

Abstract
Homocystinuria is a rare inborn error of methionine metabolism caused by cystathionine β-synthase (CBS) deficiency. The prevalence of homocystinuria in Qatar is 1:1,800 births, mainly due to a founder Qatari missense mutation, c.1006 C>T; p.R336C (p.Arg336Cys). We characterized the structure-function relationship of the p.R336C mutant protein, and investigated the effect of different chemical chaperones to restore p.R336C-CBS activity using three models: In silico, ΔCBS yeast, and CRISPR/Cas9 p.R336C knock-in HEK293T and HepG2 cell lines. Protein modeling suggested that the p.R336C induces severe conformational and structural changes, perhaps influencing CBS activity. Wildtype CBS, but not the p.R336C mutant, was able to restore the yeast growth in ΔCBS deficient yeast in a complementation assay. The p.R336C knock-in HEK293T and HepG2 cells decreased the level of CBS expression and reduce its structural stability; however, treatment of the p.R336C knock-in HEK293T cells with betaine, a chemical chaperone, restored the stability and tetrameric conformation of CBS, but not its activity. Collectively, these results indicate that the p.R336C mutation has a deleterious effect on CBS structure, stability, and activity, and using the chemical chaperones approach for treatment could be ineffective in restoring p.R336C CBS activity.
Keywords: Homocystinuria, Qatar, CBS, p.R336C mutation, in silico, in vivo models, chemical chaperones
Introduction
Homocystinuria is a rare metabolic disease caused by cystathionine beta synthase (CBS) deficiency (MIM# 236200), which is characterized by high homocysteine (Hcy) and methionine levels in blood and urine. Catastrophic mutations in CBS are considered the main cause of homocystinuria and an elevation of Hcy in urine can be the first sign for the diagnosis of homocystinuria (Mudd, Finkelstein, Irreverre, & Laster, 1964; Mudd et al., 1985). Qatar has the highest prevalence of homocystinuria in the world (1:1,800 births), making homocystinuria the most prevalent inherited monogenic disease in Qatar (Zschocke et al., 2009). The high prevalence in Qatar is attributed to the founder mutation c.1006 C>T; p.R336C (p.Arg336Cys) (El-Said et al., 2006).
Molecular genetic analyses have so far identified more than 150 different pathogenic mutations in CBS (http://cbs.lf1.cuni.cz/mutations.php). Human CBS is 63 kDa in size and resides in the cytosol as a homotetramer. CBS has three functional domains: (i) an N-terminal domain that binds heme, which was shown to be essential for the redox-linked regulation of CBS (ii) a central catalytic core that binds a cofactor called pyridoxal 5-phosphate (PLP), and (iii) a C-terminal domain that binds the allosteric effector S-adenosylmethionine (AdoMet) (Ereno-Orbea, Majtan, Oyenarte, Kraus, & Martinez-Cruz, 2014). Hcy is an intermediary amino acid that is metabolized by two different pathways. It can either be re-methylated to form methionine, or it can be condensed with serine and enter the transsulfuration pathway. CBS is the first enzyme in the transsulfuration pathway, catalyzing the condensation of Hcy with serine to form cystathionine, which can then be cleaved to form cysteine. The CBS-mediated reaction requires pyridoxine as a cofactor [reviewed in (Škovierová et al., 2016)
It is thought that the Qatari homozygous mutation renders CBS inactive. Due to CBS inactivity, Hcy accumulates, leading to homocystinuria, hypocysteinemia, and hypermethioninemia. The accumulation of Hcy in blood is strongly linked to the development of serious conditions, including cardiovascular and neurodegenerative diseases. Untreated Qatari patients usually undergo severe complications such as mental retardation, physical deformation, and death. Qatari homocystinuric patients do not respond to high doses of pyridoxine (El-Said et al., 2006; Zschocke et al., 2009). Current therapeutic approaches for Qatari patients include life-long methionine-restricted diet and cysteine supplements. However, these therapeutic approaches are not optimal, as dietary compliance can be very difficult. Consequently, there is a huge clinical, financial, and psychosocial burden requiring to develop more efficient therapeutic approaches specific for this mutation (El-Said et al., 2006; Gan-Schreier et al., 2010; Schiff & Blom, 2012; Zschocke et al., 2009)
Rather than focusing on repairing the mutation at the DNA level, which is still a challenge, research is focused on correcting the mutation at the protein level, and hence, restoring CBS enzymatic activity. Recent studies suggest that chemical chaperon (low molecular weight molecules) therapies are emerging novel concept of for protein misfolding diseases (Suzuki, 2014). Different type of chemical chaperones were shown to restore the folding errors in mutated CBS proteins (Kopecka, Krijt, Rakova, & Kozich, 2011; Singh et al., 2007; Suzuki, 2014). However, the effect of these chemical chaperons on the p.R336C mutation and the exact mechanism of action needs further investigation. In this study, we hypothesized that chemical chaperones may restore the activity and functionality of the CBS-p.R336C mutant. To achieve this, we generated and validated three models: (i) In silico, where protein modeling and simulations were performed to study the mutational consequences at the molecular level, (ii) yeast model (Saccharomyces cerevisiae), and (iii) cell culture models involving knocking-in the mutant CBS in HEK293T and HepG2 cell lines. After successful validation, these models were used to assess the effect of the p.R336C mutation on CBS expression, stability, and activity, as well as the ability of different chemical chaperones to restore CBS expression and activity.
Materials and Methods
Chemicals and reagents were purchased from Sigma Aldrich (GmBH, Germany) unless otherwise stated. These chemicals were used directly without further modifications.
Molecular modeling
Structural loop repair
From the protein data bank, we have used the full structure of human CBS (hCBS) domain (missing loop region 515–527) with the best possible high resolution at 2Å (McCorvie et al. 2014) for molecular modeling. This structure has been used in discovery studio (DS) (Accelrys Inc., San Diego, CA, USA) to model/repair the loop region, in which we used potential structural alignments of the missing residues. The repaired structure was further used in docking for original positioning of the heteroatoms heme and pyridoxal 5’-phosphate. The resultant structure is considered as wildtype of hCBS and this is used to create a mutant protein model (p.R336C) as described previously (Gajendrarao et al., 2013; Krishnamoorthy, Yacoub, & Yaliraki, 2011).
