

Abstract
Cryptococcus neoformans is an environmental yeast found worldwide that causes lethal brain infections, particularly in immunocompromised hosts. In 2016, there were 280,000 cases of cryptococcal meningitis in the HIV+ population, two-thirds of them fatal; other immunocompromised patients are also affected. The burden of cryptococcal disease and the limits of current chemotherapy create a pressing need for improved treatment. One hindrance to the development of new therapies is lack of understanding of how this pathogen breaches the barriers protecting the brain. Here we describe a tool for investigating this process. This simple in vitro blood-brain barrier (BBB) model, based on a human brain endothelial cell line grown on a permeable membrane, may be used to assay the BBB transmigration of C. neoformans or other neurotropic pathogens.
Keywords: Fungal meningitis, Cryptococcus neoformans, Cryptococcosis, Blood-brain barrier, hCMEC/D3, Transwell permeable supports
INTRODUCTION
Cryptococcus neoformans is a ubiquitous environmental yeast that is the major cause of fungal meningitis worldwide (Srikanta et al., 2014). Inhalation of infectious particles results in a lung infection that is usually asymptomatic and controlled in healthy people. Under conditions of immunocompromise, however, it can progress to pneumonia and disseminate to extrapulmonary sites, including the central nervous system (CNS). Cryptococcal meningitis (CM) is the most commonly reported clinical manifestation of cryptococcal infection and is invariably fatal without treatment; even treated patients exhibit mortality rates as high as 70%, depending on their immune status and geographical location (Rajasingham et al., 2017).
For C. neoformans to invade the CNS from the bloodstream it must interact with CNS endothelial cells (Klein et al., 2017) and traverse the blood-brain barrier (BBB). The ability to model and assay this process is central to studies of CM pathophysiology, including investigation of the factors that influence cryptococcal neurotropism and the mechanisms that promote transendothelial traversal. Here we describe an in vitro model comprised of human brain endothelial cells grown in a transwell system, which can be used for this purpose. This assay is user-friendly, cost-effective, and allows for highly controlled and reproducible conditions that can be adapted to address a variety of experimental questions.
To generate model BBB, we use a human-derived brain endothelial cell line. These cells are cultured on top of porous membranes that separate special tissue culture wells into two compartments, one of which will represent the blood (vessel lumen) and the other representing the brain tissue (outside of the vessel). Integrity of the barrier that develops in this context is assayed by measuring electrical resistance across the monolayer and transmigration of yeast is determined by sampling from the bottom compartment over time to assess colony-forming units (CFU).
CAUTION: C.neoformans is a Biosafety Level 2 (BSL-2) pathogen. Follow all appropriate guidelines and regulations for the use and handling of pathogenic microorganisms, explained in UNIT 1A.1 and APPENDIX 1B.
CAUTION:If using fresh human serum obtained from whole blood, follow all appropriate guidelines and regulations for the use and handling of human-derived materials, explained in UNIT 1A.1 and APPENDIX 1B.
BASIC PROTOCOL 1
Growth and subculture of hCMEC/D3
Although the materials and methods in this unit are specific for the hCMEC/D3 cell line, brain endothelial barriers may also be modeled using primary cells or other cell lines (Rahman et al., 2016), which would be suitable for use in the assay described here. As immortalized cell lines have unstable genomes, it is critical to monitor the passage number for each experiment; in the case of hCMEC/D3 this is usually limited to no more than 36 passages. Note also that endothelial cells are flat, large cells, so relatively few are required for seeding compared to other cell types. A confluent T25 flask yields enough cells to seed at least twelve 0.9 cm2 inserts (for 12-well plates).
Materials
Solutions and reagents
Confluent culture of hCMEC/D3 cell line (or other cell type of choice).
Rat Collagen I solution (see recipe).
1X PBS (see recipe).
0.25% Trypsin-EDTA solution.
Complete growth medium (see recipe).
25 cm2 tissue culture flasks (T25s), with vented caps.
Protocol steps
Prepare enough rat collagen I solution for the flasks to be seeded (one confluent flask is enough for one plate as shown in Figure 1). Use 3 mL per T25 flask.
