

Abstract
Background
Cell-based therapies with bone marrow-derived progenitor cells (BMDPC) lead to an improved clinical outcome in animal sepsis models. In the present study we evaluated the ability of granulocyte macrophage-colony stimulating factor (GM-CSF) to mobilize BMDPC in a lipopolysaccharide (LPS)-induced sepsis model and thereby its potential as a novel treatment strategy.
Methods
Male Wistar rats received LPS (25μg/kg/h for 4 days) intravenously and were subsequently treated with GM-CSF 12.5μg/kg (0h,24h,48h,72h). As control groups, rats were infused with sodium chloride or GM-CSF only. Clinical and laboratory parameters, proinflammatory plasma cytokines as well as BMDPC counts were analyzed. Cytokine release by isolated peripheral blood mononuclear cells from rat spleen upon incubation with LPS, GM-CSF and a combination of both were investigated in vitro.
Results
In vivo, rats receiving both LPS and GM-CSF, showed a reduced weight loss and increased mobilization of BMDPC. At the same time, this regime resulted in an increased release of proinflammatory cytokines (IL-6, IL-8) and a significantly increased mortality. In vitro, the combination of LPS and GM-CSF showed a significantly increased IL-6 release upon incubation compared to incubation with LPS or GM-CSF alone.
Conclusions
GM-CSF did not have a beneficial effect on the clinical course in our LPS-induced sepsis model. It synergistically promoted inflammation with LPS and probably thereby impaired survival.
Introduction
Septic shock with multiple organ failure is the leading cause of death in intensive care units and remains a major health problem. Despite extensive research and well-conducted clinical trials, no specific novel treatment strategy exists so far [1,2]. Since altered endothelial function and disruption of the vascular barrier is a critical step in the development of multiple organ failure [3–5], endothelial regeneration might have a beneficial effect on the course of sepsis. A potential treatment strategy could be the use of endothelial progenitor cells (EPC) [6,7], which have been shown to play a role [8–12]. An increased concentration of EPC was found in patients with sepsis and acute lung injury, which seem to be involved in endothelial and pulmonary regeneration [8,12] and correlated inversely to disease severity and mortality [8,12].
While initial research indicated that EPC are differentiated into mature endothelial cells and incorporated into the vessel wall to replace the injured endothelium, current understanding proposes different subpopulations of EPC with distinct functions [9]. The endothelial colony forming cells (ECFC) display the ability to form vessels and become part of the systemic circulation of the host animal [13]. The population of bone marrow-derived progenitor cells (BMDPC, e.g. CD34+/AC133+/KDR+), recently also referred to as proangiogenic hematopoietic cells (PHC) [14], are not incorporated into the vessel wall and lack vasculogenic features, but rather exhibit potent paracrine capacity regulating neovascularization via angiogenesis [15]. The mobilization and recruitment of EPC/BMDPC is mediated by soluble factors such as vascular endothelial growth factor (VEGF), granulocyte macrophage colony-stimulating factor (GM-CSF), erythropoietin (EPO) and angiopoetin- (ANG)-2 [16] and offers a particular therapeutic potential. There are two strategies to manipulate the microenvironment to enhance endothelial repair by increasing the number of progenitor cells in the peripheral blood of patients: 1) exogenous administration of BMDPC in form of an allogenic stem cell transplantation, 2) endogenous stimulation of BMDPC release by mobilizing factors, which minimizes immunological complications usually associated with allogenic stem cell transplantation.
In the present study we investigated the ability of GMCSF to mobilize BMDPC in a lipopolysaccharide (LPS)-induced sepsis model and thereby it’s beneficial effect on the clinical course of sepsis.
Materials and methods
Animal experiments
This study was approved by the Institutional Review Board for the care of animal subjects (University of Heidelberg, Mannheim, Germany & the Regional Council of Karlsruhe, Germany). All animals received humane care in compliance with the “Principles of Laboratory Animal Care” formulated by the National Society for Medical Research and the stipulations of the German Animal Protection Law in its current version. Twenty-four specific pathogen free male Wistar rats housed in standard conditions with food and water ad libitum were anaesthetized by intraperitoneal (IP) injection of ketamine hydrochloride (100 mg/kg) and xylazine (4 mg/kg). Anaesthesia was maintained with intravenous ketamine via an infusion pump (Braun Perfusor Secura ft, B. Braun Melsungen AG, Melsungen, Germany) at an initial rate of 20 mg/kg/h. The level of anaesthesia was assessed by pinching the paw and tail throughout the experiments. The femoral artery and vein were cannulated with a polyethylene catheter (PE-50, neoLab Heidelberg, Germany) for multiple arterial blood collection and continous intravenous infusion of LPS. Both catheters were subcutaneously tunneled, diverted in the neck and flushed with heparine. After 6 days of convalescence the animals were randomized in 4 groups (Sham, GMCSF, LPS and GMCSF+LPS, n = 6/group) and osmotic minipumps (alzet, OSMOTIC PUMP Model 2ML1, ALZET, Osmotic Pumps, Cupertino, Kalifornien, USA) were implanted subcutaneously and connected to the previously implanted venous catheter.
In the LPS and LPS+GMCSF group pumps were loaded with 0.025mg/kg bodyweight/h LPS (LPS, Escherichia coli O55:B5, Sigma-Aldrich Corporation, Saint Louis, Missouri, USA) and 3 U/ml heparine. The venous catheter was flushed using an equally concentrated fluid bolus of LPS/heparine. In the Sham and GMCSF group the pumps were loaded with isotonic saline and 3 U/ml heparine. The venous catheter was flushed using an equally concentrated fluid bolus of saline/heparine. Additionally the animals in the GMCSF and GMCSF+LPS group received subcutaneous injections of 12.5μg/kg bodyweight GM-CSF (PeproTech GmbH, Zytokine für Deutschland, Hamburg) at 0h, 24h, 48h and 72h after pump implantation. The animals were weighed at 0h and 96h after pump implantation. Arterial blood samples were collected at the same time points for blood gas analysis (Cobas b121, Roche Diagnostics GmbH, Wien, Austria), measurement of the serum chemistry panel [including aspartate aminotransferase (ASAT), alanine aminotransferase (ALAT), bilirubine, lipase, creatinine, urea, creatine kinase using the Hitachi 917 (Roche Diagnostics, Wien, Austria)], proinflammatory cytokines and flowcytometry.