Molecular dynamics simulations
The hCBS and p.R336C were used in the GROMACS simulations package for the molecular dynamics (MD) simulations. The heteroatoms and the amino acids were prepared for the simulations by applying GROMOS96 force field (Hess, Kutzner, van der Spoel, & Lindahl, 2008; Van Der Spoel et al., 2005; Van Gunsteren, 1996). The water model of SPC3 was used for solvation of the proteins in a cubic box with a size of 1.5 nm. In order to neutralize the systems, the counter ions were added, and the periodic boundary conditions were applied in all directions. The resulting systems consist of 141,443 atoms to 141,452 atoms. For handling the long-range interactions, a twin range cut-off was used: 0.8 nm for van der Walls and 1.4 nm for electrostatic interactions. The SETTLE algorithm (Shuichi Miyamoto, 1992) was applied to constrain the geometry of the water molecules, while the LINCS algorithm (Berk Hess Henk Bekker & Fraaijey, 1997) was utilized to constrain all the bond lengths. The systems were energy minimized by applying steepest descent algorithm with a tolerance of 2,000 Kj/mol/nm. The resultant structures were pre-equilibrated with 100ps simulation prior to producing MD simulation of 25ns with a time‐step of 2 fs at constant temperature (300 K), pressure (1 atm), and number of particles, without any position restraints. At every 100 ps, a coordinate file was obtained and the tools of PyMOL, DS, and GROMACS were used to analyze changes in structure, stability and molecular interactions.
Cluster analysis and surface map
To select the representative structure from MD simulations, the collected structures of 2,500 from MD simulations were all grouped into clusters according to their structural deviations. The top-ranked cluster was selected for representation as it is a frequently occurring conformation. These representative structures were used in surface mapping analysis in DS to identify changes in the binding cavities and surface conformations.
Yeast model generation
The wildtype human CBS cDNA (sequence reference # NM_001178008.2), the CBS c.1006 C>T and c.833 T>C mutant sequences were PCR amplified from pLW2:hCBS, pLW2:R336C, and pLW:I278T, respectively (L. Wang et al., 2004). The PCR-amplified cDNA fragments were cloned into a gap repair yeast cloning and expression plasmid pYEPGAPx:ΔhCBS, and were transformed into the S. cerevisiae ΔCBS yeast strain, WY35 (WY35ΔyCBS), by lithium acetate co-transformation as described in (Shan, Dunbrack Jr, Christopher, & Kruger, 2001). For simplicity, the resulted yeast strains were named as follows: (i) pLhCBS strain; contained wildtype human CBS cDNA and served as a positive control, (ii) pLR336C; contained the Qatari mutant CBS cDNA c.1006 C>T, (iii) pLI278T strain; contained human CBS cDNA c.833 T>C and served as a control, and (iv) pLΔhCBS strain; contained the empty pYEPGAPx and served as a negative control. In-frame cloning of all cDNAs was confirmed by sequencing. Note that the YEPGAPx:ΔhCBS plasmid harbors the gene required for the gap repair recombination together with a XhoI site that is flanked by a small sequence (~100 bp) of the N and C termini of the human CBS gene and also carries TRP yeast genes and ambC bacterial selection markers (Kruger & Cox, 1994).
Generation of p.R336C knock-in HEK293T and HepG2 cells using CRISPR/Cas9
Designing and cloning of CBS sgRNA in the plasmid PX459
pSpCas9 (BB)-2A-Puro (PX459) plasmid was provided by ZeClinics, Spain. This vector contains the cloning site for sgRNA sequence under U6 promoter, Cas9 sequence that can be activated by CBh promoter, and a Puromicine gene that allows successful selection of transfected cells. The vector was cut with BbsI (New England BioLabs) and loaded on 1% agarose gel, and the corresponding band was purified using the illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare). Top and bottom oligonucleotides of the sgRNA targeting the sequence of CBS were designed using Benchling design software. Sequences of sgRNA were 5’-CACCCTCTTGCGCGATCAGCATGC-3’ and 5’-AAACGCATGCTGATCGCGCAAGAG-3’ for the forward and reverse oligonucleotides, respectively. Forward and reverse sgRNA oligonucleotides were aligned through incubation at 95°C for 5 min and subsequent spontaneous heat reduction until reverse transcription. Upon sgRNA oligonucleotides alignment, the resulting double-stranded fragment was cloned into the BbsI cloning site of the vector. The ligation reaction was performed at 25°C for 1 h using 4 μL of the sgRNA diluted 1:400 in annealing buffer 0.5× and 200 ng of the digested plasmid using the DNA ligase T4 kit (Sigma Aldrich). Homemade Top10 bacteria were transformed with 4 μL of ligation reaction through heat shock method, plated in LB-agarose plates with 100 μl/mL ampicillin, and incubated overnight at 37°C. Subsequently, the colonies were picked and grown overnight in 3 mL LB-AMP. The plasmids were purified using GeneAll ExprepTM Plasmid DNA mini kit (GeneAll Biotechnology) and sent for sequencing at the in house genomic facility of the Universitat Pompeu Fabra (UPF). The primer used for sequencing is 5’-GACTATCATATGCTTACCGT-3’. Human embryonic kidney cells (HEK293T, ATCC® ACS-4500™) and human liver cancer cells (HepG2, ATCC HB-8065) were used for transfection experiments with PX459_sgCBS and ssODN_p.R336C-CBS. The sequence of the single-stranded oligodeoxynucleotide designed to introduce the p.R336C mutation in CBS (ssODN_p.R336C-CBS) was GGACAAGTGGTTCAAGAGCAACGATGAGGAGGCGTTCACCTTTGCTTGCATGTT GATCGCGCAAGAGGGGCTGCTGTGCGGTGAGTGGGTGGCGGGCA (Supp. Figure S1). HEK293T and HepG2 cells were grown in DMEM media with 4.5 g/L glucose without L-glutamine (Lonza) supplemented with 10% FBS (Gibco), 1% L-glutamine 200 mM (Lonza), and 1% penicillin/streptomycin stock 10K/10K (Lonza). Lipofectamine 3000 (ThermoFisher) was used for the transfection of HEK293T cells, while TransfeX (ATCC) was used for the transfection of the HepG2 cells. Cells were incubated with the respective transfection reagent-DNA mixes following the manufacturers’ instructions for 24 h and subsequently grown for 48 h. The drug RS1 (Sigma Aldrich) was used to improve the recombination efficiency with the genomic DNA of the cells. Transfected cells were selected using Puromycin (Ibian Tech) at a final concentration of 2 μg/mL for 3 d.
