

Abstract
The type III TGF-β receptor (TβRIII) is a TGF-β co-receptor that presents ligand to the type II TGF-β receptor to initiate signaling. TβRIII also undergoes ectodomain shedding to release a soluble form (sTβRIII) that can bind ligand, sequestering it away from cell surface receptors. We have previously identified a TβRIII extracellular mutant that has enhanced ectodomain shedding (“super shedding (SS)” – TβRIII-SS). Here we utilize TβRIII-SS to study the balance of cell surface and soluble TβRIII in the context of lung cancer. We demonstrate that expressing TβRIII-SS in lung cancer cell models induces epithelial-to-mesenchymal transition (EMT) and that these TβRIII-SS (EMT) cells are less migratory, invasive and adhesive and more resistance to gemcitabine. Moreover, TβRIII-SS (EMT) cells exhibit decreased tumorigenicity but increased growth rate in vitro and in vivo. These studies suggest that the balance of cell surface and soluble TβRIII may regulate a dichotomous role for TβRIII during cancer progression.
Keywords: TGF-β, type III TGF-β receptor, EMT, lung cancer, ectodomain shedding
Introduction
The transforming growth factor β (TGF-β) family of ligands have established roles in most human cancers, including lung cancer, with specific roles in regulating proliferation, migration, invasion and metastasis (1). TGF-β has a dichotomous role in cancer, acting as a tumor suppressor in normal cells by inhibiting proliferation and promoting apoptosis, but then functions as a tumor promoter during cancer progression by inducing epithelial-mesenchymal transition, remodeling the extracellular matrix, promoting metastasis, sustaining angiogenesis, and suppressing immunosurveillance.
TGF-β signals through a family of cell surface receptors, including the type I, type II and type III TGF-β receptors (2). The type I and type II TGF-β receptors are serine/threonine kinases, while the type III TGF-β receptor is a co-receptor. TGF-β initiates signaling by binding to the type III TGF-β (TβRIII), which presents TGF-β to the type II receptor, allowing complex formation with and phosphorylation of the type I receptor. The phosphorylation of the type I receptor induces a conformation change that releases FK506-binding protein and allows for the binding and phosphorylation of Smad2 and Smad3. Phosphorylated Smad2 and Smad3 then form a heterotrimeric complex with Smad4 that translocates to and accumulates in the nucleus, to act in concert with co-activators and co-repressors to regulate target genes (2).
The type III transforming growth factor receptor (TβRIII), also known as betaglycan, is a transmembrane homodimeric proteoglycan that is ubiquitously expressed (1–3). TβRIII is the most abundantly expressed TGF-β receptor, with ~200,000 receptors/cell, and can bind to all three isoforms of TGF-β. Interestingly, TβRIII can undergo receptor shedding to produce a soluble protein (sTβRIII) that can also bind ligand. sTβRIII is thought to sequester ligand, preventing binding to cell surface receptors and thus, blocking TGF-β-induced downstream signaling (4–6). TβRIII expression is lost or reduced in most human cancers, with loss of TβRIII correlating with disease progression, advanced stage or grade, and a poorer prognosis (7, 8). Restoring TβRIII expression decreases cancer cell motility and invasion in vitro, and reduces angiogenesis, invasion and metastasis in vivo while shRNA-mediated silencing of TβRIII expression increases cancer cell migration and invasion (7–9), supporting a role for TβRIII as a suppressor of cancer progression.
Consistent with studies in other cancer contexts, restoring TβRIII expression in lung cancer cell models decreased cancer cell migration, invasion and anchorage-dependent cell growth (10). Moreover, in breast cancer cell lines, expressing a genetic mutant of TβRIII that leads to diminished shedding capability (TβRIII-ΔShed) lead to an increase in migration and invasion while expressing a genetic mutant of TβRIII that leads to an increase in shedding (TβRIII-SS) resulted in an even greater decrease in migration, invasion and metastasis compared to expression of wild-type TβRIII (11), suggesting that the balance of cell surface TβRIII and sTβRIII is important in mediating the suppression of cancer progression. Here we investigate the role of the balance of cell surface TβRIII and sTβRIII on cancer progression in the context of lung cancer.
Results
TβRIII-SS cells undergo a gradual EMT
To investigate the significance of TβRIII ectodomain shedding in the context of lung cancer, we knocked out endogenous TβRIII in A549 and H460 lung cancer cell lines using CRISPR-Cas9 (cr-TβRIII), and then expressed either an empty vector construct (EV), wild-type (TβRIII-WT), loss of shedding (TβRIII-ΔShed) or increase in shedding “super shedding (SS)” (TβRIII-SS) TβRIII construct. Binding and crosslinking studies of these stable cell lines confirmed effective CRISPR-mediated abrogation of TβRIII expression, and restoration of wild-type, TβRIII-ΔShed and TβRIII-SS receptor expression (Supplementary Figure 1). Interestingly, expression of TβRIII-SS resulted in a phenotypic change in cells as they were passaged, from an epithelial morphology (TβRIII-SS (epi)) to a mesenchymal morphology (TβRIII-SS (EMT)), reminiscent of epithelial-to-mesenchymal (EMT) transition (Figure 1A and 1B). Cells expressing cr-TβRIII, TβRIII-WT, or TβRIII-ΔShed did not undergo a comparable EMT change after a similar number of passages (Figure 1A and 1B). This transition occurred progressively 3–12 passages after completing antibiotic selection for expression of the transfected constructs (Figure 1C and 1D). The EMT phenotype of TβRIII-SS cells was further supported by a loss of E-cadherin and a gain of N-cadherin and Slug (Figure 1E and 1F) that occurred during the phenotypic transition (Figure 1G and 1H). This data establishes that expression of TβRIII-SS, with increased production of sTβRIII, can induce EMT in these lung cancer models.
Figure 1:

TβRIII-SS induces EMT. (A, B) Representative phase contrast microscopy images of A549 (A) and H460 (B) cells stably transfected with the indicated constructs. Representative phase contrast microscopy images of A549 (C) and H460 (D) at different passages after completion of antibiotic selection. Analysis of EMT markers by Western blotting of A549 (E) and H460 (F) cell lines stably expressing the indicated constructs and are representative of three trials. Analysis of EMT markers by Western blotting of A549 (G) and H460 (H) TβRIII-SS cell lines over several passages after completion of antibiotic selection. EMT transition was observed in at least three independent experiments. Scale bar = 100μM.
TβRIII-SS (EMT) cells are less migratory and invasive
As EMT is often linked to increased migration and invasion, we performed transwell migration and invasion assays to examine the effect of increased TβRIII shedding. Surprisingly, TβRIII-SS (EMT) cells exhibited markedly decreased transwell migration and invasion relative to epithelial TβRIII-SS (epi), control (cr-NTC/EV), knock-out (cr-TβRIII/EV), wild-type (TβRIII-SS-WT) and loss of shedding (TβRIII-ΔShed). A549 (Figure 2A, 2B, 2C, and 2D) and H460 cells (Figure 2E, 2F, 2G, and 2H). In contrast, knocking out TβRIII and re-expressing TβRIII-ΔShed enhanced invasion two-fold but did not change migration in A549 cells and enhanced both migration and invasion 2-3 fold in H460 cells. TβRIII-WT expressing A549 cells also increased migration and invasion two-fold. These differences in migration and invasion were not due to differences in proliferation (Supplementary Figure 2A, 2B, and 2C). Thus, while we expected that EMT would promote migration and invasion, the TβRIII-SS-induced EMT was instead associated with inhibition of migration and invasion.
