-infiltrating lymphocytes (TIL). Particularly in metastatic melanoma, CD8+ T cell infiltration is associated with better response to immunotherapy.59 In lung cancer, T cell infiltrate and tertiary lymphoid structures were reported to be associated with a good outcome in chemotherapy patients.60 Beyond the presence of T cells, the analysis of the transcriptomic signature of the whole tumour and the deconvolution of the signals allow to identify transcriptomic signatures of activated T cells. Strong expression of effector T cell and interferon gene signatures was shown to be associated with a better response to immunotherapies targeting the PD-1/PD-L1 checkpoint in patients with NSCLC.61
The association of higher CD8+ TIL density and non-synonymous mutation burden was verified in a small cohort of patients with oncogene-addicted NSCLC.51 However, in general, the EGFR-mutated NSCLC harbour a much less inflamed tumour microenvironment compared with EGFR wild-type cancers.62 This might be induced by the expression of the immunosuppressive molecule CD73 in EGFR-mutated NSCLC.63 Indeed, given that most patients with EGFR-mutated NSCLC are never smokers, the rate of passenger mutations is lower and in consequence the TMB and the immunogenicity.
Tumour molecular alterations
Certain molecular alterations affecting the tumour cell such as the β-catenin and phosphatase and tensin homologue (PTEN) pathways have been recently linked to response to immunotherapy in melanoma. When the β-catenin pathway is activated in patients with melanoma, there is little infiltration of CD8+ T cells, whereas this infiltration is important when the β-catenin pathway is not activated.64 Similarly, the loss of anti-tumour protein PTEN is associated with a lack of response to immunotherapy in patients with melanoma.65 However, currently no data exists on the relevance of these pathways in oncogene-addicted NSCLC.
Clinical efficiency of ICI in patients with oncogene-addicted NSCLC
Beyond the already approved indications in the first-line and second-line setting of advanced NSCLC, only relatively little data is available regarding anti-PD-1 and anti-PD-L1 efficacy in patients with oncogene-addicted NSCLC. Most of the available data is for patients harbouring an EGFR mutation or ALK rearrangement, while data for the other even more rare NSCLC subtypes is mostly lacking.
The phase 1 study CA209-012 of PD-1 inhibitor nivolumab (n=52) as first-line treatment in patients with metastatic NSCLC reported an impaired overall efficacy in patients with EGFR mutation patients as compared with patients with EGFR wild-type NSCLC. The overall response rate (ORR) was only 14% in patients with EGFR mutation (ie, one of seven) versus 30% in patients with wild-type NSCLC (ie, nine of 30). Further, the PFS at 24 weeks was only 14% in patients with EGFR mutation versus 51% in wild-type NSCLC, respectively.9
Study CA209-153 is a phase 3b/4 safety trial of nivolumab in patients with advanced or metastatic squamous or non-squamous NSCLC who received at least one prior line. The EGFR mutation status was available in 549 patients and 103 patients presented with an activating EGFR mutation. However, partial response rate was 11% (n=55 patients available for response assessment) in the EGFR-mutated cohort compared with 16% (n=300 patient available for response assessment) in the EGFR wild-type cohort.10
The phase 1 KEYNOTE-001 trial found that PD-1 inhibitor pembrolizumab provides promising long-term OS benefit with a manageable safety profile for PD-L1-expressing treatment-naive advanced NSCLC, with greatest efficacy observed in patients with PD-L1 tumour proportion score (TPS) ≥50%.66 The best objective response rate based on mutation status was 16% in patients with EGFR mutation (n=19) versus 37% without mutation (n=89) and 60% in patients (n=5) with unknown EGFR status. Across all PD-L1 subgroups, patients with EGFR-mutant had a lower objective response rate than patients with EGFR wild-type tumour.11 In the 3-year follow-up of the KEYNOTE-001 trial, the median overall survival (OS) in patients with EGFR mutation was 6 months (95% CI 4.4 to 8.8) compared with 12 months (95% CI 9.2 to 14.3) in wild-type patients.67 A recent phase 2 trial investigated pembrolizumab in TKI-naive patients with PD-L1 positive EGFR-mutant NSCLC. No responses were observed in the first 11 included patients and the trial had to be discontinued.68
The phase 2 BIRCH study on PD-L1 inhibitor atezolizumab12 showed higher response rates in patients with higher expression of PD-L1 (on tumour cells (TC) ≥50%) (ORR 35% vs 26% (for all treated patients)). Overall, 13 patients with activating EGFR mutation were included and the ORR was 31% for mutant EGFR versus 23% for wild-type EGFR patients (n=103).
