

Abstract
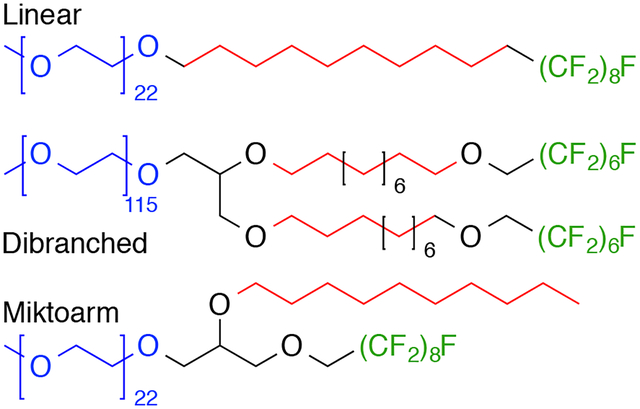
Linear, dibranched and miktoarm amphiphiles containing both hydrophobic and fluorophilic moieties were synthesized and characterized in an attempt to elucidate the relationship between semi-fluorinated amphiphile structure and aggregate behaviour in aqueous solution. For the linear and dibranched amphiphiles, there was an exponential decrease in critical aggregation concentration (CMC) and a logarithmic increase in core microviscosity with increasing length of the fluorocarbon segments; while the miktoarm architecture produced no notable trend in microviscosity or CMC. Furthermore, the linear and dibranched surfactants showed enhanced kinetic stability, dissociating more slowly in the presence of human serum than did either the dibranched or miktoarm amphiphiles. Finally, encapsulation studies with the hydrophobic drug paclitaxel (PTX) showed that the ability to solubilize and retain PTX increased with the presence and with the increasing size of the fluorocarbon moiety for both the linear and dibranched amphiphiles, while no such trend was observed for the miktoarm amphiphiles.
Keywords: Self-assembly, Fluorous, Micelles, Surfactants
Graphical Abstract
Three classes of semi-fluorinated amphiphiles were synthesized and their aggregation behaviour characterized. It was found that critical micelle concentration, microviscosity and aggregate stability were related to amphiphile architecture and the size of the fluorocarbon segment. Furthermore, it has been shown that a hydrophobic molecule – paclitaxel – can be encapsulated within the semi-fluorinated micelles derived from these polymers, thus demonstrating the potential to encapsulate hydrophobic species within a lipophilic shell without necessitating a lipophilic core.
Introduction
Polymeric micelles have found applications in fields ranging from catalysis,1,2 to antimicrobials3 to drug delivery.4,5 Especially important is designing the amphiphile structure such that specific characteristics appear in the micellar aggregate, e.g.: particle size, critical micelle concentration (CMC), microviscosity, and surface morphology. In the field of drug delivery, Torchilin has demonstrated the effectiveness of utilizing poly(ethylene glycol)ated (PEGylated) phospholipids for use as micelle-based drug delivery systems.6 These 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)] (mPEG-DSPE, Fig 1) polymeric micelles have been shown to not only efficiently load poorly water-soluble drugs but also to be effective in their delivery.7
Fig. 1.

Structures of amphiphiles presented with associated nomenclature. Mx refers to mPEG hydrophilic block, x is the average molecular weight in thousands; H# and F# refer to the number of hydrogenated and fluorinated carbon atoms, respectively. For non-linear amphiphiles, di and μ respectively specify dibranched or miktoarm architecture. The presence of an –O– indicated an ether linkage between blocks. PftB is the fluorous perfluoro-tert-butoxy group.
Previous work in our lab has led to the development of diblock, fluorinated surfactants for the solubilization8 and emulsification9 of volatile, fluorous anaesthetics. Further development of this synthetic methodology led to the preparation of semi-fluorinated mPEG-fluorocarbon-DSPE amphiphiles, which showed enhanced kinetic and thermodynamic stability with improved drug release profiles for amphotericin B compared to the commercially available, non-fluorinated analogues.10 Bates et al. have shown that multiblock amphiphiles can exhibit a wide variety of structures and behaviours, with the potential to offer unique materials for a wide variety of purposes.11 In the past year, a number of studies have looked at the aggregation of semi-fluorinated RAFT polymers in water.12–15 As it has been noted, investigation into the relationship between structure and aqueous self-assembly remains an important step in fully exploring the behavior of triphilic species.16
This paper seeks to expand on our previous work with mPEG-fluorocarbon-DSPE polymers by studying more generally how the introduction of fluorocarbon moieties to various amphiphilic architectures affects their physicochemical properties. Three polymeric architectures – linear, dibranched and miktoarm (Fig. 1) – along with various fluorinated moieties are presented. The physicochemical properties of these amphiphiles are reported along with data on kinetic stability and encapsulation ability for the micelles prepared from these surfactants. This study demonstrates that changes in amphiphile architecture and block composition can be used to modulate the physicochemical properties of a micelle, including its stability in physiological conditions and the ability to encapsulate hydrophobic molecules.
Experimental
Materials
All fluorinated compounds were obtained from SynQuest Laboratories, Inc. (Alachua, FL, USA), except perfluoro-tertbutanol that was obtained from Matrix Scientific (Columbia, SC, USA). 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (M2DSPE) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-5000] (M5DSPE) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). Paclitaxel was purchased from LC Laboratories (Woburn, MA, USA). 1,3-Bis-(1-pyrenyl)propane (P3P) was purchased from Life Technologies (Carlsbad, CA, USA). Pooled normal human serum was purchased from Innovative Research (Novi, MI, USA). All solvents were of ACS grade or higher and were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) and were used as received, unless otherwise specified. Chromatographic separations were performed using Silicycle 60 Å SiO2. Surfactants were purified by automated flash chromatography using a CombiFlash® Rf 4× system (Teledyne Isco, Lincoln, NE, USA) equipped with ELSD for compound visualization and a REDI-Sep Rf Gold C-18 silica high performance aqueous reverse phase cartridge. Products were eluted with a 10 % to 100 % MeOH in water (0.1 % formic acid) gradient. 1H- and 19F-NMR spectra were obtained on Varian Unity-Inova 400 and Unity-Inova 500 spectrometers using deuterochloroform (CDCl3) as the solvent with TMS as an internal reference.
Methods
mPEG mesylate (Mx-OMs).
To a dry 100 mL roundbottom flask charged with argon were added 50 mL DCM and 5 g monomethyl poly(ethylene glycol) alcohol (4.9 mmol M1OH/2.5 mmol M2OH/1.03 mmol M5OH). The mixture was cooled to 0°C before adding TEA (2 ml, 14.7 mmol mPEG1000-OH/1.04 mL, 7.5 mmol mPEG2000-OH/0.43 mL, 3.06 mmol mPEG5000-OH), which was allowed to stir for 30 minutes before methanesulfonyl chloride was added (1 ml, 12.2 mmol mPEG1000-OH/0.48 mL, 6.25 mmol mPEG2000-OH/0.2 mL, 2.55 mmol mPEG5000-OH). The reaction was allowed to stir overnight as it warmed to room temperature. The reaction was then diluted with 100 mL DCM and washed with 3, 50 mL aliquots saturated ammonium chloride solution, dried over magnesium sulphate and then reduced to a minimum volume under reduced pressure. The mPEG-OMs was then precipitated with cold ether, vacuum filtered, and freeze dried from 50/50 DCM/benzene to give a white, crystalline product in 67 % (M1-OMs), 86 % (M2-OMs) and 93 % (M5-OMs) yields. M1-OMs MALDI: Distribution centred on [M+Na+] = 1063.4, PDI of starting mPEG 1.27. NMR: 1H NMR (400 MHz, CDCl3): δ 4.38 (m, 2H), 3.82 (m, 1H), 3.77 (m, 2H), 3.64 (m, 89H), 3.55 (m, 2H), 3.47 (m, 1H), 3.38 (s, 3H), 3.09 (s, 3H); M2-OMs MALDI: Distribution centred on [M+Na+] = 1938.4, PDI of starting mPEG 1.10. NMR: 1H NMR (400 MHz, CDCl3): δ 4.38 (m, 2H), 3.79 (m, 2H), 3.77 (m, 2H), 3.64 (m, 165H), 3.56 (m, 2H), 3.38 (s, 3H), 3.09 (s, 3H); M5-OMs MALDI: Distribution centred on [M+Na+] = 5161.5, PDI of starting mPEG 1.05. NMR: 1H NMR (400 MHz, CDCl3): δ 3.84 − 3.44 (m, 490H), 3.38 (s, 3 H), 3.08 (s, 3H).
Linear alcohols.
HO-H10F8:
To a dry 10 mL roundbottom flask were added 1.02 mL (5.75 mmol) 9-decen-1-ol and 1.34 mL (5.0 mmol) perfluorooctyl iodide. The mixture was degassed at room temperature with argon for 45 minutes before 8.2 mg (0.05 mmol) AIBN were added and the mixture slowly heated to 80°C while being very rapidly stirred with a small stir bar. This reaction was allowed to run overnight. The reaction was then cooled to room temperature, diluted with 100 mL DCM, washed with 1, 50 mL aliquot each Na2S2O3 and brine. The organic layers were dried over MgSO4 and condensed under reduced pressure to give an off-white solid (1). This was then dissolved in 10 mL acetic acid and stirred with 0.98 g zinc powder for 24 hours open to the air. The reaction was then quenched with 200 mL saturated NaHCO3 solution and extracted with 300 mL DCM. The organic layers were then washed with 1, 100 mL aliquot each saturated NaHCO3 solution and brine and then dried over MgSO4 and concentrated under reduced pressures to give a white solid. The solid was recrystallized twice from hot toluene to give pure HO-H10F8 in 48 % yield. NMR: 1H NMR (400 MHz, CDCl3): δ 3.65 (t, J = 6.9 Hz, 2H), 2.05 (ttt, J = 18, 9.5, 2 Hz, 2H), 1.65 − 1.52 (m, 4H), 1.4 − 1.23 (m, 12H). 19F NMR (376 MHz, CDCl3): δ −81.15 (3F), −114.77 (2F), −122.32 (6F), −123.11 (2F), −123.92 (2F), −126.49 (2F).