Figure 2:

TβRIII-SS (EMT) decreases migration and invasion. Representative images of A549 cells that migrated through a transwell (A) or invaded through Matrigel coated transwell (B). Quantification of A549 transwell migration (C) and invasion (D). Representative images of H460 cells that migrated through a transwell (E) or invaded through Matrigel coated transwell (F). Quantification of H460 transwell migration (G) and invasion (H). Scale bar = 100μM. *** p < 0.001, **** p < 0.0001. Quantification represents at least three independent experiments with technical replicates within each trial. Error bars are ±S.E.M.
TβRIII-SS (EMT) cells are less adhesive
Cell migration is a tightly controlled process that can be regulated by cellular adhesion properties (12). Accordingly, to gain insight into how migration and invasion were being altered, we performed adhesion assays on plastic and different extracellular matrix (ECM) proteins. In both A549 and H460 cell lines, the TβRIII-SS (EMT) cell lines exhibited 50-75% less adhesion to plastic compared to the other cell lines (Figure 3A and 3B). The TβRIII-SS (epi) cell lines exhibited better adhesion than the TβRIII-SS (EMT) cells but still had decreased ability to adhere to plastic. To assess whether these differences in adhesion could be due to specific interactions with extracellular matrix proteins, we coated plates with fibronectin, laminin or collagen. Similar to the results on plastic, H460 TβRIII-SS (EMT) cell lines exhibited 50–75% less adhesion to these ECM proteins compared to the other cell lines (Supplementary Figure 3). This data demonstrates that EMT induced by TβRIII-SS inhibits cellular adhesion both to plastic and extracellular matrix proteins.
Figure 3:

TβRIII-SS (EMT) decreases adhesion. Quantification of A549 (A) and H460 (B) adherence to plastic. ** p< 0.01, *** p < 0.001, **** p < 0.0001. Quantification represents at least three independent experiments with technical replicates within each trial. Error bars are ±S.E.M.
TβRIII-SS (EMT) cells are more resistant to gemcitabine
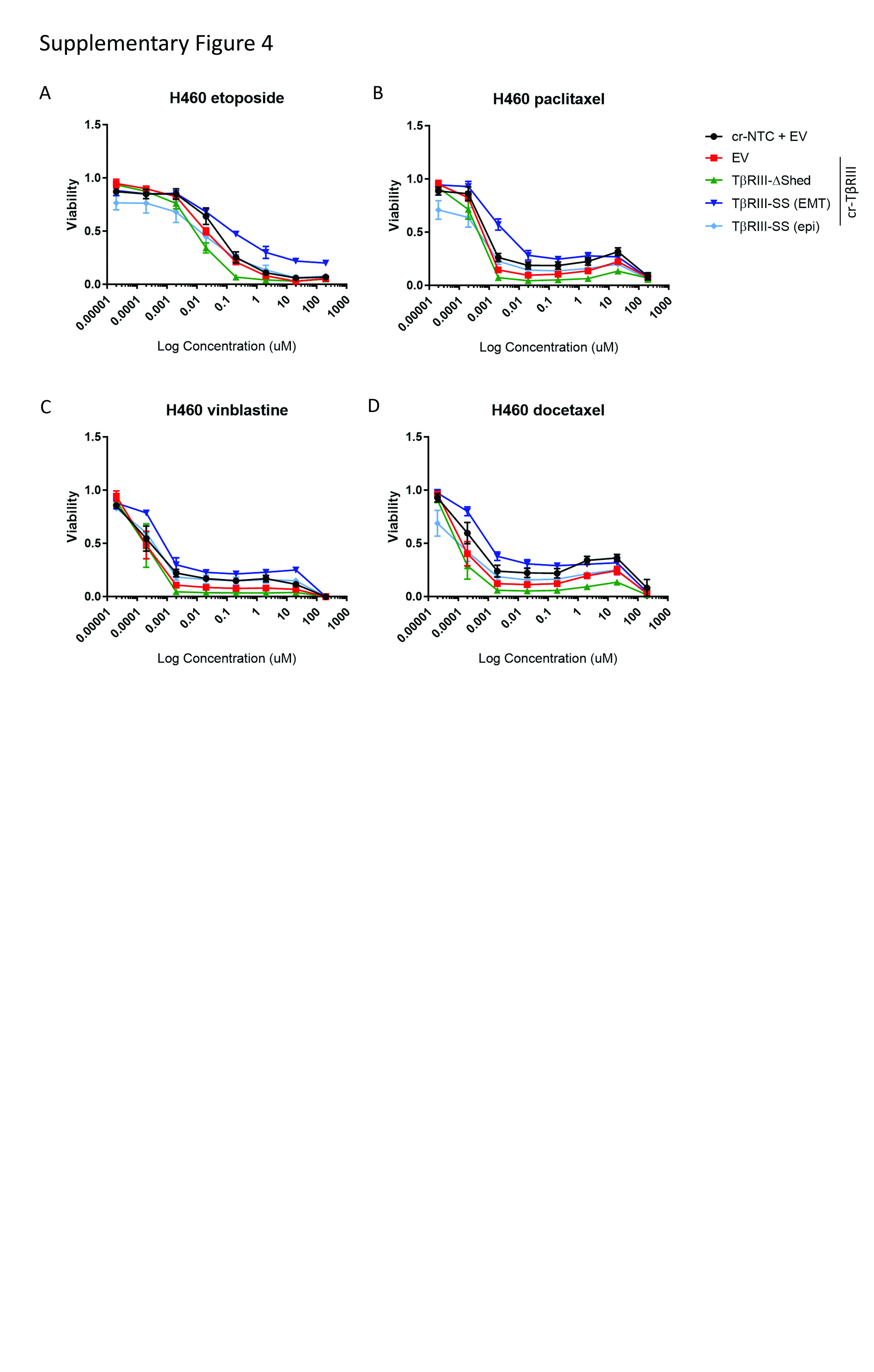
EMT has been linked to chemoresistance independent of its role in metastasis (13, 14). Accordingly, we investigated the impact of EMT on chemoresistance. Cells expressing TβRIII-SS with an EMT phenotype were more resistant to gemcitabine at concentrations greater than 200nM compared to cells with TβRIII knocked out (Figure 4A and B). Interestingly, this increase in resistance was dependent on whether the TβRIII-SS cells had undergone EMT. Cells exhibited greater than 50% viability for these TβRIII-SS (EMT) cells compared to the TβRIII-SS (epi) cells, in which only 10–20% of cell survived. Comparing the response of EMT versus epithelial TβRIII-SS to other chemotherapy reagents including docetaxel, paclitaxel, etoposide and vinblastine all suggested a trend toward increased resistance with the EMT phenotype in TβRIII cells (Supplementary Figure 4). Re-expression of wild-type TβRIII also shifted the growth rate inhibition (GR50), demonstrating increased resistance to gemcitabine treatment than cells with loss of TβRIII (cr-TβRIII/EV) (Figure 4A). Together, these data suggest that TβRIII-SS-induced EMT mediates chemoresistance.
Figure 4:

TβRIII-SS (EMT) increases gemcitabine resistance. Dose response of A549 (A) and H460 (B) cells to 10-fold dilutions of gemcitabine. Quantification represents at least three independent experiments with technical replicates within each trial. Error bars are ±S.E.M.