Assessment of CBS mutation efficiency
Genomic DNA was extracted from the HEK293T and HepG2 cells using the Extract-N-Amp Tissue PCR Kit (Sigma Aldrich), 250 ng DNA was used to perform PCR with the Expand High fidelity PCR System (Roche). The following primers were used to amplify the CBS gene: AGCTGACTTTGGCCTGAGGGAG (forward), ACATTCTGGAAGGCAGCTCGAAG (reverse). The amplified gene was purified from the agarose gel and digested with SphI to test the presence of the mutation. The mutated allele can be generated by recombination of the PX459 vector containing the sgRNA targeting CBS only (knock-out, KO), or by recombination of both the vector and the ssODN_p.R336C-CBS (knock-in, KI), with the genome of the cells. Overall mutation efficiency was measured by calculating agarose gel bands intensity with ImageJ software (Supp. Figure S1). Cells were grown in single cell suspension in 96-well plates. Each clone was expanded and screened for the presence of mutated alleles through PCR and SphI digestion (Supp. Figure S1) as described earlier. SphI-negative clones (possible p.R336C mutated) were sent for Sanger sequencing to distinguish between KI and KO homozygous and heterozygous cells (Supp. Figure S1).
For simplicity, the CRISPR-CAS p.R336C knock-in HEK293T cells were designated as p.R336C-HEK, while the CRISPRCAS p.R336C knock-in HepG2 cells were designated as p.R336C-HepG2. The wildtype cells for both cell lines were designated as wt-HEK and wt-HepG2.
Assessment of CBS mRNA expression
RNA extraction from cells was performed from 70% cell confluent 6-well plates using Trizol. Three technical replicates were performed for each sample. cDNA was obtained by reverse transcription using Invitrogen kit (Cat. No.1808030050) starting with 50 ng of RNA and following the manufacturer’s instructions. qPCR was performed using Roche kit (Cat. No. 0470751600) following the manufacturer’s instructions with an annealing temperature of 58°C and 45 amplification cycles. Three sets of primers were designed for CBS cDNA and three other sets for house-keeping GAPDH for standardization of the experiment and selecting the best primers for amplification of both genes (data not shown). The primers used for measuring the mRNA expression level of both genes were CBS (forward): CGTGATGCCAGAGAAGATGA, CBS (reverse): TTGGGGATTTCGTTCTTCAG, GAPDH (forward): CGACCACTTTGTCAAGCTCA, and GAPDH (reverse): AGGGGAGATTCAGTGTGGTG.
Cell viability and growth curve experiments
Two different models were used in this study: the yeast model and the cell culture model with two cell lines (HEK293T and HepG2). For yeast, we followed the same protocols described in (Kruger & Cox, 1995). Briefly, yeast cells from a saturated synthetic complete (SC-trp, Formedium) media overnight culture were centrifuged, washed once, and subcultured in borosilicate culture tubes. Cells were diluted to a starting OD600 of approximately 0.03 in synthetic complete (SC) media without tryptophan in presence (SC+Cys-trp) and absence of cysteine (SC-Cys-trp). Growth of different strains in both types of media was estimated as a function of OD600 at different time intervals. For HEK293T and HepG2 cells, cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco), 1% antibiotic/anti-mycotic mixture (Gibco), and 1% L-glutamine (Gibco).
Chemical chaperone treatment
HEK293T cells were treated with four chemical chaperons (sorbitol, proline, betaine, and glycerol). Briefly, cells were cultured in 6-well plates in complete media (DMEM supplemented with 10% FBS, 1% antibiotics, and 1% L-glutamine), and incubated for 16–18h at humidified conditions (37°C, 5% CO2). Cells were then treated with different concentrations of the chemical chaperones, 48h post-treatment, cells were lysed using RIPA buffer (ThermoFisher) supplemented with a protease inhibitor cocktail (ThermoFisher). Cell lysates were collected and centrifuged at 15,000 RPM for 15 min to be used for protein analysis. Protein concentration from both models was determined using BCA assay (Pierce) using bovine serum albumin (BSA) as a standard. CBS expression and conformation were assessed by Western blot analysis.
Gel electrophoresis, Western blotting, and CBS activity
To assess CBS expression in yeast, protein was extracted by a modified post-alkaline extraction procedure as previously described (Kushnirov, 2000). To assess the protein conformation, yeast extracts were prepared via mechanical lysis by sonication for 10 sec followed by 1 min on ice for four cycles at 100% amplitude (Liu, Zeng, Sun, & Han, 2013). Extracts containing 48 μg of total protein were separated on 10% native and denatured PAGE as described by (Singh & Kruger, 2009). Protein expression and conformation was assessed in mammalian cell culture model using the same standard procedure. Protein was transferred onto a polyvinylidene difluoride (PVDF) membrane (Invitrolon, Novex, life technologies) that were probed with immunopurified mouse anti-CBS pAb (Abnova) at a 1/1,000 dilution followed by a 1/5,000 dilution of goat anti-mouse IgG (H&L) secondary antibody (Abnova). Probed membranes were subjected to enhanced chemiluminescence reagent (ECL, Abcam).
CBS activity was determined using Biochrom 30+ Amino Acid Analyzer as previously described (Z. Wang, Li, Cho, & Malik, 2014), by measuring the amount of cystathionine formed per mg of CBS protein per h. The reaction mixture was composed of 200 mM Na-N, N-Bis (2-hydroxyethyl) glycine buffer (pH 8.6), 10mM DL-Hcy, 5mM L-serine, 50μM pyridoxal phosphate, 250 μM S-adenosylmethionine, and 0.03 or 0.015 mg of protein extracted from mammalian cells and yeast, respectively. The reaction mixture was incubated for 60 min at 37°C and CBS activity was expressed as nanomoles of cystathionine/mg of protein/h.
Statistical analysis
Data were represented as mean ±SD and statistical significance (taken as *p<0.05) was determined using GraphPad Prism 5. The specific statistical test used in each experiment is mentioned in the figure legends.
Results
In silico protein modeling and simulations
The modeled loop in hCBS revealed the structure and conformation of the loop region 515–527. This loop plays a key role in the substrate binding mechanism that was recently reported (McCorvie et al., 2014) (Figure 1A). The modeling of hCBS showed the importance of the location of residue Arg336, which is close to the catalytic core region. The simulation of hCBS described the stable nature of the wildtype. In contrast, the p.R336C mutation induced a large conformational shift in the secondary structural elements, which affects the functional core region (Figure 1C). These changes reduced the conformational stability of p.R336C (Figure 1Ba) and led to conformational changes (Figure 1C). As a result, the regular intra-molecular hydrogen bonds are increased (Figure 1Bb). The Arg residue is at position 336 of wt-hCBS and contributes to the local stability by forming three hydrogen bonds, on average, during the simulation (Figure 1Bc). However, in the p.R336C mutant, the Cys residue at 336 was unable to maintain the local network and stability compared to hCBS. The above changes at the integral part of the mutant bring a significant transformation to the surface with an enlargement in the near-field of the mutational spot that appears like a cavity (Figure 1D). This modeling provides a molecular basis of the mutation that could affect the function by introducing conformational changes due to the modification in the number of hydrogen bonds and the surface. However, structure-functional relationships were further studied in detail to correlate the structural changes with the phenotype using in vitro and in vivo experimental analyses.
