

Abstract
Background:
Among men with clinically low-risk prostate cancer, we have previously documented heterogeneity in terms of clinical characteristics and genomic risk scores.
Objective:
To further study the underlying tumor biology of this patient population, by interrogating broader patterns of gene expression among men with clinically low-risk tumors.
Design, setting, and participants:
Prostate biopsies from 427 patients considered potentially suitable for active surveillance underwent central pathology review and genome-wide expression profiling. These cases were compared with 1290 higher-risk biopsy cases with diverse clinical features from a prospective genomic registry.
Outcome measurements and statistical analysis:
Average genomic risk (AGR) was determined from 18 published prognostic signatures, and MSigDB hallmark gene sets were analyzed using boot-strapped clustering methods. These sets were examined in relation to clinical variables and pathological and biochemical outcomes using multivariable regression analysis.
Results and limitations:
A total of 408 (96%) biopsies passed RNA quality control. Based on AGR quartiles defined by the high-risk multicenter cases, the University of California, San Francisco (UCSF) low-risk patients were distributed across the quartiles as 219 (54%), 107 (26%), 61 (15%), and 21 (5%). Unsupervised clustering analysis of the hallmark gene set scores revealed three clusters, which were enriched for the previously described PAM50 luminal A, luminal B, and basal subtypes. AGR, but not the clusters, was associated with both pathological (odds ratio 1.34, 95% confidence interval [CI] 1.14– 1.58) and biochemical outcomes (hazard ratio 1.53, 95% CI 1.19–1.93). These results may underesti-mate within-prostate genomic heterogeneity.
Conclusions:
Prostate cancers that are homogeneously low risk by traditional characteristics demonstrate substantial diversity at the level of genomic expression. Molecular substratification of low-risk prostate cancer will yield a better understanding of its divergent biology and, in the future may help personalize treatment recommendations.
Patient summary:
We studied the genomic characteristics of tumors from men diagnosed with low-risk prostate cancer. We found three main subtypes of prostate cancer with divergent tumor biology, similar to what has previously been found in women with breast cancer. In addition, we found that genomic risk scores were associated with worse pathology findings and prostate-specific antigen recurrence after surgery. These results suggest even greater genomic diversity among low-risk patients than has previously been documented with more limited signatures.
Keywords: Active surveillance, Biomarkers, Low-risk prostate cancer, Genomics, Prognosis, Prostate cancer biopsy, Subtyping, Tumor biology
1. Introduction
Among human malignancies, prostate cancer is remarkable both for its pervasiveness and for its exceptionally variable natural history. Roughly one man in six is diagnosed in his lifetime, a high outlier incidence that belies an even higher histological prevalence as indicated by autopsy studies [1]. A large majority of prostate cancers are entirely quiescent, and would never cause any symptoms or loss of life years if undiagnosed; yet the fraction that are more aggressive still amount to the second leading cause of cancer death among men [2]. Recent molecular studies among higher-risk tumors have documented genomic heterogeneity to match prostate cancer’s clinical variability [3,4].
Clinical risk stratification of prostate cancer at diagnosis is relatively accurate in identifying cancers unlikely to progress to clinically relevant stages [5], and can be further enhanced through imaging and/or use of prognostic biomarker signatures [6]. Such approaches, however, assess aggressiveness along only one or a few biological axes, and do not allow the sort of molecular subtyping that is now standard for other tumors such as breast cancer. In fact, recent studies of high-risk prostate cancers have identified luminal and basal subtypes that echo those found in breast cancer and other cancers to a remarkable degree [7].
Active surveillance (AS) rather than immediate treat-ment is now widely endorsed as a preferred management strategy for low-risk prostate cancer [8,9] and is offered to a growing proportion of men both in the USA [10,11] and internationally [12]. An important goal of contemporary investigation into the biology of low-risk prostate cancer is to help determine which of the low-grade prostate cancers are highly aggressive and merit immediate treatment, need close AS, and could be safely followed with a less active monitoring strategy. We aimed to characterize the genomic expression patterns of tumors with relatively homoge-neous, low-risk clinical characteristics, to determine whether such tumors are characterized by similarly homogeneous expression patterns, both in terms of genomically determined clinical risk and in terms of tumor subtyping based on broader expression analysis.