-
Add 3 mL of the rat collagen I solution to each T25 flask and tap gently on the side of the flask until the whole bottom surface is covered. Incubate at 37 °C for at least 1 hour. Optional: for convenience, you can incubate overnight at 4 °C with equal results, but be sure to handle the plate carefully to keep it sterile.
Note: Collagen coating of the tissue culture flasks is required for adequate growth of the brain endothelial cells.
-
While incubating the T25 flasks, wash a confluent monolayer of hCMEC/D3 cells in a T25 by removing the medium and adding 3 mL of PBS. Swirl the flask for a few seconds, remove the PBS, add 3 mL of 0.25% Trypsin-EDTA solution, and leave the flask inside the hood for 1 – 2 minutes to detach the monolayer.
Note: hCMEC/D3 cells are extremely sensitive to this concentration of trypsin, and should detach within 2 min. If you do not see detachment after this time has elapsed, tap the flask hard on your hand and immediately proceed to step 4 to inactivate the trypsin; longer incubation will result in significant cell death. You can also use trypsin alternatives, such as the TrypLE reagents; these are gentler on the cells and still achieve complete detachment within ~5 minutes.
Immediately upon observing monolayer detachment, add 7 mL of complete growth media to inactivate the trypsin and gently pipette the cells up and down to disperse any clumps. Transfer the uniform suspension into a 15-mL conical tube and collect the cells by centrifugation in a swinging-bucket rotor (5 minutes at 600 x g, 4 °C).
Discard the supernatant and resuspend the cell pellet in 3 mL complete growth medium and count the cells (e.g. with a hemocytometer).
Remove the collagen solution from the T25 flasks prepared in step 2 by aspiration. To remove excess collagen, add 3 mL of PBS, swirl the flask for a few seconds, and remove the PBS. Place the flasks inside the hood.
For each T25 flask, add 625,000 hCMEC/D3 cells (a density of 25,000 cells/cm2) in 10 mL (total volume) of growth media. Incubate for 3 – 4 days at 37 °C in 5% CO2. At this initial density, the cells almost always reach confluence in 3 days. To limit the passage number of the cell line, it may be advantageous for the researcher to seed at a lower density. In that case, change the medium after the 3rd day.
Figure 1: Layout for a typical experiment testing two experimental conditions with experimental and control wells in triplicate.
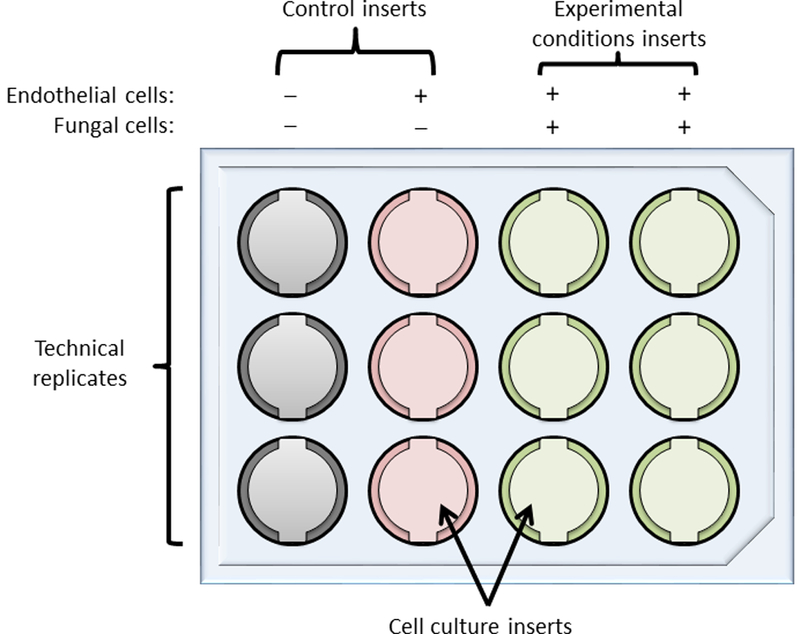
1st column, empty insert controls used to measure background values for TEER calculations. 2nd column, inserts with endothelial cells alone, to assess baseline TEER (in the absence of fungi) that can be used to correct all other TEER values. 3rd and 4th columns, inserts with both endothelial and fungal cells. The last two columns may be used to assess any alteration in TEER induced by the fungi (by comparing TEER values to those for the 2nd column) and also to measure fungal traversal under the desired experimental conditions, for example opsonized (column 3) versus non-opsonized fungal cells (column 4).