Enzyme-linked immune-sorbent assay (ELISA)
The concentrations of interleukin (IL)-6, IL-8 and vascular endothelial growth factor (VEGF) were assessed in serum using enzyme-linked immunosorbent assay kits (R&D Systems, Wiesbaden-Nordenstadt, Germany) in triplicate samples. Enzyme-linked immunosorbent assays were performed according to the manufacturer’s instructions.
Flow cytometry
Peripheral blood mononuclear cells (PBMC) were prepared by gradient centrifugation using Ficoll-Hypaque (Amersham Biosciences, Freiburg, Germany). The expression of cell-surface antigens was determined by two-color immunofluorescence staining using antibodies against CD34 –a marker protein for hematopoietic stem cells- and CD133 –a marker for progenitor cells. CD34+/CD133+-cells have been described and identified as BMDPC in the literature. In brief, one hundred microliters of PBMC (containing 1 x 106 cells) were incubated at 4°C for 30 mins with 10 μL of goat CD34 polyclonal antibodies (Santa Cruz Biotechnology, Heidelberg, Germany) and 10 μL of rabbit CD133 polyclonal antibodies (Santa Cruz Biotechnology, Heidelberg, Germany). The cells were washed three times to remove unbound antibodies and incubated at 4°C for 30 mins with the secondary antibodies swine anti-rabbit IgG- Fluorescein isothiocyanate isomer 1 (FITC) (DakoCytomation, Hamburg, Germany) and donkey anti-goat IgG (H+L)-Phycoerythrin (PE) (R&D Systems, Wiesbaden-Nordenstadt, Germany). The cells were again washed three times to remove unbound antibodies and finally resuspended in 500 μL of BD FACS lysing solution (BD Biosciences, Heidelberg, Germany). Flowcytometry analysis was performed on a FACSCalibur flow cytometer (BD Biosciences), and the data were analyzed using WinMDI 2•8 software (Scripps Research Institute, La Jolla, CA). A minimum of 300,000 events were collected. Flowcytometry analysis of each probe was performed in triplicate. The frequency of BMDPC in peripheral blood was determined by a two-dimensional side-scatter/fluorescence dot-plot analysis of the samples after appropriate gating. BMDPC counts are expressed as percentage of total PBMC in each rat.
In vitro cytokine release assay
The PBMC isolated from rat spleen were prepared by gradient centrifugation using Ficoll-Hypaque (Amersham Biosciences, Freiburg, Germany). Five specific pathogen free male Wistar rats weighting 450-500g served as organ donors. The rat spleen was first reduced to small pieces and then homogenized using a mesh (test sieve ∅100mm, Retsch GmbH, Haan, Germany). The obtained spleen cells were then diluted with phosphate buffer saline 1:4 and pipetted onto the Ficoll solution. Centrifugation was performed at 20°C with 1500 rpm for 30 minutes. After that, the interphase was extracted and purified by 3 successive centrifugation steps (each at 20°C with 1300 rpm for 15, 10 and 10 minutes). The PBMC were then seeded in 24-well plates in growth medium (Promocell, Heidelberg, Germany) and stimulated with GM-CSF (PeproTech GmbH, Zytokine für Deutschland, Hamburg) in different concentrations (20ng/ml, 125 ng/ml and 1.25μg/ml), LPS (0.25μg/ml) (Sigma, Deisenhofen, Germany) and the combination of both. After 24 hours, IL-6 and cytokine-induced neutrophil chemoattractant-1 (CINC-1, also IL-8) were measured in the supernatant using ELISA kits (R&D Systems, Wiesbaden-Nordenstadt, Germany) in triplicate samples. ELISA was performed according to the manufacturer’s instructions.
Statistical methods
All data are presented as mean ± SEM. The statistical comparisons were carried out by means of one-way ANOVA followed by Holm-Sidak’s post-hoc test as required. The survival rates determined according to Kaplan-Meier were compared with the log rank test. The corrections for multiple comparisons were ensured by the Holm-Sidak test. We considered p<0.05 to be statistically significant. Statistical analyses were performed using SigmaPlot 11.0 (Systat Software GmbH, Erkrath, Germany).
Results
Weight and blood gas analysis
Weight changes were assessed and blood gas analysis was performed at the beginning of the experiment (0h) and at the end (96h). The results are displayed in Table 1. After 96 hours, the animals in the LPS (p<0.001) and the LPS+GMCSF (p<0.001) group showed a significant weight loss compared to 0h. The weight reduction was more pronounced (p<0.001) in the LPS group compared to the LPS+GMCSF group. Oxygenation expressed as PaO2/FiO2 ratio (ratio of partial pressure arterial oxygen and fraction of inspired oxygen) was not impaired in any group. On the contrary pH was reduced in the LPS+GMCSF group compared to 0h (p<0.001) and the LPS group (p = 0.006). Ventilation was not compromised, as PaCO2 (partial pressure of carbon dioxide) was not altered. Rather we found a decreased base excess in the LPS (p<0.016) and LPS+GMCSF group p<0.001) compared to 0h. Hemoglobin was reduced in all intervention groups (Sham: p<0.006; LPS: p<0.001; GMCSF: p<0.009; LPS+GMCSF: p<0.001) compared to 0h. There was a also significant difference in hemoglobin between LPS and LPS+GMCSF (p = 0.002).
