

Abstract
This paper presents a microfluidic chip capable of isolating thermally sensitive protein-binding aptamer candidates. The chip makes use of bead-immobilized target molecules and DNA (deoxyribonucleic acid) sequences to enable a simplified chip design, in which affinity selection and PCR (polymerase chain reaction) amplification of target-binding sequences occur in temperature-controlled microchambers. Using pressure-driven flow, buffer containing single-stranded DNA molecules with randomized sequences is cycled through a series of affinity selection and PCR amplification steps on microbeads. Successive introduction of the sample to each chamber effects a process of competition whereby DNA strands with weak binding strength to target molecules are rejected in favor of strongly binding sequences. Using bead-based PCR, the amplification step was miniaturized and integrated with affinity selection, resulting in significant reductions in process time and reagent use. As a demonstration, temperature-dependent selection and amplification of single-stranded oligonucleotides that bind to human Immuno-globulin E (IgE) was performed in 4 h, a 20-fold reduction in process time as compared to conventional methods that would require approximately a week. Fluorescent binding assays then demonstrated that the desired temperature specificity was imparted to the aptamer candidates within just one round of selection, and within two rounds the aptamer candidates exhibited enhanced affinity toward IgE.
Keywords: Aptamers, Microfluidics, Proteins, SELEX, Temperature dependence
1. Introduction
Aptamers are single-stranded oligonucleotide or peptide molecules that are capable of expressing binding affinity for target molecules, in particular proteins. With a wide range of applications from sensing (Cho et al. 2009) to therapeutics (Bunka et al. 2010), the use of aptamers has experienced explosive growth in recent years. Protein-binding oligonucleotides are similar in function to antibodies, and as a result they are readily applicable in a wide range of fields (Song et al. 2012). Unlike antibodies, however, these molecules are synthetic, developed in vitro, and are more resistant to biodegradation. Once an aptamer is isolated, it can be consistently and rapidly reproduced with a wide variety of chemical modifications tailored to the desired application. The primary hindrance to the more widespread use of oligonucleotides, or oligomers, is the resource- and time-intensive nature of the in vitro protocol for discovering the target-binding sequences which are then known as aptamers.
That aptamer isolation protocol primarily consists of partitioning target-binding strands from unwanted nonbinding oligomers, followed by their chemical amplification and repetition of this process several (10–15) times (Fig. 1). This typically requires weeks or months of dedicated work by trained scientists, as well as significant material cost for the reagents required. Conventional partitioning methods, such as column- and nitrocellulose fiber-based separation, have limited separation efficiency, contributing to the time necessary to complete the process. In addition, the repeated amplification steps coupled with the necessary pipetting and analysis make the process expensive and prohibitively time-consuming. A method for rapid isolation of aptamers would greatly benefit fields such as aptamer-based therapeutics and sensing, by allowing scientists to more quickly and easily generate binding sequences against potential targets (Stoltenburg et al. 2007).
Fig. 1.
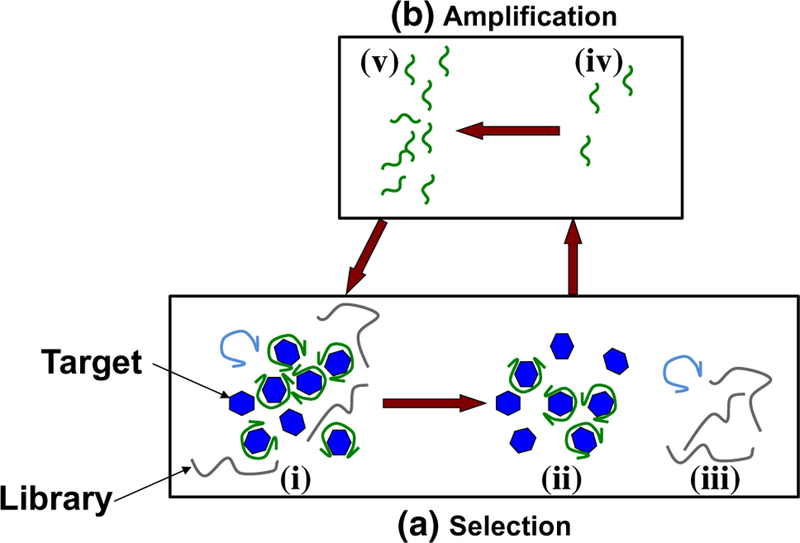
Aptamer isolation process. a A library of approximately 1014–1015 nucleic acid oligomers with randomized sequences is introduced to target molecules (i) during affinity selection. Aptamer candidates strongly bound to targets (ii) are then separated from unwanted non-binding and weakly bound nucleic acids (iii). b Aptamer candidates (iv) are then chemically amplified (v), and the process is repeated. In subsequent rounds of SELEX, these chemically amplified candidates become the new library, and the relatively weakly binding constituents are then removed during selection
Recent work has demonstrated that aptamers can be rapidly isolated by using microfluidic technology to partition desired oligomer sequences from randomized libraries (Huang et al. 2010, 2012; Hybarger et al. 2006). Microfluidic chips offer much greater surface area to volume ratios than conventional devices, which benefits surface-based reactions such as the selection of oligomers binding to surface-bound target molecules. As a result, microfluidic implementations of selection can achieve orders-of-magnitude reductions in processing time. For example, using a continuous flow of target-conjugated magnetic microbeads in a microchannel, protein-binding DNA (deoxyribonucleic acid) sequences with nanomolar dissociation constants (KD) have been developed with only a single round of selection and amplification (Lou et al. 2009). This technology has not only sped up the isolation of target-binding sequences, but allowed the development of binders with lower dissociation constants (Ahmad et al. 2011) and more customized binding traits (Oh et al. 2011). Similarly, it has been demonstrated that capillary electrophoresis (CE) can be applied to separation of target-binding oligomers to generate “smart aptamers” which have specific values of KD and other thermodynamic parameters (Drabovich et al. 2005, 2006). While these and other advances are promising, much of the microfluidic SELEX work has focused on the selection step of the aptamer isolation process (Ahn et al. 2011, 2012; Kim et al. 2011; Park et al. 2009). Despite a significant field of literature that has demonstrated integration of amplification with a variety of other functions (Zhang and Xing 2007), there are fewer examples of integrated aptamer isolation microchips with both selection and amplifica-tion stages (Hung et al. 2014; Weng et al. 2013). Some integrated prototypes suffer from a lack of miniaturization (Hybarger et al. 2006) or risk cross-contamination by providing inadequate partitioning between reaction products and reactants (Huang et al. 2010).