BASIC PROTOCOL 2
Preparation of the in vitro brain endothelial cell model
Once hCMEC/D3 cells are ready, they can be used to establish the model BBB as described in this section. Basically, this involves seeding the cells on collagen-treated membranes and allowing them to develop into a monolayer, which can be monitored by measuring TEER daily (optional once the procedure is well established). As explained above, this is a simple model using only brain endothelial cells, but this protocol can easily be adapted for more complex models of co-culture by seeding other areas of the insert or well with the corresponding additional cell type.
Materials
Solutions and reagents
Confluent culture of hCMEC/D3 cell line (or other cell type of choice).
Complete growth medium (see recipe).
Differentiation medium (see recipe).
1X PBS (see recipe).
0.25% Trypsin-EDTA solution.
Rat Collagen I solution (see recipe).
Special equipment
BD Falcon 12-well cell culture inserts, with 8 μm pore size (Corning 353182)
BD Falcon12-well culture insert companion plates (Corning 353503).
Note: Here we use 0.9 cm2-inserts for 12-well plates, but this system can be adapted for smaller inserts that fit in 24-well or even 96-well plates. For other sizes, scale based on area, not on volume, and note that TEER development is slower in the smaller inserts.
EVOM2 Epithelial voltohmmeter, with chopstick electrode (WPI).
Protocol steps
-
The inserts are supplied individually wrapped in plastic. In the hood, open the packaging and use sterile forceps to remove the inserts. Gently place the insert into one well of the 12-well plate.
Note: If using BD Falcon individual cell culture inserts, subsequent manipulations will be easier if you use BD Falcon Companion Plates, which have notches on the well edge to keep the inserts centered. This prevents liquid wicking between the sides of the insert and the well walls and provides easy pipette access to the lower chamber.
Coat the insert membrane with 300 μL of a 150 μg/mL solution of rat collagen and incubate at 37 °C for at least 1 hour (up to 2 – 3 hours, or alternatively incubate overnight at 4 °C).
While coating the inserts, harvest, wash, and count hCMEC/D3 cells as described in BASIC PROTOCOL 1.
-
Aspirate the excess collagen solution and wash by adding 300 μL of PBS and removing it by aspiration, being careful not to touch the membrane.
Note: When replacing medium or measuring TEER, be extremely careful to avoid touching the membrane or creating bubbles, particularly in the lower compartment.
-
For each insert to be seeded, prepare 500 μL of a cell suspension of 90,000 cells/mL in complete growth medium; this will yield a density of 50,000 cells/cm2. Leave one triplicate set of inserts empty (without cells) to use as a blank for TEER measurements (see Figure 1 for typical layout).
Note: It is prudent to seed more inserts than needed for the experiment to account for experimental variations, defective inserts, or accidental disruption of monolayers during handling. Also, it is essential to include blank inserts (no cells) to use for background values for TEER calculations.
Add 500 μL of the cell solution to each experimental insert and to the control that will receive no fungal cells, and 500 μL of complete growth medium to the control that will receive neither host nor fungal cells. A typical experiment layout is shown in Figure 1.
-
Add 1.5 mL of complete growth medium to the lower compartment of each well and incubate at 37 °C in 5% CO2 for 3 days.
Note: To monitor TEER development, follow the manufacturer’s instructions for use of the instrument and the recommendations above. To calculate TEER, multiply the net resistance due to the cell layer (subtracting the background resistance in the no cell control well) by the surface area of the insert in cm2. The units for TEER should be Ω*cm2. With the 12-well plate setup and materials recommended above, we typically obtain values for fully formed monolayers in the 80 Ω*cm2 (Santiago-Tirado et al., 2017).
-
Feed the cells by replacing the medium (in both the insert and the well) with fresh complete growth medium. If you remove the medium from the top chamber while the bottom chamber still contains medium, the pressure from below will disrupt the barrier. To avoid this, first aspirate the bottom medium; then aspirate the top medium; then add fresh medium to the upper compartment (insert); and finally add fresh medium to the bottom compartment (plate well). Alternatively, you can move the inserts into a new empty plate, adding medium to the bottom wells only after replacing the medium above the inserts. Incubate at 37 °C in 5% CO2 for 2 days.