Table 1. Weight and blood gas analysis.
| Group | |||||
|---|---|---|---|---|---|
| base line | Sham | LPS | GMCSF | LPS+GMCSF | |
| 0h | 96h | 96h | 96h | 96h | |
| weight loss (%) | 0 | -0.21 ± 0.8 | 18 ± 1.2 * | -0.59 ± 1.8 | 6.64 ± 1.7 */** |
| PaO2/FiO2 | 444 ± 10 | 453 ± 4 | 416 ± 2.5 | 452 ± 2 | 421 ± 12 |
| pH | 7.49 ± 0 | 7.52 ± 0 | 7.47 ± 0 | 7.51 ± 0 | 7.41 ± 0 */** |
| PaCO2 (mmHg) | 37 ± 0.7 | 34.6 ± 0.4 | 34.6 ± 0.6 | 34.7 ± 0.6 | 33.7 ± 1.2 |
| BE | 4.1 ± 0.3 | 4.38 ± 0.5 | 1.25 ± 0.6 * | 4.68 ± 0.8 | -1.56 ± 1.2 * |
| Hb (g/dl) | 14.7 ± 0.2 | 11.9 ± 0.1 * | 9.35 ± 0.2 * | 12.6 ± 0.1 * | 11.7 ± 0.5 */** |
Sham, 96h after continuous saline infusion; GM-CSF, 96h after continuous saline infusion and daily granulocyte macrophage colony-stimulating factor (GM-CSF) injection; LPS, 96h after continuous lipopolysaccharide infusion; LPS+GM-CSF, 96h after continuous LPS infusion and daily GM-CSF injection
PaO2/FiO2, ratio of partial pressure arterial oxygen and fraction of inspired oxygen; PaCO2, partial pressure of carbon dioxide; Hb, hemoglobin; BE, base excess; The results are expressed as mean simple linear regression ± SEM
*significant difference vs. base line (0h)
** significant differences vs. GM-CSF group.
Increase of clinically relevant serum parameters
After 96h we found a statistical significant increase for ASAT (p<0.001), creatinine (p<0.001), urea (p<0.001) and creatine kinase (p<0.001) in the LPS+GMCSF group compared to 0h (Table 2). In the LPS group, we found an increase of ASAT (p<0.001), creatinine (p<0.031) and urea (p<0.002).
Table 2. Clinically relevant serum parameters.
| Group | |||||
|---|---|---|---|---|---|
| base line | Sham | LPS | GMCSF | LPS+GMCSF | |
| 0h | 96h | 96h | 96h | 96h | |
| ALAT [U/L] | 55.3 ± 3.8 | 44 ± 6.9 | 62 ± 16.3 | 56.5 ± 9 | 74.5 ± 19.6 |
| ASAT [U/L] | 155 ± 24.8 | 86.2 ± 11.4 | 446 ± 113 * | 135 ± 33.4 | 492 ± 72.5 * |
| Creatinine [mg/dl] | 0.26 ± 0.0 | 0.24 ± 0.0 | 0.40 ± 0.1 * | 0.27 ± 0.0 | 0.42 ± 0.1 * |
| CK [U/L] | 173 ± 37.2 | 425 ± 87 | 447 ± 153 | 114 ± 21.7 | 1708 ± 909 * |
| Lipase [U/L] | 7.16 ± 0.23 | 7.24 ± 0.6 | 6.85 ± 0.6 | 6.74 ± 0.9 | 7.14 ± 1.0 |
| Bilirubine [mg/dl] | 0.07 ± 0.0 | 0.04 ± 0.0 | 0.14 ± 0.0 | 0.00 ± 0.0 | 0.24 ± 0.2 |
| Urea [mg/dl] | 44.9 ± 1.4 | 37.8 ± 1.5 | 122 ± 18.5 * | 46.15 ± 2.1 | 127 ± 42.4 * |
Sham, 96h after continuous saline infusion; GM-CSF, 96h after continuous saline infusion and daily granulocyte macrophage colony-stimulating factor (GM-CSF) injection; LPS, 96h after continuous lipopolysaccharide infusion; LPS+GM-CSF, 96h after continuous LPS infusion and daily GM-CSF injection
ASAT, aspartate aminotransferase; ALAT, Alanine aminotransferase; The results are expressed as mean simple linear regression ± SEM
* significant difference vs. base line (0h).
Upregulation of serum concentrations of proinflammatory cytokines
We used ELISA to detect serum concentrations of IL-6, IL-8 and VEGF in the serum of rats in all the groups after 96 hours (Table 3). For IL-6, we found significantly increased concentrations in the LPS (p<0.05) and LPS+GM-CSF (p<0.05) group compared to the base line. In addition, the IL-6 concentration in the LPS+GM-CSF group was significantly increased compared to the concentrations in the LPS group (p<0.05). For IL-8, we found similar results (Table 3). Regarding VEGF, we found significantly increased concentrations in all groups compared to the base line (p<0.05).
Table 3. Serum concentrations of proinflammatory cytokines.