TβRIII-SS (EMT) cells have decreased tumorigenicity in vitro
Since TβRIII-SS-induced EMT decreased migration, invasion and adhesion, we investigated its role in tumorigenicity by performing a soft agar colony formation assays. The colony formation assay demonstrated that TβRIII-SS (EMT) cells form 75% fewer colonies than TβRIII-SS (epi) cells (Figure 5A, 5B, 5C, and 5E)) suggesting that they were less tumorigenic. However, the colonies that formed were three times as large (Figure 5A, 5B, 5D and 5F) despite the lack of differences in proliferation (Supplementary Figure 2).
Figure 5:

TβRIII-SS (EMT) decreases in vitro tumorigenicity but increases colony size. Representative images of A549 (A) and H460 (B) soft agar colonies. A549 cells were grown for 24 days and H460 cells were grown for 16 days. The total number of colonies (C, E) and the total area of colonies (D, F). *** p < 0.001, **** p < 0.0001. Quantification represents at least three independent experiments with technical replicates within each trial. Error bars are ±S.E.M.
TβRIII-SS (EMT) cells are less tumorigenic in vivo but exhibit enhanced tumor growth rate
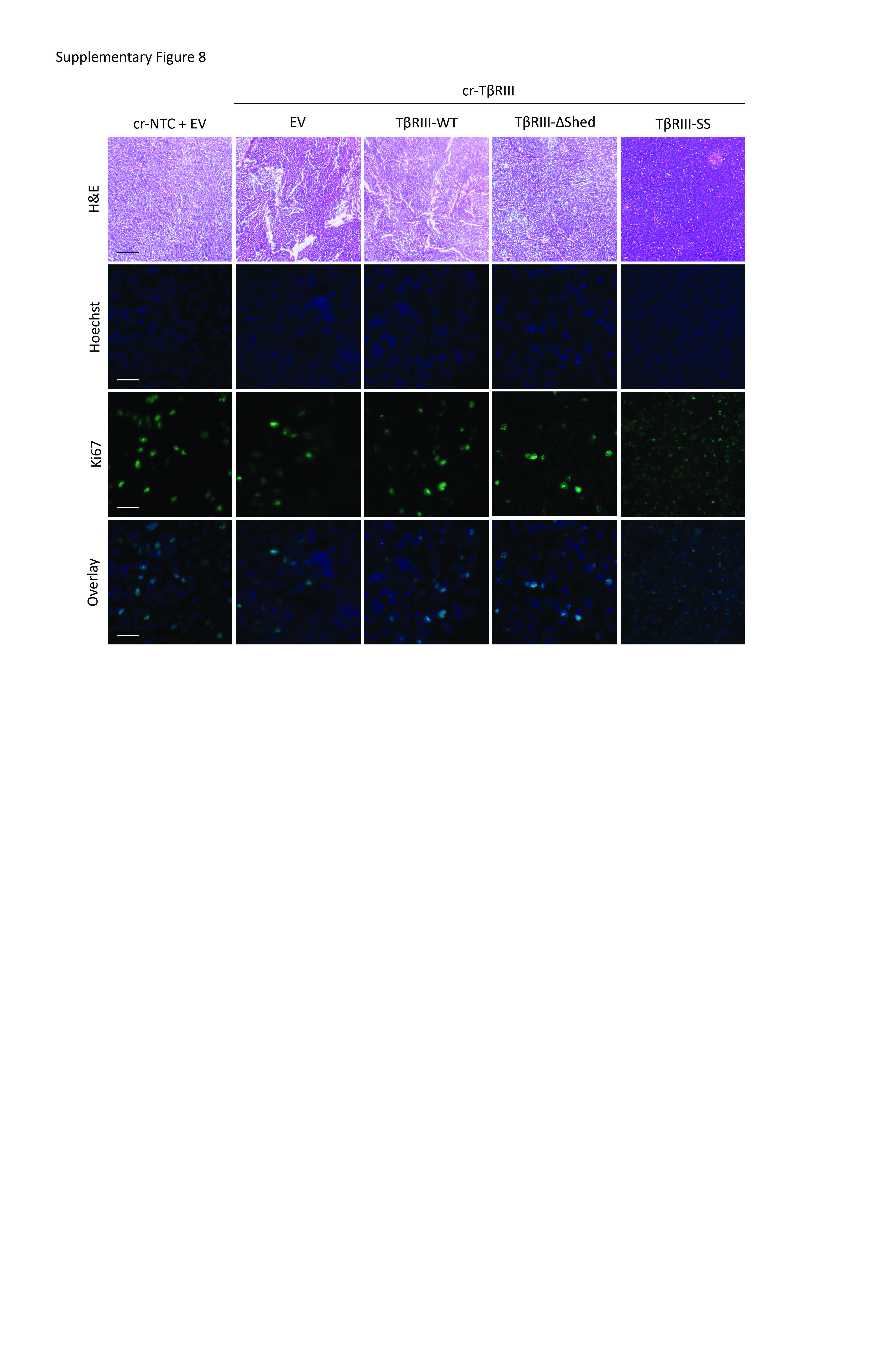
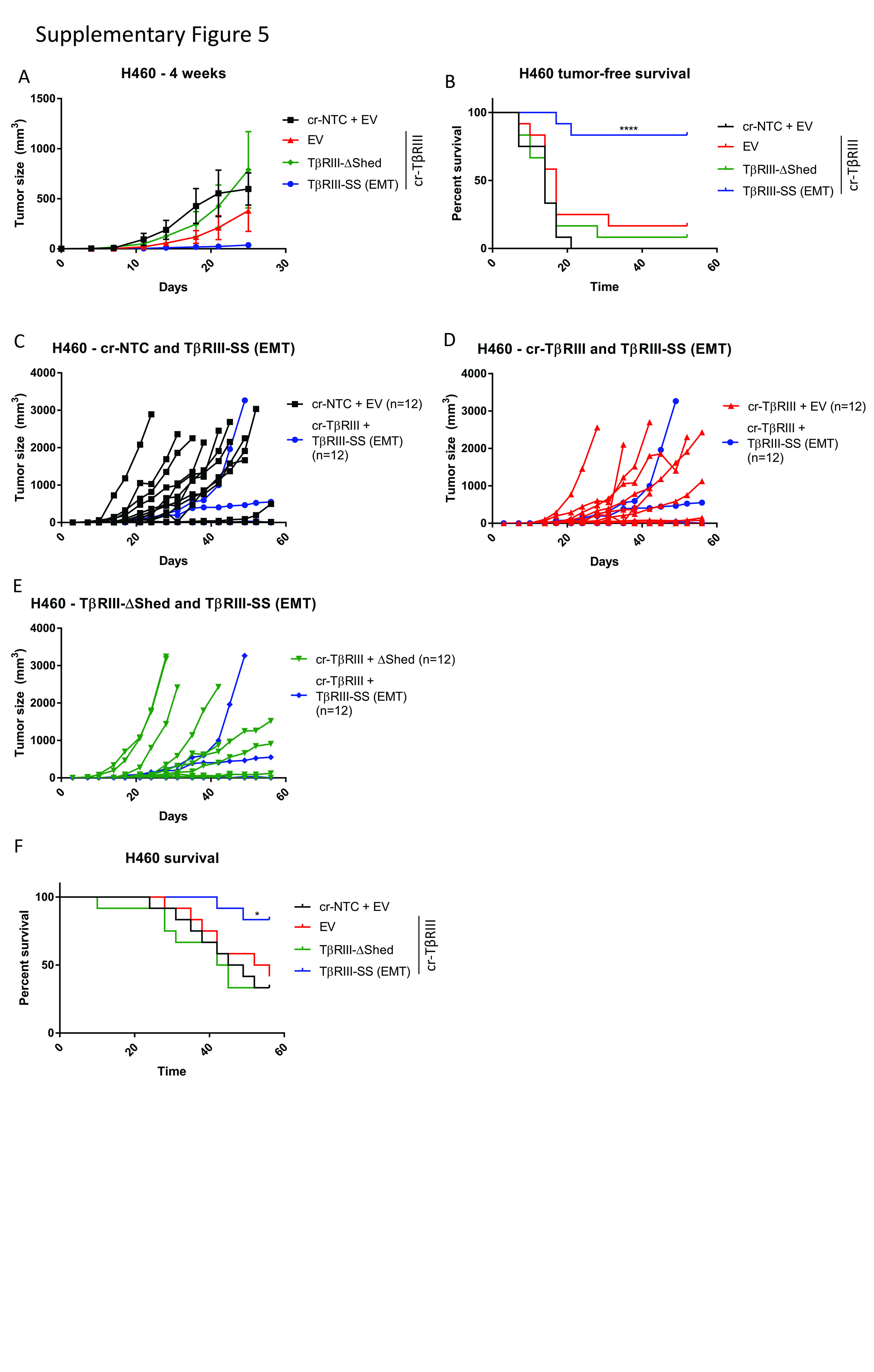
Given the striking effects of TβRIII-SS on tumorigenicity in vitro, we examined effects on tumorigenicity in vivo. We injected A549 and H460 subcutaneously into athymic mice and assessed tumor growth. The A549 TβRIII-SS (EMT) bearing mice exhibit delayed onset of tumor growth in both cell lines, with tumors not appearing until nearly 3 weeks after injection (Figure 6A and 6B). Mice injected with H460 TβRIII-SS cells took more than 6 weeks to develop tumors (Supplementary Figure 5A and 5B). The TβRIII-SS (EMT)-bearing mice had a better tumor-free survival than mice bearing the other cell lines, with 33% of the A549 TβRIII-SS (EMT) mice never developing tumors, compared to 8% of the TβRIII-WT bearing mice. All the other mice bearing cr-NTC/EV, cr-TβRIII/EV, or TβRIII-ΔShed developed tumors (Figure 6B). Similarly, 83% of the H460 mice never developed tumors while than less than 20% of the other mice were tumor-free at the end of the study (Supplementary Figure 5B). When TβRIII-SS (EMT) tumors started growing, they were initially smaller than the other tumors at the same timepoint (Figure 6A). However, once they reached a critical mass, they demonstrated a faster growth rate than the other cells (Figure 6C, 6D, 6E, 6F, and 6G). This rapid tumor growth lead to a decrease in overall survival of the mice injected with A549 TβRIII-SS cells (Figure 6H). As only 2 of the 12 mice injected with H460 TβRIII-SS (EMT) developed tumors (Supplementary Figure 5B), and these tumors had not reached critical mass by the end of study, the impact of increased growth rate on survival following tumor formation could not be assessed in this model (Supplementary Figure 5C, 5D, 5E, 5F). Palpation and caliper measurements of tumor growth was verified by bioluminescence scanning (Supplementary Figure 6 and 7). Histological examination of these tumors demonstrated a higher density of cells with smaller nuclei in the tumors derived from the TβRIII-SS cells. Further, staining with Ki67 suggested that this was associated with enhanced proliferation (Supplementary Figure 8). Together, the in vivo and in vitro data establish that TβRIII-SS-induced EMT decreases tumorigenesis but increases tumor growth rate once tumors are established.