Figure 1.
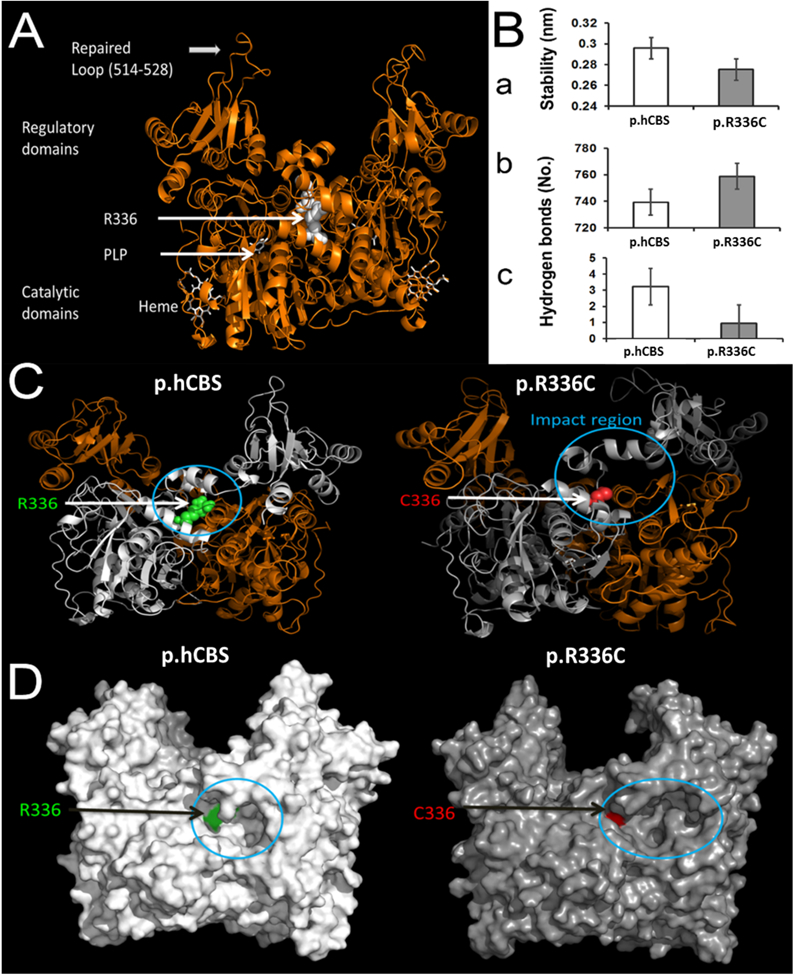
In silico model for the human CBS protein and the R336C mutant. (A) 3D structure of hCBS protein with modeled/repaired loop (515–527). The location of residue 336 (white spheres) is on the catalytic domain and it is adjacent to the catalytic core where the PLP molecule binds. PLP and Heme molecules are represented as white tubes. (B) Mutation-induced structural instability. a) Structural stability (RMS deviation) of wildtype (hCBS) and mutant R336C. b) Total number of hydrogen bonds during simulations. c) Average number of hydrogen bonds by residue 336 in wildtype and the mutant. (C) Large shift on the secondary structural elements caused by the mutation in the neighboring region. R336 and C336 are showed as spheres in green and red respectively. The impact region is highlighted with a circle near the catalytic region. (D) Surface representation of the structures of wildtype and the mutant CBS proteins. The cavity on the surface is indicated with a circle. The green and red patches on the surface through the cavity are R336 and C336 respectively.
p.R336C failed to restore the growth defect in the CBS-deficient yeast (ΔCBS)>
We used the previously established yeast model by (Kruger & Cox, 1994) to study the ability of pLR336C plasmid to restore the growth defect of ΔCBS, as indirect assessment of p.R336C enzyme activity in vivo. As shown in Figure 2A, all yeast strains were able to grow in the presence of cysteine. However, only the yeast strain carrying pLhCBS plasmid was able to grow on media lacking cysteine, while the strains carrying the pLI278T, the pLΔhCBS, and the pLR336C mutant plasmid failed to grow. The p.I278T is another deleterious CBS mutation, in which isoleucine is replaced by threonine at position 278 (Kruger & Cox, 1994, 1995), and was used as a control. These results suggested that the p.R336C mutation is lethal in yeast, and exogenous cysteine is essential to maintain growth due to the malfunctioning CBS activity.
Figure 2.

Effect of different CBS gene mutations on the growth of yeast cells. (A) Yeast stains cells were grown in selection media for 4 d at 28oC in the presence or absence cysteine. (B) Western blot analysis. Yeast cell lysates were prepared and examined for CBS expression by both denaturing and native PAGE gels followed by immunoblot. For the denaturing gel, modified alkaline protein extraction method was used using equal weights 0.03 g of cells and loading 20μl per lane. For the native-PAGE: 48μg of total protein was loaded per lane. CBS activity (underneath the native) was expressed in nmol/mg of total protein per h. (C) Quantitative measurement of CBS protein expression level by measuring the band intensity by ImageJ software using three different membranes. Data represented as mean± SD of three experiment each done in triplicate n=9. Statistical analysis for the effect between different strains was measured by two-way ANOVA followed by Bonferroni post-hoc test (*p<0.05).
p.R336C mutation affects the level CBS expression and activity in yeast
Western blot analysis of the four yeast strains under both denaturing and native conditions was performed. As shown in Figure 2B, steady-state levels of CBS in the pLR336C mutant yeast strain was lower than that in the pLhCBS strain. However, pLR336C expressed higher level of CBS protein compared to the mutant strain carrying the p.I278T mutant, which is known to affect protein stability (Singh & Kruger, 2009). Similar to hCBS, p.R336C protein seems to form a tetramer as assessed by native gel electrophoresis (Figure 2B). Whether such bands represent a functional or nonfunctional tetrameric CBS protein, our results indicate that the p.R336C mutation renders CBS non-functional. That is, CBS activity in the whole cell lysates of the pLR336C strain was lower than the detection limit (<5.0 nmol/mg/h), compared to 109.7 nmol/mg/h of the pLhCBS strain (Figure 2B). Collectively, our yeast model findings support the in silico model results, in which the p.R336C mutation might have introduced conformational changes in the structure of CBS, making it non-functional (Figure 2B)
CBS expression and activity in HEK293T and HepG2 cells
We used CRISPR/Cas9 knock-in technology to create p.R336C CBS mutation in HEK293T kidney and HepG2 liver cells (Supp. Figure S1). Due to the nature of disease pathology, we initially chose HepG2 cells as CBS is known to be expressed at a high level in the liver cells. However, at first, we failed to transfect HepG2 cells with the Cas9 plasmid. In fact, the resistance of HepG2 cells for transfection is also reported in the literature (Das et al., 2016). Thus, as a proof of concept, we decided to use, in parallel, a well-adapted laboratory cell line (HEK293T cells) that is relatively easy to transfect and capable of expressing sufficient amounts of CBS, which is well expressed in the kidney. In human and mouse, it is known that CBS is expressed predominantly in liver, pancreas, kidney, and brain [reviewed in (Kruger, 2017)]. As shown in Figure 3A and 3B, wt-HEK293T cells express high levels of CBS mRNA and protein, making them suitable for our study. Surprisingly, it turned out that wt-HEK293T was a better cell line to study CBS protein than HepG2 cells as the former cells showed a better CBS protein expression level and activity (501.1 mU/mg/h) than the wt-HepG2 cells (9.8 mU/mg/h) (Figure 3B).