| Group | |||||
|---|---|---|---|---|---|
| base line | Sham | LPS | GMCSF | LPS+GMCSF | |
| 0h | 96h | 96h | 96h | 96h | |
| IL-6 (pg/ml) | 17.2 ± 9.5 | 28.5 ± 9.2 | 617 ± 334 * | 54.2 ± 21.4 | 1158 ± 521 */** |
| IL-8 (pg/ml) | 21.8 ± 13.8 | 39.6 ± 11.9 | 554 ± 149 * | 0.0 ± 0.0 | 900 ± 420 */** |
| VEGF (pg/ml) | 5.20 ± 3.3 | 110 ± 36 * | 104 ± 40 * | 130 ± 57 * | 101 ± 31 * |
Sham, 96h after continuous saline infusion; GM-CSF, 96h after continuous saline infusion and daily granulocyte macrophage colony-stimulating factor (GM-CSF) injection; LPS, 96h after continuous lipopolysaccharide infusion; LPS+GM-CSF, 96h after continuous LPS infusion and daily GM-CSF injection
IL-6, interleukin 6; IL-8, interleukin 8; VEGF, Vascular Endothelial Growth Factor. The results are expressed as mean simple linear regression ± SEM
*significant differences vs. base line (0h)
**significant differences vs. LPS group.
Upregulation of bone marrow-derived progenitor cells
We used flowcytometry to detect CD34+/CD133+-BMDPC in the peripheral blood of rats in the sham (n = 2), LPS (n = 3), GM-CSF (n = 3) and LPS+GMCSF group at two time points (0 and 96 h). Our results showed significant differences in the groups (Fig 1). While there was no significant difference in BMDPC counts after 96 hours in the sham group (5.25±0.54 vs. 3.5±1.46, p<0.25), BMDPC counts in the LPS (9.91±1.25 vs. 22.6±1.91, p<0.004), GM-CSF (8.96±3.56 vs. 19.5±4.86, p<0.013) and LPS+GM-CSF (12.4±1.46 vs. 28.7±1.12, p<0.0001) group were significantly upregulated after 96 hours (Fig 1). In addition, BMDPC counts after 96 hours in the LPS+GM-CSF group were also significantly increased compared to BMDPC counts in the LPS (28.7±1.12 vs. 22.6±1,91, p<0.012) and GM-CSF group (28.7±1.12 vs. 19.5±4.86, p<0.016) after 96 hours (Fig 1).
Fig 1. Upregulation of bone marrow-derived progenitor cells (BMDPC).

Flowcytometry analysis of the expression of CD34+/CD133+ cells in the peripheral blood mononuclear cell (PBMC) fraction of rats treated with saline (Sham) (n = 2), lipopolysaccharide (LPS) (n = 3), granulocyte-macrophage colony-stimulating-factor (GM-CSF) (n = 3) or LPS + GM-CSF (n = 6) at two time points (0 and 96 hrs). Significant differences were found between the four groups. The results are expressed as mean simple linear regression ± SEM; p<0.05 was considered to be statistically significant.
Mortality
Although aimed as novel treatment strategy, GM-CSF application resulted in a significantly increased mortality in our sepsis-model after 96h. The survival rate of animals in the LPS+GM-CSF group was 33%, while it was 100% in all other groups (p = 0.001) (Fig 2).
Fig 2. Mortality of the investigated animals.
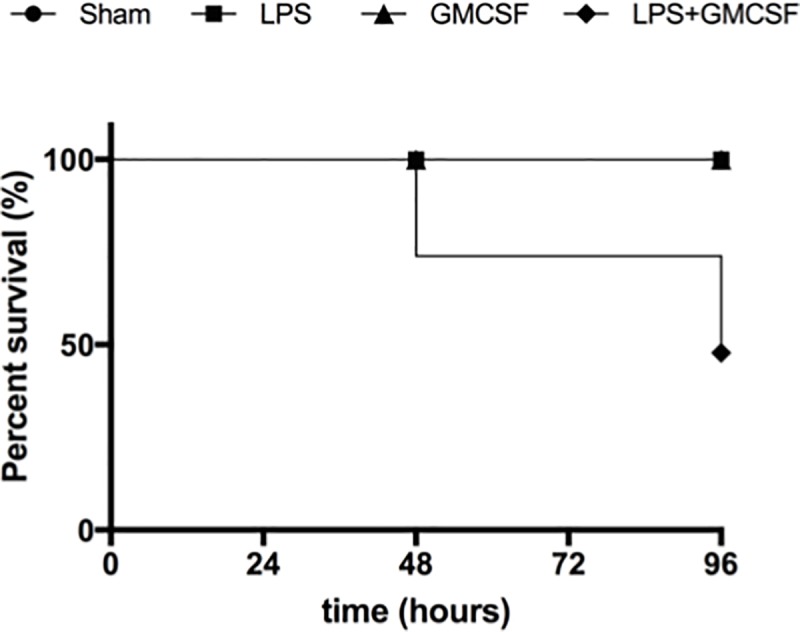
The Kaplan-Meier survival curve compares the mortality of the study groups (LPS+GM-CSF, n = 6; LPS, n, = 6; GM-CSF, n = 6; sham, n = 6). Sham, 96h after continuous saline infusion; GM-CSF, 96h after continuous saline infusion and daily granulocyte macrophage colony-stimulating factor (GM-CSF) injection; LPS, 96h after continuous lipopolysaccharide infusion; LPS+GM-CSF, 96h after continuous LPS infusion and daily GM-CSF injection.
Upregulation of cytokines in vitro
Since GM-CSF application in our in vivo-model resulted in increased inflammation markers and mortality, we set out to assess the effect of GM-CSF and LPS alone and in combination on cytokine release in vitro. We isolated PBMC from rat spleen and performed ELISA with the supernatant after 24 hour incubation. The addition of GM-CSF in three ascending dosages to the medium resulted in a moderate increase of IL-6 concentration in the supernatant, although this increase did not reach statistical significance in any of the tested GM-CSF concentrations (Fig 3). As expected, the addition of LPS caused a marked increase in IL-6 release (58.9±14.2pg/ml vs. 751±60.3pg/ml, p<0.001) compared to medium (Fig 3). The combination of GM-CSF and LPS increased IL-6 release even further and was statistically significant at the highest dose of GM-CSF compared to LPS incubation alone (751±60.3pg/ml vs. 1029±87.9pg/ml, p<0.05) (Fig 3).
Fig 3. Upregulation of cytokines in vitro.