2. Patients and methods
Paraffin-embedded prostate biopsy specimens were collected from 427 men who underwent radical prostatectomy at the University of California, San Francisco (UCSF), based on patient treatment preference, for tumors that would have been eligible for AS based on low-risk (clinical stage ≤T2N0M0, prostate-specific antigen [PSA] ≤20 ng/ml, and biopsy grade group 1) or low-volume (≤3 cores positive overall) grade group 2 tumor characteristics. Additional inclusion criteria were at least 1 mm of cancer on biopsy, prostatectomy slides available for review, and provision of informed consent under institutional review board supervision. Clinical risk was summarized using the extensively validated Cancer of the Prostate Risk Assessment (CAPRA) score [13].
Biopsies and prostatectomy slides were centrally reviewed for grade and stage (by J.P.S.), and RNA was extracted as previously described [14]. In cases with multiple positive biopsy cores, the core with the longest length of the highest-grade cancer was selected. RNA from each case was amplified and labeled using the Ovation FFPE WTA system (NuGen, San Carlos, CA, USA) and hybridized to a Human Exon 1.0 ST GeneChip (Affymetrix, Santa Clara, CA, USA).
The UCSF cases in this study, as with similar prior studies of biopsy-based genomic risk assessment, were restricted by design to low/low-intermediate risk cases. For comparison, and to provide a wider dynamic range of genomic risk, we analyzed prostate cancer cases in the Decipher Genomic Resource Information Database (GRID), a prospective, genome-wide expression registry for urological oncology (NCT02609269), which includes basic demographic and baseline clinical information. These cases were profiled as part of clinical care to facilitate a variety of treatment decisions per clinician discretion. All 1290, mostly higher-risk prostate biopsy, cases currently represented in the GRID were included. For GRID cases, the needle biopsy core with the highest grade and percentage of tumor was selected for RNA extraction and purified using the RNeasy FFPE kit (Qiagen, Valencia, CA, USA), and amplified, labeled, and hybridized as described above for the UCSF cases.
Both GRID and UCSF samples were processed in a Clinical Laboratory Improvement Amendments (CLIA/CAP)-certified laboratory (GenomeDx Biosciences, San Diego, CA, USA). Quality control, normalization and gene level summarization, and batch effect correction were performed using Affymetrix Power Tools (v 1.19.0), Single Channel Array Normalization [15], and ComBat [16], respectively.
2.1. Pathway summarization and average genomic risk
The Molecular Signatures Database (MSigDB) was queried for 50 hall-mark gene sets [17]. Non–prostate or non–cancer-related gene sets were filtered, leaving 37 gene sets for the analysis. Each hallmark set includes a variable number of genes (ranging from 32 to 200) and summarizes expression related to the given biological process. Hallmark gene sets are further grouped into seven biological process categories based on highly correlated expression profiles [17]. Hallmark gene set scores were computed by averaging the expression of each gene in the set, excluding genes not captured by the array.
A substantial number of genomic expression risk scores have previously been published, and we adapted them to the array, as previously described [18]. Eighteen prognostic signatures that achieved univariate significance for the metastasis endpoint in the study were combined into an average genomic risk (AGR) score [18] for each patient by computing the mean of their normalized scores. This AGR score, which serves as a genomic metascore, was analyzed with respect to clinical outcomes.
2.2. Clustering analysis
Patient pathway expression profiles were partitioning around medoids (PAM) clustered based on Spearman’s correlation distances. Consensus clustering [19] bootstrapped over 1000 iterations with 80% sampling of both patients and pathways was used to arrive at a robust clustering solution. Pathway cluster expression patterns were specifically exam-ined in reference to the PAM50 genomic classifier originally developed for breast cancer, and recently found to identify patterns in prostate cancer highly analogous to tumor subtypes described as “luminal A,” “luminal B,” and “basal,” which are prognostic in prostate cancer and strongly predict response to androgen deprivation therapy in particular [7]. Subset analyses focused on men with Gleason grade group 1 tumors on biopsy and on those confirmed at prostatectomy to have a “pure” Gleason grade group 1 tumor on final pathology.