The ImPOWER 150 study compared the use of bevacizumab plus atezolizumab plus carboplatin plus paclitaxel versus carboplatin plus paclitaxel plus bevacizumab.13 Eighty patients (35 in the intervention and 45 in the control arm) with EGFR mutation and 34 patients (13 in the intervention and 21 in the control arm) with ALK rearrangement were included after progression on established TKI treatment. PFS was also longer in those patients with oncogene addicted NSCLC in the intervention arm containing atezolizumab compared with the standard arm (median, 9.7 months vs 6.1 months; unstratified HR, 0.59; 95% CI 0.37 to 0.94)
ATLANTIC is a phase 2, open-label, single-arm trial studying the efficacy of durvalumab, a PD-L1 inhibitor, in pretreated NSCLC including 111 patients with EGFR or ALK alteration.14 Eligible patients had advanced NSCLC with disease progression following at least two previous systemic regimens, including platinum-based chemotherapy. Patients with EGFR or ALK alteration had received standard treatment with TKI before. Among the 111 oncogene-addicted patients, 77 presented with PD-L1 expression in at least 25% of tumour cells. The objective response rate was 12.2% (95% CI 5.7 to 21.8) in the oncogene-addicted patients with PD-L1 expression >25% of tumour cells, while patients with <25% PD-L1 expression the objective response rate was only 3.6% (95% CI 0.1 to 18.3). PFS was not different according to PD-L1 expression in the EGFR or ALK altered patients (1.9 months). In summary, the proportions of patients who achieved a response were generally lower in patients with EGFR or ALK positive NSCLC than in those with EGFR negative and ALK negative NSCLC and higher PD-L1 expression appears to enrich for response. The figure 5 shows response after durvalumab treatment in cohort 1 of the ATLANTIC trial (EGFR+/ALK+)
Figure 5.
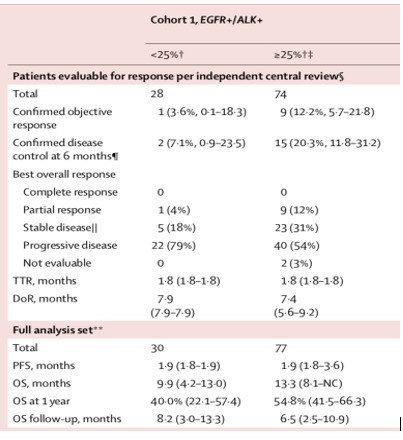
Response after durvalumab treatment in cohort 1 of the ATLANTIC trial (EGFR+/ALK+).14 Reprinted with permission from Elsevier. DOR, duration of response; OS, overall survival; PFS, progression-free survival; TTR, time to response.
A recent systematic review and meta-analysis of five randomised trials comparing ICI (nivolumab, pembrolizumab and atezolizumab) versus docetaxel in the second-line setting after chemotherapy showed an OS benefit for EGFR wild-type NSCLC with an impressive HR of 0.67 (p<0.001) for ICI compared with chemotherapy.8 However, in contrast, no OS advantage was observed for patients with EGFR-mutant NSCLC, although the small sample size needs to be considered (HR of 1.11, p=0.54). Only 12% (n=271) of the included patients indeed presented with an EGFR mutation. The major limitation of the study is that EGFR mutation was not determined by centralised testing and in 764 patients (25%) EGFR status was not assessed. The different types of mutations are also unknown.
Table 1 gives an overview on efficacy of anti-PD-1 and anti-PD-L1 in patients with wild-type NSCLC versus patients with EGFR-mutated NSCLC.
Table 1.
Anti-PD-1 and anti-PD-L1 efficacy in patients with wild-type NSCLC versus patients with EGFR-mutated NSCLC
| Study | Treatment |
EGFR-mutated n (%) |
HR OS wild-type versus mutated patients who received anti-PD-1 or PD-L1 |
| Check Mate 05744 | Nivolumab versus docetaxel | 82 (14%) | 0.66 (0.51–0.85) versus 1.18 (0.45–2.07) |
| KEYNOTE-01082 | Pembrolizumab versus docetaxel | 86 (8%) | 0.66 (0.55–0.79) versus 0.88 (0.45–1.72) |
| OAK83 | Atezolizumab versus docetaxel | 85 (10%) | 0.69 (0.57–0.83) versus 1.24 (0.71–2.18) |
| POPLAR61 | Atezolizumab versus docetaxel | 18 (6%) | 0.70 (0.47–1.04) versus 0.99 (0.29–3.40) |
| ImPOWER 15013 | Atezolizumab plus bevacizumab and chemotherapy versuschemotherapy plus bevacizumab | 80 (10%) | 0.62 (0.52–0.74) versus 0.41 (0.22–0.78) |
NSCLC, non-small cell lung cancer; OS, overall survival; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1.
The retrospective multicenter Immunotarget Cohort study reviewed data on the efficacy of ICI in 527 patients with stage IV NSCLC harbouring various activating molecular alterations including KRAS (n=252), EGFR (n=110), BRAF (n=38), MET (n=36), HER2 (n=23), ALK (n=18), RET (n=14), ROS1 (n=5) and multiple drivers (n=31).7 Outcomes by molecular subtypes are shown in table 2. Overall, EGFR-mutant patients presented with a shorter PFS after ICI-based therapy compared with KRAS-mutant patients (p<0.001). The EGFR p.T790M mutation was associated with a shorter PFS than other EGFR mutations (p=0.0001). Among patients with MET alterations, exon 14 mutations were shown to present with the highest response rate to ICI. Also, among patients harbouring an oncogene alteration, smoking status (p=0.003) and PD-L1 expression (p=0.02) were associated with PFS.
Table 2.