Peripheral blood mononuclear cells (PBMC) were isolated from rat spleen and incubated with medium only, GM-CSF in various concentrations (20ng/ml, 125ng/ml or 1.25μg/ml), LPS (0.25μg/ml) and a combination of LPS (0.25μg/ml) and the different concentrations of GM-CSF. Interleukin-6 (white bars) and Interleukin-8 (grey bars) were measured after 24 hours in the supernatant using ELISA. The results are expressed as mean simple linear regression ± SEM; * p < 0.05 vs. medium, ** p < 0.05 vs. LPS 0.25 μg/ml.
When looking at IL-8, the addition of GM-CSF resulted in a relevant increase of IL-8 for all the GM-CSF concentrations (20ng: 22.2±4.3pg/ml; 125ng: 23±4.0pg/ml; 1,25μg: 47.1±8.0pg/ml), LPS (281±24.2pg/ml) and the three different tested concentrations of LPS+GMCSF (LPS+GMCSF 20ng: 324±20.4pg/ml; LPS+GMCSF 125ng: 321±17.0pg/ml; LPS+GMCSF 1,25μg: 329±20.4pg/ml) compared to the medium (4.34±0.9pg/ml) (all p<0.001). However, there has been no statistical difference between the three different concentrations of LPS+ (Fig 3).
Discussion
In previous studies, we and others, have demonstrated that progenitor cells are increasingly mobilized from the bone marrow into the circulation in patients with sepsis [8] and acute respiratory distress syndrome (ARDS) [12] and that BMDPC transplantation in animal models of sepsis and ARDS lead to an improved clinical course and a reduced mortality [10,11]. To minimize immunological complications of allogenic BMDPC transplantation, endogenous stimulation of BMDPC release by administration of GM-CSF might be an alternative. Our findings demonstrated that administration of GM-CSF results in an increased mobilization of BMDPC in LPS-induced sepsis, but surprisingly impairs survival significantly. The animals in the LPS+GM-CSF group showed a significant weight loss, mildly increased markers of liver and kidney failure and rhabdomyolysis and an increased release of IL-6 and IL-8. In vitro, we demonstrated that incubation of isolated PBMC from rat spleen with LPS and GM-CSF simultaneously results in a significantly increased IL-6 release.
Already 25 years ago, the use of G-CSF or GM-CSF treatment in animal models of bacteraemia was investigated [17,18]. Based on studies showing that recombinant human (rh)G-CSF induce neutrophilia and modulate neutrophil proliferative and neutrophil storage pools in the newborn rat, Cairo et al. investigated the adjuvant effect of rhG-CSF given to group B streptococcus (GBS) septic newborn (less than 36 h) rats treated with and without antibiotic therapy [17]. The animals receiving rhG-CSF and antibiotics had a significantly increased survival rate after 72 hours compared to control animals and animals receiving either rhG-CSF or antibiotics only [17]. Additionally, when rhG-CSF was administered prophylactically (6 h before GBS), a similar significant synergistic effect in survival was demonstrated with G-CSF plus antibiotics versus antibiotics alone [17]. Therefore, it was concluded that either simultaneous or prophylactic pulse administration of rhG-CSF may have a synergistic and protective effect on survival in antibiotic-treated experimental GBS in the neonatal rat.
In contradiction to these results, we have found an increased mortality by GM-CSF application in our sepsis-model. We could demonstrate that animals, which received LPS and GM-CSF, released increasing amounts of pro-inflammatory cytokines and confirmed these results in vitro by incubating PBMC from rat spleen with LPS and GM-CSF, which has also lead to an increased release of proinflammatory cytokines. It has been shown in the past, that GM-CSF does induce proinflammatory cytokine release, especially IL-6 and IL-8, in macrophages [19,20]. Although also stimulating the mobilization of BMDPC, GM-CSF seems to intensify the inflammatory response induced by LPS in a synergistic manner. In this short time of clinical impairment, the increased numbers of BMDPC in the circulation cannot exert their beneficial effects and do not come to fruition.
With regard to ARDS, there are conflicting results on the effect of GM-CSF. Goodman et al. have demonstrated that plasma from atients with ARDS enhanced PBMC viability, while blocking the GM-CSF receptor significantly reduced PBMC viability in ARDS plasma, but not in normal plasma [21]. They suggested that GM-CSF receptor blockage might be a novel therapeutic approach for ARDS treatment [21]. Matuete-Bello et al. confirmed that bronchio-alveolar lavage fluid (BALF) from patients on days 1 and 3 of ARDS showed inhibition of neutrophil apoptosis, but BALF from patients at later stages of ARDS, or from patients at risk for ARDS, did not [22]. Consistently, they found increased concentrations of G-CSF and GM-CSF in BALF at early stages and decreased concentrations at later stages of ARDS [22]. However, their observations showed a significant association of higher concentrations of GM-CSF in BALF of ARDS patients with improved survival [22]. In a different study, BAL concentrations of GM-CSF, G-CSF and IL-8 were increased in ARDS patients compared to healthy individuals, but concentrations of GM-CSF were much lower than those of G-CSF and IL-8 [23]. Levels of G-CSF and IL-8, but not GM-CSF, correlated significantly with each other and with BAL neutrophil counts, and only levels of G-CSF were significantly higher in non-survivors than survivors indicating a role in the pathogenesis of ARDS [23].
In our present study, GM-CSF treatment resulted in an increased mortality of animals. Although BMDPC were increasingly mobilized in response to GM-CSF administration, they were not able to prevent the severe pro-inflammatory effects of the GM-CSF treatment. In line with these observations, levels of G-CSF mRNA correlated with severity of shock, infiltration of polymorphonuclear leukocytes (PMN), pulmonary edema and hypoxia in a rat model of hemorrhagic shock [24,25]. The same group instilled G-CSF into the lungs by intratracheal injection to determine whether increased tissue levels of G-CSF contribute to PMN recruitment and PMN-mediated injury [26]. Animals treated with G-CSF became hypoxic, hypocapnic, and alkalotic and demonstrated increased BAL fluid cellularity compared with control animals [26]. Histological examination of the lungs from G-CSF-treated rats revealed marked edema and increased PMN within the interstitium and alveoli [26]. The authors concluded that these results indicate that the presence of G-CSF alone in the lung can lead to recruitment of PMN, lung injury, and impaired pulmonary function, suggesting that local production of G-CSF may contribute to the development of lung damage and possibly ARDS in the setting of resuscitated hemorrhagic shock. In line with the hypothesis that G-CSF/GM-CSF treatment is associated with the development of lung damage, Verhoef et al. reported a case of a patient who developed ARDS during treatment with rhGM-CSF for severe transfusion-dependent refractory anemia with excess of blasts [27].