2.3. Statistical analysis
Univariate association between genomic consensus clusters and clinical variables were tested using Kruskal-Wallis and Fisher exact tests with Bonferroni corrections for multiple comparisons. AGR quartiles were calculated with respect to all GRID samples. These risk groups were verified in an independent retrospective biopsy dataset for stratification of metastasis risk using Kaplan-Meier estimates (data not shown). Adverse pathology at prostatectomy was defined by pathological grade group ≥3 or stage ≥T3a, and was analyzed using multivariable logistic regression controlling for CAPRA score and PSA density. Recurrence after surgery was defined by PSA >0.2 with verification or any secondary treatment, analyzed using multivariable Cox proportional hazards regression adjusting for CAPRA score and PSA density. Analyses were performed in R v3.3.3, and all p values were two tailed.
3. Results
Of the 427 UCSF cases, 408 (96%) had sufficient RNA. Table 1 summarizes the demographic and clinical characteristics of the UCSF and GRID patients. Compared with the UCSF AS-eligible patients, GRID patients were older and had higher PSAs, much less grade group 1 disease (37% of GRID vs 74% of UCSF patients), and higher-volume, higher-risk prostate cancer at diagnosis.
Table 1.
– Clinical characteristics of the UCSF and GRID cohorts
| Variables | Values | UCSF (N, %) | GRID (N, %) |
|---|---|---|---|
| 408 (24) | 1290 (76) | ||
| Patient age at biopsy | Median (Q1, Q3) | 59 (54, 64) | 68 (62, 73) |
| % Positive biopsy cores | Median (Q1, Q3) | 20 (13, 33) | 33 (17, 50) |
| PSA | Median (Q1, Q3) | 5.5 (4.3, 7.5) | 6.4 (4.7, 9.3) |
| Clinical stage | T1 | 232 (57) | 719 (56) |
| T2 | 176 (43) | 196 (15) | |
| T3 | 15 (1.2) | ||
| T4 | 3 (0.23) | ||
| Unknown | 357 (28) | ||
| Gleason grade group | 1 | 301 (74) | 482 (37) |
| 2 | 107 (26) | 445 (35) | |
| 3 | 175 (14) | ||
| 4 | 109 (8.4) | ||
| 5 | 79 (6.1) | ||
| CAPRA | 0–2 | 308 (76) | 334 (26) |
| 3–5 | 92 (23) | 389 (30) | |
| 6+ | 150 (12) | ||
| Unknown | 8 (2.0) | 417 (32) | |
| PSA density | Median (Q1, Q3) | 0.17 (0.11, 0.24) | – |
CAPRA = Cancer of the Prostate Risk Assessment; GRID = Decipher Genomic Resource Information Database; PSA = prostate-specific antigen; UCSF = University of California, San Francisco.
Figure 1 presents a heat map summarizing average expression in each patient for each of 37 hallmark gene sets. Patients (UCSF and GRID) were sorted by increasing AGR. Most of the prognostic signature scores summarized into the AGR correlated closely with each other. Gene sets with a significant positive correlation (p < 0.05) with high AGR included cell cycle/proliferation gene sets (MYC targets, G2 M checkpoint, E2F targets), immune response (tumor necrosis factor [TNF] signaling via nuclear factor kappa B [NFKB], IL2 STAT5 signaling), angiogenesis, upregulation of Kras signaling, cellular stress, and metabolism (unfolded protein response, oxidative phosphorylation). Gene sets negatively associated with AGR included those involved in steroid signaling (androgen and estrogen response, choles-terol homeostasis), cell-cell interactions (apical surface, apical junction), downregulation of Kras signaling, apoptosis, and metabolism (xenobiotic and fatty acid metabolism).
Fig. 1.
