

Abstract
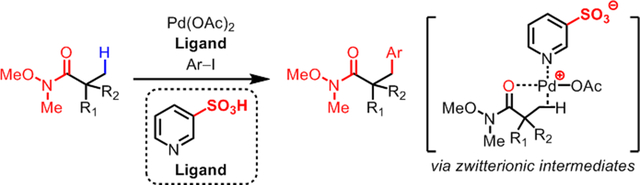
We report the development of Pd(II)-catalyzed C(sp3)–H arylation of Weinreb amides. This work demonstrates the first example of using Weinreb amide as a directing group for transition metal-catalyzed C(sp3)–H activation. Both the inductive effect and the potential bidentate coordination mode of the Weinreb amides pose a unique challenge for this reaction development. A pyridinesulfonic acid ligand is designed to accommodate the weak, neutral coordinating property of Weinreb amides via preserving the cationic character of Pd center through zwitterionic assembly of Pd/ligand complexes. DFT studies of the C–H cleavage step indicate that the superior reactivity of 3-pyridinesulfonic acid ligand compared to pyridine, Ac-Gly-OH, and ligandless conditions originates from the stabilization of overall substrate-bound Pd species.
Keywords: Weinreb Amide, Ligand Design, C(sp3)–H Activation, Palladium, Pyridinesulfonic Acid
Graphical Abstract
Chelation-assisted, Pd-catalyzed C–H functionalization has been extensively developed in the past two decades.1 One of the major advances include developing conditions and ligands to accommodate synthetically versatile weak-coordinating substrates (e.g. carbonyl compounds2). Despite these achievements, using native functional groups or common protecting groups as directing groups for C(sp3)–H activation is still highly limited.3 Traditionally, nitrogen-based, strong-coordinating groups have been most widely used as directing groups for C(sp3)–H activation, presumably due to the facile cyclometalation process that forms stable palladacycle intermediates.4,5 On the other hand, utilizing oxygen-based neutral-coordinating groups such as tertiary amides, esters, and ketones to direct C(sp3)–H activation remains a significant challenge.6 These weak-coordinating functionalities have neither the basicity of nitrogen nor the anionic character of carboxylates/amidates that can stabilize the substrate-to-catalyst binding. Our group has shown that mono-protected amino acid (MPAA) ligands with Pd catalyst can enable the functionalization of C(sp2)–H bonds with a wide range of neutral carbonyl directing groups (Scheme 1, top).2l The significant ligand effect observed for MPAA prompted us to develop a new class of ligands that can enable C(sp3)–H functionalization on these weak-coordinating substrates using Pd catalysis. Herein, we report the discovery of 3-pyridinesulfonic acid as a uniquely enabling ligand for Pd-catalyzed C(sp3)–H arylation using Weinreb amide as the directing group (Scheme 1, bottom). To the best of our knowledge, this work demonstrates the first example of Pd-catalyzed C(sp3)–H activation directed by a Weinreb amide group.
Scheme 1.

Pd-catalyzed C–H Functionalization Using Neutral, Carbonyl Directing groups
The broad synthetic utility of Weinreb amides as synthons for ketone/aldehyde synthesis7 prompted us to develop β-arylation of Weinreb amides. However, using Weinreb amide as directing group poses two unique challenges compared to simple amides (Scheme 1, bottom): First, the inductive effect of –OMe weakens the binding of the directing group with the Pd catalyst. Second, the unproductive bis-coordination with –OMe and carbonyl group can decelerate the catalysis. We postulated that these challenges can be addressed when an appropriate ligand is used to activate the Pd catalyst. However, examination of the corresponding pre-transition state structures suggests that the privileged pyridine/quinoline-type ligands8 would be incompatible with neutral-coordinating substrates, due to the lack of a free coordination site necessary for the agostic interaction with C–H bonds on the predominant Pd species A (Figure 1, top). Also, the dissociation of an acetate (or any counteranion) from complex A would be disfavored owing to the high energy associated with cationic complex B. To address this issue, we envisioned that installing a non-coordinating anion, such as sulfonate, into a pyridine ligand may promote the dissociation of an acetate anion from anionic complex C via electrostatic repulsion and thus stabilize the active Pd intermediate D as a zwitterionic species (Figure 1, bottom). Furthermore, the preserved cationic character of the Pd center would facilitate the binding of our weak-coordinating substrates. It is noteworthy that Carrow and coworkers recently reported that anionic thioether ligands can promote Pd-catalyzed C(sp2)–H olefination of electron-rich heterocycles.9
Figure 1.