Figure 3.

CBS expression in HEK293T and HepG2 at mRNA and protein levels. (A) qRT-PCR quantification of the expression of CBS mRNA in HEK293T and HepG2. CBS/GAPDH (Y-axis) ratio was used to measure fold change in the level of CBS expression. (B&D) Western blots of wildtype and mutant CBS proteins in HEK293T and HepG2. 40 μL of cell lysates were loaded in 10% denaturing (B) and native PAGE. Note: CBS activity was measured as described in the “Materials and methods” section. (D). GAPDH was used as a loading control. (C) Relative band intensity analyzed by ImageJ software using GAPDH as a control. Data represented as mean± SD (n=3). Statistical analysis was carried out with one-way ANOVA followed by Tukey’s Multiple Comparison Tests (*p<0.05).
p.R336C mutation affects CBS mRNA expression, structure, and activity in mammalian cells
To study the effect of c.1006 C>T mutation (p.R336C) on CBS gene expression, the level of mRNA expression in both wildtype and mutant HEK293T and HepG2 cells was measured in relation to GAPDH mRNA levels (Figure 3A). There was no significant difference in the mRNA expression level of CBS between the wt-HEK and the p.R336C-HEK cells. However, in HepG2 cells, c.1006 C>T mutation significantly decreased mRNA expression level of CBS.
Next, we examined the effect of p.R336C mutation on CBS protein level in both cell lines by denatured Western blot analysis. CBS protein expression (as a monomer) was significantly lower in p.R336C-HEK cells compared to wt-HEK cells (Figure 3B & 3C). Notably, CBS monomer was not detected in p.R336C-HepG2 cells compared to wt-HepG2 cells, suggesting that p.R336C mutation has a dramatic effect on CBS protein expression level in both cell lines. Furthermore, the native gel showed the presence of the CBS tetrameric active conformation in wt-HEK cells, which was absent in p.R336C-HEK cells (Figure 3D). These findings were congruent with the CBS activity results, in which CBS activity was below detection limit (<5 mU/mg/h) in p.R336C-HEK and p.R336C-HepG2 cells compare to wt-HEK and wt-HepG2 cells (501.1 and 9.8 mU/mg/h, respectively) (Figure 3B). These results suggest that p.R336C mutation leads to significant destabilization of CBS in mammalian cells.
Betaine restores CBS conformation, but not activity, in HEK293T cells
Since CBS protein was only detectable in p.R336C-HEK293T cells and not in p.R336C-HepG2 cells, we tested the ability of the chemical chaperons to restore the p.R336C mutant activity in HEK293T cells. As shown in Figure 4, CBS protein was undetectable in untreated p.R336C-HEK cells due to its degradation. However, the four chemical chaperons, betaine (0.5M and 0.8M), sorbitol (0.4M and 0.6M), proline (0.1M and 0.4M), and glycerol (0.05M and 0.1M), were capable of retrieving CBS protein in the p.R336C-HEK cells (Figure 4). Yet, the retrieved CBS protein did not always have the right tetrameric conformation. Although sorbitol, proline, and glycerol protected CBS from degradation, they were unable to restore the active tetrameric conformation. Hence, CBS activity was not assessed in p.R336C-HEK cells after treatment with sorbitol, proline, and glycerol. Interestingly, betaine not only protected the CBS protein from undergoing degradation, but also restored the tetrameric conformation (Figure 4). Surprisingly, however, CBS activity was below the detection limit (<5.0 mU/mg/h) in betaine-treated p.R336C-HEK cells. These results indicate that betaine can re-establish the tetrameric conformation of p.R336C CBS, but not its activity, in HEK293T cells.
Figure 4.

Restoring CBS protein conformation in p.R336C-HEK cells by treatments with different chemical chaperons. Cell lysates were prepared and examined for CBS conformation using 10% native PAGE gels followed by immunoblotting. 40 μg of protein sample was loaded per lane. This experiment were repeated three times and similar results were obtained. CBS activity was measured as described in the “Materials and methods” section. CBS activity in sorbitol-, proline-, and glycerol-treated HEK293 was not assessed as these chaperones did not restore the tetrameric conformation of CBS
Discussion
One of the most severe homocystinuria phenotypes is due to the p.R336C mutation, which was shown to be non-responsive to the pharmaceutical therapeutic approaches. Therefore only low methionine diet can help the patients harboring this mutation. Furthermore, little is known about this mutant in terms of structural conformation, in vitro, and in vivo responses to different treatments. For instance, clinically, supplementation with high dose of pyridoxine (vitamin B6) was shown to lower the plasma level of Hcy in patients with the p.I278T (Kluijtmans et al., 1999; J. P. Kraus et al., 1999), but not the p.R336C mutation (El-Said et al., 2006), hence the need for additional strategies to lower plasma Hcy in patients harboring the p.R336C mutation. Here, we used the previously characterized CBS mutation, p.I278T, as a negative control for the yeast growth and rescue experiments (Figure 2).
We constructed an in silico model that revealed the structural and conformational differences between hCBS and its p.R336C mutant (Figure 1). The p.R336C mutation causes severe secondary structural changes to the functional core region. The near-field of the mutational spot on the surface is modified and looks like a cavity (Figure 1). This cavity might provide a binding site for a single or multiple substrates. It is possible that this cavity exhibits a new access route for the substrates to the functional core, which seems to be different from previous report (McCorvie et al., 2014). However, further studies with target substrates are needed to understand the entry/exit mechanism in order to design selective drugs that could retrieve CBS function. Our in silico model shows that p.R336C is also a buried mutation that confers lower structural stability compared to the hCBS. These similarities between the p.R336C and the p.I278T mutants explain the reason of using p.I278T as a control in our study. Overall, our molecular modeling data suggest that the p.R336C mutation induced conformational rearrangements at the functional core region, leading to loss of CBS activity.