HO-H10-O-F3 and HO-H10-O-F6:
To a dry roundbottom, at 0°C argon, were added 25 mL dry DCM, 9-decen-1-ol (2.5 mL, 13 mmol) and TEA (4.3 mL, 31 mmol). This sitrred for 30 minutes before methanesulfonyl chloride (1.3 mL, 16 mmol) was added dropwise. After running overnight, the reaction was diluted with 50 mL DCM and then washed with 3, 50 mL aliquots of saturated ammonium chloride solution. The organic layers were dried over MgSO4 and condensed under reduced pressure to give 3.21 g (quantitative yield) of yellow oil. NMR: 1H NMR (400 MHz, CDCl3): δ 5.81 (ddt, J = 16.5, 10, 6.5 Hz, 1H), 4.99 (ddt, J = 16.5, 2, 1Hz, 1H), 4.93 (ddt, J = 10, 2, 1Hz, 1H), 4.22 (t, J = 7 Hz, 2H), 3.00 (s, 3H), 2.04 (qt, J = 6.5, 1 Hz, 2H), 1.60 (q, J = 7 Hz, 2H), 1.41 − 1.29 (m, 10H).
To a 100 mL oven-dried roundbottom, under argon, were added 35 mL of THF and 761 mg (31 mmol) of NaH. The suspension was cooled to 0°C over the course of 10 minutes before 28 mmol of semi-fluorinated alcohol were added (3.25 mL 1H,1H-perfluorobutan-1-ol (F3H1-OH)/5.8 mL (28 mmol) of 1H,1H-perfluoroheptan-1-ol (F6H1-OH)) was added dropwise over the course of 1 hour. Then 3.20 g (13 mmol) of 9-decen-1-yl methane sulfonate were added (as a solution in 10 mL of anhydrous THF). This was then warmed slowly to reflux and allowed to react for 24 hours. The reaction was then allowed to cool and diluted with 100 mL of DCM. This was washed with 3, 50 mL aliquots of saturated NH4Cl solution and dried over MgSO4 and condensed under reduced pressure to give an opaque, yellow liquid. The product was then purified by column chromatography (4% ethyl acetate in hexanes) to give 3.83 g (86% yield F3) and 5.28 g (83 % yield F6) of product as a clear liquid. 10-(1H,1H-perfluorobutoxy)dec-1-ene NMR: 1H NMR (400 MHz, CDCl3): δ 5.81 (ddt, J = 16.5, 10, 6.5 Hz, 1H), 4.99 (ddt, J = 16.5, 2, 1Hz, 1H), 4.93 (ddt, J = 10, 2, 1Hz, 1H), 3.90 (tt, J = 14, 2 Hz, 2H), 3.58 (t, J = 7 Hz, 2H), 2.04 (qt, J = 6.5, 1 Hz, 2H), 1.60 (q, J = 7 Hz, 2H), 1.41 − 1.29 (m, 10H). 19F NMR (376 MHz, CDCl3): δ −81.51 (3F), −121.09 (2F), −128.28 (2F). 10-(1H,1H-perfluoroheptoxy)dec-1-ene NMR: 1H NMR (400 MHz, CDCl3): δ 5.81 (ddt, J = 16.5, 10, 6.5 Hz, 1H), 4.99 (ddt, J = 16.5, 2, 1Hz, 1H), 4.93 (ddt, J = 10, 2, 1Hz, 1H), 3.91 (tt, J = 14, 2 Hz, 2H), 3.59 (t, J = 7 Hz, 2H), 2.04 (qt, J = 6.5, 1 Hz, 2H), 1.59 (q, J = 7 Hz, 2H), 1.39 − 1.29 (m, 10H). 19F NMR (376 MHz, CDCl3): δ −81.26 (3F), −120.03 (2F), −122.69 (2F), −123.29 (2F), −123.91 (2F), −126.59 (2F).
To an oven-dried round-bottom flask was added BH3-THF (1.0 M, 16.5 mmol). The solution was diluted with 10 mL of dry THF and then cooled to 0°C. The semi-fluorinated alkene ether (10-(1H,1H-perfluorobutoxy)dec-1-ene (3.83 g, 11.2 mmol)/10-(1H,1H-perfluoroheptoxy)dec-1-ene (5.28 g, 10.8 mmol)) was added dropwise and the reaction was allowed to stir at room temperature for 16 h. The reaction was cooled to 10°C followed by addition of NaOH solution (3 M, 20 mL). Hydrogen peroxide (30 wt. % in water, 6 mL) was added at 10°C. The reaction mixture was stirred at 50°C for 2 h and then cooled to room temperature. Ether (20 mL) was added and the organic phase was washed with water (20 mL), brine (20 mL), and dried over MgSO4 and condensed under reduced pressure to give 3.9 g (98 % yield HO-H10-O-F3), 4.5 g (80 % yield HO-H10-O-F6) of clear oil. HO-H10-O-F3 NMR: 1H NMR (400 MHz, CDCl3): δ 3.90 (tt, J = 14, 2 Hz, 2H), 3.64 (t, J = 7 Hz, 2H), 3.58 (t, J = 7 Hz, 2H), 1.60 (septet, J = 7 Hz, 4H), 1.41 − 1.29 (m, 12H). 19F NMR (376 MHz, CDCl3): δ −81.51 (3F), −121.08 (2F), −128.28 (2F). HO-H10-O-F6 NMR: 1H NMR (400 MHz, CDCl3): δ 3.92 (tt, J = 14, 2 Hz, 2H), 3.64 (t, J = 7 Hz, 2H), 3.59 (t, J = 7 Hz, 2H), 1.58 (septet, J = 7 Hz, 4H), 1.36 − 1.29 (m, 12H). 19F NMR (376 MHz, CDCl3): δ −81.29 (3F), −120.07 (2F), −122.71 (2F), −123.32 (2F), −123.93 (2F), −126.66 (2F).
Dibranched alcohols.
HO-diH10, HO-diH10-O-F3 and HO-diH10-O-F6:
To an oven dried 25 mL round-bottom flask were added 22.7 mmol alcohol (3.59 g HO-H10, 8.1 g HO-H10-O-F3, 11.5 g HO-H10-O-F6) and 0.51 g (9.08 mmol) of crushed KOH. This was allowed to stir at room temperature until all of the KOH dissolved. Epichlorohydrin (0.42 g, 4.54 mmol) was then added and the reaction was head to 120°C and allowed to react overnight, stirring vigorously. The reaction was then allowed to cool and diluted with 100 mL brine and extracted with 3, 100 mL aliquots DCM. The organic layers were then dried over MgSO4 and concentrated under reduced pressure. The resulting oil was then distilled under reduced pressure (20 mmHg at 200°C) to remove excess starting alcohol. The remaining oil was then purified by flash chromatography (twice, once with 17% EtOAc in hexanes and then again in 10% hexanes in DCM) to give semi-solid product (2.7 g 32 % yield HO-diH10/0.550 g, 16 % yield HO-diH10-O-F3/1.852 g, 43 % yield HO-diH10-O-F6). HO-diH10 NMR: 1H NMR (400 MHz, CDCl3): δ 3.94 (p, J = 6.4 Hz, 1H), 3.47 (A of ABX, JAB = 9.9 Hz, JAX = 4.2 Hz, 2H), 3.45 (t, J = 6.7 Hz, 4H), 3.45 (B of ABX, Jab = 9.9 Hz, Jbx = 6.4 Hz, 2H), 2.66 (broad s, 1H), 1.55 − 1.50 (m, 4H), 1.23 (broad s, 28H), 0.84 (t, J = 7Hz, 6H). HO-diH10-O-F3 NMR: 1H NMR (400 MHz, CDCl3): δ 3.93 (pentet, J = 5 Hz, 1H), 3.90 (tt, J = 14, 2 Hz, 5H), 3.58 (t, J = 7 Hz, 4H), 3.47 (t, J = 7 Hz, 4H), AB of an ABX with signals at 3.45 and 3.47 (JAB = 10 Hz, JAX = JBX = 5 Hz, 4 H), 2.49 (broad singlet, 1H), 1.58 (sextet, J = 7 Hz, 8H), 1.41 − 1.29 (m, 24H). 19F NMR (376 MHz, CDCl3): δ −81.51 (3F), −121.08 (2F), −128.28 (2F). HO-diH10-O-F6 NMR: 1H NMR (400 MHz, CDCl3): 3.91 (2 signals, 1 the obscured x of an ABX and 1 tt, J = 14, 2 Hz, 5H), 3.59 (t, J = 6.6 Hz, 4H), 3.46 (t, J = 6.7 Hz, 4H), AB of an ABX with signals at 3.48 and 3.43 (JAB = 10 Hz, JAX = 11 Hz, JBX = 9 Hz, 4H), 1.58 (sextet, J = 7 Hz, 8H), 1.39 − 1.24 (m, 24H). 19F NMR (376 MHz, CDCl3): δ −81.37 (6F), −120.11 (4F), −122.76 (4F), −123.36 (4F), −123.37 (4F), −126.69 (4F).
Miktoarm alcohols.