Design Principle of Pyridinesulfonic acid Ligand.
To test our design principle, we chose Weinreb amide 1a as our model substrate for β-C–H arylation (Table 1). Under various conditions, we observed only trace amounts of product without using any ligand. Next, based on our hypothesis, we tested pyridine ligands bearing a sulfonate group at the 2- (L1), 3- (L2), and 4- (L3) positions. While L1 only gave trace amount of products, we were delighted to find that using L2 and L3 as ligands promoted arylation of 1a in high yields. To further support our design principle, we tested various classes of ligands that are known to promote Pd-catalyzed C–H activation. In accordance with our initial hypothesis, pyridine/quinoline-based ligands (L4-L7) only gave trace amounts of products. While pivalic acid10 (L8) showed similarly poor performance, MPAA ligands (L9-L10) afforded the products in 21 and 22% yields. Our recent finding of using electron-deficient 2-pyridones as ligands for C(sp3)–H11 functionalization prompted us to try L11 as a ligand, but only slight improvement was observed compared to using no ligand. Interestingly, nicotinic acid (L12) and isonicotinic acid (L13) were completely ineffective ligands. We presume that the much stronger coordinating ability of carboxylate compared to sulfonate prevents the formation of the zwitterionic Pd/ligand assembly. Finally, we conducted simple control experiments to show that L2-L3 were not merely serving as sulfonic acid additives. First, using the sulfonic acid L14 only gave 8% yield of the arylated product. Second, using L4 and L14 together completely inhibited the reaction, showing the importance of having both the pyridine and the sulfonic acid moiety in the same molecule. Overall, Table 1 shows the uniquely enabling property of pyridinesulfonic acid ligands (L2, L3) and the importance of synergistic matching of the ligand and the directing group.12
Table 1.
Evaluation of Ligands for C(sp3)–H Arylation of Weinreb Amidea,b
 |
Conditions: 1a (0.1 mmol), Pd(OAc)2 (10 mol%), Ligand (12 mol%), methyl 4-iodobenzoate (0.2 mmol), AgOAc (0.2 mmol), HFIP (0.5 mL), 70 °C, under air, 24 h.
The yield was determined by 1H NMR analysis of the crude product using CH2Br2 as the internal standard.
80 °C.
We decided to use the commercially available and inexpensive L2 as the optimal ligand to evaluate the substrate scope of the arylation of Weinreb amides (Table 2). First, substrates with simple alkyl chains afforded the arylated products in good yields (2a-2d). Next, Weinreb amides containing heteroatoms (O, N, F) gave the corresponding products in moderate to good yields (2e-2g). Unfortunately, Weinreb amides with α-H did not yield any products (2h, 2i), presumably due to the lack of Thorpe-Ingold effect. However, it was interesting to find that the corresponding N,N-dimethylamide gave 2j in 34% yield. Weinreb amides derived from quaternary cyclopropanecarboxylic acids gave products 2k-2m in excellent yields. While an α-H containing cyclopropane substrate gave product 2n in only 18% yield, again the corresponding N,N-dimethylamide gave much higher 87% yield of 2o. Cyclopropane substrates bearing various aryl groups on the α-position were reactive (2p-2r) with exclusive selectivity towards cyclopropyl C(sp3)–H bond over C(sp2)–H bond. Moderate to good yields were obtained with cyclopropane substrates bearing aryl groups on more remote positions (2s-2v).
Table 2.