CBS was reported to have several conformations, with the tetrameric and the higher multimeric forms; the correctly folded and active forms. Hence, any other form with misfolded conformation will force CBS to exist in an inactive state. In addition, it was previously reported that several CBS mutations cause a sharp decline in the resultant active tetramer compared to wildtype CBS, which was due to improper folding (Kozich et al., 2010). Our findings in the yeast model showed the presence of tetrameric CBS protein in the pLR336C strain that resembled the one in the pLhCBS strain (Figure 2). However, the pLR336C strain failed to grow in the absence of exogenous cysteine, and CBS activity in the p.R336C mutant strain was below the detection limit, which explains the inability of p.R336C CBS enzyme to support the growth of yeast and confirms our in silico and the mammalian cell model findings.
As previously reported (Singh et al., 2007; Singh & Kruger, 2009), we showed that the p.I278T mutation leads to destabilization and degradation of CBS in yeast. However, this was not the case with the p.R336C mutation (Figure 2B). The different patterns of expression and migration between p.I278T and p.R336C in the denatured and native gels suggest that these two mutations may behave differently in our yeast model. Furthermore, the differences in the migration (stability) pattern of the p.R336C in yeast and mammalian cell models suggests that heterologous expression systems behave differently with regards to modeling missense mutations. We believe that although p.R336C CBS protein may exist as a stable tetramer in p.R336C mutant yeast strain or in p.R336C-HEK cells in response to betaine treatment, these tetrameric forms are most likely misassembled and are not identical to the wt-hCBS conformation, which may explain the loss of p.R336C CBS protein activity.
Our study is the first of its kind to use the knock-in technology to examine the effect of the p.R336C mutation in mammalian systems. The p.R336C mutation, which was successfully introduced in the HEK293T and HepG2 cells (Figure 3), had a more profound effect on CBS assembly in both cell lines than in the yeast model, whereby the properly assembled, functional tetrameric form of CBS was missing. The qRT-PCR results showed that p.R336C mRNA expression in both cell lines (Figure 3A). We do understand that it might be hard to explain the reduction of mRNA levels harboring missense mutations. However, we constantly got these data, and this could be explained due changes in the stability of mRNA caused by p.R336C, which might lead to partial degradation of mRNA, however testing for this is out of the scope of this work.
Yet, proper assembly of CBS protein seems to fail, at least in p.R336C-HEK cells (Figure 3D). Maclean et al. showed 40 mU/mg when cells were harvested at 70% confluence and about 15 mU/mg when cells were quiescent (Maclean et al., 2002), compared to 20 mU/mg in wt-HepG2 cells in this study (Figure 3). These differences could be due to the variations in methodology used, in Maclean et al. study, the CBS activity was measured by the radioisotope assay using [14C]-serine (Jan P. Kraus, 1987); however, in this study, we used a non-radioactive method using Biochrom 30+ Amino Acid (L. Wang et al., 2004).
CBS is mainly synthesized in the liver. Thus, we expected that HepG2 would be the ideal cell line to assess the effects of the p.R336C mutation. Yet, it turned out that CBS activity in the wt-HepG2 was relatively low (9.8 mU/mg/h) compared to the wt-HEK293T (501.1 mU/mg/h) (Figure 3). It is known that CBS activity positively correlates with the rate of cell proliferation in humans and yeast (Blank, Gajjar, Belyanin, & Polymenis, 2009). Therefore, it was not surprising that the wt-HepG2 cells had lower CBS activity than wt-HEK293T, as the doubling time of wt-HEK293T cells (16–24 h) is about three times shorter than that of wt-HepG2 cells (48–72 h) (Amann et al., 2009; López-Terrada, Cheung, Finegold, & Knowles, 2009; Soni & Lai, 2011).
Casique and colleagues over-expressed two CBS mutants (p.T87N and p.D234N) in HEK293 cells using pcDNA4A/HisA (Casique, Kabil, Banerjee, Martinez, & De Lucca, 2013). They found lower soluble expression of CBS in HEK-293 cells: 19% and 23% in p.T87N and p.D234N mutants, respectively, compared to wt-CBS. Although they demonstrated excellent CBS protein expression in wt-HEK293 cells using anti-human CBS antibodies, they reported that they were not able to detect endogenous CBS expression in HEK-293 cells, which contradicts our observation of high expression and activity of endogenous CBS (Figure 3). This could be due to the variation among the HEK293 cell lines used in both studies. It should be noted that Casique et al. did not specify the HEK293 cell lineage used in their study. Interestingly, Lin and colleagues reported genomic and copy number variations among different lineages of HEK293 cells (e.g. 293T, 293S, and 293SG) (Lin et al., 2014). Furthermore, Casique and colleagues did not measure CBS activity in wt-HEK293 cells, but only in HEK293 cells transfected with mutated CBS. They could not rule out the possibility that recombinant (mutated CBS) and endogenous CBS subunits do not co-assemble in the transfected HEK-293 cells, and hence, they were unable to detect endogenous CBS in these cells.
Out of the four chemical chaperones, only betaine was able to prevent the degradation of the p.R336C mutant form of CBS in HEK293T cells, allowing the detection of the tetrameric form of CBS (Figure 4). Yet, and despite the restoration of the tetrameric form of CBS, betaine failed to restore CBS activity (Figure 4). Our findings were consistent with what previously reported by Kopecka et al. (2011), who studied the effect of selected chaperones on 27 different CBS mutants. Of which, four mutants showed increased formation of CBS tetramers, without increase in the CBS activity (Kopecka et al., 2011). In conclusion, our results suggest that the p.R336C mutation could be resistant to different chemical chaperones.
We have used different types of chaperone treatments at different concentrations in both yeast and tissue culture models other than the one shown in figure 4, such as DMSO, ethanol, heat, and even combinations of different treatments. All of these treatments were not able to rescue the conformation of the p.R336C CBS mutant protein as betaine did (data not shown). However, we cannot rule out that other chaperone treatments that are available in the market could be able to rescue the p.R336C CBS conformation and activity defects. In addition, our study emphasizes the need for further investigation to improve chaperone treatment in order to benefit the Qatari patients. One approach would be to use fibroblasts from Qatari CBS patients as a disease model and to test the potential effects of a wider range of chaperones on cells isolated from patients. This study is already underway.
Different treatment approaches, such as gene therapy or CBS enzyme replacement therapy (Bublil et al., 2016) could be more promising than chemical chaperone approach for treating Qatari patients. Other treatment options include the use of protein stabilizers or proteasome inhibitors. For instance, in a recent study, proteasome inhibitors were reported to rescue the phenotype in p.S466L and p.I278T mutant mice (Gupta et al., 2013). In addition, bortezomib was reported to restore the growth and CBS activity in p.I278T mutant yeast as well as in mice (Singh, Gupta, Honig, Kraus, & Kruger, 2010). Hence, proteasome inhibitors are considered potential future candidates to be tested with our p.R336C mutation.