HO-μF8H10, HO-μF8H18, HO-μPftBH18:
Alcohol (decanol 5.012 g, 31.66 mmol/octadecanol 1.609 g, 5.95 mmol) was dissolved in anhydrous DCM (50.0 ml) and flask flushed with Ar. TEA (decanol (8.80 ml, 63.1 mmol)/octadecanol (1.3 mL, 9.33 mmol)) was added to solution and flask cooled in ice bath. MsCl (decanol (3.70 ml, 47.6 mmol)/octadecanol (0.6 mL, 7.72 mmol)) was then added via syringe, dropwise, and reaction stirred under Ar overnight, allowing the ice bath to warm to room temperature. Reaction was then stopped and washed with 4, 100 ml aliquots of aqueous NH4Cl, dried over MgSO4 and condensed under reduced pressure. Yield: 7.413 g decyl methane sulfonate (99 %), 1.973 g octadecyl methane sulfonate (95 %). Decyl methane sulfonate: 1H NMR (400 MHz, CDCl3): δ 4.22 (t, J = 6.6 Hz, 2H), 3.00 (s, 3H), 1.75 (p, J = 6.7 Hz, 2H), 1.42 (t, J = 7.5 Hz, 2H), 1.26 (m, 12H), 0.88 (t, J = 6.8 Hz, 3H). Octadecyl methane sulfonate: 1H NMR (400 MHz, CDCl3): δ 4.16 (t, J = 6.4 Hz, 2H), 2.95 (s, 3H), 1.69 (p, J = 6.8 Hz, 2H), 1.37–1.12 (m, 30H), 0.83 (t, J = 6.8 Hz, 3H).
2-phenyl-1,3-dioxan-5-ol (2):
glycerol (24.31 g, 264.0 mmol) and benzaldehyde (28.03 g, 264.1 mmol) were dissolved in anhydrous toluene (70 ml) and flask flushed with argon. Para-toluenesulfonic acid monohydrate (115.1 mg, 0.61 mmol) was added and flask fitted with Dean-Stark trap and heated to reflux. After 72 hours, reaction was cooled to room temperature and washed with sodium bicarbonate (100 ml), brine (100 ml), dried over MgSO4, and remaining toluene was placed in freezer overnight to crystallize out product. White crystals were then collected by filtration and dried under vacuum to yield 4.487 g (24.90 mmol, 9 % yield). NMR: 1H NMR (400 MHz, CDCl3): δ 7.48 (m, 2H), 7.36 (m, 3H), 5.50 (s, 1H), 4.12 (dd, J = 12.0, 1.4 Hz, 2H), 4.02 (dd, J = 12.0, 1.3 Hz, 2H), 3.54 (dt, J = 10.6, 1.5 Hz, 1H), 3.36 (d, J = 10.5 Hz, 1H).
5-(decyloxy)-2-phenyl-1,3-dioxane (3a) and 5-(octadecyloxy)-2-phenyl-1,3-dioxane (3b):
2-phenyl-1,3-dioxan-5-ol (2) (3a 3.686 g, 20.46 mmol/3b 4.860 g, 26.97 mmol) was dissolved in anhydrous toluene (3a 80 ml/3b 130 mL) and crushed KOH (5a 2.30 g, 41.0 mmol/5b 3.08 g, 54.9 mmol) added. Reaction fitted with Dean-Stark trap and heated to reflux for 6 hours. Reaction then cooled, and alkyl methane sulfonate (decyl methane sulfonate 7.413 g, 31.36 mmol/octadecyl methane sulfonate 5.218 g, 14.97 mmol) added as solution in toluene (20 ml). Reaction fitted with condenser and heated to reflux for 5 days. Reaction was then cooled to room temperature, diluted with 100 mL water, extracted with 3, 100 mL aliquots of ether, dried over MgSO4 and condensed under reduced pressure. Crude oil purified by flash column (5% ethyl acetate in hexanes) to obtain 3.309 g 5-(decyloxy)-2-phenyl-1,3-dioxane (10.33 mmol, 51 %)(3a), 8.060 g 5-(octadecyloxy)-2-phenyl-1,3-dioxane (3b) (18.63 mmol, 70 %). 3a: 1H NMR (400 MHz, CDCl3): δ 7.50 (m, 2H), 7.33 (m, 3H), 5.54 (s, 1H), 4.31 (dd, J = 12.4, 1.2 Hz, 2H), 4.02 (dd, J = 12.4, 1.6 Hz, 2H), 3.53 (t, J = 6.8 Hz, 2H), 3.24 (t, J = 2.0 Hz, 1H), 1.65 (p, J = 6.8 Hz, 2H), 1.28 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 3b: 1H NMR (400 MHz, CDCl3): δ 7.51 (m, 2H), 7.33 (m, 3H), 5.54 (s, 1H), 4.32 (dd, J = 12.5, 1.4 Hz, 2H), 4.03 (dd, J = 12.5, 1.8 Hz, 2H), 3.54 (t, J = 6.7 Hz, 2H), 3.25 (p, J = 1.8 Hz, 1H), 1.65 (p, J = 7.6 Hz, 2H), 1.25 (m, 30H), 0.88 (t, J = 6.9 Hz, 3H).
3-(benzyloxy)-2-(decyloxy)propan-1-ol (4a) and 3-(benzyloxy)-2-(octadecyloxy)propan-1-ol (4b):
3a (7.745 g, 24.17 mmol) 3b (903.1 mg, 2.087 mmol) was dissolved in anhydrous DCM to achieve 400 mmol L−1 concentration and the flask flushed with argon. Reaction cooled in ice bath, and 1 M DIBAL (4a 48.3 ml/4b 4.2 mL) was added dropwise over 20 minutes and reaction stirred overnight, allowing reaction to warm to room temperature. Reaction was quenched dropwise with 0.5 M NaOH (4a 30 ml/4b 3 mL), then diluted with 0.5 M NaOH (10 ml) and extracted with 2, 50 mL aliquots DCM. Combined organics were washed with 2, 100 mL aliquots Rochelle’s salt, 100 mL brine, dried over MgSO4 and condensed under reduced pressure. The crude oil was purified by column chromatography (0–5% methanol in DCM) to yield 6.01 g 3-(benzyloxy)-2-(decyloxy)propan-1-ol (4a) (18.6 mmol, 77 %), 650 mg 3-(benzyloxy)-2-(octadecyloxy)propan-1-ol (4b) (1.50 mmol, 72 %). 4a 1H NMR (400 MHz, CDCl3): δ 7.36–7.26 (m, 5H), 4.54 (AB quartet, 2H), 3.74 (m, 1H), 3.66–3.48 (m, 6H), 2.10 (dd, J = 5.7, 6.9 Hz, 1H), 1.57 (p, J = 7.0 Hz, 2H), 1.26 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 4b 1H NMR (400 MHz, CDCl3): δ 7.35–7.24 (m, 5H), 4.53 (AB quartet, 2H), 3.72 (m, 1H), 3.64–3.48 (m, 6H), 2.27 (t, J = 5.0 Hz, 1H), 1.57 (p, J = 7.0 Hz, 2H), 1.26 (m, 32H), 0.88 (t, J = 6.8 Hz, 3H).
3-(benzyloxy)-2-(decyloxy)propyl methanesulfonate (5a) and 3-(benzyloxy)-2-(octadecyloxy)propyl methanesulfonate (5b):
4 (4a 6.01 g, 18.6 mmol/4b 215.4 mg, 0.495 mmol) was dissolved in anhydrous DCM to achieve a 50 mmol L−1 concentration and the flask was flushed with Ar. TEA (5a 5.20 mL/5b 0.14 mL) was added and reaction cooled in ice bath. MsCl (5a 2.20 ml/5b 0.06 mL) was added dropwise and reaction was stirred under Ar overnight, allowing ice bath to warm to room temperature. Reaction was then diluted with DCM (50 ml) and washed with 3 aliquots saturated NH4Cl solution, dried over MgSO4 and condensed under reduced pressure to give pale yellow oil. (5a 7.102 g, 95 % yield/5b 0.253 g, quantitative yield). 5a 1H NMR (400 MHz, CDCl3): δ 7.33 (m, 5H), 4.54 (dd, J = 12.1, 2.3 Hz, 2H), 4.39 (dd, J = 10.9, 3.8 Hz, 1H), 4.27 (dd, J = 10.8, 5.7 Hz, 1H), 3.70 (p, J = 4.7 Hz, 1H), 3.55 (m, 4H), 3.00 (s, 3H), 1.56 (p, J = 6.8 Hz, 2H), 1.28 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 5b 1H NMR (500 MHz, CDCl3): δ 7.41–7.33 (m, 5H), 4.60 (dd, J = 14.8, 12.1 Hz, 2H), 4.44 (dd, J = 10.9, 3.8 Hz, 1H), 4.33 (dd, J = 10.9, 5.6 Hz, 1H), 3.76 (m, 1H), 3.62 (m, 4H), 3.04 (s, 3H), 1.62 (p, J = 6.8 Hz, 2H), 1.32 (m, 32H), 0.95 (t, J = 7.0 Hz, 3H).