Immune checkpoint inhibitor efficacy outcomes in various molecular alterations
| Driver | n | Best response (%) | PFS | OS | ||||
| CR/PR | SD | PD | Median (months) | 6 month PFS (%) | 1 year PFS (%) | Median (months) | ||
| BRAF | 38 | 28.1 | 28.1 | 43.8 | 3 | 35 | 19 | 13.6 |
| KRAS | 252 | 27.2 | 23.1 | 49.8 | 3.2 | 39 | 26 | 13.5 |
| ROS1 | 5 | 20 | 0 | 80 | NA | NA | NA | NA |
| MET | 36 | 15.6 | 34.4 | 50 | 3.4 | 33 | 23 | 18.4 |
| EGFR | 110 | 11 | 18 | 71 | 2 | 16 | 6 | 8.8 |
| HER2 | 23 | 9.5 | 28.6 | 61.9 | 3.5 | 34 | 17 | 10 |
| RET | 14 | 7.1 | 21.4 | 71.4 | 2.2 | 16 | 8 | 6.5 |
| ALK | 18 | 0 | 21.4 | 78.6 | 2.1 | 16 | 8 | 17 |
Reprinted with permission from American Society of Clinical Oncology. Copyright 2018. All rights reserved. Mazieres J, Drilon AE, Mhanna LJ et al. Efficacy of immune-checkpoint inhibitors in non-small cell lung cancer patients harbouring activating molecular alterations (ImmunoTarget). J Clin Oncol 2018; 36, suppl:abstr 9010.
CR, complete response; NA, not available; PFS, progression-free survival; PR, partial response.
Radiological methods for response evaluation during ICI treatment in NSCLC
Response Evaluation Criteria in Solid Tumours (RECIST) and immune-related response criteria (irRC) are size-based response assessment methods. However, during ICI treatment, lesions can initially increase in size due to an influx of immune cells. When subsequent radiological follow-up shows a decrease in tumour size after initial increase in size or even lesion frequency, this pattern of response is called ‘pseudoprogression’. It is associated with a favourable response to immunotherapy. However, early during treatment pseudoprogression cannot be radiologically discriminated from tumour progression. The resolution limitation of CT, ranging typically around 1 mm,69 can even cause the appearance of ‘new lesions’ as a result of pseudoprogression. Lesions just below the resolution limitation of the CT can grow due to immune cell influx and become large enough to be visible on a CT. With RECIST, this would be classified as progressive disease, while with irRC, the diameters of the new lesion(s) are added to the sum of all diameters and when this sum remains below 20% increase as compared with the baseline value, the response will be classified as stable disease (figure 6). In melanoma, it was estimated that conventional RECIST underestimates the benefit of single agent PD-1 treatment with pembrolizumab in approximately 15% of patients and that the use of irRC better classifies patients according to survival benefit and prevents premature cessation of a potentially successful treatment.70
Figure 6.
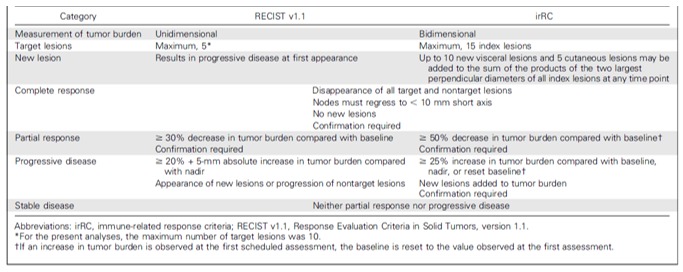
Comparison of key differences in Response Evaluation Criteria in Solid Tumours (RECIST) V.1.1 and immune-related response criteria (irRC). Reprinted with permission from American Society of Clinical Oncology, Copyright 2016. All rights reserved.70
Positron emission tomography (PET)-CT using 18F-fluorodeoxyglucose (18F-FDG) (FDG-PET) visualises and quantifies glucose metabolism of tumour lesions. Metabolic responses after chemotherapy have been associated with favourable outcome in terms of PFS and can precede size-based responses.71 72 A FDG-PET study in patients with NSCLC treated with atezolizumab showed that FDG-PET 6 weeks after treatment initiation is able to classify patients according to survival benefit.73 However, FDG-PET did not seem to outperform the predictive value of CT. This could be due to the nature of FDG-PET, not discriminating between metabolic activity of immune cells and tumour cells. After 6 weeks of PD-L1 directed treatment, it is likely that tumour FDG uptake is the result of a mixture of an increase in FDG metabolism due to influx of immune cells and a decrease due to tumour cell death, hampering response evaluation. Therefore, an earlier time point after treatment initiation aiming at quantifying the metabolic activity of the influx of immune cells and preceding tumour cell death might be a better discriminator of responders and non-responders.
The balance between the a priori probabilities of progression and pseudoprogression should guide treatment decision. For patients with molecularly driven NSCLC, response rates are generally low to very low and therefore progression according to RECIST and irRC is expected to represent real tumour progression in most cases and rarely pseudoprogression. In patients with molecularly driven NSCLC, RECIST-based response evaluation is expected to be the best discriminator between patients who derive benefit from PD-L1 directed therapy and those who do not. Treatment beyond progression is therefore currently not advised, as well as the use of irRC.
Sequence of treatments in molecularly mutated NSCLC: is there a place for ICI?