Supplementary Material
Acknowledgment
The authors would like to thank the following for their help in the technical work of the grant: Dr. Hanan Rizk, Ms. Layla Mohammed, Ms. Salma Yones, and Ms. Enas AlAbsi. We express our gratitude to the facility and technical team of research computing at Texas A&M University in Qatar for providing supercomputing for calculations.
Funding
This work was supported by NPRP grant # (NPR7-355-3-088). This report (publication) was made possible by NPRP grant # (NPR7-355-3-088) from the Qatar national research fund (a member of Qatar foundation). Additional support for S.G and W.D.K. is from NIH (DK101404). The statements made herein are solely the responsibility of the authors.
Footnotes
Ethical compliance
None. No human or animal samples were used in this study.
Conflict of interest statement
None to declare.
References
- Amann T, Bataille F, Spruss T, Mühlbauer M, Gäbele E, Schölmerich J, … Hellerbrand C (2009). Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer science, 100(4), 646–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess Berk, Bekker Henk, B. HJC, & Fraaijey Johannes G. E. M.(1997). LINCS: A Linear Constraint Solver for Molecular Simulations. J Comput Chem, pp. 1463–1472. [DOI] [PubMed] [Google Scholar]
- Blank HM, Gajjar S, Belyanin A, & Polymenis M (2009). Sulfur metabolism actively promotes initiation of cell division in yeast. PLoS One, 4(11), e8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bublil EM, Majtan T, Park I, Carrillo RS, Hulkova H, Krijt J, … Kraus JP (2016). Enzyme replacement with PEGylated cystathionine beta-synthase ameliorates homocystinuria in murine model. J Clin Invest, 126(6), 2372–2384. doi: 10.1172/jci85396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casique L, Kabil O, Banerjee R, Martinez JC, & De Lucca M (2013). Characterization of two pathogenic mutations in cystathionine beta-synthase: different intracellular locations for wild-type and mutant proteins. Gene, 531(1), 117–124. doi: 10.1016/j.gene.2013.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J, Han JW, Choi YJ, Song H, Cho SG, Park C, … Kim JH (2016). Cationic lipid-nanoceria hybrids, a novel nonviral vector-mediated gene delivery into mammalian cells: investigation of the cellular uptake mechanism. Sci Rep, 6, 29197. doi: 10.1038/srep29197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Said MF, Badii R, Bessisso MS, Shahbek N, El-Ali MG, El-Marikhie M, … Zschocke J (2006). A common mutation in the CBS gene explains a high incidence of homocystinuria in the Qatari population. Hum Mutat, 27(7), 719. doi: 10.1002/humu.9436 [DOI] [PubMed] [Google Scholar]
- Ereno-Orbea J, Majtan T, Oyenarte I, Kraus JP, & Martinez-Cruz LA (2014). Structural insight into the molecular mechanism of allosteric activation of human cystathionine beta-synthase by S-adenosylmethionine. Proc Natl Acad Sci U S A, 111(37), E3845–3852. doi: 10.1073/pnas.1414545111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajendrarao P, Krishnamoorthy N, Kassem H, Moharem-Elgamal S, Cecchi F, Olivotto I, & Yacoub MH (2013). Molecular modeling of disease causing mutations in domain C1 of cMyBP-C. PLoS One, 8(3), 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego-Villar L, Hannibal L, Haberle J, Thony B, Ben-Omran T, Nasrallah GK, … Blom HJ (2017). Cysteamine revisited: repair of arginine to cysteine mutations. J Inherit Metab Dis, 40(4), 555–567. doi: 10.1007/s10545-017-0060-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan-Schreier H, Kebbewar M, Fang-Hoffmann J, Wilrich J, Abdoh G, Ben-Omran T, … Al Khal AL (2010). Newborn population screening for classic homocystinuria by determination of total homocysteine from Guthrie cards. The Journal of pediatrics, 156(3), 427–432. [DOI] [PubMed] [Google Scholar]
- Gupta S, Wang L, Anderl J, Slifker MJ, Kirk C, & Kruger WD (2013). Correction of cystathionine beta-synthase deficiency in mice by treatment with proteasome inhibitors. Hum Mutat, 34(8), 1085–1093. doi: 10.1002/humu.22335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, & Lindahl E (2008). GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput, 4(3), 435–447. doi: 10.1021/ct700301q [DOI] [PubMed] [Google Scholar]
- Kluijtmans LA, Boers GH, Kraus JP, van den Heuvel LP, Cruysberg JR, Trijbels FJ, & Blom HJ (1999). The molecular basis of cystathionine β-synthase deficiency in Dutch patients with homocystinuria: effect of CBS genotype on biochemical and clinical phenotype and on response to treatment. The American Journal of Human Genetics, 65(1), 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecka J, Krijt J, Rakova K, & Kozich V (2011). Restoring assembly and activity of cystathionine beta-synthase mutants by ligands and chemical chaperones. J Inherit Metab Dis, 34(1), 39–48. doi: 10.1007/s10545-010-9087-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich V, Sokolova J, Klatovska V, Krijt J, Janosik M, Jelinek K, & Kraus JP (2010). Cystathionine beta-synthase mutations: effect of mutation topology on folding and activity. Hum Mutat, 31(7), 809–819. doi: 10.1002/humu.21273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus JP (1987). Cystathionine β-synthase (human) In Methods in Enzymology (Vol. 143, pp. 388–394): Academic Press. [DOI] [PubMed] [Google Scholar]
- Kraus JP, Janosik M, Kozich V, Mandell R, Shih V, Sperandeo MP, … Gaustadnes M (1999). Cystathionine beta-synthase mutations in homocystinuria. Hum Mutat, 13(5), 362–375. doi: 10.1002/(SICI)1098-1004(1999)13:5<362::AID-HUMU4>3.0.CO;2-K [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy N, Yacoub MH, & Yaliraki SN (2011). A computational modeling approach for enhancing self-assembly and biofunctionalisation of collagen biomimetic peptides. Biomaterials, 32(30), 7275–7285. [DOI] [PubMed] [Google Scholar]