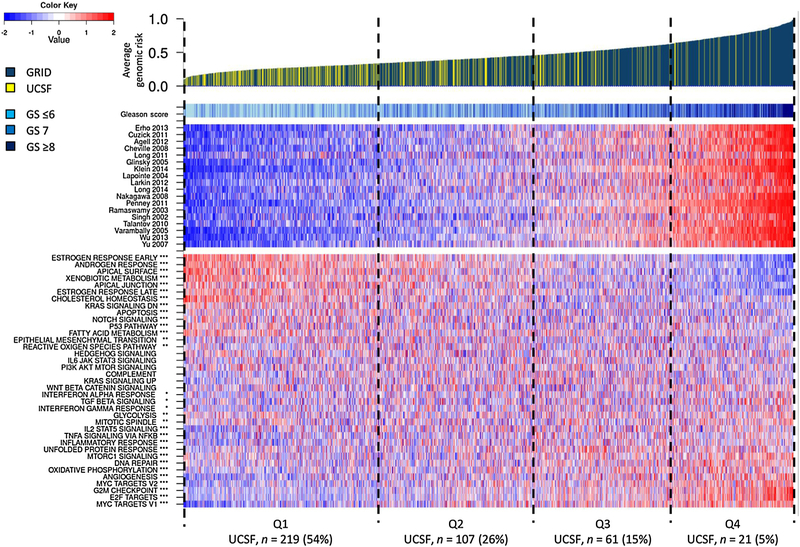
– Heatmap of UCSF and GRID patients (n = 1698) ordered by increasing AGR. The map indicates the following (from top to bottom): (1) the average genomic risk colored by the study the patient is part of, (2) Gleason score, (3) normalized scores for 18 prognostic signatures, and (4) hallmark gene set scores and their correlations to AGR indicated. The patients are broken up into quartiles based on the GRID reference set, and the number (%) of UCSF patients in each quartile is annotated. UCSF patients are associated with lower AGR; however, some UCSF patients are also found in the highest-risk quartile. AGR = average genomic risk; GRID = Decipher Genomic Resource Information Database; GS = Gleason score; UCSF = University of California, San Francisco. * p < 0.05. ** p < 0.01. *** p < 0.001.
Supplementary Figure 1 presents a similar heat map including only the UCSF cases, with very similar overall findings. Of the UCSF cases, 54%, 26%, 15%, and 5%were sorted into each quartile of AGR with thresholds defined based on the GRID cases. Among grade group 1 and 2 cancers, 2.0% and 14%, respectively, were in the top quartile of AGR, and 13% and 20%, respectively, were in the third quartile. Likewise, 2.1% and 9.2% of tumors with the lowest clinical risk, as defined by CAPRA 0–1 tumors, were in the top and third quartiles, respectively.
A second heat map presented in Figure 2 illustrates the results of an unsupervised clustering analysis of the hallmark gene sets for the UCSF cases. Three clusters of cases clearly emerge from this analysis. Cluster 1 (from left) is driven by gene sets associated with migration and invasion, and immune response (IL2 STAT5 signaling, IL6 JAK STAT3 signaling, TNFA signaling via NFKB, interferon alpha, gamma and inflammatory response, complement). The second cluster is enriched by gene sets for cell cycle/ proliferation (G2 M checkpoint, MYC, and E2F targets), DNA repair, canonical beta-catenin signaling, and metabolism (oxidative phosphorylation, glycolysis, fatty acid metabo-lism, mTORC1 signaling, PI3K signaling via AKT to mTORC1). The third cluster is driven by androgen and estrogen signaling (androgen and estrogen response, cholesterol homeostasis), mitotic spindle, p53, Hedgehog, Notch signaling and Kras downregulation, xenobiotic metabolism, reactive oxygen species, and apical surface gene sets. Supplementary Figure 2 summarizes the extent of overlap across the various gene sets, indicating their relative independence in contributing to the cluster definitions.
Fig 2.

– Heatmap of the UCSF patients (n = 408) consensus clustered based on the expression score of 37 hallmark gene sets. The patients tend to cluster into three distinct groups, which are loosely associated with PAM50’s basal, luminal A, and luminal B subtypes. CAPRA = Cancer of the Prostate Risk Assessment; PSA = prostate specific antigen; UCSF = University of Calfornia, San Francisco.
Next, we evaluated the three clusters for enrichment of previously described subtypes [20]. Cluster 1 was enriched with triple negative tumors (ERG–, ETS–, and SPINK1) [20] and cluster 2 with ERG+ tumors. Likewise, cluster 1 was enriched with basal-like tumors, whereas the other clusters were almost exclusively luminal like [21]. A related analysis based on PAM50 subtypes found cluster 1 to be enriched with basal, cluster 2 with luminal B, and cluster 3 with luminal A subtypes (Supplementary Table 1) [7]. Supplementary Figures 3 and 4 present similar clustering analyses including both UCSF and GRID cases, and restricted to grade group 1 cases, again identifying the same three clusters similar to basal, luminal B, and luminal A classifications. Supplementary Figure 4C indicates that substantial hetero-geneity exists in cancer-related gene pathway expression even among men with pathologically confirmed Gleason grade group 1 tumors.