((2-(decyloxy)-3-(1H,1H-perfluorononyloxy)propoxy) methyl)benzene (6a) and ((2-(octadecyloxy)-3-(1H,1H-perfluorononyloxy)propoxy)methyl)benzene (6b):
5 (5a 3.160 g, 7.9 mmol/5b 1.79 g, 3.49 mmol) was dissolved in anhydrous benzotrifluoride, F8H1-OH (6a 5.21 g, 11.57 mmol/6b 3.14 g, 6.98 mmol) added, and flask flushed with Ar. NaH slowly added (6a 667 mg, 28 mmol/6b 335 mg, 14 mmol), and reaction heated to reflux for 3 days. Reaction was quenched dropwise with H2O and further diluted with water and DCM and layers separated. Organics dried over MgSO4 and condensed under vacuum. Purified by column chromatography (5% ethyl acetate in hexanes) to obtain pure product (6a 3.985 g, 67 % yield/6b 2.29 g, 76 % yield). 6a 1H NMR (400 MHz, CDCl3): δ 7.33 (m, 5H), 4.54 (s, 2H), 4.00 (t, J = 13.9 Hz, 2H), 3.76 (dd, J = 10.4, 4.0 Hz, 1H), 3.68 (dd, J = 10.4, 5.6 Hz, 1H), 3.61 (p, J = 4.7 Hz, 1H), 3.54 (m, 4H), 1.56 (p, J = 6.8 Hz, 2H), 1.28 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 19F NMR (376 MHz, CDCl3): δ −81.19 (3F), −120.22 (2F), −122.38 (6F), −123.12 (2F), −123.80 (m, 2F), −126.52 (2F). 6b 1H NMR (400 MHz, CDCl3): δ 7.33 (m, 5H), 4.54 (s, 2H), 3.99 (t, J = 14.4 Hz, 2H), 3.77 (dd, J = 10.4, 4.2 Hz, 1H), 3.69 (dd, J = 10.4, 5.5 Hz, 1H), 3.61 (p, J = 4.9 Hz, 1H), 3.54 (m, 4H), 1.55 (p, J = 7.0 Hz, 2H), 1.28 (m, 32H), 0.88 (t, J = 6.8 Hz, 3H). 19F NMR (376 MHz, CDCl3): δ −81.31 (3F), −120.28 (2F), −122.44 (6F), −123.19 (2F), −123.85 (2F), −126.60 (2F).
((3-(perfluoro-tert-butyloxy)-2-(octadecyloxy)propoxy) methyl)benzene (6c):
5.604 g (10.93 mmol) 5b was dissolved in 50 mL anhydrous DMF. 3.64 g (13.28 mmol) Potassium perfluoro-tert-butoxide was added, and reaction heated to 120°C. After 24 hours reaction was stopped, cooled to room temperature, diluted with H2O and extracted with ether. The ethereal layer was dried over MgSO4 and condensed under vacuum. Crude oil purified by column chromatography (DCM) to yield 6.48 g (91 % yield) white solid. 1H NMR (400 MHz, CDCl3): δ 7.41–7.33 (m, 5H), 4.60 (dd, J = 14.8, 12.1 Hz, 2H), 4.44 (dd, J = 10.9, 3.8 Hz, 1H), 4.33 (dd, J = 10.9, 5.6 Hz, 1H), 3.76 (m, 1H), 3.62 (m, 4H), 3.04 (s, 3H), 1.62 (p, J = 6.8 Hz, 2H), 1.32 (m, 32H), 0.95 (t, J = 7.0 Hz, 3H). 19F NMR (376 MHz, CDCl3): δ −70.7.
HO-μH10F8, HO-μH18F8 and HO-μH18PftB:
6 (6a 3.679 g, 4.8 mmol/6b 2.265 g, 2.6 mmol/6c 0.108 g, 0.2 mmol) was dissolved in anhydrous DCM to achieve 20 mmol L−1 concentration and anisole (HO-μH10F8 2.10 ml, 19.8 mmol/HO-μH18F8 1.14 mL, 10.45 mmol/HO-μH18PftB 0.08 mL, 0.74 mmol) was added. Flask was flushed with Ar and cooled in ice bath. AlCl3 (HO-μH10F8 1.951 g, 14.63 mmol/HO-μH18F8 1.045 g, 7.84 mmol/HO-μH18PftB 0.076 g, 0.57 mmol) was added and reaction was stirred under Ar. After (HO-μH10F8 18 hours/HO-μH18F8 1.5 hours/HO-μH18PftB 1 hour) reaction was quenched dropwise with 0.5 M HCl, and further diluted with 0.5 M HCl and layers separated. Organic layer was washed with H2O, brine, dried over MgSO4 and condensed under reduced pressure. Crude oil was purified by column chromatography, ((HO-μH10F8 10–40 %/HO-μH18F8 10 %/HO-μH18PftB 0–5 %) ethyl acetate in hexanes) to give pure product (HO-μH10F8 2.875 g, 89 % yield/HO-μH18F8 1.400 g, 69 % yield/HO-μH18PftB 0.091 g, 98 % yield). HO-μH10F8 1H NMR (400 MHz, CDCl3): δ 4.00 (t, J = 13.7 Hz, 2H), 3.73 (m, 3H), 3.57 (m, 4H), 2.00 (t, J = 6.1 Hz, 1H), 1.57 (p, J = 7.1 Hz, 2H), 1.26 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 19F NMR (376 MHz, CDCl3): δ −81.25 (3F), −120.16 (2F), −122.42 (6F), −123.16 (2F), −123.82 (2F), −126.57 (2F). HO-μH18F8 1H NMR (400 MHz, CDCl3): δ 3.99 (t, J = 13.8 Hz, 2H), 3.73 (m, 3H), 3.52 (m, 4H), 2.11 (t, J = 5.9 Hz, 1H), 1.57 (p, J = 7.2 Hz, 2H), 1.26 (m, 32H), 0.88 (t, J = 6.8 Hz, 3H). 19F NMR (376 MHz, CDCl3): δ −81.18 (3F), −120.12 (2F), −122.39 (6F), −123.13 (2F), −123.79 (2F), −126.53 (2F). HO-μH18PftB 1H NMR (400 MHz, CDCl3): δ 4.05 (d, J = 4.0 Hz, 2H), 3.71 (q, J = 6.0 Hz, 1H), 3.57 (m, 4H), 2.32 (t, J = 5.6 Hz, 1H), 1.56 (p, J = 7.2 Hz, 2H), 1.25 (m, 30H), 0.87 (t, J = 6.8 Hz, 3H). 19F NMR (376 MHz, CDCl3): δ −70.80.
Linear and dibranched amphiphiles.
General procedure: To a dry 100 mL flask charged with argon were added 50 mL benzotrifluoride (BTF) and 4.0 mmol alcohol. The mixture was cooled on ice and 5.0 mmol NaH were added. This was allowed to stir for 30 minutes before adding 2.0 mmol mPEG-OMs. The reaction was then heated to reflux and allowed to react for 7 days. The reaction was cooled, diluted with 100 mL DCM and washed with 150 mL NH4Cl solution, 50 mL brine and dried over MgSO4. The organics were then concentrated to a minimum volume under reduced pressure and the surfactants precipitated upon addition of 500 mL cold ether. The solid was collected by vacuum filtration and then purified by reverse-phase chromatography. The product was then freeze dried from 50/50 DCM/Benzene to give a powdery solid.
M1H10:
60 % Yield, MALDI: Distribution centred on [M+Na+]= 1255.2, 1H NMR (400 MHz, CDCl3): δ 3.85 − 3.81 (m, 1H), 3.68 − 3.61 (m, 91H), 3.58–3.54 (m, 5H), 3.44 (t, J = 7 Hz, 2H), 3.38 (s, 3H), 1.57 (pentet, J = 7.3 Hz, 2H), 1.33 − 1.16 (m, 14H), 0.878 (t, J = 6.9 Hz, 3H).
M1F13:
58 % Yield, MALDI: Distribution centred on [M+Na+]= 1749.5, 1H NMR (400 MHz, CDCl3): δ 4.04 (t, J = 14 Hz, 2H), 3.46 − 3.46 (m, 106H), 3.38 (s, 3H). 19F NMR (376 MHz, CDCl3): δ −81.10 (3F), −120.13 (2F), −122.01 (16F), −123.01 (2F), −123.79 (2F), −126.45 (2F).
M1H10-O-F3:
52 % Yield, MALDI: Distribution centred on [M+Na+] = 1581.6, 1H NMR (400 MHz, CDCl3): δ 3.90 (tt, J = 13.7, 1.7 Hz, 2H), 3.84 − 3.81 (m, 1H), 3.75 − 3.71 (m, 1H), 3.68 − 3.61 (m, 95H), 3.60 − 3.54 (m, 6H), 3.44 (t, J = 6.9 Hz, 2H), 3.38 (s, 3H), 1.58 (sextet, J = 7.1 Hz, 4H), 1.35 − 1.22 (m 12H). 19F NMR (376 MHz, CDCl3): δ −81.39 (3F), −121.12 (2F), −128.20 (2F).
M1H10-O-F6:
42 % Yield, MALDI: Distribution centred on [M+Na+] = 1600.2, 1H NMR (400 MHz, CDCl3): δ 3.92 (tt, J = 13.3, 2.2 Hz, 2H), 3.70 – 3.62 (m, 95H), 3.61 − 3.54 (m, 9H), 3.44 (t, J = 6.6 Hz, 2H), 3.38 (s, 3H), 1.63–1.52 (m, 4H), 1.33 − 1.23 (m, 12H). 19F NMR (376 MHz, CDCl3): δ −81.28 (3F), −120.07 (2F), −122.73 (2F), −123.34 (2F), −123.95 (2F), −126.64 (2F).
M1H10F8:
79 % Yield, MALDI: Distribution centred on [M+Na+] = 1670.8, 1H NMR (400 MHz, CDCl3): δ 3.86–3.80 (m, 1H), 3.76 − 3.54 (m, 98H), 3.45 (t, J = 6.7 Hz, 2H), 3.38 (s, 3H), 2.05 (ttt, J = 19, 8.2, 2 Hz, 2H), 1.57 (septet, J = 6.7 Hz, 2H), 1.42 − 1.20 (m, 10H). 19F NMR (376 MHz, CDCl3): δ −81.19 (3F), −114.74 (2F), −122.36 (6F), −123.16 (2F), −123.96 (2F), −126.57 (2F).
M5diH10:
70 % Yield, MALDI: Distribution centred on [M+Na+] = 5120.8, 1H NMR (400 MHz, CDCl3): δ 3.82 (dd, J = 6.4, 4.6 Hz, 2H), 3.76 (dd, J = 6.5, 4.7 Hz, 2H), 3.70 – 3.59 (m, 530H), 3.56–3.53 (m, 3H), 3.50 − 3.45 (m, 6H), 3.42 (t, J = 6.8 Hz, 4H), 3.38 (s, 3H), 1.55 (pentet, J = 7.8 Hz, 4H), 1.34 − 1.21 (m, 28H), 0.88 (t, J = 6.3 Hz, 6H).