Figure 6:

TβRIII-SS (EMT) decreases in vivo tumorigenicity but increases tumor growth rate. (A) A549 cells were subcutaneously injected in athymic mice and tumor growth was assessed via caliper measurement twice a week. Mean tumor volume was quantified. 4-week timepoint highlights differences in early tumor growth. (B) Mice were palpated twice a week for tumors and tumor-free was survival plotted. (C, D, E, F) Tumor size of individual mice over time comparing TβRIII-SS (EMT)-injected mice with other groups. (G) Area under the curve calculation to assess tumor growth rate tumors. Mice without tumors were excluded. (H) Mice were sacrificed when tumors reached 2000mm3 or reached humane endpoints, and survival was plotted. n = 12 for each cohort of mice. * p< 0.05, ** p < 0.01, **** p < 0.0001. Error bars are ±S.E.M.
TβRIII-SS (EMT) cells have decreased canonical but increased non-canonical TGF-β signaling
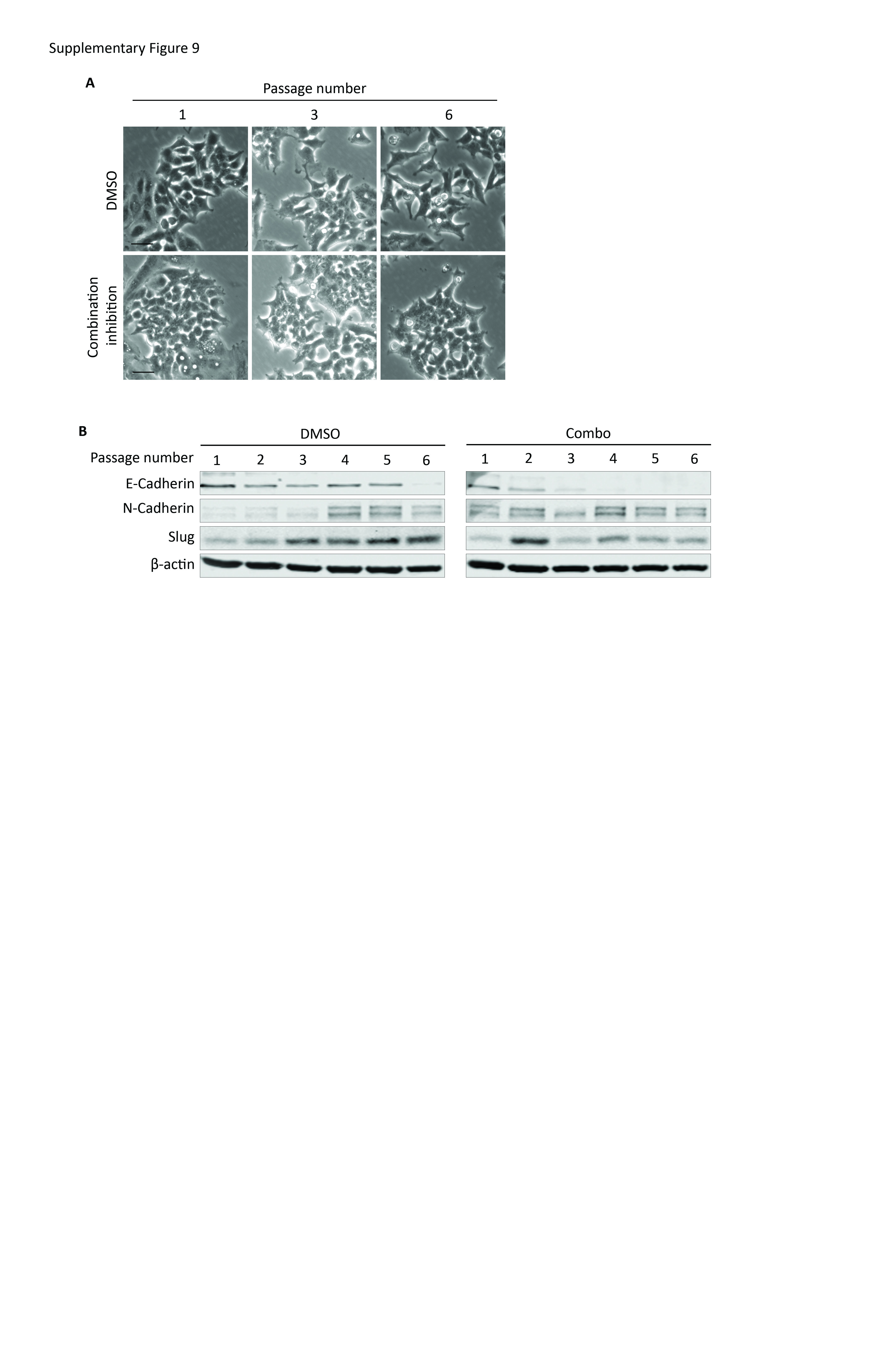
To gain insight into how TβRIII-SS might regulate cancer cell biology, we analyzed effects on both canonical and non-canonical TGF-β signaling pathways. Both A549 and H460 TβRIII-SS (EMT) cells lines demonstrated decreased canonical Smad2 and Smad3 signaling compared to both the parental cr-NTC/EV and the TβRIII-SS (epi) cell lines with TGF-β stimulation. Interestingly, there was also a decrease in total Smad2 and Smad3 for the TβRIII-SS (EMT) cells. In contrast, TβRIII-SS (EMT) cell demonstrated increased non-canonical signaling with activation of ERK½, AKT and p38 at baseline and with TGF-β stimulation compared to the TβRIII-SS (epi) lines (Figure 7A and B). To investigate the role of elevated non-canonical signaling, we inhibited these pathways with pharmacologic inhibitors. Pharmacologic inhibition of ERK, AKT and p38 in TβRIII-SS (epi) significantly delayed transition to the mesenchymal phenotype and blunted the sustained induction of Slug (Supplementary Figure 9A and B). These data support a role for TβRIII-SS-induced EMT in inhibiting canonical but promoting non-canonical TGF-β signaling, with the effects on non-canonical TGF-β signaling potentially mediating the effects through sustained induction of Slug.
Figure 7:

TβRIII-SS (EMT) decreases canonical and increases non-canonical signaling. Western blot analysis of canonical and non-canonical TGF-β signaling pathways in A549 (A) and H460 (B) cells. Results are representative of at least three independent experiments with technical replicates within each trial.
Discussion
Here we have demonstrated for the first time that expression of TβRIII-SS, which increases soluble TβRIII production, can induce epithelial-to-mesenchymal transition (EMT) in a cell autonomous manner, decreasing cell migration, invasion, adhesion, gemcitabine sensitivity and tumorigenesis (Figure 8). While we used lung cancer cell lines as our model system, these results may be applicable to other cancers as TβRIII undergoes ectodomain shedding in other cancer contexts. The role of TβRIII-SS in inducing EMT was unexpected as TGF-β is a predominant driver of