C(sp3)–H Arylation of Weinreb Amides: Substrate Scopea,b,c
 |
Conditions: 1a-1v (0.1 mmol), Pd(OAc)2 (10 mol%), L2 (12 mol%), methyl 4-iodobenzoate (0.2 mmol), AgOAc (0.2 mmol), HFIP (0.5 mL), 80 °C, under air, 24 h.
Isolated yield.
The numbers in parentheses indicate the ratio of mono:di.
The high reactivity of the cyclopropane scaffold prompted us to examine the scope of aryl iodides with 1k as the substrate (Table 3). Aryl iodides with electron-withdrawing groups (3a-3c) and halides (3d-3f) at the para- position smoothly underwent the reaction to give products in high yields. While the reaction could tolerate p-Me group (3g), p-OMe group was not tolerated. Instead, aryl iodide with p-OTs group gave 3h in a very good yield. Substitution at the meta- and ortho- position of aryl iodides were well-tolerated with electron-withdrawing (3i-3l, 3o-3p) and electron-donating functional groups (3m-3n). A di-substituted aryl iodide also gave 3q in a good yield. Finally, heteroaryl iodides based on furan (3r), thiophene (3s), and pyridine (3t) also proved to be suitable coupling partners. To further test the utility of the developed β-C(sp3)–H arylation protocol, we conducted the reaction on gram scale of 1k with 5 mol% Pd(OAc)2, and 1.85g (95% yield) of product 2k was isolated (Scheme 2, top). We were also delighted to find that reducing the catalyst loading to 1 mol% and increasing the reaction time can afford 2k in a synthetically useful yield (Scheme 2, bottom). Without using any ligand, 16% of 2k was obtained. With MPAA L9 as the ligand, 30% of 2k was obtained, again showcasing the superior ligand effect with L2. We believe that in this case the ligand effect is not as dramatic as with 1a due to the innate reactivity of the cyclopropane substrate 1k.
Table 3.
C(sp3)–H Arylation of Weinreb Amides: Aryl Iodide Scopea,b
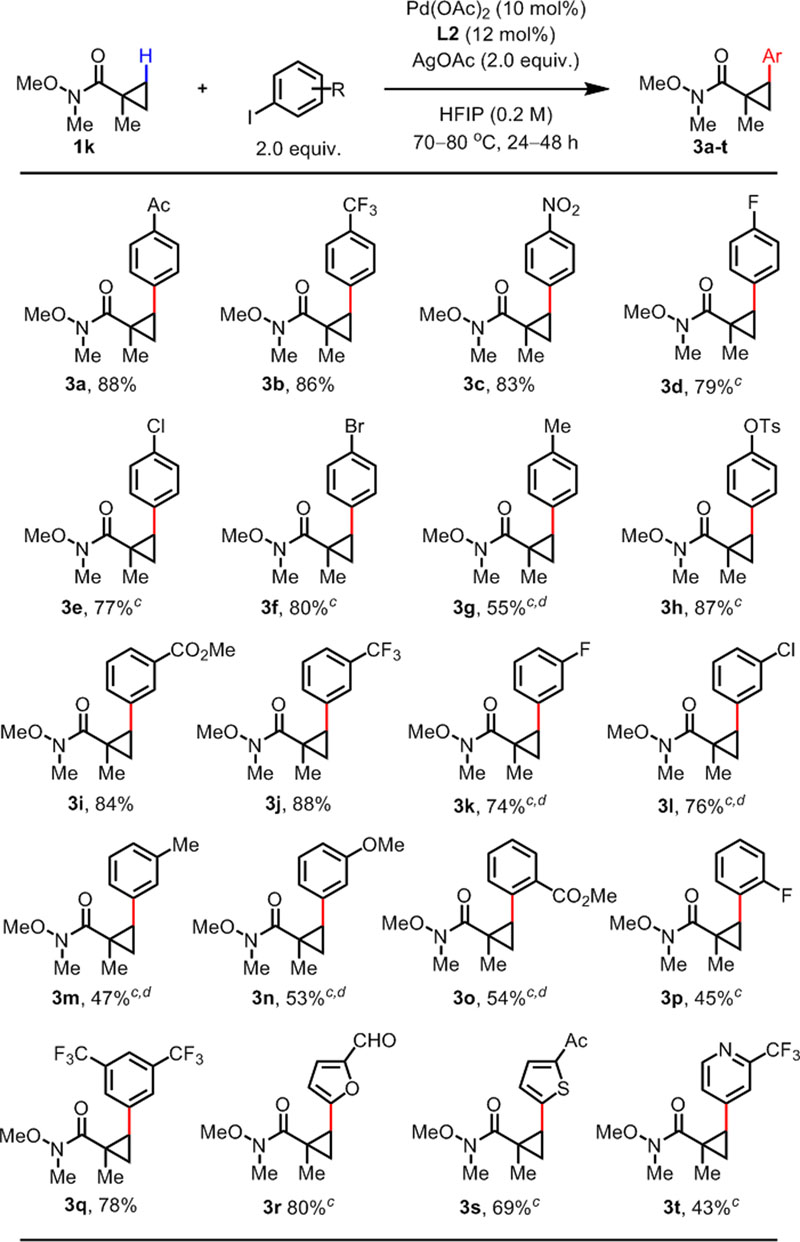 |
Conditions: 1k (0.1 mmol), Pd(OAc)2 (10 mol%), L2 (12 mol%), Ar–I (0.2 mmol), AgOAc (0.2 mmol), HFIP (0.5 mL), 70 °C, under air, 24 h.
Isolated yield.
80 °C.
48 h.
Scheme 2.

Gram Scale Reaction / 1 mol% Pd Loading
While our manuscript was being prepared, ligand-free β-arylation of N-alkyl pivalamides was reported by Lu and coworkers.6 Thus, we found it necessary to evaluate and compare Lu and coworkers’ ligand-free protocol (Conditions A) with our ligand-enabled protocol (Conditions B) on identical substrates and coupling partners (Table 4). First, Weinreb amide 1a showed sluggish reactivity under Conditions A albeit the high reaction temperature. In contrast, our protocol gave 2a in high yield, and the reaction is completely enabled by the ligand L2. Second, α-H-containing