Note: Full monolayer establishment on a 12-well plate insert is usually achieved in 5 days (steps 7 and 8), with an additional day of growth in differentiation medium (step 9) yielding higher TEER.
Stimulate differentiation by replacing the medium, as described in step 8, with differentiation medium. Incubate overnight and the cells will be ready for assay the next day.
BASIC PROTOCOL 3
Preparation of C. neoformans cells for transmigration assay
Opsonization of the fungi is required to obtain significant transmigration of free C. neoformans (KN99 lineage) across the brain endothelia (Chang et al., 2004; Chen et al., 2003; Santiago-Tirado et al., 2017). The protocol below describes this procedure.
Materials
Solutions and reagents
KN99α, or other relevant C. neoformans strain(s).
Yeast peptone dextrose (YPD) liquid medium (see recipe).
YPD solid medium (see recipe).
17 × 100 mm sterile culture tubes (one per fungal strain to be tested)
EBM-2 basal media (Lonza).
Opsonizing agent, either fresh or frozen human serum (see SUPPORT PROTOCOL 1).
Protocol steps
-
Resuspend a single colony of KN99α or other strain to be tested in 5 mL of YPD in a culture tube and incubate overnight in a 30 °C shaker at 240 rpm.
Note: Here we use C. neoformans strain KN99, which is derived from the reference strain H99 but is amenable to genetic manipulations by mating (Nielsen et al., 2003). These protocols may be used to study other genetic lineages of C. neoformans, such as JEC20 and JEC21 (Kwon-Chung et al., 1992), or for C. gattii strains.
-
The next morning, assess culture density by any convenient method, such as cell counting or using a spectrophotometer. The goal is to obtain cultures in late log-phase (~2×107/mL or OD600 ~1, depending on your instrument), with all strains to be used grown to similar density.
Note: If needed, allow the cells to grow longer or dilute and then grow for another 1–2 hours to achieve the desired density.
Collect the cells by centrifugation (5 minutes at 3,000 x g, RT) in a swinging-bucket rotor and wash once with 5 mL PBS.
Discard the supernatant, resuspend the cell pellet in 1 mL PBS and dilute a small sample of the cells as needed for accurate cell counting, using a hemacytometer or automated cell counting device.
Quickly thaw fresh human serum (400 μL/108 fungal cells) at 37 °C and adjust it to 40% with prewarmed PBS.
Pipette 108 cryptococcal cells of each strain to be tested into a new 1.5 mL microfuge tube, fill with PBS, and spin (8,000 x g for, 5 minutes, RT) in a fixed-angle rotor microcentrifuge. Aspirate the liquid. (You may scale this procedure down as appropriate.)
Resuspend the pellet in 1 mL pre-warmed 40% fresh human serum solution. Be sure to disperse any clumps by pipetting.
Incubate with end-over-end rotation at 37 °C for 30 minutes to opsonize the cells.
Collect the cells by centrifugation (5 minutes at 8,000 x g, RT) in a fixed-angle rotor microcentrifuge and replace the liquid with 1 mL EBM-2 basal media.
Remove a small aliquot to confirm the cell density.
SUPPORT PROTOCOL 1
Isolation of serum from human blood
Blood is composed of three fractions: cellular blood, plasma, and serum. Cellular blood contains erythrocytes, leukocytes, and platelets; plasma is whole blood minus cellular blood; and serum is plasma minus coagulation factors. Serum include antibodies and complement proteins, which are the basis for opsonization of foreign organisms in the blood. Serum is easily isolated from whole blood by allowing the blood to clot, sedimenting the clot by centrifugation, and collecting the supernatant (the serum). The following support protocol describes the procedure for isolating serum from 60 mL of whole blood, collected from healthy donors by a trained professional using procedures approved by an institutional review board (IRB), including informed consent. Alternatively, you may purchase human serum from a commercial source.
Materials
Solutions and reagents
10% bleach
Special equipment
Glass tubes (18 mm x 150 mm or larger; 3 tubes per 60 mL of whole blood)
2 mL cryogenic tubes
Liquid nitrogen and cryogenic vessel
Protocol steps
-
Dispense 20 mL of blood into each of three sterile glass tubes.