M5diH10-O-F3:
87 % Yield, MALDI: Distribution centred on [M+Na+] = 5695.7, 1H NMR (400 MHz, CDCl3): δ 3.90 (tt, J = 13.8, 1.7 Hz, 4H), 3.82 (dd, J = 5.8, 4.6 Hz, 2H), 3.71 (dd, J = 6.3, 4.9 Hz, 2H), 3.71 − 3.53 (m, 453H), 3.50 − 3.41 (m, 12H), 3.38 (s, 3H), 1.63 − 1.51 (m, 8H), 1.39 − 1.23 (m 28H). 19F NMR (376 MHz, CDCl3): δ −81.39 (6F), −121.096 (4F), −128.200 (4F).
M5diH10-O-F6:
45 % Yield, MALDI: Distribution centred on [M+Na+] = 6173.0, 1H NMR (400 MHz, CDCl3): δ 3.917 (tt, J = 13.2, 2.8 Hz, 4H), 3.75 – 3.79 (m, 6H), 3.68 − 3.45 (m, 480H), 3.42 (t, J = 7 Hz, 4H), 3.38 (s, 3H), 1.61 − 1.53 (m, 8H), 1.33 − 1.27 (m, 26H); 19F NMR (376 MHz, CDCl3): δ −81.13 (6F), −119.94 (4F), −122.66 (4F), −123.21 (4F), −123.82 (4F), −126.50 (4F).
All amphiphiles are at most as polydisperse as the mPEG-OH from which they are synthesized. MALDI spectra for starting mPEG-OMs and synthesized polymers can be found in the supporting information.
Miktoarm amphiphiles.
Typical procedure: Alcohol and M-OMs were dissolved in 20–75 mL BTF to achieve 20 mM concentrations. Flask flushed with Ar, NaH added (to achieve 40 mM concentration), and flask heated to reflux. After 5 days reaction was cooled to room temperature and quenched dropwise with H2O. The organics were dried over MgSO4. Solvents evaporated under reduced pressure, and crude polymer purified by reverse phase chromatography. Solid was lyophilized to give white, fluffy product.
M1μH10F8:
89 % Yield, MALDI: Distribution centred on [M+Na+] = 1715.0, 1H NMR (400 MHz, CDCl3): δ 4.02 (t, J = 14 Hz, 2H), 3.81 (m, 1H), 3.75 (dd, J = 10.4, 3.6 Hz, 2H), 3.67–3.62 (m, 80H), 3.59–3.51 (m, 7H), 3.46 (m, 1H), 3.38 (s, 3H), 1.57 (p, J = 7.2 Hz, 2H), 1.26 (m, 16H), 0.88 (t, J = 6.8 Hz, 3H). 19F NMR (400 MHz, CDCl3): δ −81.14 (3F), −120.18 (2F), −122.36 (6F), −123.09 (2F), −123.77 (2F), −126.48 (2F).
M2μH18F8:
58 %Yield, MALDI: Distribution centred on [M+Na+] = 2586.5, 1H NMR (400 MHz, CDCl3): δ 4.03 (t, J = 13.9 Hz, 2H), 3.82 (m, 2H), 2.76 (dd, J = 10.5, 3.6 Hz, 1H), 3.60–3.50 (m, 144H), 3.55 (m, 6H), 3.47 (m, 2H), 3.38 (s, 3H), 1.55 (p, J = 6.8 Hz, 2H), 1.26 (m, 26H), 0.88 (t, J = 7.0 Hz, 3H). 19F NMR (400 MHz, CDCl3): δ −81.27 (3F), −120.28 (2F), −122.49 (6F), −123.22 (2F), −123.89 (2F), −126.65 (2F).
M2μH18PftB:
42 % Yield, MALDI: Distribution centred on [M+Na+] = 2184.7, 1H NMR (400 MHz, CDCl3): δ 4.14 (m, 1H), 4.04 (m, 1H), 3.79 (m, 1H), 3.69–3.59 (m, 144H), 3.55 (m, 4H), 3.50 (m,1H), 3.38 (s, 3H), 1.54 (p, J = 7.2 Hz, 2H), 1.27 (m, 32H), 0.88 (t, J = 6.8 Hz, 3H). 19F NMR (400 MHz, CDCl3): δ −70.66.
M5μH18PftB:
50 % Yield, MALDI: Distribution centred on [M+Na+] = 5503.8, 1H NMR (400 MHz, CDCl3): δ 4.13 (m, 1H), 4.03 (m, 1H), 3.82 (m, 2H), 3.70–3.60 (m, 386H), 3.55 (m, 5H), 3.47 (m, 2H), 3.38 (s, 3H), 1.54 (p, J = 7.1 Hz, 2H), 1.28 (m, 32H), 0.88 (t, J = 6.8 Hz, 3H). 19F NMR (400 MHz, CDCl3): δ −71.15.
All amphiphiles are at most as polydisperse as the mPEG-OH from which they are synthesized. MALDI spectra for starting mPEG-OMs and synthesized polymers can be found in the supporting information.
Physicochemical Characterization
Micelle preparation – solvent evaporation method (SEM).
Polymer is dissolved in MeOH or ACN to a desired concentration. 1 mL of polymer solution and additive (e.g. PTX in ACN) are added to a 25 mL roundbottom flask and rotated for 5 minutes at 60°C on a rotary evaporator, no vacuum, and then the solvent was removed in vacuo with rotation for 15 minutes. The film was then dispersed with Millipore water heated to 60°C and filtered with a 0.45-μm nylon filter.
Particle size determination by dynamic light scattering (DLS).
Micelles were prepared by solvent evaporation, polymer solution concentration 1 mg mL−1 in MeOH. Particle sizes of polymeric aggregates were analysed by dynamic light scattering (NICOMP 380ZLS, Particle Sizing Systems, Santa Barbara, CA). The surfactant solution was measured directly without dilution and analysed. Each particle size analysis was run at room temperature and repeated in triplicate with the number of scans of each run determined automatically by the instrument according to the concentration of the solution. The data was analysed using NICOMP analysis and reported as volume weighted average diameters.
Critical aggregation concentration (CMC) determination – Surface Tensiometry.
Surfactant was dissolved in Millipore water to a concentration of 1 mM and concentrations down to 1 nM were prepared by serial dilution and transferred to 20 mL disposable scintillation vials. After solutions were made, the samples were heated in a water bath at 40°C with sonication for 2–3 hours and allowed to equilibrate for 24 hours. Surface tensions were measured on a KSV sigma 701 tensiometer (KSV Instruments, Helsinki, Finland) equipped with a Julabo F12-MC circulator for constant temperature control. Custom round rod made of platinum with a diameter of 1.034 nm with wetted length of 3.248 mm was used. The rod was submerged in absolute alcohol and flame dried with a Bunsen burner for 4 seconds. This was repeated after 4 minutes. The rod was then hung on the instrument and allowed to cool to room temperature without touching any surface. Before running the experimental samples, the surface tension of millipore water was measured as a control to confirm the rod was fully cleaned and surface tension was within 1 mN/m of 78.2 mN/m. The surface tension measurements began with the least concentrated solution and proceeded to successively more concentrated solutions. The surface tension at each concentration was measured in quadruplet and average recorded. The critical micelle concentration value was determined from the crossover point of two lines: the baseline of minimal surface tension and the slope where surface tension showed linear decline; error determined by weighted least squares analysis.
Critical aggregation concentration (CMC) determination – Pyrene Fluorescence.
Based on the methodology developed by Torchilin et al.17 Micelles were prepared by the solvent evaporation method. 200 μL micelle solutions in MeOH ranging in concentration from 10−4.5 to 10−8 M were delivered to 25 mL roundbottom flasks with 100 μL 10 mg mL−1 pyrene solution. The thin films were dispersed with 2 mL 60°C PBS and filtered with a 0.45-μm nylon filter. The fluorescence analysis was carried out on an AMINCO-Bowman Series 2 spectrometer with excitation at 339 nm, emission at 390 nm and a spectral window of 370 – 400 nm. The analysis was carried out in triplicate and the average CMC and standard deviation are reported.
Microviscosity measurement.
Surfactant solutions of 0.2 mmol L−1 in MeOH and a 2.7 ng mL−1 1,3-bis-(1-pyrenyl)propane (P3P) in chloroform were prepared. Micelle solutions were then prepared via the solvent evaporation method with 67 μL of the P3P solution added and solutions stored in amber vials. The fluorescence analysis was carried out on an AMINCO-Bowman Series 2 spectrometer with excitation at 333 nm, emission at 378 nm and a spectral window of 350 – 500 nm.
Föster resonance energy transfer (FRET) stability.
Surfactant solutions of 1 mg mL−1 in methanol and 0.1 mg mL−1 of 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI) and 3,3’-dioctadecyloxacarbocyanine perchlorate (DiO) in methanol were prepared. Micellar solutions were then prepared by the solvent evaporation method. For blank micelles only polymer solution is used; for FRET loaded micelles, 46 μL DiI and 44 μL of DiI are added.
Three analytic samples were then prepared (the two components were mixed just before analysis: 50 μL of FRET dye loaded micelles and 950 μL PBS (used to set sensitivity), 50 μL empty micelles and 950 μL human serum (used to correct for baseline), 150 μL FRET dye loaded micelles and 2.85 mL human serum (mixed gently before first measurement and vigorously before each measurement, every 15 minutes, thereafter. The fluorescence analysis was carried out on an AMINCO-Bowman Series 2 spectrometer with excitation at. The FRET ratio calculated was the ratio of I565/(I501+I565).
Paclitaxel (PTX) encapsulation measurements.