EMT (15, 16) and soluble TβRIII has been thought to function largely to sequester TGF-β. Thus, one would hypothesize that expression of TβRIII-SS would decrease TGF-β bioavailability and subsequent EMT induction. However, this EMT induction may not be TGF-β dependent as TGF-β-induced EMT occurs in 24–72 hours (17) while the TβRIII-SS-induced EMT took much longer, suggesting an indirect mechanism, including potential epigenetic rewiring of the cells. Known epigenetic modifiers of EMT include polycomb repressive complexes, histone deacetylases and histone demethylases (18, 19). As these effects may be either on a few specific genes, or more likely through alteration of genome wide epigenetic configurations, this remains an active area of investigation. Our TβRIII-SS construct exhibits increased levels of soluble TβRIII, decreased levels of cell surface TβRIII and increased amount of the transmembrane and cytoplasmic remnant left after ectodomain cleavage. To evaluate the contribution of soluble TβRIII, we treated cr-TβRIII/EV cells with conditioned media from either cr-TβRIII/EV or TβRIII-SS (EMT) cells. Although the study was limited by nutrient depletion from the conditioned media, which led to minor phenotypic changes in the cells treated with the cr-TβRIII/EV conditioned media, the conditioned media from TβRIII-SS (EMT) cells was able to induce EMT, suggesting that soluble TβRIII may be sufficient to mediate these effects (data not shown). The respective contribution of each of these components to the phenotypes described here remains an active area of investigation.
Figure 8:

Model of TβRIII-SS function. TβRIII-SS induces epithelial-to-mesenchymal transition. TβRIII-SS (EMT) then decreases migration and invasion, potentially through decreased adhesion. TβRIII-SS also increases gemcitabine resistance, decreases tumorigenicity but increases tumor growth rate, potentially through inhibiting canonical TGF-β signaling and promoting non-canonical TGF-β signaling.
Although EMT is classically associated with increased migration, invasion and metastasis (20), recent studies have demonstrated that EMT is not necessary for metastasis but instead is critical for inducing chemoresistance (13, 14). Our TβRIII-SS (EMT) cells have decreased transwell migration and invasion, in vitro surrogate assays for metastasis, in comparison to their epithelial counterparts, lending support to potential disconnect between EMT and metastasis. However, the decreased adherence of TβRIII-SS (EMT) cells may explain this decrease in migration and invasion since cells need to be able to cyclically adhere and detach from the extracellular matrix for effective motility and invasion (21). Although our TβRIII-SS (EMT) cells do not support the classical idea that EMT promotes motility, our studies do support a role for EMT in mediating chemoresistance, as we demonstrate that TβRIII-SS (EMT) cells are more resistant to gemcitabine than the TβRIII-SS (epi) cells, along with a similar trend with other chemotherapy agents. Thus, assessing sTβRIII levels in the tumor microenvironment might be a method for predicting response to therapy. Future studies will focus on investigating the role of soluble TβRIII in metastasis and in vivo drug resistance.