We have developed a fully integrated microchip for isolation of aptamer candidates with temperature-dependent target-binding characteristics. The microchip makes use of a bead-based protocol for both affinity selection and PCR amplification, permitting precise control of buffer and temperature conditions. Target-coated microbeads provide a means of surface-based affinity selection in a microchamber, and primer-coated microbeads act as a surface for solid-phase capture and amplification of candidate binding sequences. The coupling of target molecules and DNA to microbeads has also allowed the use of straightforward pressure-driven flow for reagent introduction, washing, and elution steps, greatly simplifying the device design. This has in turn simplified device operation, enabling an approach in which reagent use and error-prone pipetting steps are minimized and overall process time is reduced from several hours or days to just 4 h. Through integration of affinity selection and PCR amplification, our device is capable of performing multiple rounds of temperature-sensitive enrichment in a rapid manner (2 h per round) and with minimal user intervention. Within a single round, the desired temperature-dependent characteristics of target binding were observed, and after two rounds of closed-loop aptamer isolation, the binding affinity to the target molecule (at 37 °C) was found to have greatly increased (KD = 83.9 nM). The microfluidic device was thus shown to allow highly effective and efficient isolation of aptamer candidates, with equilibrium binding strength that varies with desired temperature dependence.
2. Materials and methods
2.1. Integrated temperature‑controlled bead‑based microfluidic selection
The process of aptamer isolation consists of repeated rounds of affinity selection and PCR amplification of Microfluid Nanofluid (2015) 19:795–804 target-binding nucleic acid oligomers (Fig. 1). In affinity selection, a randomized library of single-stranded DNA oligomers is exposed to the target molecule (Fig. 1i), and desired binding strands (Fig. 1ii) are partitioned from undesired nonbinders (Fig. 1iii). Strongly binding molecules are then isolated (Fig. 1iv) and duplicated (Fig. 1v), and the process is repeated. In this case, target protein molecules are immobilized on polymeric microbeads via covalent attachment; the microbeads are packed in a microchamber whose temperature is maintained via on-chip temperature control (below). Affinity selection is then performed by flowing solutions of the oligomer library and buffer through the bead-packed chamber at a desired temperature set point. Solutions of the oligomer library are exposed to the beads, allowed to incubate, and then removed via washing. Nonbinding and weakly binding oligomers are then removed by washing with pure buffer. Target-binding oli-gomers are released from the target at a different, binding-disrupting temperature set point for amplification. This approach allows complete control over ambient conditions for the affinity selection, including pH, buffer conditions, temperature, and wash flowrate, thereby affording a high degree of control over binding affinity of the evolved aptamer candidates. In addition, the immobilization of the target molecule simplifies intermediate steps in the protocol, such as changing buffers between selection and amplification. This offers simplicity of chip design and facilitates the integration of affinity selection in an aptamer isolation microchip.
2.2. Integrated bead‑based polymerase chain reaction
Target-binding oligomers obtained by affinity selection are then chemically amplified by polymerase chain reaction (PCR), using a bead-based protocol described in detail elsewhere (Hilton et al. 2012). Briefly, polymeric microbe-ads are functionalized with reverse primers and held in a second chamber of the microchip. Target-binding ssDNA oligomers from affinity selection are transferred to and captured by the beads via hybridization with reverse primers. Reagents necessary for PCR can then be introduced into the chamber and the chamber temperature cycled, again via on-chip closed-loop temperature control, to generate an exponentially larger number of duplicate copies of the captured target-binding ssDNA. These duplicate copies of ssDNA (i.e., the unamplified target-binding oligomers) are released from the beads via thermal denaturation for a new round of affinity selection.
This bead-based PCR scheme drastically simplifies the manipulation of ssDNA in an integrated microdevice. That is, the capture, amplification, and retrieval of target-binding ssDNA, each of which must occur in a different buffer in the aptamer selection process, can be efficiently performed on microbead surfaces. Additionally, bead-based PCR enables an efficient process control scheme whereby the generation of product DNA can be detected by fluorescent optical microscopy. Forward primers contain a carboxyflu-orescein moiety at the 5′ terminus, and as PCR incorporates these into bead-bound dsDNA (double-stranded DNA), more fluorescent labels accumulate on the bead surfaces and can be detected by microscopy. This increase in fluo-rescent intensity can be used as an indicator that PCR has produced enough DNA to proceed with SELEX, or alternatively to debug the selection process by monitoring the amount of DNA eluted from the microbeads.
2.3. Design and fabrication
The aptamer isolation microchip consists primarily of two microchambers for affinity selection and PCR amplification, respectively, on a glass substrate (Fig. 2). Between these two chambers is a serpentine microchannel that acts as a passive diffusion-based mixer. Fluid flow in the chip is pressure driven, with no combination of flow path and average flow-rate (between 1 and 10 μL/min) generating a pressure drop greater than 0.5 psi prior to bead introduction. Flowrates for the bead-packed chambers were calculated as in our previous work (Nguyen et al. 2009) and also found to produce negligible pressure drop. The entire chip is coated with Parylene C to prevent microbubble formation and adsorption of nucleic acids or PCR reactants. Previous work in poly-dimethylsiloxane (PDMS)-based PCR amplification has indicated that Parylene C prevents microbubble formation by sealing microcracks formed at the PDMS–glass interface and providing a barrier against the migration of moisture through the bulk PDMS (Shin et al. 2003). The microchambers are designed to maximize thermal responsivity, there-fore decreasing process time, but are placed apart to ensure thermal isolation between process chambers.