- Kruger WD (2017). Cystathionine beta-synthase deficiency: Of mice and men. Mol Genet Metab, 121(3), 199–205. doi: 10.1016/j.ymgme.2017.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger WD, & Cox DR (1994). A yeast system for expression of human cystathionine beta-synthase: structural and functional conservation of the human and yeast genes. Proc Natl Acad Sci U S A, 91(14), 6614–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger WD, & Cox DR (1995). A yeast assay for functional detection of mutations in the human cystathionine beta-synthase gene. Hum Mol Genet, 4(7), 1155–1161. [DOI] [PubMed] [Google Scholar]
- Kushnirov VV (2000). Rapid and reliable protein extraction from yeast. Yeast, 16(9), 857–860. doi: 10.1002/1097-0061(20000630)16:9<857::AID-YEA561>3.0.CO;2-B [DOI] [PubMed] [Google Scholar]
- Lawson-Yuen A, & Levy HL (2006). The use of betaine in the treatment of elevated homocysteine. Mol Genet Metab, 88(3), 201–207. doi: 10.1016/j.ymgme.2006.02.004 [DOI] [PubMed] [Google Scholar]
- Lin YC, Boone M, Meuris L, Lemmens I, Van Roy N, Soete A, … Callewaert N (2014). Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat Commun, 5, 4767. doi: 10.1038/ncomms5767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Zeng XA, Sun DW, & Han Z (2013). Disruption and protein release by ultrasonication of yeast cells. Innovative Food Science & Emerging Technologies, 18, 132–137. doi: 10.1016/j.ifset.2013.02.006 [DOI] [Google Scholar]
- López-Terrada D, Cheung SW, Finegold MJ, & Knowles BB (2009). Hep G2 is a hepatoblastoma-derived cell line. Human pathology, 40(10), 1512–1515. [DOI] [PubMed] [Google Scholar]
- Maclean KN, Janosik M, Kraus E, Kozich V, Allen RH, Raab BK, & Kraus JP (2002). Cystathionine beta-synthase is coordinately regulated with proliferation through a redox-sensitive mechanism in cultured human cells and Saccharomyces cerevisiae. J Cell Physiol, 192(1), 81–92. doi: 10.1002/jcp.10118 [DOI] [PubMed] [Google Scholar]
- McCorvie TJ, Kopec J, Hyung SJ, Fitzpatrick F, Feng X, Termine D, … Yue WW (2014). Inter-domain communication of human cystathionine beta-synthase: structural basis of S-adenosyl-L-methionine activation. J Biol Chem, 289(52), 36018–36030. doi: 10.1074/jbc.M114.610782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello JP, Petaja-Repo UE, Bichet DG, & Bouvier M (2000). Pharmacological chaperones: a new twist on receptor folding. Trends Pharmacol Sci, 21(12), 466–469. [DOI] [PubMed] [Google Scholar]
- Mudd SH, Finkelstein JD, Irreverre F, & Laster L (1964). Homocystinuria: an enzymatic defect. LIVER, 1(1), 1. [DOI] [PubMed] [Google Scholar]
- Mudd SH, Skovby F, Levy HL, Pettigrew KD, Wilcken B, Pyeritz RE, … Cerone R (1985). The natural history of homocystinuria due to cystathionine β-synthase deficiency. American journal of human genetics, 37(1), 1. [PMC free article] [PubMed] [Google Scholar]
- Schiff M, & Blom HJ (2012). Treatment of inherited homocystinurias. Neuropediatrics, 43(6), 295. [DOI] [PubMed] [Google Scholar]
- Schwab U, Torronen A, Toppinen L, Alfthan G, Saarinen M, Aro A, & Uusitupa M (2002). Betaine supplementation decreases plasma homocysteine concentrations but does not affect body weight, body composition, or resting energy expenditure in human subjects. Am J Clin Nutr, 76(5), 961–967. [DOI] [PubMed] [Google Scholar]
- Shan X, Dunbrack RL Jr, Christopher SA, & Kruger WD (2001). Mutations in the regulatory domain of cystathionine β–synthase can functionally suppress patient-derived mutations in cis. Human molecular genetics, 10(6), 635–643. [DOI] [PubMed] [Google Scholar]
- Shuichi Miyamoto PA (1992). Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem, 13: 952–962. [Google Scholar]
- Singh LR, Chen X, Kozich V, & Kruger WD (2007). Chemical chaperone rescue of mutant human cystathionine beta-synthase. Mol Genet Metab, 91(4), 335–342. doi: 10.1016/j.ymgme.2007.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh LR, Gupta S, Honig NH, Kraus JP, & Kruger WD (2010). Activation of mutant enzyme function in vivo by proteasome inhibitors and treatments that induce Hsp70. PLoS Genet, 6(1), e1000807. doi: 10.1371/journal.pgen.1000807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh LR, & Kruger WD (2009). Functional rescue of mutant human cystathionine beta-synthase by manipulation of Hsp26 and Hsp70 levels in Saccharomyces cerevisiae. J Biol Chem, 284(7), 4238–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Škovierová H, Vidomanová E, Mahmood S, Sopková J, Drgová A, Červeňová T, … Lehotský J (2016). The molecular and cellular effect of homocysteine metabolism imbalance on human health. International journal of molecular sciences, 17(10), 1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni M, & Lai F (2011). Cell-Based Co-transfection Microarrays for Use with HEK293T Cells on a Poly d-Lysine-Coated Polystyrene Microplate In Cell-Based Microarrays ( 13–25): Springer. [DOI] [PubMed] [Google Scholar]
- Suzuki Y (2014). Emerging novel concept of chaperone therapies for protein misfolding diseases. Proc Jpn Acad Ser B Phys Biol Sci, 90(5), 145–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, & Berendsen HJ (2005). GROMACS: fast, flexible, and free. J Comput Chem, 26(16), 1701–1718. doi: 10.1002/jcc.20291 [DOI] [PubMed] [Google Scholar]
- Van Gunsteren WF, et al. (1996). Biomolecular Simulation: The GROMOS 96 Manual and User Guide. VDF Hochschulverlag AG an der ETH Zürich; 1–1042. [Google Scholar]
- Wang L, Jhee KH, Hua X, DiBello PM, Jacobsen DW, & Kruger WD (2004). Modulation of cystathionine beta-synthase level regulates total serum homocysteine in mice. Circ Res, 94(10), 1318–1324. doi: 10.1161/01.RES.0000129182.46440.4a [DOI] [PubMed] [Google Scholar]
- Wang Z, Li J, Cho J, & Malik AB (2014). Prevention of vascular inflammation by nanoparticle targeting of adherent neutrophils. Nat Nanotechnol, 9(3), 204–210. doi: 10.1038/nnano.2014.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zschocke J, Kebbewar M, Gan‐Schreier H, Fischer C, Fang‐Hoffmann J, Wilrich J, … Lindner M (2009). Molecular neonatal screening for homocystinuria in the Qatari population. Human mutation, 30(6), 1021–1022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.