In addition to mediating chemoresistance, TβRIII-SS also influences tumorigenicity and tumor growth, further establishing the complex role that TβRIII plays in tumor biology. Both our soft agar and xenograft studies demonstrate that TβRIII-SS (EMT) cells were less tumorigenic but grew faster once established despite similar in vitro proliferation rates, suggesting that TβRIII-SS can initially suppress tumorigenesis but once tumors develop and a critical mass has been reached, TβRIII-SS promotes tumor growth through enhanced proliferation. Thus, similar to the dichotomous role of the TGF-β signaling pathway during cancer initiation/progression (3), TβRIII-SS also has a dichotomous role. Our signaling data suggests that the TβRIII-SS (EMT) cells have decreased canonical TGF-β signaling through Smad2 and Smad3 and increased signaling through non-canonical pathways including ERK, p38 and AKT. This decrease in canonical TGF-β signaling suggests that sTβRIII from the TβRIII-SS (EMT) line can effectively sequester ligand away from the cell surface, preventing downstream signaling and the pro-tumorigenic effects of TGF-β. This could potentially explain the loss of tumorigenicity of established cancer cells demonstrated here. Meanwhile, the up-regulation of ERK and AKT are linked to dysregulated cell cycle control and increased survival (22–25) and unchecked activation of these pathways may allow the TβRIII-SS (EMT) cells to avoid apoptosis and attain uncontrolled tumor growth once tumors form. Although p38 activation’s role in apoptosis is cell type dependent, studies suggest that p38 is activated in lung cancer, leading to prevention of apoptosis (26, 27). Thus, p38 activation could also contribute to cell survival and the rapid growth. This baseline increase in ERK, AKT and p38 phosphorylation and the lack of further activation upon TGF-β stimulation suggests that these pathways are already maximally activated or that a TGF-β-independent mechanism is activating these pathways. However, ERK, AKT and p38 are often activated during EMT (15), and sTβRIII’s induction of EMT could be driving their activation. Consistent with this hypothesis, ERK, AKT and p38 were elevated only in the TβRIII-SS (EMT) model and not the epithelial TβRIII-SS (epi) model and simultaneous pharmacologic inhibition of ERK, AKT and p38 significantly delayed transition of TβRIII-SS (epi) cells to a mesenchymal phenotype and blunted sustained Slug induction. The relative contribution of decreased canonical signaling and elevated non-canonical signaling to the effects of TβRIII-SS on tumor biology demonstrated here remains to be further explored.
Although our work focuses on the role of TβRIII-SS in cancer cells, enhanced shedding of cell surface TβRIII on other cells within the tumor environment including fibroblasts, immune cells and endothelial cells could also lead to increased levels of sTβRIII in the tumor microenvironment. TβRIII found on these other cells is especially notable since although TβRIII is often decreased or lost in cancer (4), sTβRIII generated from these other cells could contribute to the overall levels in the tumor environment and can influence tumorigenesis, growth and metastasis. In vitro, TβRIII shedding can be modulated by pervanadate, a tyrosine phosphatase inhibitor, and plasmin and can be inhibited by tissue inhibitors of metalloproteinases (TIMPs). Overexpression of membrane-type 1 matrix metalloprotease also leads to cell autonomous TβRIII cleavage and sTβRIII generation (28). However, specific physiological proteases that target TβRIII shedding within the tumor microenvironment are still unknown and specific binding protease binding sites within TβRIII have not been identified. Ongoing work in our lab have suggested that neutrophil elastase and cathepsin G secreted from activated neutrophils can cleave cell surface TβRIII (unpublished data). Neutrophil infiltration and extracellular matrix remodeling, which often involves matrix metalloproteinases (MMPs) occur in the lung cancer microenvironment (29, 30), and their activity may also generate sTβRIII. How cells in the tumor microenvironment regulate shedding of TβRIII and how this contributes to remodeling the tumor microenvironment remains to be explored.
While TβRIII-SS was identified in a structure/function screen (11), and mutations that alter TβRIII shedding have not been identified, as there are numerous proteases in the tumor microenvironment, the elevated levels of sTβRIII with TβRIII-SS may well be recapitulated in the tumor microenvironment. Studies in melanoma and breast cancer have found that elevated circulating sTβRIII levels in plasma correlated to better survival (31, 32). However, the levels of sTβRIII in the tumor microenvironment are currently unknown. As we have demonstrated a critical role for TβRIII in metastasis, tumorigenesis and tumor growth and current studies have suggested the importance of TGF-β and EMT for responses to chemotherapy or checkpoint inhibitors (13, 14, 33, 34), investigating sTβRIII levels and its impact on metastasis, survival and response to chemotherapy and checkpoint inhibitors is of interest. Disitertide (P144) is a synthetic peptide based on TβRIII that acts as a soluble TβRIII mimetic intended to sequester TGF-β. Disitertide is currently undergoing clinical trials to treat fibrosis (35, 36). Early studies of these drugs in the context of glioblastoma, thymoma and melanoma have suggested a role in decreasing tumorigenicity and enhancing immunotherapy efficacy (37, 38). However, since sTβRIII also seems to have dichotomous roles, in which it first decreases tumorigenesis but then later supports tumor growth, it is vital to have a better understanding of the role of sTβRIII in carcinogenesis and tumor progression will allow us to better harness and combine therapeutic strategies to inhibit tumor growth and metastasis both in lung cancer and other types of cancer.
The role of soluble TβRIII in the complex tumor microenvironment has been difficult to study because of challenges of modeling differences in sTβRIII levels in vivo. It is also challenging to study the role of endogenous proteases in the tumor microenvironment since the extracellular matrix is a rich source of a variety of proteases, and TβRIII may be the target of multiple proteases. However, since TβRIII-SS is a NAAIRS mutagenesis variant of TβRIII, we hope to study the role of sTβRIII in tumor cancer initiation, progression and spontaneous metastasis by making a genetically engineered mouse model with this mutant TβRIII-SS variant. This model would also facilitate study the interaction between sTβRIII and the tumor microenvironment. Altering levels of sTβRIII in the microenvironment can impact the bioavailability of TGF-β, which has numerous roles in immunosurveillance, angiogenesis and ECM remodeling. TGF-β suppresses the immune system by inhibiting cytotoxic T cells, regulatory T cells NK cells, and dendritic cells. Moreover, it also is a critical mediator of angiogenesis, which is needed to sustain tumor growth. TGF-β also remodels the extracellular matrix by inducing MMP production and myofibroblast differentiation (3). Current studies have emphasized the importance of TGF-β in anti-tumor immune responses by demonstrating that combining TGF-β inhibitors with PD-L1 checkpoint inhibitors was more effective at promoting anti-tumor immunity and decreasing tumor growth and metastasis than checkpoint inhibitor alone (33, 34). Similarly, modulating TβRIII levels by altering shedding can also impact TGF-β bioavailability and activity and be critical to determining responsiveness to checkpoint inhibitor therapy. A genetically engineered TβRIII-SS mouse model would enable understanding of the role of sTβRIII in the complex tumor microenvironment of an immunocompetent model.
In summary, we have demonstrated that TβRIII-SS decreases tumorigenesis by inducing EMT. We have also demonstrated that TβRIII-SS may have dichotomous roles since it also acts as a tumor promoter by supporting tumor growth. This suggests a critical role for the cell autonomous effects of TβRIII/sTβRIII on tumor characteristics behavior and the potential paracrine roles it plays in the tumor microenvironment. In turn, the microenvironment may also modify sTβRIII levels through cleavage of cell surface TβRIII. This further adds to previous work in breast cancer that suggests that soluble TβRIII can prevent pro-tumorigenic downstream effects (11). Further exploring the mechanism through which TβRIII-SS induces EMT and influences biology and identifying the proteases that enhance soluble TβRIII production will lead to better understanding of strategies to manipulate the TGF-β pathway to develop better therapeutic approaches.
Materials and Methods
Cell culture and reagents
A549, H460 and 293FT cell lines were obtained from the American Type Culture Collection (ATCC) and authenticated using short tandem repeat analysis and mycoplasma negative. The cell lines were cultured in RPMI-1650 supplemented with 1mM sodium pyruvate, 10mM HEPES, 2.25g/L glucose and 10% FBS. 293FT cell lines were cultured in Dulbecco’s modified Eagle’s minimum essential medium supplemented with 10% FBS. All cells were grown at 37°C in 5% CO2. Selumetinib and LY294002 were purchased form Selleckchem (Houston, TX, USA), and SB20290 was purchased form Sigma-Aldrich (St. Louis, MO, USA).