Fig. 2.
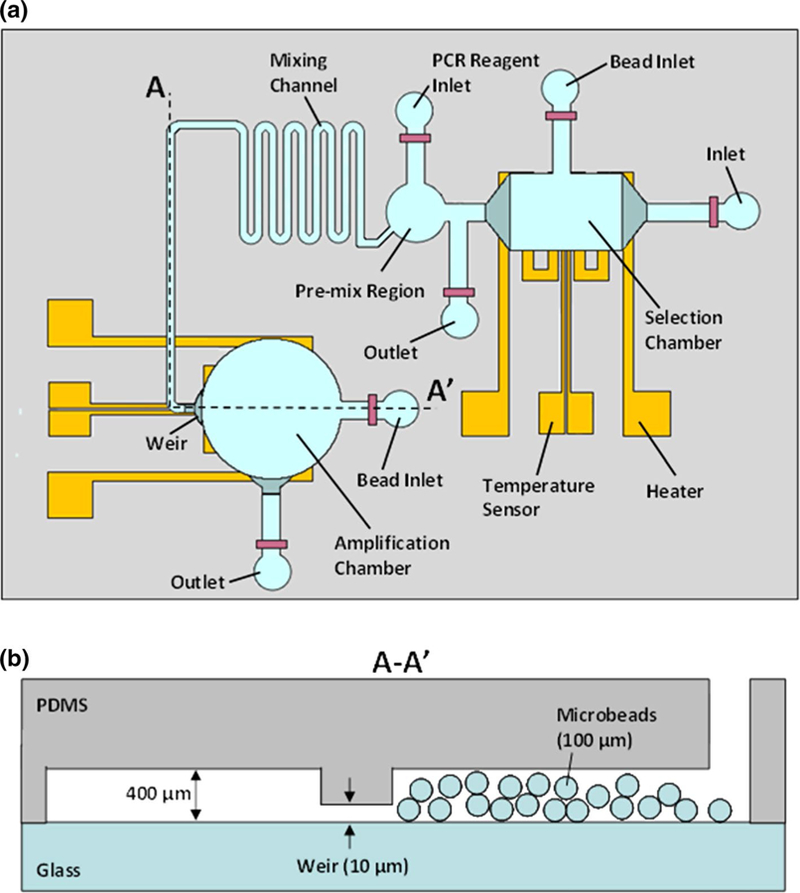
Schematics of the aptamer isolation microchip. a Top view showing microchambers for affinity selection and PCR amplification. These chambers are connected via a serpentine diffusion-based mixing channel and integrated with thin-film resistive heaters and temperature sensors. b Cross-sectional view showing a weir-like restriction in which openings of 10 μm height retain microbeads roughly 100 μm in diameter
The affinity selection chamber (Fig. 2a) is of cuboid shape 4 μL in volume, 400 μm in height with 2.3 mm-long sides. The chamber is placed over patterned chrome/gold thin-film resistive heaters and sensors. The heater is 3.145 cm long by 300 μm wide, and 200 nm thick with a nominal resistance of 20 Ω, while the temperature sensor is 0.55 cm long by 40 μm wide, and 200 nm thick with a nominal resistance of 100 Ω. The chamber temperature is controlled on-chip in closed loop using the heater (whose layout allows generation of a sufficiently uniform temperature field) and the temperature sensor (which is based on a linear temperature–resistance relationship). The chamber is connected through a channel 400 μm in height to its bead inlet for insertion and removal of target-coated microbe-ads approximately 100 μm in diameter. Two weir-like flow restrictions 10 μm in height on opposite sides of the chamber allow fluid to flow while retaining the microbeads.
The amplification chamber (Fig. 2a) is of cylindrical shape 4 μL in volume, 400 μm in height, and 3600 μm in diameter. The associated bead inlet, connected to the chamber via a channel 400 μm in height, allows insertion of polymeric microbeads approximately 100 μm in diameter that are then retained by two weir-like flow restrictions (again 10 μm high). Target-binding oligomers and PCR reagents combine in the premix region, become mixed by passive diffusion in the mixing channel, and then enter the amplification chamber for PCR amplification. As with the selection chamber, chrome/gold resistors beneath the chamber act as an integrated heater and temperature sensor for on-chip, closed-loop temperature control. The heater is 4.635 cm long by 300 μm wide, 200 nm thick with a nominal resistance of 60 Ω. The temperature sensor is 0.8 cm long by 40 μm wide, 200 nm thick with a nominal resistance of 120 Ω.
The locations of the microchambers with respect to one another were chosen after multiple experimental iterations, in which conflicting design requirements were balanced. Closer placement of the chambers would create a large degree of thermal cross-talk, complicating operation of the chip as a closed-loop SELEX device. However, as discussed above, flow paths must be limited so as not to introduce large hydrodynamic pressures during operation. In addition to mixing PCR reactants, the serpentine mixer also behaves as a thermal shunt providing an outlet for excess heat generated by the high (95 °C) temperatures used during PCR. When backfilled with water prior to PCR amplification, the mixer channel prevents the loss of amplification reactants by evaporation.
The overall microchip design consists of PDMS micro-channels bonded to a glass substrate having electrical components, with fluid flow and electrical patterns generated using contact lithography. Positive lithography using S1800 series (Dow Chemical, Newark, DE) photoresist and wet etching defines patterns for resistors and contact pads on a gold-coated glass substrate. These are then passivated with a 1 μm coating of SiO2 generated using plasma-enhanced chemical vapor deposition (Oxford Instruments, Concord, MA), with 500 μm thick pieces of silicon used as shadow masks to define openings for electrical contacts. Negative lithography using SU-8 2000 (Microchem, Newton, MA) photoresist on silicon wafers defines molds for microchannels. PDMS prepolymer (Robert McKeown, Somerville, NJ) is then mixed at a 10:1 ratio of base to curing agent, poured over the molds, and heated at 75 °C for 25 min to generate PDMS microchannels and chambers. The PDMS is then bonded to the glass substrate following 10 min of exposure to UV-generated ozone (Uvocs, Lansdale, PA) and heated to 75 °C for 30 min to complete the bonding. Finally, the entire chip is coated with Parylene C via chemical vapor deposition (Specialty Coating Systems, Indianapolis, IN), with scotch tape used as a shadow mask to define openings for electrical contacts. A fabricated and packaged microchip is shown in Fig. 3.