The introduction of mutation-specific TKI revolutionised the treatment of patients with NSCLC harbouring an oncogene addiction. However, at some point, resistance occurs in almost all patients. Durable responses can be observed in patients treated with ICI; however, the response in patients with oncogene-addicted NSLCC was shown to be impaired compared with wild-type patients.8–11 To offer patients with oncogene addiction the chance of immune induced long-term control of their disease, new combinations are most certainly the only clinical possibility. Here, the ATLANTIC trial proposed that the quartet combination therapy could be a promising approach in patients’ progression on all available generations of oncogene-specific TKI.14 Given the potential negative impact of the oncogene on the activity of the inflammatory tumour microenvironment, the combination of TKI with ICI is of potential clinical interest. Certainly, side effects would increase as seen by the combination of ICI with TKI in other entities like melanoma.74–77 However, in theory, the rapid antigen release through dying tumour cells by the TKI could enhance the inflammatory response. First phase I trials combining ICI with EGFR mutation directed TKI show acceptable side effect profiles.78–81 Several clinical trials investigating the combination of TKI with ICI are currently recruiting (eg, NCT02364609, NCT01454102, NCT01998126).
Conclusion
Lung cancer is becoming a more diverse disease with regard to management with a wide range of targets and treatment options. Recent clinical data on ICI in NSCLC harbouring activating mutations reviewed at the round-table discussion and summarised in this article shows overall low efficacy, although interpretations have to be drawn carefully due to the limited amount of data available. However, promising subgroups needing further clinical investigation were identified. For example, the clinical activity of durvalumab late line salvage therapy in patients with EGFR-mutated NSCLC showing PD-L1 expression in ≥25% of tumour cells is encouraging.14 Here, the clinical challenge is to further understand the biological drivers of inflammation in NSCLC and to identify subgroups driving the benefit as well as defining the optimal treatment sequence with established TKI. In addition, further research is needed to address the heterogeneity of EGFR-mutant lung cancer and to assess whether changes in the biology of the disease following different lines of therapy might increase the sensitivity to ICI.
To offer patients with oncogene addiction the chance of immune induced long-term control of their disease, new combinations are probably the only clinical possibility. A combination strategy as recently analysed in the ImPOWER 150 study13 including platin-based chemotherapy, bevacizumab and atezolizumab showed a clinically meaningful efficacy also in patients with oncogene-addicted NSCLC progressing on all available generations of TKI. However, several other trials reported no significant increase in response rate or PFS in patients with oncogene-addicted NSCLC.9–11 67 Given the potential negative impact of the oncogene on the activity of the inflammatory tumour microenvironment, the combination of TKI with ICI is of potential clinical interest. Several clinical trials investigating the combination of TKI with ICI are currently recruiting (eg, NCT02364609, NCT01454102, NCT01998126). Due to the overall low clinical efficiency of ICI in patients with oncogene-addicted NSCLC based on the so far available data from prospective clinical trials, the round-table panel concluded that ICI should currently only be considered after exhaustion of targeted therapies including standard and salvage chemotherapies in these patients.
Acknowledgments
This is to acknowledge Dr Christiane Rehwagen’s work in organising the round table and her medical writing support.
Footnotes
Contributors: All authors contributed to and reviewed the final manuscript. The roundtable agenda was conceived by Christoph Zielinski and Matthias Preusser.
Funding: This initiative is sponsored by Astra Zeneca through the provision of an unrestricted educational grant. Astra Zeneca has had no influence over the content other than a review of the paper for medical accuracy. The participants/authors received an honorarium for their participation in the round table from BMJ.
Competing interests: ASB: Travel support: Daiichi Sankyo, Bristol-Meyers Squibb, Roche, Amgen, Merck, AbbVie; Research Support: Daiichi Sankyo; Advisory Board: Roche. BB: Travel support: Bristol-Meyers Squibb, Roche, Merck; Research support: Roche, Pfizer, Novartis; Advisory Board: Roche, Novartis; Speaker: AstraZeneca, Pfizer, Roche. AJdL: Related to this work: none; in general: Advisor for AstraZeneca, BMS, Boehringer, Pfizer, MSD, Roche; research grants from AstraZeneca, BMS, Merck-Serono, MSD, Roche. JM: Travel support: MSD, MSD, Roche; Research support: Roche, Astra-Zeneca; Advisory Board: Roche, MSD, BMS, Takeda, Astra-Zeneca, Pharmamar, Boehringer. NN: Personal financial interests (speaker’s fee and/or advisory boards): MSD, Qiagen, Biocartis, Incyte, Roche, BMS, MERCK, Thermofisher, Boehringer Ingelheim, Astrazeneca, Sanofi, Eli Lilly; Institutional financial interests (financial support to research projects): MERCK, Sysmex, Thermofisher, QIAGEN, Roche, Astrazeneca, Biocartis; non-financial interests: President, International Quality Network for Pathology (IQN Path); President Elect, Italian Cancer Society (SIC). MP: Personal fees from BMS, Astra Zeneca, Pierre Fabre, Roche, Novartis and Takeda. MP: Received honoraria for lectures, consultation or advisory board participation from the following for-profit companies: Bristol-Myers Squibb, Novartis, Gerson Lehrman Group (GLG), CMC Contrast, GlaxoSmithKline, Mundipharma, Roche, Astra Zeneca, AbbVie, Lilly, Medahead, Daiichi Sankyo, Merck Sharp & Dome. FR: Travel support: Roche, Merck, Pfizer. Scientific advisor: Genomic Health, Roche, Guardant Health, Merck, Pfizer, Bristol-Meyers Squibb, Abbvie, Astra Zeneca, Novartis. JW: Advisory boards and lecture fees: Abbvie, AstraZeneca, BMS, Boehringer-Ingelheim, Chugai, Ignyta, Lilly, MSD, Novartis, Pfizer, Roche; research support (to institution): BMS, MSD, Novartis, Pfizer. CZ: Roche, Novartis, BMS, MSD, Imugene, Ariad, Pfizer, Merrimack/Shire, Merck KGaA, Fibrogen, AstraZeneca, Tesaro, Gilead, Servier, Eli Lilly, Amgen.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; internally peer reviewed.