alkyl amide 1j showed no reactivity under Conditions A, but Conditions B can provide the product 2j albeit in low yield. Lastly, N-alkyl pivalamide 1w gave products 2w in 55% yield under Conditions A. Remarkably, our protocol can reduce the reaction temperature to 40 °C and still obtain 84% isolated yield of 2w. Overall, the results indicate that the pyridinesulfonic acid ligand effect is significant not only in Weinreb amides, but also in general amide substrates.
Table 4.
Comparison with Ligand-free Protocolsa
 |
The numbers in parentheses indicate the ratio of mono:di.
The yield was determined by 1H NMR analysis of the crude product using CH2Br2 as the internal standard.
Isolated yields.
40 °C, 48 h. For experimental details, see Supporting Information.
In order to achieve insight for the observed ligand effect with L2, a DFT study of the C(sp3)–H cleavage step (via CMD mechanism) with 1a was conducted (Figure 2). Quasi-harmonic Gibbs free energies of the substrate-bound-Pd complex (Int), C(sp3)–H-bound Pd complex (preTS), transition state for C(sp3)–H cleavage (TS), and the final palladacycle (Pdt) were calculated and compared for selected ligands. In each case, using AcO− (i.e. no ligand), L2, L4, and L9 as the ligand were compared. Also, various conformations of both the amide substrate and the ligand binding were considered, and the pathways with the lowest TS free energies are presented in Figure 2 (For full details, see Supporting Information). First, the reaction pathway with simple pyridine L4 had the highest energy overall, showing the instability of the cationic species (Int A, preTS A, TS A) compared to neutral species. Next, the calculated free energy values of the Pd species on the ligandless pathway (preTS B, TS B) are too high to be reached under our reaction temperature (70 °C), which complies with the trace amount of product formed under these conditions. Also, the lower free energy for the pathway with L9 compared to ligandless pathway agrees with the minor ligand effect observed with L9. Lastly, the lowest TS free energy with L2 is nearly 5 kcal/mol lower than that with L9, which also qualitatively matches with the superior reactivity observed with L2. These results show that L2 enables the reaction via significantly lowering the free energy values of the corresponding substrate-bound Pd species (Int D, preTS D, and TS D). It also implies that the weak-coordinating ability of carbonyl moiety renders the binding of substrate to Pd center generally undesirable (i.e. preTS B, TS B have high energies), and the cationic character of Pd center generated with L2 is essential to accommodate such weak binding.