Fig. 3.

Photograph of a fabricated and packaged aptamer isolation microchip. The microfluidic chambers and channels have been filled with blue dye to aid visualization
Target molecules used for this aptamer isolation protocol were human immunoglobulin E (IgE), an antibody associated with allergic response. The randomized oli-gomer library is an 80-base single-stranded DNA molecule consisting of a 40 base central randomized region flanked by two 20 base priming regions. The buffer used for selection was phosphate-buffered saline modified (PBSM) with 40 mM MgCl2. The library/buffer system was designed based on previous work targeting IgE for development of a temperature-sensitive aptamer (Wiegand et al. 1996). Buffer conditions are an important design consideration: The ionic content is partially responsible for target-sequence binding, so to maintain consistent binding strength the buffer conditions used in applications of the evolved binding sequence must in general replicate those used during the selection process.
2.4. Materials
The following materials were used for the microchip aptamer isolation procedure. Human IgE was purchased from Athens Research, Athens, GA. All DNA was obtained in lyophilized form from Integrated DNA Technologies, Coralville, IA. The purchased library sequence is as follows: 5′-C TAC CTA CGA TCT GAC TAG CNN NNN NNN NNN NNN NNN NNN NNN NNN NNN NNN NNN NNN NNG CTT ACT CTC ATG TAG TTC C-3′. The forward primers used were 5′-C TAC CTA CGA TCT GAC TAG C-3′, with or without a 5′ carboxyfluorescein modification. The reverse primers used were 5′-G GAA CTA CAT GAG AGT AAG C-3′, with a 5′ dual-biotin attachment. In each case, an inert spacer molecule is incorporated between the sequence and the terminal modification. Microbeads used for sequence selection were NHS-activated Sepha-rose beads obtained from Bio-Rad laboratories. Microbe-ads used for bead-based PCR were Ultralink streptavidin-coated beads obtained from Thermo Scientific Pierce Protein Research Products, Rockford, IL. Materials used in microfabrication included S1800 photoresist (Dow Chemical, Midland, MI), SU-8 2000 (Microchem, Newton, MA), PDMS prepolymer (Dow Chemical), and Dix C Parylene prepolymer (Uniglobe Kisko, White Plains, NY). PCR rea-gents included GoTaq Flexi PCR Mix (Promega, Madison, WI), Taq enzyme (Promega), and deoxynucleotide triphosphates (dNTPs, Promega).
The reactants used for on-chip amplification of candidate sequences consisted of the following: 5 × PCR Buffer (2 μL), 25 mM MgCl2 (0.6 μL), 10 mM dNTPs (0.4 μL), 50 μg/mL BSA (0.4 μL), microbeads (0.5 μL), water (5.6 μL), 25 μM forward primer (0.4 μL), and enzyme (0.1 μL). The reactants were degassed at −0.4 psig for 30 min in a darkened container prior to adding enzyme and proceeding with on-chip amplification.
2.5. Experimental protocol
Prior to experimentation, the resistive temperature sensors were calibrated in an environmental chamber with a resistance temperature detector probe. Baseline temperature versus resistance readings were also taken prior to every experiment. NHS-activated microbeads were coated with IgE by washing them with 1 μM IgE 3× in a filtration column followed by incubation for 5 h. Unbound ester molecules were then passivated by incubating the beads for 1 h in Tris buffer. Following another wash in pure PBS, the beads were suspended and stored in PBS. Streptavi-din-coated microbeads were coated with reverse primers by incubation with 1 μM dual-biotin-labeled primers for 30 min immediately prior to DNA capture.
The temperature of the selection chamber was set to 37 °C using closed-loop feedback programming in Lab-VIEW and maintained during library incubation and removal of nonbinding oligomers. Having introduced the target-coated beads to the selection chamber and washed them with PBSM, three 30 μL samples of 10 μM oligomer library DNA (in PBSM) were flowed through the chamber at 5 μL/min, and each aliquot was retained following washing. Ten 30 μL samples of pure PBSM were then flowed through the chamber at 5 μL/min, and each was retained afterward for analysis. At this point, the amplification chamber was filled with reverse-primer-coated microbeads. The selection chamber temperature was then set to 57 °C, and four 30 μL samples of PBSM were introduced at 1 μL/min. These were subsequently directed to wash through the amplification chamber, over the reverse-primer-coated microbeads. Following each 30 μL wash, the aliquot was retrieved and stored, and PCR reagents were introduced to the amplification chamber, and the chamber temperature cycled to perform PCR amplification of the candidate aptamer sequences directly onto the microbeads. The microbeads were subsequently washed with PBS buffer and examined using fluorescent microscopy to visualize surface DNA. Target ssDNA was then eluted by raising the chamber temperature to 95 °C and flushing with pure PBSM.
Fluorescent binding analysis was conducted using a selection chamber on a fresh microchip. Following device calibration, the chamber was filled with IgE-coated micro-beads. Enriched library ssDNA was normalized to 1 μM concentration by measuring the concentration with UV/VIS spectroscopy and adding PBSM buffer. This sample was introduced to the chamber maintained at 37 °C, and the fluorescent intensity was captured via a digital camera attached to the fluorescent microscope. The experiment was repeated with random library also labeled with an identical fluorophore, purchased from IDT. Temperature dependence of binding was measured in similar fashion, by incubating a 1 μM concentration of enriched library with the micro-beads and then continuously flowing PBSM at 2 μL/min while varying the chamber temperature.
Cloning of enriched libraries was performed using the TOPO Cloning protocol, with plasmid DNA purified using the PureLink Quick protocol. Pools of enriched DNA were amplified using conventional PCR and unlabeled primer sequences for 25 cycles. Sequences were then cloned into the TOPO vector, and DNA sequences were harvested from E.coli and purified prior to sequencing. Sequencing was performed by the DNA Sequencing Lab at the Columbia University Protein Core Facility.