References
- 1. Barlesi F, Mazieres J, Merlio J-P, et al. . Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative thoracic intergroup (IFCT). The Lancet 2016;387:1415–26. 10.1016/S0140-6736(16)00004-0 [DOI] [PubMed] [Google Scholar]
- 2. Novello S, Barlesi F, Califano R, et al. . Metastatic non-small-cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up†. Ann Oncol 2016;27:v1–27. 10.1093/annonc/mdw326 [DOI] [PubMed] [Google Scholar]
- 3. Gandhi L, Rodríguez-Abreu D, Gadgeel S, et al. . Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. N Engl J Med 2018;378:2078–92. 10.1056/NEJMoa1801005 [DOI] [PubMed] [Google Scholar]
- 4. Gandara DR, Li T, Lara PN, et al. . Acquired resistance to targeted therapies against oncogene-driven non–small-cell lung cancer: approach to subtyping progressive disease and clinical implications. Clinical Lung Cancer 2014;15:1–6. 10.1016/j.cllc.2013.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schadendorf D, Hodi FS, Robert C, et al. . Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. JCO 2015;33:1889–94. 10.1200/JCO.2014.56.2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qin H, Wang F, Liu H, et al. . New advances in immunotherapy for non-small cell lung cancer. Am J Transl Res 2018;10:2234–45. [PMC free article] [PubMed] [Google Scholar]
- 7. Mazieres J, Drilon AE, Mhanna L, et al. . Efficacy of immune-checkpoint inhibitors (ICI) in non-small cell lung cancer (NSCLC) patients harboring activating molecular alterations (ImmunoTarget). JCO 2018;36(15_suppl). 10.1200/JCO.2018.36.15_suppl.9010 [DOI] [Google Scholar]
- 8. Lee CK, Man J, Lord S, et al. . Clinical and molecular characteristics associated with survival among patients treated with checkpoint inhibitors for advanced non-small cell lung carcinoma. A systematic review and meta-analysis. JAMA Oncol 2018;4:210–6. 10.1001/jamaoncol.2017.4427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gettinger S, Rizvi NA, Chow LQ, et al. . Nivolumab monotherapy for first-line treatment of advanced non-small cell lung cancer. J Clin Oncol 2016;34:2980–7. 10.1200/JCO.2016.66.9929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bauer T, McCleod M, Chadler J, et al. . An ongoing phase IIIb/IV safety trial of nivolumab in patients with advanced or metastatic non-small cell lung cancer who progressed after receiving > = 1 prior systemic regime. ASCO meeting, 2015. [Google Scholar]
- 11. Hellmann M. Efficacy of pembrolizumab in key subgroups of patients with advanced NSCLC. 16th World Conference on Lung Cancer, 2015. [Google Scholar]
- 12. Carcereny E, Felip E, Reck M, et al. . Updated efficacy results from the birch study: first-line atezolizumab therapy in PD-L1 –selected patients with advanced NSCLC. World Conference on Lung Cancer, 2017. [Google Scholar]
- 13. Socinski MA, Jotte RM, Cappuzzo F, et al. . Atezolizumab for first-line treatment of metastatic non-squamous NSCLC. N Engl J Med 2018;378:2288–301. 10.1056/NEJMoa1716948 [DOI] [PubMed] [Google Scholar]
- 14. Garassino MC, Cho B-C, Kim J-H, et al. . Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): an open-label, single-arm, phase 2 study. The Lancet Oncology 2018;19:521–36. 10.1016/S1470-2045(18)30144-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. NCCN Clinical Practice Guidelines in Oncology (NCCN guidelines): non-small cell lung cancer, version 5, 2018. Available: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf
- 16. Kerr KM, Bubendorf L, Edelman MJ, et al. . Second ESMO consensus conference on lung cancer: pathology and molecular biomarkers for non-small-cell lung cancer. Ann Oncol 2014;25:1681–90. 10.1093/annonc/mdu145 [DOI] [PubMed] [Google Scholar]
- 17. Kerr KM, López-Ríos F. Precision medicine in NSCLC and pathology: how does ALK fit in the pathway? Ann Oncol 2016;27(suppl_3):iii16–24. 10.1093/annonc/mdw302 [DOI] [PubMed] [Google Scholar]
- 18. Lim C, Tsao MS, Le LW, et al. . Biomarker testing and time to treatment decision in patients with advanced nonsmall-cell lung cancer. Ann Oncol 2015;26:1415–21. 10.1093/annonc/mdv208 [DOI] [PubMed] [Google Scholar]
- 19. Leighl NB, Rekhtman N, Biermann WA, et al. . Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: american Society of clinical oncology endorsement of the College of American Pathologists/International association for the study of lung cancer/association for molecular pathology guideline. J Clin Oncol 2014;32:3673–9. 10.1200/JCO.2014.57.3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Büttner R, Gosney JR, Skov BG, et al. . Programmed Death-Ligand 1 immunohistochemistry testing: a review of analytical assays and clinical implementation in Non–Small-Cell lung cancer. J Clin Oncol 2017;35:3867–76. 10.1200/JCO.2017.74.7642 [DOI] [PubMed] [Google Scholar]