Note: Apply universal precautions for bloodborne pathogens work throughout this procedure. Dispose of all blood contaminated materials and needles in appropriate approved containers for hazardous materials and sharps. Since the blood should clot for serum collection, no special collection tubes are required. Alternatively, commercially available red-topped (no additive) tubes may be used. By dividing into three tubes, we increase the surface area exposed to the environment, fostering coagulation while minimizing cell lysis, which can contaminate the serum preparation.
-
Place parafilm tightly over the top of each tube and poke a few holes with a 21-gauge needle.
Note: This will minimize the risk of contamination while waiting for the blood to clot (next step).
Place tubes in a 37 °C / 5% CO2 incubator for 30 minutes to allow blood to clot. Alternatively, tubes can be placed at 4 °C overnight.
-
Transfer the fluid from around the clotted blood to 50 mL-conical tubes.
Note: Any clotted material moved in error at this stage will be removed in the next step.
Centrifuge 15 minutes at 1,800 x g, 4 °C in tabletop swinging-bucket centrifuge.
-
Carefully remove the clear serum, avoiding any clot material or cells at the bottom, and aliquot 400 μL into 2 mL cryotubes.
Note: This volume was chosen to facilitate reconstitution into 40% serum later by adding 600 μL of PBS, but may be modified for convenience of the researcher.
Flash freeze in a liquid nitrogen vessel and store at −80 °C.
BASIC PROTOCOL 4
In vitro brain endothelial cell transmigration assay
In BASIC PROTOCOL 2, a brain endothelial monolayer is established, with its development monitored by TEER. In BASIC PROTOCOL 3, the C. neoformans cells were treated for maximal interaction with the endothelial cells. The protocol below details the assay for transmigration of free C. neoformans cells across a brain endothelial monolayer as in (Santiago-Tirado et al., 2017).
Materials
Solutions and reagents
Opsonized fungal cells (from BASIC PROTOCOL 3).
Differentiation medium (see recipe).
YPD solid medium (see recipe).
1X PBS (see recipe).
Special equipment
EVOM2 Epithelial voltohmmeter, with chopstick electrode (WPI).
BD Falcon 12-well culture insert companion plates (Corning 353503; one per timepoint).
Protocol steps
-
Dilute fungal cells to 2 × 106 cells/mL in differentiation media pre-warmed to 37 °C, preparing enough cell suspension for all of the experimental inserts (500 μL per insert). Keep the cells at 37 °C while you check the monolayers.
Note: The recommended ratio in this protocol of 106 fungal cells/insert represents approximately 7 – 8 fungal cells per hCMEC/D3 cell. This ratio yields efficient transmigration efficiency under the conditions described, without disrupting the monolayer.
-
Immediately before starting the experiment, measure the TEER to ensure that all of the inserts have well-developed, uninterrupted monolayers (see BASIC PROTOCOL 2).
Note: Although TEER measurements during monolayer development are optional, TEER must in all cases be measured at the beginning of the transmigration experiment to ensure integrity of the monolayer. This is imperative since inserts may (rarely) be defective or the barrier disrupted during experimental manipulations, which will alter results.
-
Carefully move the inserts into a new (empty) 12-well companion plate, aspirate the medium, and add 500 μL of the fungal cell suspension to each insert that should receive fungal cells (see Figure 1). Add pre-warmed differentiation medium alone to control inserts that will receive neither cell type or only hCMEC/D3 cells (the control inserts).
Note: Keep the remainder of the fungal cell suspension to confirm the inoculum by spreading appropriate dilutions (e.g. 10 μl of a 1:100 dilution) on YPD agar and counting colony forming units (CFU).
Promptly add 1.5 mL of differentiation medium to the bottom chamber of each well. Be sure to use pre-warmed solutions to prevent cell shock and disruption of the monolayer.
Return the plate to the 37 °C incubator with 5% CO2 and note the start time.
After 2 hr, remove the plate from the incubator and carefully move the inserts into a fresh plate containing 1.5 mL pre-warmed differentiation media. Measure the TEER and return the fresh plate to the incubator.
Spot aliquots from all wells of the original lower chamber plate (from which the inserts were removed in step 6 above) on YPD agar for CFU quantification; spot control wells also.
Repeat steps 6 and 7 at 4 hours and 6 hours after the start of the experiment.