Figure 2.

Computed qh-G (T=343.15 K) Free Energy Profiles for the C(sp3)–H activation step at the M06/SDD, 6–311++G(d,p)(SMD)//ωB97X-D/SDD,6–31+G(d)(SMD) level of theory
In summary, a Weinreb amide-directed β-C(sp3)–H arylation using Pd catalysis has been developed. The reaction is enabled by the commercially available 3-pyridinesulfonic acid as the ligand. Computational studies indicate that the origin of ligand effect is the overall stabilization of the neutral substrate-bound Pd species, which is likely due to the enhanced cationic character of the Pd center. This is in accordance with our initial design principle of an anionic pyridine ligand that can stabilize the cationic Pd center via a zwitterionic assembly. Based on our finding that our new ligand can enable C(sp3)–H activation of α-H containing alkyl amides, our future work will focus on expanding the substrate scope towards more general substrates and different transformations.
Supplementary Material
ACKNOWLEDGMENT
We gratefully acknowledge The Scripps Research Institute and the NIH (NIGMS, 2R01GM084019) for financial support. We acknowledge the TSRI High Performance Computing Resources and the University of Oxford Advanced Research Computing facility.13 We also thank Dr. Curtis E. Moore for X-Ray crystallographic analysis and Dr. Jason S. Chen for analytical support. H.P. thanks the Korea Foundation for Advanced Studies for a predoctoral fellowship and N.C. thanks The Scripps Research Institute for the AYRIU scholarship.
Footnotes
Supporting Information. Experimental procedures and spectral data for all new compounds and full computational data (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).For selected reviews on chelation-assisted Pd-catalyzed C–H functionalization:Chen X; Engle KM; Wang DH; Yu JQ Palladium(II)‐Catalyzed C–H Activation/C–C Cross‐Coupling Reactions: Versatility and Practicality. Angew. Chem. Int. Ed 2009, 48, 5094–5115.Lyons TW; Sanford MS Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chem. Rev 2010, 110, 1147–1169.Engle KM; Mei T-S; Wasa M; Yu J-Q Weak Coordination as a Powerful Means for Developing Broadly Useful C–H Functionalization Reactions. Acc. Chem. Res 2012, 45, 788–802.Rouquet G; Chatani N Catalytic Functionalization of C(sp2)–H and C(sp3)–H Bonds by Using Bidentate Directing Groups. Angew. Chem. Int. Ed 2013, 52, 11726–11743.Daugulis O; Roane J; Tran LD Bidentate, Monoanionic Auxiliary-Directed Functionalization of Carbon–Hydrogen Bonds. Acc. Chem. Res 2015, 48, 1053–1064.He J; Wasa M; Chan KSL; Shao Q; Yu J-Q Palladium-Catalyzed Transformations of Alkyl C–H Bonds. Chem. Rev 2017, 117, 8754–8786.