Conflicting results in regard to the effect of G-CSF/GM-CSF have also been described for sepsis. In pre-clinical models of sepsis in rats, GM-CSF correlated positively with the survival outcome [28]. A meta-analysis investigating the effects of G-CSF and GM-CSF therapy in non-neutropenic patients with sepsis identified twelve randomized control trials (RCT) with 2.380 patients [29]. There was no significant 28-day mortality, in-hospital mortality or adverse events when G-CSF or GM-CSF were compared with placebo [29]. However, G-CSF or GM-CSF therapy significantly increased the reversal rate from infection. Another meta-analysis investigating the effects of G-CSF and GM-CSF therapy in sepsis included four RCTs with 154 patients [30]. In accordance with the previous study, there was no significant difference in 28-day mortality or rate of adverse events as well as no significant difference in length of stay in hospital or intensive care units (ICU) and sepsis-related organ failure assessment score between the GM-CSF treatment and traditional therapy [30]. Therefore, there is no current evidence supporting the routine use of GM-CSF in septic patients. A recent randomized trial in very preterm small-for-gestational age (SGA) babies investigated whether the administration of GM-CSF could prevent neonatal sepsis and produce differences in survival free of severe disability [31]. The authors found no significant differences in health outcomes or health and social care costs between the trial groups at two years of age [31]. Marginally, more children receiving GM-CSF were reported to have cough and had signs of chronic respiratory disease, though this was not reflected in bronchodilator use or need for hospitalisation for respiratory disease [31]. Therefore, they also concluded that the administration of GM-CSF to very preterm SGA babies is not associated with improved or more adverse outcomes [31].
The limitations of this study are the limited number of experiments and the lack of GM-CSF receptor blockade studies. The results from this study remain therefore associative and loosely mechanistic. To interpret the results of our study within a clinical context and with respect to the design of future in vivo studies, we also have to discuss the limitations of our experimental set-up. Our endotoxemia model induced by LPS is obviously not identical with an infection caused by a pathogen, in which LPS leads to an activation of the innate immune system to eliminate the pathogen. Also, the survival rate of the animals in the LPS group was hundred percent, which already shows, that the endotoxemia did only cause a mild sepsis. We can only speculate that GM-CSF application could lead to different results in a model of severe sepsis. Another limitation of our set-up relates to the application of GM-CSF simultaneously to the induction of sepsis, which does not reflect the clinical setting, where an application is only possible after sepsis has been manifested. A respective prolonged set-up might have revealed different result. Also, we have not tested a treatment strategy based on G-CSF, which could have also led to a different outcome, since previous studies showed a differing impact by GM-CSF and G-CSF [21]. Another important aspect is, that augmented cytokine production might not be the only reason that might affect survival rate in our model. The available data in our study is unfortunately limited and discussing other influencing factors would only be speculation. Taken together, the interpretation of our results with respect to clinical implications must be made carefully. However, we suggest to test alternative growth factors to enhance endogenous BMDPC mobilization.
Conclusions
We have demonstrated that GM-CSF administration not only results in an increased mobilization of BMDPC, but surprisingly impairs survival significantly in our experimental model. There is indication that GM-CSF administration amplifies the LPS-induced release of proinflammatory cytokines, which could also be confirmed in vitro. Fig 4 gives an overview of our findings in comparison to the previous literature [32–41]. Due to our limited results, we would suggest that GM-CSF does not qualify as a novel treatment option to increase endogenous mobilization of BMDPC to improve the clinical course in sepsis. Future studies will have to investigate other mobilizing factors to increase BMDPC numbers endogenously.
Fig 4. New findings of this study in comparison with previous literature.

The new findings of our current study are highlighted and an overview of the findings of previous studies in regard to BMDPC mobilization and GM-CSF application in sepsis is presented. The references to the studies are found in brackets.