Micelles solutions were prepared in triplicate using the solvent evaporation method. PTX stock solution was generated by dissolving PTX in ACN, aided by sonication, at a concentration of 1 mg mL−1. Surfactant was dissolved in ACN to give a final concentration of 2.4×10−3 mol L−1. One mL of surfactant solution was then mixed with 230 μL PTX solution. The sample was then centrifuged at 12,000 rpm for 5 minutes and filtered through 0.45-μm nylon syringe filter to remove any insoluble precipitate. A 100-μL aliquot of micelle solution was mixed with 900 μL of ACN and the remaining micelle solution was allowed to sit for 24 hours. The sample was then recentrifuged at 12,000 rpm for 5 minutes and filtered through 0.45-μm nylon syringe filter to remove any insoluble precipitate. A 100-μL aliquot of micelle solution was mixed with 900 μL of ACN. The paclitaxel loaded in the micelle was quantified by reverse phase HPLC. The HPLC system used was a Shimadzu prominence HPLC system (Shimadzu, Japan), consisting of a LC-20AT pump, SIL-20 AC HT autosampler, CTO-20 AC column oven and an SPD-M20A diode array detector. 20 μL of the mixture was injected into a C18 column (Agilent XDB-C8, 4.6 Å × 150 mm), eluting with an isocratic mixture of 25 % water in acetonitrile. The run time was 7 min, the flow rate was 1.0 mL min−1 and the detection was set at 227 nm.
Results and Discussion
The synthesis of both linear and dibranched alcohols starts with 9-decen-1-ol. Two methodologies were developed: radical and anionic syntheses (Scheme 1). The radical synthesis utilizes methodology developed by Brace.18 In an atom transfer radical addition, perfluorooctyl iodide is added to 9-decen-1-ol with AIBN as a radical initiator. Subsequent reduction by zinc in acetic acid then removes the iodide (Scheme 1).
Scheme 1.

Synthesis of linear and dibranched alcohols.
This methodology worked in moderate to good yields for producing F8H10-OH and F6H10-OH, not shown. F8H10-OH worked well in subsequent Williamson ether syntheses, but the smaller F6H10-OH decomposed under the basic reaction conditions. F6H10-OH suffered from HF elimination across the CF2-CH2, a potential pitfall when using semi-fluorinated molecules under basic conditions.19
To circumvent the basic-instability of the radical-synthesis products, an anionic synthesis was developed that produced more stable alcohols for further synthesis (Scheme 1). A semi-fluorinated alkene ether is prepared by Williamson ether synthesis using 9-decen-1-yl methane sulfonate and 1H,1H-perfluorobutan-1-ol (F3-O-H10-OH) or 1H,1H-perfluoroheptan-1-ol (F6-O-H10-OH). Only 1H,1H-perfluoro-alcohols were found to proceed in high yields, as 1H,1H,2H,2H-perfluoroalcohols suffered from complete HF elimination in lieu of any SN2 reaction.20 Hydroboration oxidation of the semi-fluorinated alkene yields the linear semi-fluorinated alcohols in good overall yield. The linear alcohols can then be used to form dibranched alcohols by a concomitant SN2 reaction and ring opening of epichlorohydrin (Scheme 1). This represents a more robust methodology for the synthesis of linear (and subsequently dibranched) alcohols than the radical mechanism because these alcohols did not suffer from base-induced decomposition.
The miktoarm alcohols were prepared starting from glycerol (Scheme 2). Acetal protection of the two primary alcohols proceeds in low yield due to the mixture of 1,2- and 1,3-protection products.21 Subsequent alkylation of the secondary alcohol was then found to proceed best using alkyl methane sulfonates in a mixture of KOH and toluene at reflux. Reductive ring opening with DIBALH affords 3-benzyloxy-2-alkoxy-1-propanols. Mesylation and Williamson ether synthesis with semi-fluorinated alcohols 1H,1H-perfluorononan-1-ol (F8H1-OH) or perfluoro-tert-butanol (PftB-OH) followed by deprotection gave the miktoarm alcohols (Scheme 2).
Scheme 2.

Synthesis of miktoarm alcohols.
The linear, dibranched and miktoarm alcohols were coupled to mono-methoxy capped poly(ethylene glycol) methane sulfonate (Scheme 3). The reactions were found to proceed best in benzotrifluoride (BTF) rather than THF, as higher temperatures could be achieved for reflux. Purification of the produced amphiphiles was achieved by reverse-phase chromatography.
Scheme 3.

PEGylation of semi-fluorinated alcohols.
For the hydrophilic head group, the size of the mPEG was selected so as to favor micelle formation. In our lab we have seen that diblock amphiphiles with mPEG1000 form micellar aggregates thus mPEG1000 was selected as a hydrophilic group for the linear amphiphiles. Similarly, it was found that mPEG1000 with dibranched hydrophobic tails yielded random aggregates,9 as such mPEG2000 and mPEG5000 were selected for coupling to the miktoarm and dibranched alcohols to ensure micelle formation.
Each amphiphile was analysed by MALDI mass spectrometry to determine average molecular weight (Table 1 and Table 2). To determine the critical micelle concentration (CMC), the concentration above which aggregates begin to form, surface tension analysis was used. Surfactant solutions were prepared in deionized water at concentrations from 1 mM to 1 nM. The CMC was then determined as the cross over of two lines and the error was determined by weighted, least-squares analysis (Table 1 and Table 2). M5diH10-O-F6 produced inconclusive results by surface tension measurements and was thus analysed by pyrene encapsulation fluorescence.17 The inconclusive surface tension measurements are possibly due to slow kinetics in the equilibration of polymer chains. Solutions above the CMC were analysed by dynamic light scattering (DLS) (Table 1 and Table 2). Most of the surfactants showed small (<20 nm), narrow size-distributions consistent with spherical micelles. Note that cryogenic transmission electron microscopy (cryo-TEM) is typically used to verify the morphology of the aggregates formed. For the amphiphiles presented here, cryo-TEM failed due to the highly solvated nature of the PEG (rendering it indistinguishable from the surrounding vitrified water),22 and the relatively small size of the hydrophobic blocks. Core microviscosity for each amphiphile was determined by encapsulation and fluorescence analysis of 1,3-bis-(1-pyreneyl)propane. The microviscosity is presented as the ratio of the monomer fluorescence (Im) over the eximer fluorescence (Ie), with higher Im/Ie ratios corresponding to greater microviscosities (Table 1 and Table 2).23
Table 1.
Physicochemical data of linear and dibranched amphiphiles.
| Amphiphile | Mol. Wt. (g mol−1) | pCMC (−log(M) ± S.D.) | Ave Size (nm) | Microviscosisty (Im/Ie) |
|---|---|---|---|---|
| M5diH10 | 5097.8 | 3.92 ± 0.14 | 16.1 ± 1.6 | 3.64 ± 0.23 |
| M5diH10-O-F3 | 5672.7 | 4.44 ± 0.18 | 21.6 ± 5.9 | 6.76 ± 0.13 |
| M5diH10-O-F6 | 6150.0 | 5.05 ± 0.03 | 20.3 ± 6.5 | 6.83 ± 0.17 |
| M1H10 | 1232.2 | 3.01 ± 0.25* | 149.7 ± 36.0 | 2.75 ± 0.11 |
| M1H10-O-F3 | 1558.6 | 3.82 ± 0.08 | 9.5 ± 1.3 | 5.99 ± 0.32 |
| M1H10-O-F6 | 1577.2 | 4.53 ± 0.12 | 10.6 ± 1.0 | 6.26 ± 0.19 |
| M1H10F8 | 1647.8 | 5.85 ± 0.06 | 10.7 ± 1.2 | 6.81 ± 0.11 |
| M1F13 | 1726.5 | 6.08 ± 0.13 | 11.9 ± 1.3 | 3.40 ± 0.18 |
CAC reported. M1H10 does not form spherical micelles, but rather it randomly aggregates to yield a variety of assemblies of various sizes
Table 2.
Physicochemical data of miktoarm amphiphiles.
| Amphiphile | Mol. Wt.1 (g mol−1) | pCMC (-log(M) ± S.D.) | Ave Size (nm) | Microviscosity (Im/Ie) |
|---|---|---|---|---|
| M1μH10F8 | 1692.0 | 5.51 ± 0.05 | 10.8 ± 1.0 | 5.08 ± 0.06 |
| M2μH18F8 | 2563.5 | 4.61 ± 0.07 | 11.5 ± 1.5 | 5.38 ± 0.08 |
| M2μH18PftB | 2161.7 | 4.62 ± 0.42 | 12.5 ± 1.5 | 4.43 ± 0.08 |
| M2DSPE | 2805.5 | 4.90 ± 0.17 | 13.9 ± 1.6 | 5.60 ± 0.22 |
| M5μH18PftB | 5480.4 | 5.22 ± 0.22 | 19.6 ± 2.7 | 4.25 ± 0.07 |
| M5DSPE | 5801.1 | 5.38 ± 0.10 | 18.6 ± 2.8 | 5.23 ± 0.03 |
The first trend to notice is the effect of the fluorinated moieties on the average particle size (Table 1). For these linear amphiphiles, the addition of the fluorous block was essential for micelle formation, with M1H10 showing the formation of random aggregates by DLS and M1H10-O-F3 showing a DLS consistent with small, spherical micelles. Further changes in the length of the fluorocarbon block do not show significant changes in average particle size going from M1H10-O-F3 to M1H10F8. The dibranched amphiphiles show a very similar trend (Table 1). While there is a noticeable change between M5diH10 and M5diH10-O-F3, M5diH10-O-F3 and M5diH10-O-F6 show similar particle sizes. It can be seen that in a linear arrangement of moieties, the introduction of a fluorocarbon segment has an effect on particle size, but changes of the fluorocarbon block size have little to no effect among these ternary amphiphiles.