Among the UCSF cases, the clusters analogous to luminal A (cluster 3), basal (cluster 1), and luminal B (cluster 2) cancers had median (interquartile range) AGR scores of 0.27 (0.22–0.32), 0.34 (0.28–0.42), and 0.41 (0.31–0.49), respectively (p < 0.001; Fig. 3). On the contrary, aside from a statistically significant but clinically small difference in percent of biopsy cores involved and stage, there were no differences in standard clinical parameters or clinical risk stratification by CAPRA score across the three subtypes (Table 2).
Fig. 3.

– Boxplot showing the AGR for each of the clusters from Figure 2. Patients in cluster 3 are found to be at the lowest AGR, while patients in cluster 2 are at the highest AGR (p < 0.001). AGR = average genomic risk.
Table 2.
– Clinical characteristics of genomic cluster groups
| Variables | Values | Cluster 1 | Cluster 2 | Cluster 3 | p value |
|---|---|---|---|---|---|
| 142 (35) | 134 (33) | 132 (32) | |||
| Patient age at biopsy | Median (Q1, Q3) | 60 (55, 65) | 59 (53, 64) | 58 (53, 62) | 0.5 |
| % Positive biopsy cores | Median (Q1, Q3) | 18.8 (12.1, 30) | 25 (16.7, 34.8) | 16.7 (12.5, 28.6) | 0.008 |
| Preop PSA | Median (Q1, Q3) | 5.48 (4.15, 7.73) | 5.50 (4.18, 7.10) | 5.78 (4.59, 7.82) | 1 |
| Clinical stage | T1 | 85 (60) | 61 (46) | 86 (65) | 0.03 |
| T2 | 57 (40) | 73 (54) | 46 (35) | ||
| Gleason grade group | 1 | 105 (74) | 94 (70) | 102 (77) | |
| 2 | 37 (26) | 40 (30) | 30 (23) | 1 | |
| CAPRA | 1 | 52 (37) | 44 (33) | 46 (35) | |
| 2 | 58 (41) | 57 (43) | 51 (39) | 1 | |
| 3 | 24 (17) | 17 (13) | 25 (19) | ||
| 4–5 | 5 (3.5) | 12 (9.0) | 9 (6.8) | ||
| Unknown | 3 (2.1) | 4 (3.0) | 1 (0.8) | ||
| PSA density | Median (Q1, Q3) | 0.17 (0.12, 0.25) | 0.16 (0.12, 0.24) | 0.17 (0.11, 0.24) | 1 |
| Average genomic risk | Median (Q1, Q3) | 0.34 (0.28, 0.42) | 0.41 (0.31, 0.49) | 0.27 (0.22, 0.32) | <0.001 |
CAPRA = Cancer of the Prostate Risk Assessment; PSA = prostate-specific antigen.
Adverse pathology at prostatectomy was identified in 105 (26%) of cases. On logistic regression for this outcome of adverse pathology, both CAPRA and AGR were indepen-dently prognostic, but the three genomic clusters were not (Table 3). AGR was more predictive of adverse pathology than any of the individual signatures reflected in the AGR. Biochemical recurrence was identified in 30 of 357 of men with median follow-up time of 39 mo among censored patients. For this outcome, on proportional hazards regres-sion, only AGR (not CAPRA or genomic clusters) was prognostic (Table 3).
Table 3.
– Genomic risk and clusters as predictors of outcomes
| Adverse pathology |
Biochemical recurrence |
|||
|---|---|---|---|---|
| OR (95% CI) | p value | HR (95% CI) | p value | |
| CAPRA | 1.54 (1.14–2.07) | 0.004 | 1.14 (0.72–1.80) | 0.6 |
| PSA density | 1.16 (0.92–1.45) | 0.2 | 1.32 (0.99–1.63) | 0.06 |
| AGR | 1.23 (1.03–1.47) | 0.02 | 1.58 (1.21–2.03) | 0.001 |
| CAPRA | 1.65 (1.23–2.22) | <0.001 | 1.20 (0.75–1.90) | 0.4 |
| PSA density | 1.14 (0.9–1.43) | 0.3 | 1.28 (0.95–1.60) | 0.1 |
| Genomic cluster 1 | REF | REF | ||
| Genomic cluster 2 | 1.19 (0.61–2.28) | 0.6 | 1.17 (0.45–2.96) | 0.7 |
| Genomic cluster 3 | 1.78 (0.98–3.23) | 0.06 | 0.95 (0.35–2.46) | 0.9 |
AGR = average genomic risk; CI = confidence interval; CAPRA = Cancer of the Prostate Risk Assessment; HR = hazard ratio; OR = odds ratio; REF = reference. Adverse pathology outcomes are based on multivariable logistic regression and biochemical recurrence outcomes are based on Cox proportional hazards regression.