Acknowledgments
We thank Jutta Schulte for her skillful technical and Krista Rafat for her editorial assistance.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This study was supported by a research grant from the German Research Foundation (DFG) (G.B.). N.R. was supported by a research scholarship from the Postdoc-Program of the University of Heidelberg. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Ware LB, Matthay MA. The Acute Respiratory Distress Syndrome. N Engl J Med. 2000;342(18):1334–49. 10.1056/NEJM200005043421806 [DOI] [PubMed] [Google Scholar]
- 2.Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med. 2006;27(4):337–49. 10.1055/s-2006-948288 [DOI] [PubMed] [Google Scholar]
- 3.Sakr Y, Dubois M-J, De Backer D, Creteur J, Vincent J-L. Persistent microcirculatory alterations are associated with organ failure and death in patients with septic shock. Crit Care Med. 2004;32(9):1825–31. [DOI] [PubMed] [Google Scholar]
- 4.Peters K, Unger RE, Brunner J, Kirkpatrick CJ. Molecular basis of endothelial dysfunction in sepsis. Cardiovasc Res. 2003. October 15;60(1):49–57. 10.1016/s0008-6363(03)00397-3 [DOI] [PubMed] [Google Scholar]
- 5.Mutunga M, Fulton B, Bullock R, Batchelor A, Gascoigne A, Gillespie JI, et al. Circulating endothelial cells in patients with septic shock. Am J Respir Crit Care Med. 2001;163(1):195–200. 10.1164/ajrccm.163.1.9912036 [DOI] [PubMed] [Google Scholar]
- 6.Lee JW, Fang X, Gupta N, Serikov V, Matthay MA. Allogeneic human mesenchymal stem cells for treatment of E. coli endotoxin-induced acute lung injury in the ex vivo perfused human lung. Proc Natl Acad Sci. 2009;106(38):16357–62. 10.1073/pnas.0907996106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee JW, Krasnodembskaya A, McKenna DH, Song Y, Abbott J, Matthay MA. Therapeutic effects of human mesenchymal stem cells in ex vivo human lungs injured with live bacteria. Am J Respir Crit Care Med. 2013;187(7):751–60. 10.1164/rccm.201206-0990OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rafat N, Hanusch C, Brinkkoetter PT, Schulte J, Brade J, Zijlstra JG, et al. Increased circulating endothelial progenitor cells in septic patients: Correlation with survival. Crit Care Med. 2007. July;35(7):1677–84. 10.1097/01.CCM.0000269034.86817.59 [DOI] [PubMed] [Google Scholar]
- 9.Rafat N, Tönshoff B, Bierhaus A, Beck GC. Endothelial progenitor cells in regeneration after acute lung injury: Do they play a role? Am J Respir Cell Mol Biol. 2013;48(4):399–405. 10.1165/rcmb.2011-0132TR [DOI] [PubMed] [Google Scholar]
- 10.Rafat N, Kowanetz G, Krebs J, Tsagogiorgas C, Betzen C, Ghezel-Ahmadi V, et al. Therapeutic Effects of Bone Marrow-derived Progenitor Cells in Lipopolysaccharide-induced Acute Respiratory Distress Syndrome. J Pulm Respir Med. 2014;4:2. [Google Scholar]
- 11.Rafat N, Dacho C, Kowanetz G, Betzen C, Tönshoff B, Yard B, et al. Bone marrow-derived progenitor cells attenuate inflammation in lipopolysaccharide-induced acute respiratory distress syndrome. BMC Res Notes. 2014. January;7(1):613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burnham EL, Taylor WR, Quyyumi AA, Rojas M, Brigham KL, Moss M. Increased circulating endothelial progenitor cells are associated with survival in acute lung injury. Am J Respir Crit Care Med. 2005;172(7):854–60. 10.1164/rccm.200410-1325OC [DOI] [PubMed] [Google Scholar]
- 13.Melero-Martin JM, Khan ZA, Picard A, Wu X, Paruchuri S, Bischoff J. In vivo vasculogenic potential of human blood-derived endothelial progenitor cells. Blood. 2007. June 1;109(11):4761–8. 10.1182/blood-2006-12-062471 [DOI] [PubMed] [Google Scholar]
- 14.Richardson MR, Yoder MC. Endothelial progenitor cells: Quo Vadis? J Mol Cell Cardiol. 2011;50(2):266–72. 10.1016/j.yjmcc.2010.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medina RJ, CL O’Neill, Sweeney M, Guduric-Fuchs J, Gardiner TA, Simpson DA, et al. Molecular analysis of endothelial progenitor cell (EPC) subtypes reveals two distinct cell populations with different identities. BMC Med Genomics. 2010;3(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urbich C, Heeschen C, Aicher A, Sasaki K, Bruhl T, Farhadi MR, et al. Cathepsin L is required for endothelial progenitor cell-induced neovascularization. Nat Med. 2005;11(2):206–13. 10.1038/nm1182 [DOI] [PubMed] [Google Scholar]
- 17.Cairo MS, Mauss D, Kommareddy S, Norris K, van de Ven C, Modanlou H. Prophylactic or simultaneous administration of recombinant human granulocyte colony stimulating factor in the treatment of group B streptococcal sepsis in neonatal rats. Pediatr Res. 1990;27(10):612–6. [DOI] [PubMed] [Google Scholar]
- 18.Bermudez LE, Martinelli JC, Gascon R, Wu M, Young LS. Protection against gram-negative bacteremia in neutropenic mice with recombinant granulocyte-macrophage colony-stimulating factor. Cytokine. 1990;2(4):287–93. [DOI] [PubMed] [Google Scholar]
- 19.Bergamini A, Bolacchi F, Bongiovanni B, Cepparulo M, Ventura L, Capozzi M, et al. Granulocyte-macrophage colony-stimulating factor regulates cytokine production in cultured macrophages through CD14-dependent and -independent mechanisms. Immunology. 2000. October;101(2):254–61. 10.1046/j.1365-2567.2000.00117.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khameneh HJ, Isa SABM, Min L, Nih F, Ruedl C. GM-CSF signalling boosts dramatically IL-1production. PLoS One. 2011;6(7):e23025 10.1371/journal.pone.0023025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodman E, Stricker P, Velavicius M, Fonseca R, Kleinstein E, Lavery R, et al. Role of Granulocyte-Macrophage Colony-Stimulating Factor and Its Receptor in the Genesis of Acute Respiratory Distress Syndrome Through an Effect on Neutrophil Apoptosis. Arch Surg. American Medical Association; 1999. October 1;134(10):1049–54. [DOI] [PubMed] [Google Scholar]