Upon analysis of Table 1 trends between CMC and the number of fluorocarbons can be observed. As shown in Fig. 2, there is a linear relationship between pCMC (-log(M)) and the number of fluorinated carbon atoms. Linear relationships between CMC (log(M)) and the number of −CH2–24 or −CF2–25 has been reported previously. Here, the interesting result is the difference in slope between the linear and dibranched surfactants (Fig. 2). There are two differences between the two classes of amphiphiles that could account for this. The first is the difference in PEG block size (M1 compared to M5). To evaluate the effect of PEG size, the pCMC of the linear M1H10F8 (5.85) was compared to that of the linear M5H10F8 (5.60). There is a difference in CMC; however, the linear fit for the dibranched amphiphiles predicts a pCMC of 4.67, almost a full order of magnitude different. As such, it seems that the difference in PEG size is not the determining factor. The second difference is that of hydrophobic architecture itself. The difference between the linear and dibranched series can be explained by the differences in hydrophobic architecture. The linear polymers have a much smaller base hydrophobic block (H10) than the dibranched polymers (diH10). Thus the addition of each fluorocarbon to the linear architecture has a larger effect, whereas the addition of fluorocarbons to the larger dibranched hydrophobic block has a diminished effect. This difference in the effectiveness of each fluorocarbon on the CMC thus explains the difference in slope for Fig. 2, a result of the size of the hydrocarbon block size relative to the fluorocarbon block size.
Fig. 2.

Relationship between linear and dibranched amphiphile number of fluorocarbons and pCMC (-log(M) ± S.D., n = 4).
Microviscosity measurements of the triblock surfactants show dramatically different behaviour than either the hydrocarbon M1H10/M5diH10 or fluorocarbon M1F13 diblock amphiphiles (Table 1). The diblock surfactants show similar Im/Ie ratios, while the smallest triblock amphiphiles – M1H10-O-F3 and M5diH10-O-F3 – show a sharp increase in microviscosity. Typically, increases in microviscosity are associated with more crystalline micelles resulting from either chain entanglement or tighter packing of unimers.4 The triblock amphiphiles are believed to show higher microviscosities, compared to the diblock surfactants, due to hydrophobic phase segregation of the hydrocarbon and fluorocarbon blocks, which results in tighter unimer packing within the micelles.
The higher rigidity of the triblock polymeric micelles has important implications in terms of kinetic stability and encapsulation. In Fig. 3, the microviscosities for all hydrocarbon-diblock and triblock amphiphiles are plotted against the number of fluorocarbons in each amphiphile. This plot shows a logarithmic increase in microviscosity with the number of fluorocarbons. Hence the initial addition of a small number of fluorocarbons causes the most dramatic increase in microviscosity (Fig. 3) compared to the diblock analogues. This is the result of phase segregation. Each subsequent fluorocarbon, however, does not lead to further phase-segregation but merely diminishes unimer dynamics.
Fig. 3.

Relationship between microviscosity (Im/Ie ratio ± S.D, n = 3) and the number of fluorocarbons for linear and dibranched amphiphiles.
A third general surfactant design (Scheme 2) in which a pure hydrocarbon is used as one branch and a pure fluorocarbon is used as the second branch (miktoarm surfactants) was seen as an alternative that would allow for a wider variety of surfactant design. Furthermore, the miktoarm architecture offered a distinctly different design, which in reference to the linear and dibranched amphiphiles already presented could offer an interesting comparison. The miktoarm semi-fluorinated surfactants’ physicochemical data are summarized in Table 2.
One of the most immediate trends to notice is the relatively small change that is observed in the CMC among non-fluorinated polymeric amphiphiles (M2DSPE and M5DSPE) and fluorinated miktoarm analogues (M2μH10F8, M2μH18PftB, M5μH18PftB). The parallel arrangement of fluorocarbon and hydrocarbon chains does not lead to the decrease in CMC that was observed for linear and dibranched amphiphiles. This could be due to the compartmental segregation of fluorocarbon and hydrocarbon blocks, which increases unfavourable hydrocarbon-fluorocarbon interfacial interactions.26
For the miktoarm amphiphiles there is no consistent trend in average particle size associated with the changes in hydrocarbon or fluorocarbon moieties. There are noticeable differences in particle size, which can be attributed solely to the changes in the size of the mPEG block. This follows logically as the change in the size of the hydrophilic block is the greatest change occurring among the various miktoarm surfactants.
The microviscosity among the various miktoarm amphiphiles differs little. The exceptions to this trend are the M2μH18PftB and M5μH18PftB surfactants. Here spherical perfluoro-tert-butanoxy is used, which interferes with chain packing and lowers the microviscosity compared to F8H1-OH.
The use of fluorocarbon segments was investigated not only for their potential effects on thermodynamic stability, CMC, micelle formation, or microviscosity of aggregates, but also for their potential to improve kinetic stability in vivo. Micelles have found widespread use in the development of drug delivery and of central importance is the lifetime of micelles in vivo.27 For this application, better results have been observed with long circulating particles.28
Using methodology developed by Chen et al. the dissociation of micelles in human serum was monitored through Förster Resonance Energy Transfer (FRET)29 (Fig. 4), FRET data for all polymers can be found in the Supporting Information. It was found that non-fluorinated micelles (M5diH10 and M5DSPE) rapidly dissociated in vitro when exposed to human serum (observed by a decrease in FRET ratio). For fluorinated micelles, both the architecture and block composition are important. The linear surfactants showed a dramatic increase in stability with only the addition of an F3 block and even greater stability with an F8 (an F6 block fell in between F3 and F8). This trend follows the microviscosity trend observed (Table 1) and suggests that the more crystalline (more viscous) micelles are more resistant to dissociation. For dibranched triblock amphiphiles, only M5diH10-O-F6 shows any increase in stability (Fig. 4), while M5diH10-O-F3 (not shown) does not show any increase in stability over M5diH10. This is likely due to the less efficient packing of the dibranched hydrophobic amphiphiles – compared to the linear amphiphiles. The less efficient packing contributed to a smaller effect of each fluorocarbon, in the dibranched amphiphiles, on the kinetic stability.
Fig. 4.

FRET stability of representative amphiphiles
The relationship between microviscosity and kinetic stability is further underlined by the results of the miktoarm surfactants, which did not show such dramatic improvements in stability when comparing M2μH18PftB versus M2μH18F8. The less viscous M2μH18PftB micelle demonstrates much more rapid dissociation, mirroring the trend in microviscosity and supporting the idea that the M2μH18PftB micelles pack less efficiently due to the spherical nature of the fluorous moiety. Measurement of M1μH10F8 particle stability was especially difficult and was not shown. This polymer failed to encapsulate the FRET dyes. As such, after numerous trials, only one run showed any FRET ratio above the 0.400 minimum, which, however, decreased very quickly. Given the poor encapsulation ability no comparison could be made, using this methodology, between the linear M1H10F8 and the miktoarm M1μH10F8.
Given the possible structure of the linear/dibranched (fluorocarbon core-hydrophobic intermediate shell-hydrophilic corona)16,30 and miktoarm (compartmental structure) amphiphiles,26 encapsulation of paclitaxel (PTX), a model hydrophobic species, was investigated. It is known that hydrophobic molecules can be encapsulated in classical hydrocarbon-based micelles by sequestration within the hydrophobic core.31 Here PTX encapsulation was investigated to probe the effect of a fluorous core (linear/dibranched amphiphiles) or a mixed hydrocarbon/fluorocarbon core (miktoarm amphiphiles) on the solubilization of hydrophobic species.
Paclitaxel was chosen as the model hydrophobic drug because it is not solubilized to any extent in a purely fluorophilic micelle (Fig. 5). This allows for the direct interrogation of how PTX is encapsulated into a hydrophobic shell, in the case of the linear and dibranched amphiphiles, or a mixed core, in the case of the miktoarm amphiphiles. PTX loaded micelles were prepared by the solvent evaporation method (SEM), which we found to give more reproducible results than other methods and to give higher drug loading.32 All encapsulation data are presented as the average percent weight of the encapsulated PTX in linear and dibranched amphiphiles (Fig. 5) and miktoarm amphiphiles (Fig. 6). Equimolar amounts of surfactants were used in all encapsulation studies to allow for direct comparisons.
Fig. 5.

Percent weight encapsulation (mean ± S.D., n = 3) by linear and dibranched amphiphiles
Fig. 6.

Percent weight encapsulation (mean ± S.D., n = 3) of PTX by miktoarm and DSPE amphiphiles
Each of the linear amphiphiles is able to initially encapsulate the same amount of PTX on a percent weight basis (Fig. 5), despite varying the fluorocarbon length. However, after 24 hours – when poorly encapsulated PTX is lost33,34 – an increase in the PTX remaining encapsulated in the micelles can be seen with increasing size of the fluorocarbon moiety. The increase in PTX remaining encapsulated after 24 hours trends with the increase in microviscosity. This suggests that more PTX is retained for a longer period of time in M1H10F8 due to its less dynamic, higher microviscosity aggregates. A similar inclination is also seen in the dibranched amphiphiles. Here, the higher microviscosity of M5diH10-O-F3 and M5diH10-O-F6 is directly related to the higher encapsulation of and retention of PTX after 24 hours (Fig. 5) compared to M5diH10. Note that M1H10 was not studied for its encapsulation ability because it forms random aggregates.