Incubate the YPD agar plates at 30 °C for 2 – 3 days, or until colonies are visible and can be counted.
-
Record results as absolute CFU in the lower chamber; you may also wish to normalize this value to CFU in the original inoculum (from step 9 of BASIC PROTOCOL 3). These values represent the fungi that crossed the model BBB at the measured time point.
Note: No growth should be seen from the control wells that did not receive fungi, this would suggest cross-contamination of wells during TEER measurements.
REAGENTS AND SOLUTIONS
Complete growth medium:
Fetal bovine serum (FBS; 2.5%), penicillin-streptomycin (1%), hydrocortisone (1.4μM), ascorbic acid (5μg/mL), chemically defined lipid concentrate (1%), HEPES (10mM), bFGF (1ng/mL), in EBM-2 basal media. Store at 4 °C for up to one month.
We use this recipe as described in the original report for this cell line [4]. However, many researchers have used the EGM-MV BulletKit from Lonza, with similar or better growth (Fazakas et al., 2011; Forster et al., 2008; Sabiiti & May, 2012; Vu et al., 2013). Commercial distributors of this line also recommend other media, but we have not tested these.
Differentiation medium:
Fetal bovine serum (FBS; 1%), penicillin-streptomycin (1%), hydrocortisone (1.4μM), ascorbic acid (5μg/mL), chemically defined lipid concentrate (1%), HEPES (10mM), in EBM-2 basal media. This is the same as complete growth medium but with only 1% FBS and no growth factors). Store at 4 °C for up to a month.
Growth in this medium leads to a significant increase in TEER for hCMEC/D3 cells (Forster et al., 2008).
Rat Collagen I:
From a commercially available high concentration (5 – 10 mg/mL) solution of rat collagen in acetic acid, dilute in molecular biology grade, sterile, distilled H2O, to a final protein concentration of 150 μg/mL. Use distilled H2O to avoid gelling of the solution and facilitate pipetting. The stock solution can be kept at 4 °C until expiration date. The diluted, working solution should be used immediately and any leftovers discarded.
YPD liquid medium:
Mix Bacto yeast extract (10 g/L), Bacto peptone (20 g/L), and glucose (20 g/L), in H2O and autoclave. Keep at RT indefinitely.
YPD solid medium:
Mix Bacto yeast extract (10 g/L), Bacto peptone (20 g/L), glucose (20 g/L), and Bacto agar (20 g/L) in H2O. Autoclave, cool to 60 °C and pour into sterile petri dishes (typically 100 mm, but larger ones can be used). Store at RT until media starts to dry, or at 4 °C for up to several months.
1X Phosphate-buffered saline (PBS):
Mammalian cells are sensitive to trace amounts of detergents, so it is critical that the PBS not contain these contaminants. You may either purchase 1X or 10X stock solutions of Dulbecco’s PBS, or, using detergent-free containers, make a 1X solution by mixing the following in ultrapure water: 8 g/L NaCl, 200 mg/L KCl, 1.44 g/L Na2HPO4, and 240 mg/L KH2PO4. After complete dissolution of salts, adjust the pH to 7.2 with HCl. Dispense into convenient volumes and sterilize by autoclaving. Store at RT indefinitely.
COMMENTARY
Background Information
In 2016, invasive fungal infections collectively killed 1.6 million people (Bongomin et al., 2017). Of these infections, the ones that affect the central nervous system (CNS) have the highest morbidity and mortality. One of these is the ubiquitous environmental fungus Cryptococcus neoformans, which causes ~200,000 deaths yearly in the HIV+ population, and increasing burden in other patient groups (O’Halloran et al., 2017). C. neoformans is acquired by inhalation, but the fatal pathology occurs in the brain, making the mechanism of this dissemination a critical area of research in cryptococcal pathogenesis.
Of the roughly 50 species of Cryptococcus, only C. neoformans and C. gattii are significant pathogens of humans. C. neoformans causes 95% of cases of cryptococcosis in immunosuppressed individuals, generally manifested as cryptococcal meningitis (CM). C. gattii, on the other hand, causes 90% of the disease in immunocompetent individuals, mostly as a complex respiratory infection.