Generation of constructs and stable cell lines
sgRNA targeting TβRIII and a non-targeting control guide were selected from the GeCKO library (cr-TβRIII #1: GATTATCGAGGCGTCCAGCG; cr-TβRIII #2: GTCCTGAATCTCCGCACTGC; cr-NTC: ACGGAGGCTAAGCGTCGCAA) and subcloned into the lentiCRISPRv2 vector (gift from Feng Zhang, Addgene plasmid # 52961) (39). Single-cell clones were isolated and deletion verified through TβRIII binding and crosslinking. Cells then underwent transfection with overexpression constructs of TβRIII-WT, TβRIII-ΔShed and TβRIII-SS genetic mutants, which were previously described (11). Cells subsequently underwent selection and passaging in parallel and thus, all cell lines were used at the same passage number post transfection and selection. Luciferase positive cell lines were generated using pLenti-PGK-Blast-V5-Luc (w528-1) (gift from Eric Campeau, Addgene plasmid #19166) (40). Stable cell lines were generated using X-tremeGENE 9 transfection reagent (Sigma-Aldrich) and psPAX2 packaging and pMD2.G envelop plasmids according to manufacturer’s directions. 2ug/mL of puromycin was used to select for positive constructs.
Binding and crosslinking
TβRIII was identified through binding and crosslinking of iodinated TGF-β1 (Perkin Elmer, Waltham, MA, USA), as previously described (7). Briefly, iodinated TGF-β1 was incubated with either adherent cells or cell medium at 4°C before being crosslinked with DSS. An antibody against the extracellular domain of TβRIII (R&D Systems, Minneapolis, MN, USA) was used to immunoprecipitate TβRIII. The sample was run on an SDS-PAGE gel, and signal was visualized using a phosphorimager.
Transwell migration and invasion
Transwell migration was assessed by seeding 2.5 × 104 A549 cells or 1.5 × 105 H460 cells in complete medium with 1% serum in the upper chamber of a transwell with 8.0 μM pores (Corning, Corning, NY, USA) and allowed to migrate for 24 hours at 37°C toward complete medium with 10% serum. Transwell invasion was assessed in a similar manner by seeding 1 × 105 A549 cells or 5 × 105 H460 cells in the upper chamber of a Matrigel-coated transwell with 8.0 μM pores (BD Biosciences, Franklin Lakes, NJ), and invasion was assessed after incubation at 37°C for 24 hours. Colonies were counted using ImageJ.
Proliferation
Click-iT™ Plus EdU Incorporation Assay (Invitrogen, Carlsbad, CA, USA) was used to assess proliferation and assay was performed according to manufacturer’s directions. Briefly, A549 and H460 cells were seeded at 3×105 cells and 4×105 cells respectively in 6 well dishes. After 24 hours, A549 and H460 cells were incubated with EdU at a final concentration of 5 μM and 10 μM. Cells were then fixed with 4% paraformaldehyde and resuspended in saponin-based permeabilization and wash reagent. Cells were incubated with the Click-iT™ cocktail (PBS, copper protectant, Alexa Fluor™ 488 picolyl azide, and reaction buffer additive) for 30 minutes at room temperature and then resuspended in wash reagent. 4’,6-diamidino-2-phenylindole (DAPI) was added to each sample at a final concentration of 0.2 μg/mL. A549 and H460 cells were analyzed on a BD Bioscience FACSCanto II flow cytometer (BD Biosciences). A total of 10,000 events were analyzed from each sample. Cells were analyzed using FlowJo software (Tree Star, Inc., Ashland, OR, USA) and gated to remove dead cells and debris.
Adhesion assay
5 × 104 A549 or H460 cells were plated per well in a 96-well plate and allowed to incubate at 37°C and adhere for 30 minutes. After incubation, cells were washed with PBS, fixed with 5% glutaraldehyde for 30 minutes at room temperature or overnight at 4°C, stained with 0.01% crystal violet, and solubilized using 10% acetic acid. Absorbance of the solubilized crystal violet was then measured. If extracellular matrix proteins were used, 96-well dishes were pre-coated overnight with 5ug/mL of either fibronectin (EMD Millipore, Burlington, MA, USA), laminin (Invitrogen) or collagen (BD Biosciences).
Colony formation assay
1.8% bacto-agar, 2X DMEM, 10% FBS and 1% penicillin-streptomycin was plated on the bottom layer of a 6-well plate. Upper agar was mixed in a 1:1 ratio with 1 × 105 A549 or H460 cells before plating. Wells were hydrated with complete media every 3-5 days. A549 cells were grown for 24 days and H460 cells were grown for 16 days. Cells were then stained with crystal violet and imaged. Colonies were counted using ImageJ.
Chemotoxicity assay
2.5 × 103 A594 or H460 cells in 96-well opaque plates and allowed to proliferate for 24 hours before treating with 10-fold dilutions of docetaxel (Cayman Chemical, Ann Arbor, MI, USA) #11637), paclitaxel (Cayman Chemical #10461), etoposide (Cayman Chemical #12092), vinblastine (Cayman Chemical #11762), and gemcitabine (Duke Pharmacy Store Room, Durham, NC, USA). Cells were treated with chemotherapy reagents for 48 hours, and cell viability was assessed using CellTiter-Glo (Promega, Madison, WI, USA) and normalized to an untreated control.
Western blotting
Cells were serum-starved overnight and treated with 100pM TGF-β for 60 minutes. Cells were lysed in 2× Laemmli sample buffer, and the proteins were resolved on a 10% SDS-PAGE gel. All primary antibodies (Slug #9585, p-Smad2 #3108, Smad2 #3103, p-Smad3 #9520, Smad3 #9523, p-ERK1/2 #9101, ERK1/2 #9102, p-AKT #4058, AKT #4691, p-p38 #4631, p38 #9212) were purchased from Cell Signaling (Danvers, MA, USA) except for β-actin (Sigma, AA5441) and N-cadherin (#610921) and E-cadherin (#610182) (BD Biosciences). Anti-mouse (#5470) and anti-rabbit (#5151) secondary antibodies were purchased from Cell Signaling. Blots were scanned using the LI-COR Odyssey (LI-COR Biosciences, Lincoln, NE, USA).
Xenograft
2 × 106 A549 cells or 2.5 × 105 H460 cells expressing luciferase were injected subcutaneously into the right flank of 6-week old female athymic nude mice (Duke Breeding Core, Durham, NC, USA). Tumors were measured with calipers twice a week, and tumor volume was calculated as 0.5 × width2 × length. Mice were injected with 100uL of 15mg/mL luciferin and scanned weekly using an IVIS Kinetic (Caliper Life Sciences, Waltham, MA, USA) to assess bioluminescence. Tumors and any metastatic lesions were harvested, and necropsy was performed when tumors reached 2000 mm3 or at end of study. n=12 mice for each cell line for both A549 and H460 experiments were used for a power of 80%. All mice were 6-week old female athymic nude mice so no randomization was performed. Study complies with ethical regulations and approved by the Duke Institutional Animal Care and Use Committee (IACUC). Blinding was accomplished by assigning each mouse a unique identifier without annotation of experimental group. Only this identifier was known to investigators during caliper measurements.