4. Discussion
In the current analysis, we conducted the most compre-hensive expression profiling study to date focused on newly diagnosed, relatively low-risk prostate cancer. These cases, characterized by low PSA, low stage, and either grade group 1 or low-volume grade group 2, would be considered at least potentially eligible for AS at our institution (with the men understanding clearly that AS for them is more likely to imply deferred rather than avoided treatment) [9,22]. We identified very substantial genomic heterogeneity within this cohort. About 15% of low-risk cases-even those defined as lowest risk based on either Gleason group or multivari-able CAPRA score-are characterized by higher-risk genomic features, with approximately 2% of grade group 1 cases found at the top of the genomic risk range.
More importantly, we identified three distinct cancer subtypes at the genomic expression level, which correspond with similar subtypes previously described for breast, lung, and bladder cancers. These subtypes had minimal differ-ences in clinical risk profiles and did not predict outcome, suggesting that the subclassification reflects biological distinctions not reflected in clinical parameters, or in the genomic risk signatures summarized in the AGR score. AGR, by contrast, was the variable most strongly associated with both pathological and biochemical outcomes. We stress, however, that AGR is not intended to serve as yet another genomic predictor of oncological outcomes for clinical practice, but rather as a metascore that reflects an overall summary of genomic risk and aggressiveness, as reflected in multiple previously validated scores.
We found strong positive correlations for cell cycle/ proliferation gene sets and high AGR, and an inverse correlation for this surrogate for androgen signaling. Using hallmarks of oncology gene set analysis, we identified clusters quite similar to those described by the PAM50 classification. PAM50 was originally developed for women with breast cancer, and recently, Zhao et al [7] analyzed the PAM50 breast cancer subtypes in several large cohorts of high-risk radical prostatectomy tumors. The authors found that luminal B tumors had the worst oncological out-comes, similar to our findings, in which cluster 2 patients had the highest AGR for metastasis. We found in cluster 1 higher levels of invasion/migration as well as immune response gene sets, suggestive of higher levels of immune infiltration or inflammatory response in these basal-like tumors. Finally, we observed that cluster 3 (enriched with luminal A) tumors had higher levels of androgen response but lower levels of immune response gene sets. These subtypes may ultimately predict responses to emerging targeted and immunological therapies for progressive prostate cancer.
Low-risk prostate cancer rarely progresses to a clinically meaningful stage over at least the 1st decade of follow-up [23,24], but at least a quarter of biopsies indicating low-risk features in fact undersample the tumor in terms of stage or grade [25]. More importantly, with extended follow-up, the risk of progression to metastasis or cancer-specific mortali-ty is not insubstantial for men on AS [24,26]. While many of these adverse outcomes likely could have been avoided with closer monitoring and/or earlier treatment, it is not always clear what the triggers for intervention should have been, even in retrospect [27]. Moreover, the burden of surveillance is significant, and men whose tumors have negligible biological potential for metastasis are still subjected to repeated PSA checks, biopsies, and other interventions, with ongoing anxiety of the attendant, risks of infection, and opportunities for overtreatment [9].
Emerging tests based on assessment of RNA expression in paraffin-embedded prostate biopsy tissue have shown promise for improving prognostic assessments over clinical parameters alone, and have clearly shown genomic heterogeneity among tumors that are relatively homogeneous clinically [14,28]. These tests are now endorsed to aid in treatment decision making by the National Comprehensive Cancer Network prostate cancer guideline. Nearly all the other published signatures reflected in the AGR, likewise, are based on genes originally selected to predict PSA recurrence, metastasis, and other relatively distal clinical events. However, these existing assays are based on measurement of relatively small numbers of genes in one or a few pathways, and therefore provide relatively limited insight into the extent of biological heterogeneity. For this reason, among others, existing tests are independently prognostic but cannot clearly guide treatment selection.
Moreover, most genomic studies on the molecular variability of prostate cancer have focused on advanced disease and, not surprisingly, have found great heterogeneity among heavily treated progressive cancers. Much less prior work has focused on untreated, early-stage clinically localized tumors. The Cancer Genome Atlas is a notable exception, but even in this cohort of 333 cases, in which the full spectrum of clinical risk was represented, only 65 (20%) of the cases had grade group 1 disease [3]. Moreover, this study focused primarily on mutations, chromosomal rearrangements, and methylation events [4]. Both studies also performed limited RNA analysis, focusing on expression directly related to identified DNA changes.