3. Results and discussion
3.1. Characterization of thermal and fluidic control
We first calibrated the integrated temperature sensors and tested the functionality of the mixer and amplification chamber. The chip was placed in an environmental chamber, and its temperature varied while the resistance of the temperature sensor was measured. From the linear relationship between temperature and resistance, the temperature coefficient of resistance was measured for both the selection chamber (Fig. 4a) and amplification chamber (Fig. 4b), enabling future resistance measurements to be correlated to on-chip temperature. The on-chip mixer was designed to fully mix reagents with library DNA. As shown in Fig. 5, the mixer fully mixes a clear stream of buffer with a stream of green-dyed PCR rea-gents. The amplification chamber was tested for its ability to properly amplify DNA by performing bead-based PCR of a sample of library DNA. We previously characterized bead-based microfluidic PCR in an integrated device (Hilton et al. 2012). Sample loss and bubble generation were minimized by coating the chip with Parylene C and degassing reagents prior to temperature cycling. A LabVIEW digital PID controller was programmed to achieve and maintain set point temperatures by measuring the resistance of the on-chip resistive temperature sensor and applying voltage to the resistive heater. Fluorescent and UV/VIS microscopy confirmed the generation of product DNA using on-chip amplification.
Fig. 4.

Characterization of the microchip temperature sensors. The linear relationship between temperature and resistor resistance for the a selection chamber and b amplification chamber resistors allows determination of a temperature coefficient of resistance, in this case 1.554 × 103 °C−1 and 1.613 × 103 °C−1, respectively
Fig. 5.
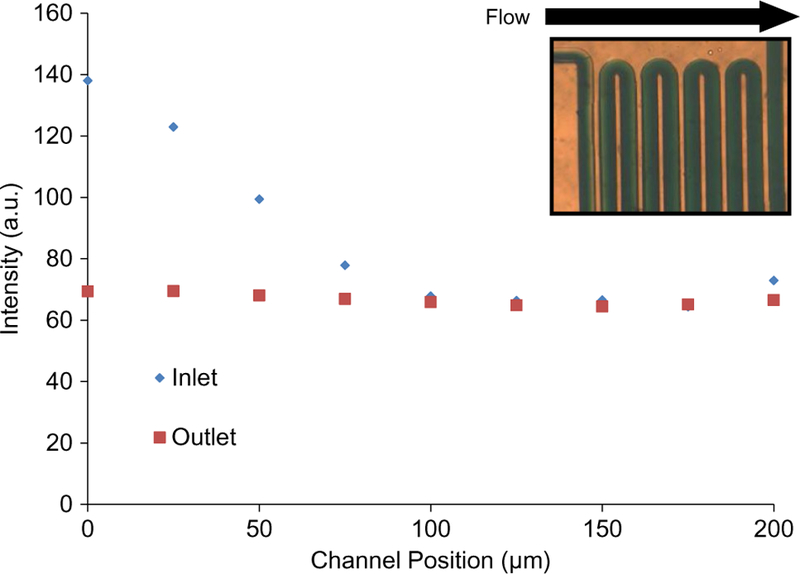
Characterization of the passive mixer. The mixer (inset photo) fully mixes a stream of buffer and dye at a flowrate of 5 μL/min. An analysis of pixel intensity at the inlet and outlet indicates that the unmixed inlet flow has become uniform and mixed at the outlet
3.2. Characterization of microfluidic isolation of aptamer candidates
The operation of the microfluidic aptamer isolation chip consists of two functional steps: affinity selection and PCR amplification. The selection protocol serves to partition non-binding and weakly bound nucleic acids in the randomized library from the few oligomers which express temperature-dependent binding affinity to the target molecule. To illustrate the effectiveness of this microfluidic approach, two rounds of temperature-controlled microfluidic aptamer isolation were performed using the microchip to generate a pool of enriched oligomers which express temperature-dependent binding affinity toward a target protein, IgE.
Initially, the entire DNA oligomer library was incubated with the target-coated microbeads at the binding temperature, 37 °C. Selection was then effected by maintaining the microchamber temperature and washing the microbeads with aliquots of pure buffer solution to elute undesired oli-gomers. As samples of buffer were washed over the beads and recovered from the chip, each sample retained the unwanted nucleic acids. Following the selection step during the first round of aptamer isolation, samples of each buffer wash were amplified using PCR and visualized using slab-gel electrophoresis (Fig. 6). As indicated by the intensity of the intercalating fluorescent dye, the concentration of oli-gomers in each sample (identically amplified) decreases as the beads continue to be washed. This reflects the increasing average binding strength of the aptamer candidates that remain on the target-coated microbeads. The slight increase in intensity from lane 4 to lane 5 is an artifact of the gel image and can be neglected; these results are representative of multiple experiments performed under identical conditions in which the device was operated under consistent experimental conditions (flowrate, temperature, buffer, etc.).
Fig. 6.