- 21. JCM W, Massie C, Garcia-Corbacho J, et al. . Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 2017;17:223–38. [DOI] [PubMed] [Google Scholar]
- 22. Normanno N, Maiello MR, Chicchinelli N, et al. . Targeting the EGFR T790M mutation in non-small-cell lung cancer. Exp Opin Ther Targets 2017;21:159–65. 10.1080/14728222.2017.1272582 [DOI] [PubMed] [Google Scholar]
- 23. Mok TS, Wu Y-L, Ahn M-J, et al. . Osimertinib or platinum–pemetrexed in EGFR T790M–positive lung cancer. N Engl J Med 2017;376:629–40. 10.1056/NEJMoa1612674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oxnard GR, Thress KS, Alden RS, et al. . Association between plasma genotyping and outcomes of treatment with Osimertinib (AZD9291) in advanced non-small-cell lung cancer. JCO 2016;34:3375–82. 10.1200/JCO.2016.66.7162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chabon JJ, Simmons AD, Lovejoy AF, et al. . Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun 2016;7 10.1038/ncomms11815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Blakely CM, Watkins TBK, Wu W, et al. . Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet 2017;49:1693–704. 10.1038/ng.3990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ortiz-Cuaran S, Scheffler M, Plenker D, et al. . Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin Cancer Res 2016;22:4837–47. 10.1158/1078-0432.CCR-15-1915 [DOI] [PubMed] [Google Scholar]
- 28. Fassunke J, Müller F, Keul M, et al. . Overcoming EGFRG724S-mediated osimertinib resistance through unique binding characteristics of second-generation EGFR inhibitors. Nature Communications 2018;9 10.1038/s41467-018-07078-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burrell RA, Swanton C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol Oncol 2014;8:1095–111. 10.1016/j.molonc.2014.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tang Z-H, Lu J-J. Osimertinib resistance in non-small cell lung cancer: mechanisms and therapeutic strategies. Cancer Lett 2018;420:242–6. 10.1016/j.canlet.2018.02.004 [DOI] [PubMed] [Google Scholar]
- 31. Kobayashi S, Boggon TJ, Dayaram T, et al. . EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 2005;352:786–92. 10.1056/NEJMoa044238 [DOI] [PubMed] [Google Scholar]
- 32. SG W, Liu YN, Tsai MF, et al. . The mechanism of acquired resistance to irreversible EGFR tyrosine kinase inhibitor-afatinib in lung adenocarcinoma patients. Oncotarget 2016;7:12404–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Costa DB, Schumer ST, Tenen DG, et al. . Differential responses to erlotinib in epidermal growth factor receptor (EGFR)-mutated lung cancers with acquired resistance to gefitinib carrying the L747S or T790M secondary mutations. JCO 2008;26:1182–4. 10.1200/JCO.2007.14.9039 [DOI] [PubMed] [Google Scholar]
- 34. Balak MN, Gong Y, Riely GJ, et al. . Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res 2006;12:6494–501. 10.1158/1078-0432.CCR-06-1570 [DOI] [PubMed] [Google Scholar]
- 35. Bean J, Riely GJ, Balak M, et al. . Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res 2008;14:7519–25. 10.1158/1078-0432.CCR-08-0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. HA Y, Arcila ME, Rekhtman N, et al. . Analysis of tumour specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013;19:2240–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sequist LV, Waltman BA, Dias-Santagata D, et al. . Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3 10.1126/scitranslmed.3002003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Westover D, Zugazagoitia J, Cho BC, et al. . Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol 2018;29(suppl_1):i10–19. 10.1093/annonc/mdx703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. HA Y, Suzawa K, Jordan E, et al. . Concurrent alterations in EGFR-mutant lung cancers associated with resistance to EGFR kinase inhibitors and characterization of mTOR as a mediator of resistance. Clin Cancer Res 2018;24:3108–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ricordel C, Friboulet L, Facchinetti F, et al. . Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann Oncol 2018. [DOI] [PubMed] [Google Scholar]
- 41. Soria J-C, Ohe Y, Vansteenkiste J, et al. . Osimertinib in untreated EGFR -mutated advanced non-small-cell lung cancer. N Engl J Med 2018;378:113–25. 10.1056/NEJMoa1713137 [DOI] [PubMed] [Google Scholar]