There are important limitations to this analysis which must be acknowledged. All the UCSF cases underwent a central pathology review, whereas the GRID cases were read and submitted by many different pathologists. RNA processing was slightly different for the UCSF and GRID cases, but quality control results were substantively similar between the two. Cases were all microdissected (without laser capture) to enrich for malignant glands. The sampled tumor specimen and resultant transcriptomic profile may therefore represent a complex mixture including other nonmalignant cells from the immediate tumor microenvironment, such as stromal, benign, and immune infiltrate (if present). Therefore, these results could also reflect heterogeneity in overall cellular composition of tumor and not only of the malignant cells in lower-risk prostate cancer. However, all three commercial RNA-based assays are based on similar microdissection without laser capture, suggesting that these techniques are at least partially robust to variation in local cell type populations.
The AGR score is only a surrogate for clinical outcomes, but it is based on multiple scores that have been validated for such outcomes in many prior studies, and we believe that averaging the existing scores reduces the potential for bias in reflecting overall genomic risk. The analysis of biochemical outcomes is limited by small event numbers and a relatively narrow range of risk. Only one biopsy per case was processed. We acknowledge that there may be within-patient heterogeneity in terms of AGS and/or tumor subtype classifications. Indeed, the consistency or variability of gene expression even within normal prostate tissue is not well defined. However, the success of all existing tissue-based biopsy tests is based on the presumption that the selected biopsy can accurately reflect the biopsy of the cancer overall. Adverse pathology is an imperfect surrogate for distal clinical outcomes, but has been used in multiple prior genomic studies. The biochemical recurrence analyses in the present manuscript are further limited by a relatively low event rate, reflecting the low-risk clinical characteristics.
5. Conclusions
The diversity that we observed, in terms of both the AGR spectrum and subtyping across the luminal A/luminal B/ basal spectrum, was present both in the clinically diverse GRID cohort and in the much more clinically homogeneous UCSF AS-eligible cohort. While we cannot yet confirm the differential response to androgen deprivation therapy observed for luminal B versus basal and luminal A tumors, we believe that these data underscore the potential utility of a taxonomy and nomenclature that considers molecular characteristics and biological potential for progression at least as strongly as the organ of origin and traditional histology. With future studies planned in our prospective AS cohort, we hope to demonstrate that such molecular substratification can help stratify men not only to surveillance versus immediate treatment, but also to more or less intensive surveillance strategies.
Supplementary Material
Acknowledgments
Funding/Support and role of the sponsor: The Department of Defense Prostate Cancer Research Program (Grant W81XWH-13-2-0074) and GenomeDx Biosciences had a role in the design and conduct of the study and collection, management, and analysis of the data.
Financial disclosures: Matthew R. Cooperberg certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: Nicholas Erho, Nick Fishbane, Jonathan Lehrer, Mohammed Alshalalfa, Tyler Kolisnik, Jijumon Chelliserry, Jennifer Margrave, Maria Aranes, Marguerite du Plessis, Christine Buerki, and Elai Davicioni are employees of GenomeDx Biosciences.