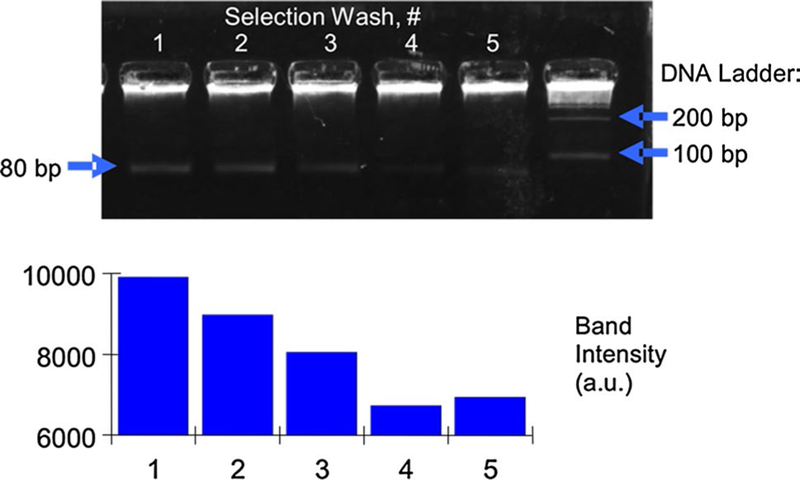
Gel electrophoresis of buffer samples containing weakly bound oligomers washed away from target molecules during the selection step of the first round of microfluidic aptamer isolation. The decreasing fluorescent intensity with continuing washes indicates that the affinity selection process is removing weakly bound oligomers, leaving strongly bound oligomers to eventually be eluted, captured, and amplified
Following removal of unwanted oligomers during selection, desired strands were eluted by raising the micro-chamber temperature to 57 °C and again washing with pure buffer. This aliquot of buffer, containing oligomers expressing temperature-dependent binding affinity to IgE, was driven to the amplification chamber and incubated with primer-coated microbeads. There, the primers captured the oligomers onto the microbeads where it was amplified via PCR. The elution–capture–amplification step was confirmed with fluorescent-based measurements since the forward (in-solution) primers used during PCR include a fluorescent label. Following amplification, fluorescent bead intensity was monitored via fluorescent microscopy to confirm the generation of labeled dsDNA on the bead surfaces. This elution–capture–amplification procedure was completed three times to ensure that all desired IgE-binding DNA was captured from the selection chamber. As shown in Fig. 7, an analysis of the fluorescent bead intensity following the first round of aptamer isolation shows that oli-gomers from the initial aliquot of buffer were successfully amplified on the bead surfaces, but processing of further aliquots resulted in no detectable DNA. This indicates that not only was amplification successful, but that the elution–capture–amplification process efficiently captured the desired oligomers in the first aliquot of buffer.
Fig. 7.
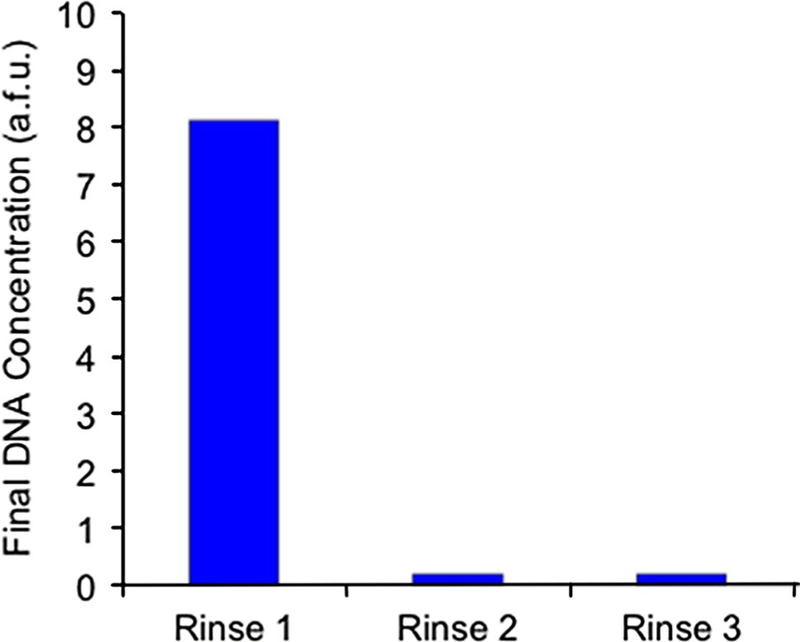
Fluorescent analysis of bead-based PCR amplification of candidate aptamers. Target-coated microbeads with strongly bound oligomers were washed multiple times, and the graph indicates the fluorescent intensity of microbeads following 25 cycles of PCR amplification of the eluent. The relative intensity of beads after each wash indicates that the starting DNA concentration was highest in the first eluted sample
Following the first round of temperature-controlled aptamer isolation, the temperature dependence of the IgE-aptamer candidate binding was analyzed. A microchamber filled with fresh IgE-coated microbeads was exposed to 1 μM of enriched fluorescently labeled oligomers, while being observed under a fluorescent microscope, until the fluorescent signal saturated. Maintaining the chamber temperature at 37 °C, fresh buffer was flowed through the chamber to ensure that binding equilibrium had been reached. The chamber temperature was then varied while flowing buffer solution, and the fluorescent intensity measured as a function of temperature. As shown in Fig. 8, the concentration of surface-bound DNA varies strongly as a function of temperature, indicating the effectiveness of the temperature-controlled affinity selection.
Fig. 8.
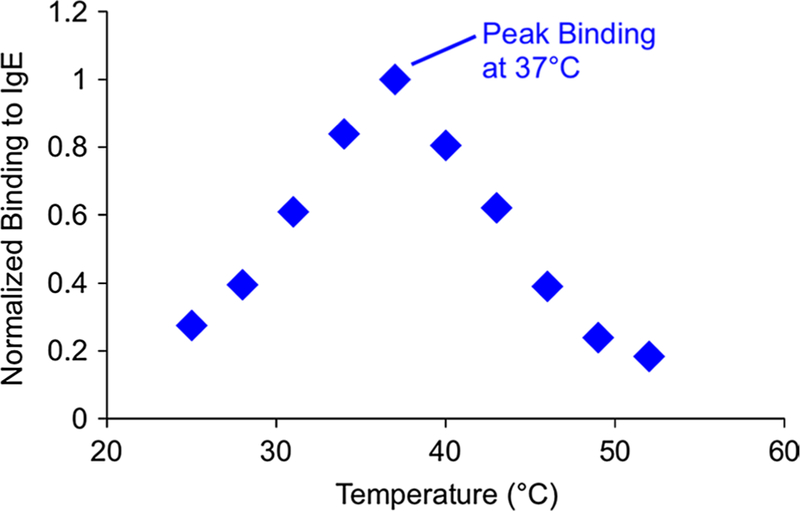
Temperature dependence of IgE-aptamer candidate binding
Following two closed-loop rounds of temperature-specific aptamer isolation, binding affinity between target molecules and candidate sequences was analyzed using a fluorescent assay in which purified, fluorescently labeled aptamer candidates in the concentration range of 6.25–150 nmol/L were incubated with target-coated microbe-ads and maintained at 37 °C (Stoltenburg et al. 2005). The amount of protein-bound ssDNA was determined by meas-uring the fluorescent intensity of the microbeads following incubation and washing with buffer. As shown in Fig. 9, the enriched pool of aptamer candidates exhibited a dissociation constant (KD) of 83.9 nM, indicating that the isolation process generated a sample with strong binding affinity to IgE.