- (2).For selected examples of directed C(sp2)–H functionalization using free carbonyl directing groups:Murai S; Kakiuchi F; Sekine S; Tanaka Y; Kamatani A; Sonoda M; Chatani N Efficient catalytic addition of aromatic carbon-hydrogen bonds to olefins. Nature 1993, 366, 529–531.Kakiuchi F; Sato T; Igi K; Chatani N; Murai S The Ruthenium-Catalyzed Addition of β C–H Bonds in Aldehydes to Olefins. Chem. Lett 2001, 30, 386–387.Satoh T; Miura M; Nomura M Multiple arylation of carbonyl compounds via palladium catalysis. J. Organomet. Chem 2002, 653, 161–166.Gandeepan P; Parthasarathy K; Cheng C-H Synthesis of Phenanthrone Derivatives from sec-Alkyl Aryl Ketones and Aryl Halides via a Palladium-Catalyzed Dual C–H Bond Activation and Enolate Cyclization. J. Am. Chem. Soc 2010, 132, 8569–8571.Patureau FW; Besset T; Glorius F Rhodium‐Catalyzed Oxidative Olefination of C–H Bonds in Acetophenones and Benzamides. Angew. Chem. Int. Ed 2011, 50, 1064–1067.Xiao B; Gong T-J; Xu J; Liu Z-J; Liu L Palladium-Catalyzed Intermolecular Directed C–H Amidation of Aromatic Ketones. J. Am. Chem. Soc 2011, 133, 1466–1474.Mo F; Trzepkowski LJ; Dong G Synthesis of ortho‐Acylphenols through the Palladium‐Catalyzed Ketone‐Directed Hydroxylation of Arenes. Angew. Chem. Int. Ed 2012, 51, 13075–13079.Shan G; Yang X; Ma L; Rao Y Pd‐Catalyzed C–H Oxygenation with TFA/TFAA: Expedient Access to Oxygen‐Containing Heterocycles and Late‐Stage Drug Modification. Angew. Chem. Int. Ed 2012, 51, 13070–13074.Wang Y; Li C; Li Y; Yin F; Wang XS Rhodium‐Catalyzed C–H Olefination of Aryl Weinreb Amides. Adv. Synth. Catal 2013, 355, 1724–1728.Yang F; Ackermann L Ruthenium-Catalyzed C–H Oxygenation on Aryl Weinreb Amides. Org. Lett 2013, 15, 718–720.Kim J; Chang S Iridium-Catalyzed Direct C–H Amidation with Weakly Coordinating Carbonyl Directing Groups under Mild Conditions. Angew. Chem. Int. Ed 2014, 53, 2203–2207.Li G; Wan L; Zhang G; Leow D; Spangler J; Yu J-Q Pd(II)-Catalyzed C–H Functionalizations Directed by Distal Weakly Coordinating Functional Groups. J. Am. Chem. Soc 2015, 137, 4391–4397.Wang Y; Zhou K; Lan Q; Wang X-S Pd(ii)-catalyzed C–H arylation of aryl and benzyl Weinreb amides. Org. Biomol. Chem 2015, 13, 353–356.Shang R; Ilies L; Nakamura E Iron-Catalyzed Ortho C–H Methylation of Aromatics Bearing a Simple Carbonyl Group with Methylaluminum and Tridentate Phosphine Ligand. J. Am. Chem. Soc 2016, 138, 10132–10135.Bu Q; Rogge T; Kotek V; Ackermann L Distal Weak Coordination of Acetamides in Ruthenium(II)‐Catalyzed C–H Activation Processes. Angew. Chem. Int. Ed 2018, 57, 765–768.
- (3).For selected examples of Pd-catalyzed C(sp3)–H functionalization of free carboxylic acids:Giri R; Maugel N; Li J-J; Wang D-H; Breazzano SP; Saunder LB; Yu J-Q Palladium-Catalyzed Methylation and Arylation of sp2 and sp3 C–H Bonds in Simple Carboxylic Acids. J. Am. Chem. Soc 2007, 129, 3510–3511.Chen G; Zhuang Z; Li G-C; Saint-Denis TG; Xiao Y; Joe CL; Yu J-Q Ligand‐Enabled β‐C–H Arylation of α‐Amino Acids Without Installing Exogenous Directing Groups. Angew. Chem., Int. Ed 2017, 56, 1506–1509.Zhu Y; Chen X; Yuan C; Li G; Zhang J; Zhao Y Pd-catalysed ligand-enabled carboxylate-directed highly regioselective arylation of aliphatic acids. Nat. Commun 2017, 8, 14904.Ghosh KK; van Gemmeren M Pd-Catalyzed β-C(sp3)–H Arylation of Propionic Acid and Related Aliphatic Acids. Chem. Eur. J 2017, 23, 17697–17700.