Statistical Analysis
All assays were performed at least in triplicate with technical replicates within each trial as appropriate. One-way analysis of variance (ANOVA) followed by a Tukey’s post-hoc test to determine significance among groups and a Mantel-Cox log-rank test was used to determine significance for Kaplan-Meier graphs. Statistical significance was defined as a p-value less than 0.05. Variance was similar between groups that were being statistically compared. Graphpad Prism 7 software was used for analysis (GraphPad Software, La Jolla, CA). Epithelial-to-mesenchymal transition of TβRIII-SS (epi) cells were observed at least as three independent biological replicates in both the A549 and H460 cell lines.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by the following grants from the National Institutes of Health/National Cancer Institute: F30CA196162 (J.J.H.), T32-GM007171 (Medical Scientist Training Program; J.J.H), and R01CA135006 (G.C.B).
Footnotes
Conflict of interest:
The authors declare that they have no conflicts of interest.
References
- 1.Massague J TGFbeta in Cancer. Cell. 2008;134(2):215–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Massague J TGFbeta signalling in context. Nat Rev Mol Cell Biol 2012;13(10):616–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang JJ, Blobe GC. Dichotomous roles of TGF-beta in human cancer. Biochem Soc Trans 2016;44(5):1441–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gatza CE, Oh SY, Blobe GC. Roles for the type III TGF-beta receptor in human cancer. Cell Signal. 2010;22(8):1163–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopez-Casillas F, Wrana JL, Massague J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell. 1993;73(7):1435–44. [DOI] [PubMed] [Google Scholar]
- 6.Vilchis-Landeros MM, Montiel JL, Mendoza V, Mendoza-Hernandez G, Lopez-Casillas F. Recombinant soluble betaglycan is a potent and isoform-selective transforming growth factor-beta neutralizing agent. Biochem J 2001;355(Pt 1):215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong M, How T, Kirkbride KC, Gordon KJ, Lee JD, Hempel N, et al. The type III TGF-beta receptor suppresses breast cancer progression. J Clin Invest 2007;117(1):206–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turley RS, Finger EC, Hempel N, How T, Fields TA, Blobe GC. The Type III Transforming Growth Factor-{beta} Receptor as a Novel Tumor Suppressor Gene in Prostate Cancer. Cancer research. 2007;67(3):1090–8. [DOI] [PubMed] [Google Scholar]
- 9.Hempel N, How T, Dong M, Murphy SK, Fields TA, Blobe GC. Loss of betaglycan expression in ovarian cancer: role in motility and invasion. Cancer research. 2007;67(11):5231–8. [DOI] [PubMed] [Google Scholar]
- 10.Finger EC, Turley RS, Dong M, How T, Fields TA, Blobe GC. TbetaRIII suppresses non-small cell lung cancer invasiveness and tumorigenicity. Carcinogenesis. 2008;29(3):528–35. [DOI] [PubMed] [Google Scholar]
- 11.Elderbroom JL, Huang JJ, Gatza CE, Chen J, How T, Starr M, et al. Ectodomain shedding of TbetaRIII is required for TbetaRIII-mediated suppression of TGF-beta signaling and breast cancer migration and invasion. Mol Biol Cell. 2014;25(16):2320–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huttenlocher A, Sandborg RR, Horwitz AF. Adhesion in cell migration. Curr Opin Cell Biol 1995;7(5):697–706. [DOI] [PubMed] [Google Scholar]
- 13.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527(7579):472–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014;15(3):178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res 2009;19(2):156–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell. 2005;16(4):1987–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nature medicine. 2013;19(11):1438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skrypek N, Goossens S, De Smedt E, Vandamme N, Berx G. Epithelial-to-Mesenchymal Transition: Epigenetic Reprogramming Driving Cellular Plasticity. Trends Genet 2017;33(12):943–59. [DOI] [PubMed] [Google Scholar]
- 20.Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166(1):21–45. [DOI] [PubMed] [Google Scholar]
- 21.Guan X Cancer metastases: challenges and opportunities. Acta Pharm Sin B. 2015;5(5):402–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J. 2010;277(1):2–21. [DOI] [PubMed] [Google Scholar]
- 23.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22(56):8983–98. [DOI] [PubMed] [Google Scholar]
- 24.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med 2005;9(1):59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chambard JC, Lefloch R, Pouyssegur J, Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta. 2007;1773(8):1299–310. [DOI] [PubMed] [Google Scholar]
- 26.Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res 2005;15(1):11–8. [DOI] [PubMed] [Google Scholar]
- 27.Greenberg AK, Basu S, Hu J, Yie TA, Tchou-Wong KM, Rom WN, et al. Selective p38 activation in human non-small cell lung cancer. Am J Respir Cell Mol Biol 2002;26(5):558–64. [DOI] [PubMed] [Google Scholar]
- 28.Velasco-Loyden G, Arribas J, Lopez-Casillas F. The shedding of betaglycan is regulated by pervanadate and mediated by membrane type matrix metalloprotease-1. J Biol Chem 2004;279(9):7721–33. [DOI] [PubMed] [Google Scholar]
- 29.Stamenkovic I Extracellular matrix remodelling: the role of matrix metalloproteinases. J Pathol 2003;200(4):448–64. [DOI] [PubMed] [Google Scholar]
- 30.Treffers LW, Hiemstra IH, Kuijpers TW, van den Berg TK, Matlung HL. Neutrophils in cancer. Immunol Rev 2016;273(1):312–28. [DOI] [PubMed] [Google Scholar]
- 31.Hanks BA, Holtzhausen A, Evans KS, Jamieson R, Gimpel P, Campbell OM, et al. Type III TGF-beta receptor downregulation generates an immunotolerant tumor microenvironment. J Clin Invest 2013;123(9):3925–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jurisic D, Erjavec I, Trkulja V, Dumic-Cule I, Hadzibegovic I, Kovacevic L, et al. Soluble type III TGFbeta receptor in diagnosis and follow-up of patients with breast cancer. Growth Factors. 2015;33(3):200–9. [DOI] [PubMed] [Google Scholar]
- 33.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–43. [DOI] [PubMed] [Google Scholar]
- 35.Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov 2012;11(10):790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dotor J, Lopez-Vazquez AB, Lasarte JJ, Sarobe P, Garcia-Granero M, Riezu-Boj JI, et al. Identification of peptide inhibitors of transforming growth factor beta 1 using a phage-displayed peptide library. Cytokine. 2007;39(2):106–15. [DOI] [PubMed] [Google Scholar]
- 37.Gallo-Oller G, Vollmann-Zwerenz A, Melendez B, Rey JA, Hau P, Dotor J, et al. P144, a Transforming Growth Factor beta inhibitor peptide, generates antitumoral effects and modifies SMAD7 and SKI levels in human glioblastoma cell lines. Cancer letters. 2016;381(1):67–75. [DOI] [PubMed] [Google Scholar]
- 38.Llopiz D, Dotor J, Casares N, Bezunartea J, Diaz-Valdes N, Ruiz M, et al. Peptide inhibitors of transforming growth factor-beta enhance the efficacy of antitumor immunotherapy. International journal of cancer Journal international du cancer. 2009;125(11):2614–23. [DOI] [PubMed] [Google Scholar]
- 39.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11(8):783–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, et al. A versatile viral system for expression and depletion of proteins in mammalian cells. PloS one. 2009;4(8):e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.