Fig. 9.
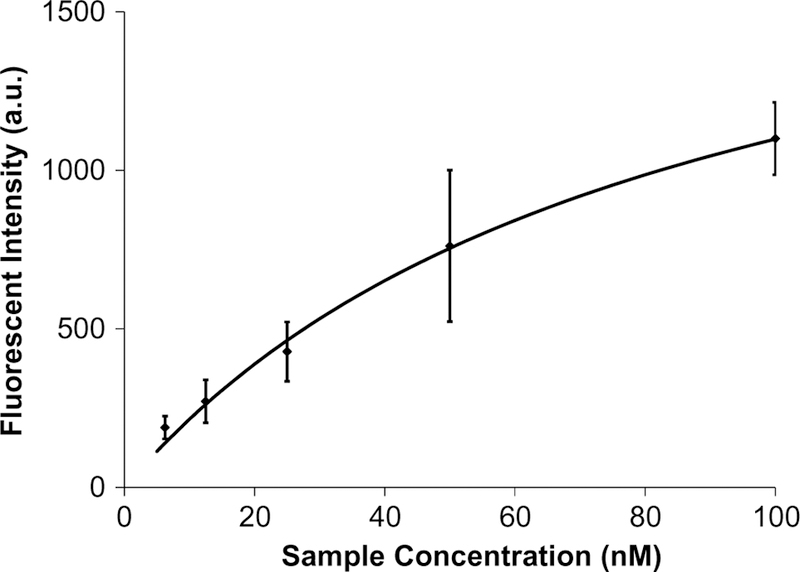
Analysis of IgE-aptamer candidate binding after two rounds of aptamer isolation. IgE-coated microbeads were exposed to fluorescently labeled aptamer candidates, and the fluorescence intensity of the beads was measured. This signal corresponded to the surface concentration of DNA and was used to estimate a KD of 83.9 nM
Following amplification, protein-binding aptamer candidates were eluted as ssDNA into pure buffer solution and collected for further analysis. A 2 µL sample was amplified off-chip using unlabeled primers and cloned using the TOPO vector (Invitrogen). Individual bacterial colonies containing plasmids were harvested, cultured, and then subjected to DNA extraction. Twenty-four clones were chosen at random for sequencing, and a comparison of the sequences indicated that the library had not converged to a binding sequence. However, alignment analysis indicated a 39 base region with increased agreement among each base (Mcwilliam et al. 2009). As the fluorescent analysis concluded that the library showed increased affinity to the targets, it is likely that further selection will result in at least one but possibly several target-binding sequences.
4. Conclusions
Protein-binding oligomers, such as aptamers, have applications in a wide variety of fields, but their use has been hindered by the long development times and significant costs associated with generating binding sequences. Attempts have been made to address these challenges, previously by robotic automation (Cox and Ellington 2001), or more recently by use of microchips to improve selection protocols (Ahn et al. 2012; Lou et al. 2007, 2009; Park et al. 2009), or by integrating the selection and amplification procedures (Huang et al. 2010; Hybarger et al. 2006). This work aimed to integrate the entire process for the isolation of temperature-dependent target-binding sequences on a single chip.
We have constructed a closed-loop microfluidic device and demonstrated that it can be used to perform temperature-controlled isolation of protein-binding aptamer candidates. Our microfluidic approach reduces the sample size to the microliter scale and integrates the required processes on-chip, thus reducing the required reagents and process time by orders of magnitude compared to conventional protocols. The bead-based affinity selection and PCR amplification protocol successfully separate weakly binding nucleic acids from strongly bound candidates, and allows on-chip amplification and collection of target DNA. The chip-based aptamer isolation protocol was shown to enrich a randomized library of DNA for temperature-sensitive binding to IgE. After two rounds of isolation, the DNA library showed greatly enhanced binding but minimal consensus, indicating that further selection may result in multiple target-binding sequences. The on-chip selection and amplification required only 4 hours and approximately 5 microliters of PCR reagents, a significant improvement over conventional aptamer isolation methods.
Acknowledgments
We gratefully acknowledge financial support from the National Science Foundation (Award Nos. CBET-0854030, CBET-1033288, and CBET-1026591), the National Institutes of Health (Award No. 8R21 GM104204–03), the Alternatives Research Development Foundation, and the Chinese Academy of Sciences SAFEA International Innovation Team program.