- 22.Matute-Bello G, Liles WC, Radella F, Steinberg KP, Ruzinski JT, Hudson LD, et al. Modulation of neutrophil apoptosis by granulocyte colony-stimulating factor and granulocyte/macrophage colony-stimulating factor during the course of acute respiratory distress syndrome. Crit Care Med. 2000. January;28(1):1–7. [DOI] [PubMed] [Google Scholar]
- 23.Aggarwal A, Baker CS, Evans TW, Haslam PL. G-CSF and IL-8 but not GM-CSF correlate with severity of pulmonary neutrophilia in acute respiratory distress syndrome. Eur Respir J. 2000. May;15(5):895–901. [DOI] [PubMed] [Google Scholar]
- 24.Hierholzer C, Kelly E, Tsukada K, Loeffert E, Watkins S, Billiar TR, et al. Hemorrhagic shock induces G-CSF expression in bronchial epithelium. Am J Physiol. 1997;273:L1058–64. 10.1152/ajplung.1997.273.5.L1058 [DOI] [PubMed] [Google Scholar]
- 25.Hierholzer C, Kelly E, Billiar TR, Tweardy DJ. Granulocyte colony-stimulating factor (G-CSF) production in hemorrhagic shock requires both the ischemic and resuscitation phase. Arch Orthop Trauma Surg. 1997;116(3):173–6. [DOI] [PubMed] [Google Scholar]
- 26.Hierholzer C, Kelly E, Lyons V, Roedling E, Davies P, Billiar TR, et al. G-CSF instillation into rat lungs mediates neutrophil recruitment, pulmonary edema, and hypoxia. J Leukoc Biol. 1998;63(2):169–74. 10.1002/jlb.63.2.169 [DOI] [PubMed] [Google Scholar]
- 27.Verhoef G, Boogaerts M. Treatment with granulocyte-macrophage colony stimulating factor and the adult respiratory distress syndrome. Am J Hematol. 1991. April;36(4):285–7. [DOI] [PubMed] [Google Scholar]
- 28.Gao M, Zhang L, Liu Y, Yang M, Wang N, Wang K, et al. Use of blood urea nitrogen, creatinine, interleukin-6, granulocyte- macrophage colony stimulating factor in combination to predict the severity and outcome of abdominal sepsis in rats. Inflamm Res. 2012;61(8):889–97. 10.1007/s00011-012-0481-3 [DOI] [PubMed] [Google Scholar]
- 29.Bo L, Wang F, Zhu J, Li J, Deng X. Granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) for sepsis: A meta-analysis. Crit Care. 2011;15(1):R58 10.1186/cc10031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song F, Liu YL, Yang KH, Ma L. [Granulocyte-monocyte colony-stimulating factor for the treatment of sepsis: a meta analysis]. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2011;23(5):294–8. [PubMed] [Google Scholar]
- 31.Marlow N, Morris T, Brocklehurst P, Carr R, Cowan FM, Patel N, et al. A randomised trial of granulocyte-macrophage colony-stimulating factor for neonatal sepsis: Outcomes at 2 years. Arch Dis Child Fetal Neonatal Ed. 2013. January;98(1):F46–53. 10.1136/fetalneonatal-2011-301470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho H-J, Kim H-S, Lee M-M, Kim D-H, Yang H-J, Hur J, et al. Mobilized Endothelial Progenitor Cells by Granulocyte-Macrophage Colony-Stimulating Factor Accelerate Reendothelialization and Reduce Vascular Inflammation After Intravascular Radiation. Circulation. 2003. December 9;108(23):2918–25. 10.1161/01.CIR.0000097001.79750.78 [DOI] [PubMed] [Google Scholar]
- 33.Spitzer G, Adkins D, Mathews M, Velasquez W, Bowers C, Dunphy F, et al. Randomized comparison of G-CSF + GM-CSF vs G-CSF alone for mobilization of peripheral blood stem cells: effects on hematopoietic recovery after high-dose chemotherapy. Bone Marrow Transplant. 1997. December 18;20(11):921–30. 10.1038/sj.bmt.1700999 [DOI] [PubMed] [Google Scholar]
- 34.Gazitt Y. Comparison between granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor in the mobilization of peripheral blood stem cells. Curr Opin Hematol. 2002. May;9(3):190–8. [DOI] [PubMed] [Google Scholar]
- 35.Spight D, Trapnell B, Zhao B, Berclaz P, Shanley TP. Granulocyte-macrophage-colony-stimulating factor-dependent peritoneal macrophage responses determine survival in experimentally induced peritonitis and sepsis in mice. Shock. 2008. October;30(4):434–42. 10.1097/SHK.0b013e3181673543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toda H, Murata A, Oka Y, Uda K, Tanaka N, Ohashi I, et al. Effect of granulocyte-macrophage colony-stimulating factor on sepsis-induced organ injury in rats. Blood. 1994. May 15;83(10):2893–8. [PubMed] [Google Scholar]
- 37.Carr R, Modi N, Doré C. G-CSF and GM-CSF for treating or preventing neonatal infections. Cochrane database Syst Rev. 2003. July 21;(3):CD003066 10.1002/14651858.CD003066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Basu S, Dunn AR, Marino MW, Savoia H, Hodgson G, Lieschke GJ, et al. Increased tolerance to endotoxin by granulocyte-macrophage colony-stimulating factor-deficient mice. J Immunol. 1997. August 1;159(3):1412–7. [PubMed] [Google Scholar]
- 39.Asakura E, Hanamura T, Umemura A, Yada K, Yamauchi T, Tanabe T. Effects of Macrophage Colony-Stimulating Factor (M-CSF) on Lipopolysaccharide (LPS)-induced Mediator Production from Monocytes in vitro. Immunobiology. 1996. August;195(3):300–13. 10.1016/S0171-2985(96)80047-7 [DOI] [PubMed] [Google Scholar]
- 40.Tiegs G, Barsig J, Matiba B, Uhlig S, Wendel A. Potentiation by granulocyte macrophage colony-stimulating factor of lipopolysaccharide toxicity in mice. J Clin Invest. 1994. June 1;93(6):2616–22. 10.1172/JCI117274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brissette WH, Baker DA, Stam EJ, Umland JP, Griffiths RJ. GM-CSF rapidly primes mice for enhanced cytokine production in response to LPS and TNF. Cytokine. 1995. April;7(3):291–5. 10.1006/cyto.1995.0035 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.