- 42. Niederst MJ, Hu H, Mulvey HE, et al. . The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res 2015;21:3924–33. 10.1158/1078-0432.CCR-15-0560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Le X, Puri S, Negrao MV, et al. . Landscape of EGFR-dependent and -independent resistance mechanisms to Osimertinib and continuation therapy beyond progression in -Mutant NSCLC. Clin Cancer Res 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Borghaei H, Paz-Ares L, Horn L, et al. . Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med 2015;373:1627–39. 10.1056/NEJMoa1507643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 2017;377:2500–1. 10.1056/NEJMc1713444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252–64. 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Paz-Ares L, Luft A, Vicente D, et al. . Pembrolizumab plus chemotherapy for squamous Non–Small-Cell lung cancer. N Engl J Med 2018;379:2040–51. 10.1056/NEJMoa1810865 [DOI] [PubMed] [Google Scholar]
- 48. Reck M, Rodríguez-Abreu D, Robinson AG, et al. . Pembrolizumab versus chemotherapy for PD-L1–Positive Non–Small-Cell lung cancer. N Engl J Med 2016;375:1823–33. 10.1056/NEJMoa1606774 [DOI] [PubMed] [Google Scholar]
- 49. Mansfield AS, Aubry MC, Moser JC, et al. . Temporal and spatial discordance of programmed cell death-ligand 1 expression and lymphocyte tumor infiltration between paired primary lesions and brain metastases in lung cancer. Ann Oncol 2016;27:1953–8. 10.1093/annonc/mdw289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Evans M, O’Sullivan B, Hughes F, et al. . The clinicopathological and molecular associations of PD-L1 expression in non-small cell lung cancer: analysis of a series of 10,005 cases tested with the 22C3 assay. Pathol Oncol Res 2018;375 10.1007/s12253-018-0469-6 [DOI] [PubMed] [Google Scholar]
- 51. Lan B, Ma C, Zhang C, et al. . Association between PD-L1 expression and driver gene status in non-small-cell lung cancer: a meta-analysis. Oncotarget 2018;9:7684–99. 10.18632/oncotarget.23969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ota K, Azuma K, Kawahara A, et al. . Induction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin Cancer Res 2015;21:4014–21. 10.1158/1078-0432.CCR-15-0016 [DOI] [PubMed] [Google Scholar]
- 53. Haratani K, Hayashi H, Tanaka T, et al. . Tumor immune microenvironment and nivolumab efficacy in EGFR mutation-positive non-small-cell lung cancer based on T790M status after disease progression during EGFR-TKI treatment. Ann Oncol 2017;28:1532–9. 10.1093/annonc/mdx183 [DOI] [PubMed] [Google Scholar]
- 54. Hata A, Katakami N, Nanjo S, et al. . Programmed death-ligand 1 expression and T790M status in EGFR -mutant non-small cell lung cancer. Lung Cancer 2017;111:182–9. 10.1016/j.lungcan.2017.07.022 [DOI] [PubMed] [Google Scholar]
- 55. Vormehr M, Diken M, Boegel S, et al. . Mutanome directed cancer immunotherapy. Curr Opin Immunol 2016;39:14–22. 10.1016/j.coi.2015.12.001 [DOI] [PubMed] [Google Scholar]
- 56. Yarchoan M, Johnson BA, Lutz ER, et al. . Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer 2017;17:209–22. 10.1038/nrc.2016.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McGranahan N, Furness AJS, Rosenthal R, et al. . Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016;351:1463–9. 10.1126/science.aaf1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hellmann MD, Ciuleanu T-E, Pluzanski A, et al. . Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med 2018;378:2093–104. 10.1056/NEJMoa1801946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tumeh PC, Harview CL, Yearley JH, et al. . PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71. 10.1038/nature13954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Germain C, Gnjatic S, Tamzalit F, et al. . Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med 2014;189:832–44. 10.1164/rccm.201309-1611OC [DOI] [PubMed] [Google Scholar]
- 61. Fehrenbacher L, Spira A, Ballinger M, et al. . Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (poplar): a multicentre, open-label, phase 2 randomised controlled trial. The Lancet 2016;387:1837–46. 10.1016/S0140-6736(16)00587-0 [DOI] [PubMed] [Google Scholar]
- 62. Dong Z-Y, Zhang J-T, Liu S-Y, et al. . EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. OncoImmunology 2017;6:e1356145 10.1080/2162402X.2017.1356145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Streicher K, Higgs BW, Wu S, et al. . Increased CD73 and reduced IFNG signature expression in relation to response rates to anti-PD-1(L1) therapies in EGFR-mutant NSCLC. JCO 2017;35(15_suppl). 10.1200/JCO.2017.35.15_suppl.11505 [DOI] [Google Scholar]
- 64. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015;523:231–5. 10.1038/nature14404 [DOI] [PubMed] [Google Scholar]