References
- Ahmad KM, Oh SS, Kim S, McClellen FM, Xiao Y, Soh HT (2011) Probing the limits of aptamer affinity with a microfluidic SELEX platform. PLoS One doi: 10.1371/journal.pone.0027051 [DOI] [PMC free article] [PubMed]
- Ahn JY, Jo M, Dua P, Lee DK, Kim S (2011) A sol–gel-based micro-fluidics system enhances the efficiency of RNA aptamer selection. Oligonucleotides 21:93–100. doi: 10.1089/oli.2010.0263 [DOI] [PubMed] [Google Scholar]
- Ahn J-Y et al. (2012) Sol-gel derived nanoporous compositions for entrapping small molecules and their outlook toward aptamer screening. Anal Chem 84:2647–2653. doi: 10.1021/ac202559w [DOI] [PubMed] [Google Scholar]
- Bunka DHJ, Platonova O, Stockley PG (2010) Development of aptamer therapeutics. Curr Opin Pharmacol 10:557–562. doi: 10.1016/j.coph.2010.06.009 [DOI] [PubMed] [Google Scholar]
- Cho EJ, Lee JW, Ellington AD (2009) Applications of aptamers as sensors. In: Annual review of analytical chemistry, vol 2, annual review of analytical chemistry Annual Reviews, Palo Alto, pp 241–264. doi: 10.1146/annurev.anchem.1.031207.112851 [DOI] [PubMed] [Google Scholar]
- Cox JC, Ellington AD (2001) Automated selection of anti-protein aptamers. Bioorgan Med Chem 9:2525–2531 [DOI] [PubMed] [Google Scholar]
- Drabovich A, Berezovski M, Krylov SN (2005) Selection of smart aptamers by equilibrium capillary electrophoresis of equilibrium mixtures (ECEEM). J Am Chem Soc 127:11224–11225. doi: 10.1021/ja0530016 [DOI] [PubMed] [Google Scholar]
- Drabovich AP, Berezovski M, Okhonin V, Krylov SN (2006) Selection of smart aptamers by methods of kinetic capillary electro-phoresis. Anal Chem 78:3171–3178. doi: 10.1021/ac060144h [DOI] [PubMed] [Google Scholar]
- Hilton J, Nguyen T, Barbu M, Pei R, Stojanovic M, Lin Q (2012) Bead-based polymerase chain reaction on a microchip. Micro-fluid Nanofluid doi: 10.1007/s10404-012-0993-8 [DOI] [PMC free article] [PubMed]
- Huang C-J, Lin H-I, Shiesh S-C, Lee G-B (2010) Integrated micro-fluidic system for rapid screening of CRP aptamers utilizing systematic evolution of ligands by exponential enrichment (SELEX). Biosens Bioelectron 25:1761–1766. doi: 10.1016/j.bios.2009.12.029 [DOI] [PubMed] [Google Scholar]
- Huang C-J, Lin H-I, Shiesh S-C, Lee G-B (2012) An integrated microfluidic system for rapid screening of alpha-fetoprotein-specific aptamers. Biosens Bioelectron 35:50–55. doi: 10.1016/j.bios.2012.02.024 [DOI] [PubMed] [Google Scholar]
- Hung LY, Wang CH, Hsu KF, Chou CY, Lee GB (2014) An on-chip Cell-SELEX process for automatic selection of high-affinity aptamers specific to different histologically classified ovarian cancer cells. Lab Chip 14:4017–4028. doi: 10.1039/c4lc00587b [DOI] [PubMed] [Google Scholar]
- Hybarger G, Bynum J, Williams R, Valdes J, Chambers J (2006) A microfluidic SELEX prototype. Anal Bioanal Chem 384:191–198. doi: 10.1007/s00216-005-0089-3 [DOI] [PubMed] [Google Scholar]
- Kim TK, Lee SW, Ahn JY, Laurell T, Kim SY, Jeong OC (2011) Fabrication of microfluidic platform with optimized fluidic network toward on-chip parallel systematic evolution of ligands by exponential enrichment process. Jpn J Appl Phys doi: 10.1143/jjap.50.06gl05 [DOI]
- Lou X et al. (2009) Micromagnetic selection of aptamers in microfluidic channels. Proc Natl Acad Sci 106:2989–2994. doi: 10.1073/pnas.0813135106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X et al. (2007) Microfluidic screening of aptamer libraries. Paper presented at the μTAS 2007, Paris, France, 11 October 2007 [Google Scholar]
- Mcwilliam H et al. (2009) Web services at the European bioinformatics institute-2009. Nucleic Acids Res doi: 10.1093/nar/gkp302 [DOI] [PMC free article] [PubMed]
- Nguyen T, Pei R, Stojanovic M, Lin Q (2009) An aptamer-based microfluidic device for thermally controlled affinity extraction. Microfluid Nanofluid doi: 10.1007/s10404-008-0322-4 [DOI]
- Oh SS, Ahmad KM, Cho M, Kim S, Xiao Y, Soh HT (2011) improving aptamer selection efficiency through volume dilution, magnetic concentration, and continuous washing in micro-fluidic channels. Anal Chem 83:6883–6889. doi: 10.1021/ac201269f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-M, Ahn J-Y, Jo M, Lee D-K, Lis JT, Craighead HG, Kim S (2009) Selection and elution of aptamers using nanoporous sol-gel arrays with integrated microheaters. Lab Chip 9:1206–1212 [DOI] [PubMed] [Google Scholar]
- Shin YS et al. (2003) PDMS-based micro PCR chip with parylene coating. J Micromech Microeng 13:768–774 [Google Scholar]
- Song K-M, Lee S, Ban C (2012) Aptamers and their biological applications. Sensors 12:612–631. doi: 10.3390/s120100612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoltenburg R, Reinemann C, Strehlitz B (2005) FluMag-SELEX as an advantageous method for DNA aptamer selection. Anal Bio-anal Chem 383:83–91. doi: 10.1007/s00216-005-3388-9 [DOI] [PubMed] [Google Scholar]
- Stoltenburg R, Reinemann C, Strehlitz B (2007) SELEX—A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol Eng 24:381–403. doi: 10.1016/j.bioeng.2007.06.001 [DOI] [PubMed] [Google Scholar]
- Weng C-H, Hsieh IS, Hung L-Y, Lin H-I, Shiesh S-C, Chen Y-L, Lee G-B (2013) An automatic microfluidic system for rapid screening of cancer stem-like cell-specific aptamers. Microfluid Nanofluid 14:753–765. doi: 10.1007/s10404-012-1095-3 [DOI] [Google Scholar]
- Wiegand TW, Williams PB, Dreskin SC, Jouvin MH, Kinet JP, Tas-set D (1996) High-affinity oligonucleotide ligands to human IgE inhibit binding to Fc epsilon receptor I. J Immunol 157:221–230 [PubMed] [Google Scholar]
- Zhang CS, Xing D (2007) Miniaturized PCR chips for nucleic acid amplification and analysis: latest advances and future trends. Nucleic Acids Res 35:4223–4237. doi: 10.1093/nar/gkm389 [DOI] [PMC free article] [PubMed] [Google Scholar]