References
- [1].Jahn JL, Giovannucci EL, Stampfer MJ. The high prevalence of undiagnosed prostate cancer at autopsy: implications for epidemi-ology and treatment of prostate cancer in the prostate-specific antigen-era. Int J Cancer 2015;137:2795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- [3].Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015;163:1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fraser M, Sabelnykova VY, Yamaguchi TN, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017;541:359–64. [DOI] [PubMed] [Google Scholar]
- [5].Cooperberg MR, Hinotsu S, Namiki M, et al. Risk assessment among prostate cancer patients receiving primary androgen deprivation therapy. J Clin Oncol 2009;27:4306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Leapman MS, Nguyen HG, Cooperberg MR. Clinical utility of bio-markers in localized prostate cancer. Curr Oncol Rep 2016;18:30. [DOI] [PubMed] [Google Scholar]
- [7].Zhao SG, Chang L, Erho N, et al. Luminal and basal subtyping of prostate cancer is prognostic and predicts response to androgen deprivation therapy. JAMA Oncol 2017;3:1663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ganz PA, Barry JM, Burke W, et al. National Institutes of Health State-of-the-Science Conference: role of active surveillance in the management of men with localized prostate cancer. Ann Intern Med 2012;156:591–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chen RC, Rumble RB, Loblaw DA, et al. Active surveillance for the management of localized prostate cancer (Cancer Care Ontario guideline): American Society of Clinical Oncology clinical practice guideline endorsement. J Clin Oncol 2016;34:2182–90. [DOI] [PubMed] [Google Scholar]
- [10].Womble PR, Montie JE, Ye Z, et al. Contemporary use of initial active surveillance among men in Michigan with low-risk prostate cancer. Eur Urol 2015;67:44–50. [DOI] [PubMed] [Google Scholar]
- [11].Cooperberg MR, Carroll PR. Trends in management for patients with localized prostate cancer, 1990–2013. JAMA 2015;314:80–2. [DOI] [PubMed] [Google Scholar]
- [12].Loeb S, Folkvaljon Y, Curnyn C, Robinson D, Bratt O, Stattin P. Uptake of active surveillance for very-low-risk prostate cancer in Sweden. JAMA Oncol 2017;3:1393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Brajtbord JS, Leapman MS, Cooperberg MR. The CAPRA score at 10 years: contemporary perspectives and analysis of supporting studies. Eur Urol 2017;71:705–9. [DOI] [PubMed] [Google Scholar]
- [14].Klein EA, Cooperberg MR, Magi-Galluzzi C, et al. A 17-gene assay to predict prostate cancer aggressiveness in the context of Gleason grade heterogeneity, tumor multifocality, and biopsy undersam-pling. Eur Urol 2014;66:550–60. [DOI] [PubMed] [Google Scholar]
- [15].Piccolo SR, Sun Y, Campbell JD, Lenburg ME, Bild AH, Johnson WE. A single-sample microarray normalization method to facilitate per-sonalized-medicine workflows. Genomics 2012;100:337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007;8:118–27. [DOI] [PubMed] [Google Scholar]
- [17].Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ross AE, Johnson MH, Yousefi K, et al. Tissue-based genomics augments post-prostatectomy risk stratification in a natural history cohort of intermediate- and high-risk men. Eur Urol 2015;69:157–65. [DOI] [PubMed] [Google Scholar]
- [19].Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 2010;26:1572–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tomlins SA, Alshalalfa M, Davicioni E, et al. Characterization of 1577 primary prostate cancers reveals novel biological and clini-copathologic insights into molecular subtypes. Eur Urol 2015;68:555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang D, Park D, Zhong Y, et al. Stem cell and neurogenic gene-expression profiles link prostate basal cells to aggressive prostate cancer. Nat Commun 2016;7, 10798.s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Welty CJ, Cowan JE, Nguyen H, et al. Extended followup and risk factors for disease reclassification in a large active surveillance cohort for localized prostate cancer. J Urol 2015;193:807–11. [DOI] [PubMed] [Google Scholar]
- [23].Wilt TJ, Brawer MK, Jones KM, et al. Radical prostatectomy versus observation for localized prostate cancer. N Engl J Med 2012;367:203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hamdy FC, Donovan JL, Lane JA, et al. 10-Year outcomes after monitoring, surgery, or radiotherapy for localized prostate cancer. N Engl J Med 2016;375:1415–24. [DOI] [PubMed] [Google Scholar]
- [25].Conti SL, Dall’era M, Fradet V, Cowan JE, Simko J, Carroll PR. Pathological outcomes of candidates for active surveillance of prostate cancer. J Urol 2009;181:1628–33, discussion 1633–4. [DOI] [PubMed] [Google Scholar]
- [26].Klotz L, Vesprini D, Sethukavalan P, et al. Long-term follow-up of a large active surveillance cohort of patients with prostate cancer. J Clin Oncol 2015;33:272–6. [DOI] [PubMed] [Google Scholar]
- [27].Tosoian JJ, Mamawala M, Epstein JI, et al. Intermediate and longer-term outcomes from a prospective active-surveillance program for favorable-risk prostate cancer. J Clin Oncol 2015;33:3379–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cooperberg MR, Simko JP, Cowan JE, et al. Validation of a cell-cycle progression gene panel to improve risk stratification in a contem-porary prostatectomy cohort. J Clin Oncol 2013;31:1428–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.