- 65. Peng W, Chen JQ, Liu C, et al. . Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov 2016;6:202–16. 10.1158/2159-8290.CD-15-0283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hui R, Garon EB, Goldman JW, et al. . Pembrolizumab as first-line therapy for patients with PD-L1-positive advanced non-small cell lung cancer: a phase 1 trial. Ann Oncol 2017;28:874–81. 10.1093/annonc/mdx008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Leighl NB, Hellmann MD, Hui R, et al. . KEYNOTE-001: 3-year overall survival for patients with advanced NSCLC treated with pembrolizumab. ASCO 2017. [Google Scholar]
- 68. Lisberg A, Cummings A, Goldman JW, et al. . A phase II study of pembrolizumab in EGFR-mutant, PD-L1+, tyrosine kinase inhibitor naïve patients with advanced NSCLC. J Thorac Oncol 2018;13:1138–45. 10.1016/j.jtho.2018.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tsukagoshi S, Ota T, Fujii M, et al. . Improvement of spatial resolution in the longitudinal direction for isotropic imaging in helical CT. Phys Med Biol 2007;52:791–801. 10.1088/0031-9155/52/3/018 [DOI] [PubMed] [Google Scholar]
- 70. Hodi FS, Hwu W-J, Kefford R, et al. . Evaluation of immune-related response criteria and RECIST v1.1 in patients with advanced melanoma treated with pembrolizumab. JCO 2016;34:1510–7. 10.1200/JCO.2015.64.0391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hoekstra CJ, Stroobants SG, Smit EF, et al. . Prognostic relevance of response evaluation using [ 18 F]-2-Fluoro-2-Deoxy-D-Glucose Positron Emission Tomography in patients With Locally Advanced Non–Small-Cell Lung Cancer. JCO 2005;23:8362–70. 10.1200/JCO.2005.01.1189 [DOI] [PubMed] [Google Scholar]
- 72. Sheikhbahaei S, Mena E, Yanamadala A, et al. . The value of FDG PET/CT in treatment response assessment, follow-up, and surveillance of lung cancer. Am J Roentgenol 2017;208:420–33. 10.2214/AJR.16.16532 [DOI] [PubMed] [Google Scholar]
- 73. Fredrickson J, Funke R. Utility of FDG-PET in immunotherapy: results from a phase II study of NSCLC patients undergoing therapy with the PD-L1 inhibitor atezolizumab (MPDL3280A). Society of Nuclear Medicine and Molecular Imaging Annual Meeting, 2016. p. Abstract 134. [Google Scholar]
- 74. Ribas A, Hodi FS, Callahan M, et al. . Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med 2013;368:1365–6. 10.1056/NEJMc1302338 [DOI] [PubMed] [Google Scholar]
- 75. Ribas A, Butler M, Lutzky J, et al. . Phase I study combining anti-PD-L1 (MEDI4736) with BRAF (dabrafenib) and/or MEK (trametinib) inhibitors in advanced melanoma. JCO 2015;33(15_suppl):3003 10.1200/jco.2015.33.15_suppl.3003 [DOI] [Google Scholar]
- 76. Hershman DL, Hodi FS, Lawrence DP, et al. . Pembrolizumab (pembro) in combination with dabrafenib (D) and trametinib (T) for BRAF-mutant advanced melanoma: phase 1 KEYNOTE-022 study. J Clin Oncol 2016;34. [Google Scholar]
- 77. Minor DR, Puzanov I, Callahan MK, et al. . Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment Cell Melanoma Res 2015;28:611–2. 10.1111/pcmr.12383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gibbons DL, Chow LQ, Kim D-W, et al. . 57O Efficacy, safety and tolerability of MEDI4736 (durvalumab [D]), a human IgG1 anti-programmed cell death-ligand-1 (PD-L1) antibody, combined with gefitinib (G): A phase I expansion in TKI-naïve patients (pts) with EGFR mutant NSCLC. J Thorac Oncol 2016;11 10.1016/S1556-0864(16)30171-X [DOI] [Google Scholar]
- 79. Gettinger S, Chow LQ, Borghaei H, et al. . Safety and response with nivolumab (anti-PD-1; BMS-936558, ONO-4538) plus erlotinib in patients (PTS) with epidermal growth factor receptor mutant (EGFR MT) advanced non-small cell lung cancer (NSCLC). Int J Radiat Oncol Biol Phys 2014;90:S34–S35. 10.1016/j.ijrobp.2014.08.210 [DOI] [Google Scholar]
- 80. Rudin C, Cervantes A, Dowlati A, et al. . P3.02c-046 safety, clinical activity and biomarker results from a phase Ib study of erlotinib plus Atezolizumab in advanced NSCLC. J Thorac Oncol 2017;12:S1302–S1303. 10.1016/j.jtho.2016.11.1841 [DOI] [Google Scholar]
- 81. Planchard D, Barlesi F, Gomez-Roca C, et al. . Phase I, safety, tolerability and preliminary efficacy study of tremelimumab (TREM) in combination with gefitinib (GEF) in EGFR-mutant (EGFR-mut) NSCLC (GEFTREM). Ann Oncol 2016;27(suppl_6). 10.1093/annonc/mdw383.45 [DOI] [Google Scholar]
- 82. Herbst RS, Baas P, Kim D-W, et al. . Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. The Lancet 2016;387:1540–50. 10.1016/S0140-6736(15)01281-7 [DOI] [PubMed] [Google Scholar]
- 83. Rittmeyer A, Barlesi F, Waterkamp D, et al. . Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (oak): a phase 3, open-label, multicentre randomised controlled trial. The Lancet 2017;389:255–65. 10.1016/S0140-6736(16)32517-X [DOI] [PMC free article] [PubMed] [Google Scholar]