The linear and dibranched amphiphile encapsulation data show that an inner hydrophobic core is not necessary for stable encapsulation of hydrophobic species such as PTX. Moreover, as the fluorous inner-core increases in size, the micelles show enhanced retention ability as a result of the more tightly packed micellar structure, vis-à-vis microviscosity. The linear arrangement of hydrocarbon and fluorocarbon segments shows both enhanced kinetic stability and enhanced ability to retain hydrophobic species over time. In the case of the miktoarm amphiphiles (Fig. 6), trends that relate the amount of PTX encapsulated to the microviscosity can be observed. The less viscous micelle (M2μH18PftB) retains less PTX than the more viscous (M2μH18F8). The more noticeable trend, however, is the overall reduction in both initial and retained PTX encapsulated when the semi-fluorinated miktoarm amphiphiles are compared to the commercially available M2DSPE and M5DSPE. Perhaps unsurprisingly, this suggests that one of the key parameters in encapsulation and retention of hydrophobic species is the hydrophobic carrying capacity – which is analogous to the number of hydrocarbons in the aggregate. For example, M2μH18F8 has half the number of hydrocarbons in the core compared to M2DSPE and the percent weight encapsulation of PTX after 24 hours for M2μH18F8 (1.89 % wt.) is slightly more than half of that for M2DSPE (3.21 % wt.). Interestingly, it seems that sufficiently high microviscosity can compensate for a reduction in hydrophobic carrying capacity as seen in comparing M1H10F8 (2.00 % wt. PTX encapsulated after 24 hours) and M2μH18F8. M1H10F8 performs better than M2μH18F8 despite having 55 % of the number of methylene groups of M2μH18F8.
It should be noted that compared to the mPEG-DSPE polymeric surfactants, all triphilic amphiphiles presented here retain significantly less PTX than they initially encapsulate. The loss of PTX over time is associated with two factors. First, hydrophobic molecules that are poorly encapsulated – in the mPEG corona rather than in the hydrophobic core – are lost rapidly.33,34 Second, overall stability of the micellar aggregate is critical, with more stable, less dynamic micelles showing greater overall retention of encapsulated species.35 For the polymers presented here, all possess much smaller hydrophobic blocks than mPEG-DSPE – the largest M2μH18F8 has 50 % of the hydrophobic groups in mPEG-DSPE. As such, there is less space to encapsulate PTX. However, despite the overall smaller hydrophobic carry capacity, the linear and dibranched triblock polymers show high microviscosities, which correlate well with the size of the fluorocarbon block. As the microviscosity, and therefore the aggregate stability, increases so does the overall retention of PTX.
Ultimately, when one directly compares the linear (M1H10F8) and miktoarm (M1μH10F8) architectures several points stand out. Both M1H10F8 and M1μH10F8 have very similar CMC’s and particle sizes. Their point of divergence is microviscosity. M1H10F8 has a microviscosity (6.81, Table 1) 36 % greater than that of M1μH10F8 (5.01, Table 2). This results in a higher net retention of PTX by M1H10F8 than M1μH10F8. Given the poor encapsulation of the FRET dyes by M1μH10F8, no direct comparison can be made on architecture stability differences, but one can infer from the performance of M2μH18F8 (which has a higher microviscosity) that M1μH10F8 would dissociate far more rapidly than M1H10F8.
Conclusions
The peculiar properties of fluorocarbons offer a means of not only improving micellar kinetic and thermodynamic stability, but also a way of improving and probing encapsulation behaviour. Herein the syntheses for the preparation of linear, dibranched, and miktoarm semi-fluorinated amphiphiles have been presented. Each class of polymer was studied in an attempt to elucidate relationships between the ternary amphiphile structure and the resulting physicochemical behaviour of the aggregates. For linear and dibranched amphiphiles, the introduction of fluorocarbon moieties significantly increases their thermodynamic stability and microviscosity; while for miktoarm amphiphiles, the more important factors are the shape of the fluorocarbon block and the size of the hydrophobic and hydrophilic segments. For the linear surfactants alone, the introduction of fluorocarbon blocks leads to significantly greater kinetic stability, which was correlated with higher microviscosity. All triblock amphiphiles that formed small, narrow size distributions in water that were consistent with micelles, were studied for their ability to encapsulate PTX. Overall, the semi-fluorinated surfactants retained less of the PTX initially encapsulated as compared to the phospholipid-based mPEG2000DSPE and mPEG5000DSPE polymers. This is certainly a direct result of the smaller hydrophobic block of the studied semi-fluorinated polymers compared to that present in mPEG-DSPE. Taken together, the results for the three classes of semi-fluorinated polymers show that changes in architecture and composition of the amphiphiles blocks can be rationally used to modulate the physicochemical properties of a micelle, including stability in physiological conditions and encapsulation of hydrophobic molecules.
Supplementary Material
Acknowledgements
The research described in this article was partially supported by the National Institutes of Health (grant # GM079375 to SM) and by the University of Wisconsin-Madison School of Pharmacy. The authors would like to thank Dr. Glen Kwon for the use of DLS and HPLC instrumentation.
References
- 1.Lipshutz BH, Isley NA, Fennewald JC, Slack ED, Angew. Chem. Int. Ed, 2013, 52, 10942–10958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Isley NA, Gallou F, Lipshutz BH, J. Am. Chem. Soc, 2013, 135, 17707–17710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caillier L, de Givenchy ET, Levy R, Vandenberghe Y, Geribaldi S, Guittard F, J. Colloid Interf. Sci, 2009, 332, 201–207. [DOI] [PubMed] [Google Scholar]
- 4.Allen C, Maysinger D, Eisenberg A, Colloids Surf., B, 1999, 16, 3–27. [Google Scholar]
- 5.Torchilin VP, Adv. Drug. Deliv. Rev, 2012, 64, 302–315. [DOI] [PubMed] [Google Scholar]
- 6.Trubetskoy VS, Torchilin VP, Adv. Drug Deliv. Rev, 1995, 16, 311–320. [Google Scholar]
- 7.Weissig V, Whiteman KR, Torchilin VP, Pharm. Res, 1998, 15, 1552–1556. [DOI] [PubMed] [Google Scholar]
- 8.Hoang KC, Mecozzi S, Langmuir, 2004, 20, 7347–7350. [DOI] [PubMed] [Google Scholar]
- 9.Parlato MC, Jee J-P, Teshite M, Meozzi S, J. Org. Chem, 2011, 76, 6584–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jee J-P, McCoy A, Mecozzi S, Pharm. Res, 2012, 29, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bates FS, Hillmyer MA, Lodge TP, Bates CM, Delaney KT, Fredrickson GH, Science, 2012, 336, 434–440. [DOI] [PubMed] [Google Scholar]
- 12.Chen J, Li J-J, Luo Z-H, J. Polym. Sci., Part A: Polym. Chem, 2013, 51, 1107–1117. [Google Scholar]
- 13.Chen Y, Zhang Y, Wang Y, Sun C, Zhang C, J. Appl. Polym. Sci, 2013, 127, 1485–1492. [Google Scholar]
- 14.Zhou Y-N, Luo Z-H, Chen J-H, AIChE J, 2013, 59, 3013–3033. [Google Scholar]
- 15.Alaimo D, Beigbeder A, Dubois P, Broze G, Jérôme C, Grignard B, Polym. Chem, 2014, 5, 5273–5282. [Google Scholar]
- 16.Amado E, Kressler J, Soft Matter, 2011, 7, 7144–7149. [Google Scholar]
- 17.Torchilin VP, Levchenko TS, Whiteman KR, Yaroslavov AA, Tsatsakis AM, Rizos AK, Michailova EV, Shtilman MI, Biomaterials, 2001, 22, 3035–3044. [DOI] [PubMed] [Google Scholar]
- 18.Brace NO, J. Fluor. Chem, 1999, 93, 1–25. [Google Scholar]
- 19.Rocaboy C, Rutherford D, Bennett BL, Gladysz JA. J. Phys. Org. Chem, 2000, 13, 596–603. [Google Scholar]
- 20.Tucker WB, Mecozzi S, J. Fluor. Chem, 2013, 156, 26–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Juaristi E, Antúnez S, Tetrahedron, 1992, 48, 5941–5950. [Google Scholar]
- 22.Johansson E, Lundquist A, Zuo S, Edwards K, BBA – Biomembranes, 2007, 1768, 1518–1525. [DOI] [PubMed] [Google Scholar]
- 23.Zana R, J. Phys. Chem, 1999, 103, 9117–9125. [Google Scholar]
- 24.Meguro K, Takasawa Y, Kawahashi N, Tabata Y, Ueno M, J. Colloid Interface Sci, 1981, 83, 50–56. [Google Scholar]
- 25.Kunieda H, Shinoda K, J. Phys. Chem, 1976, 80, 2468–2470. [Google Scholar]
- 26.Li Z, Hillmyer MA, Lodge TP, Langmuir, 2006, 22, 9409–9417. [DOI] [PubMed] [Google Scholar]
- 27.Torchilin VP, J. Control. Release, 2001, 73, 137–172. [DOI] [PubMed] [Google Scholar]
- 28.Torchilin VP, Pharm. Res, 2007, 1, 1–16. [DOI] [PubMed] [Google Scholar]
- 29.Chen H, Kim S, He W, Wang H, Low PS, Park K, Chang J-X, Langmuir, 2008, 24, 5213–5217. [DOI] [PubMed] [Google Scholar]
- 30.Moughton AO, Hillmyer MA, Lodge TP, Macromolecules, 2012, 45, 2–19. [Google Scholar]
- 31.Akiba I, Terada N, Hashida S, Sakurai K, Langmuir, 2010, 26, 7544–7551. [DOI] [PubMed] [Google Scholar]
- 32.Aliabadi HM, Elhasi S, Mahmud A, Gulamhusein R, Mahdipoor P, Lavasanifar A, Int. J. Pharm, 2007, 329, 158–165. [DOI] [PubMed] [Google Scholar]
- 33.Vakil R, Kwon GS, Langmuir, 2006, 22, 9723–9729. [DOI] [PubMed] [Google Scholar]
- 34.Li L, Tan YB, J. Colloid Interf. Sci, 2008, 317, 326–331. [DOI] [PubMed] [Google Scholar]
- 35.Gao Z, Lukyanov AN, Singhal A, Torchilin VP, Nano Lett, 2002, 2, 979–982. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.