- (4).For selected examples of Pd-catalyzed C–H functionalization using pyridine/imine-based directing groups:Desai LV; Hull KL; Sanford MS Palladium-Catalyzed Oxygenation of Unactivated sp3 C–H Bonds. J. Am. Chem. Soc 2004, 126, 9542–9543.Dick AR; Hull KL; Sanford MS A Highly Selective Catalytic Method for the Oxidative Functionalization of C–H Bonds. J. Am. Chem. Soc 2004, 126, 2300–2301.Zaitsev VG; Shabashov D; Daugulis O Highly Regioselective Arylation of sp3 C–H Bonds Catalyzed by Palladium Acetate. J. Am. Chem. Soc 2005, 127, 13154–13155.
- (5).For selected examples of cyclopalladation using nitrogen-based directing group:Cope AC; Siekman RW Formation of Covalent Bonds from Platinum or Palladium to Carbon by Direct Substitution. J. Am. Chem. Soc 1965, 87, 3272–3273.Fahey DR The coordination-catalyzed ortho-halogenation of azobenzene. J. Organomet. Chem 1971, 27, 283–292.Constable AG; McDonald WS; Sawkins LC; Shaw BL Palladation of dimethylhydrazones, oximes, and oxime O-allyl ethers: crystal structure of [Pd3(ON=CiPrPh)6]. J. Chem. Soc., Chem. Commun 1978, 1061–1062.Fuchita Y; Hiraki K; Uchiyama T Metallation of aliphatic carbon atoms. Part 1. Synthesis and characterization of the cyclopalladated complexes of 2-neopentylpyridine. J. Chem. Soc., Dalton Trans 1983, 897–899.
- (6).While we were preparing this manuscript, β-arylation of pivalamides was reported:Zhao R; Lu W Palladium-Catalyzed β-Arylation of Amide via Primary sp3 C–H Activation. Organometallics 2018, 37, 2188–2192.
- (7).Nahm S; Weinreb SM N-methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett 1981, 22, 3815–3818.Singh J; Satyamurthi N; Aidhen IS The Growing Synthetic Utility of Weinreb′s Amide. J. Prakt. Chem 2000, 342, 340–347.
- (8).Wasa M; Chan KSL; Zhang X-G; He J; Miura M; Yu J-Q Ligand-Enabled Methylene C(sp3)–H Bond Activation with a Pd(II) Catalyst. J. Am. Chem. Soc 2012, 134, 18570–18572.He J; Li S; Deng Y; Fu H; Laforteza BN; Spangler JE; Homs A; Yu J-Q Ligand-Controlled C(sp3)–H Arylation and Olefination in Synthesis of Unnatural Chiral α–Amino Acids. Science 2014, 343, 1216–1220.
- (9).Gorsline BJ; Wang L; Ren P; Carrow BP C–H Alkenylation of Heteroarenes: Mechanism, Rate, and Selectivity Changes Enabled by Thioether Ligands. J. Am. Chem. Soc 2017, 139, 9605–9614. [DOI] [PubMed] [Google Scholar]
- (10).Lafrance M; Fagnou K Palladium-Catalyzed Benzene Arylation: Incorporation of Catalytic Pivalic Acid as a Proton Shuttle and a Key Element in Catalyst Design. J. Am. Chem. Soc 2006, 128, 16496–16497. [DOI] [PubMed] [Google Scholar]
- (11).Zhu R-Y; Li Z-Q; Park HS; Senanayake CH; Yu J-Q Ligand-Enabled γ-C(sp3)–H Activation of Ketones. J. Am. Chem. Soc 2018, 140, 3564–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Engle KM; Yu J-Q Developing Ligands for Palladium(II)-Catalyzed C–H Functionalization: Intimate Dialogue between Ligand and Substrate. J. Org. Chem 2013, 78, 8927–8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Richards A 2015. University of Oxford Advanced Research Computing, Zenodo.10.5281/zenodo.22558.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.