The BBB is a diffusion barrier that prevents toxic agents in the blood, from ions to pathogens, from entering the CNS, maintaining a safe environment for the brain function (Daneman & Prat, 2015). In vivo, the BBB is a multicomponent unit, known as the neurovascular unit, made up of endothelial cells, astrocytic end-feet, pericytes, and various extracellular matrices known as the basal, or basement, membrane. However, endothelial cells that play the most important role in construction of the BBB and provide most of the barrier function thanks to expression of tight junctions between them. Hence, simple models of monoculture using endothelial cells, like the one described here, are widely used due to their ease of usage, cost-effectiveness, and reproducibility.
The model detailed here uses the hCMEC/D3 human cell line, which was obtained by transducing normal human microvascular endothelial cells with the large T antigen oncogene from SV40 (Weksler et al., 2005). These cells exhibit multiple properties that make them an excellent option for this assay: they are of human origin; express most of the markers associated with normal brain or meningeal endothelial cells; maintain these properties for at least 35 passages; and are commercially available. These cells have been used to show that C. neoformans can cross such barriers either alone or as a passenger within migrating leukocytes (Sabiiti & May, 2012; Santiago-Tirado et al., 2017; Sorrell et al., 2016; Vu et al., 2009; Vu et al., 2013).
In vitro models like this one have long been used to study the dynamics, development, and permeability of the blood-brain barrier (BBB); other applications include the transmigration of pathogens. Inclusion of other cell types, such as astrocytes and pericytes (Helms et al., 2016), would yield a model that most closely resembles the actual neurovascular unit. BASIC PROTOCOLS 3 and 4 would still be applicable to these more complex in vitro models.
Critical Parameters
For the most reproducible results, consistency in experimental setup is of the utmost importance. As noted below in the troubleshooting guide, consistent materials are critical; for example, the membranes of 12-well format inserts from different distributors might vary in size and pore density. It is important to use at least 8 μm pore size inserts because C. neoformans cells are 5 – 6 μm in length and surrounded by a rigid cell wall. Handle the cell culture inserts and plates aseptically inside the hood at all times.
To measure TEER across monolayers grown on large inserts in 12-well plates (typically used for experiments other than high-throughput applications), you need to use a “chopstick” style electrode. For smaller inserts, used in plates for high throughput screening applications, fixed, stand-alone electrodes can be used. This type of electrode has the advantage that it fits neatly on top of the filter well, giving stable and reproducible readings. Once the cells are seeded on the inserts, temperature variations will have a large effect on TEER readings, so pre-warm all solutions to 37 °C and limit the length of time that the plate is out of the incubator as much as possible. Extreme care must be employed when handling the electrodes or replacing the media, to avoid touching the membrane or creating bubbles, particularly in the lower compartment.
The passage number of the hCMEC/D3 cells is another critical parameter, because as these cells age they begin to they lose endothelial marker expression and functionality, and barrier development is not as robust. Also critical to reproducibility is how TEER is measured, including the choice of instrument, the effect of temperature (lower temperature increases TEER), and avoiding cross-contamination.
Additional experimental parameters that may alter results include the age of the fungal colony used, the choice of serum source (because commercial serum may vary between lots), and the ratio of serum to cell number in opsonization (which may impact the number of fungi that interact with the hCMEC/D3 cells). For the latter this protocol suggests using 1 mL of 40% serum per 108 fungal cells, which we found to give optimal phagocytosis after a 30-minute incubation, but this may need optimization for specific protocols in individual laboratories. In any even the the ratio selected should be kept constant throughout the experiments. An alternative to serum opsonization is to use mouse monoclonal antibodies against the capsule that surrounds the fungal cell (Feldmesser et al., 2000; Mukherjee et al., 1995), although the difference in species (mouse antibody versus human cell line) should be kept in mind, as well as the type of fungal cell being assayed (e.g. for acapsular mutants, antibody opsonization would not work).
Troubleshooting
Understanding Results
In this unit, we describe the protocols and methods necessary to perform a transmigration assay of C. neoformans across a simple BBB model. With this assay it is possible to rigorously test experimental hypotheses about the behavior of C. neoformans, in terms of crossing the barrier and how this is influenced by changes in host cells or conditions. This method may be applied to other fungi and neuropathogens as well.
The interaction of C. neoformans with brain microvascular endothelial cells, at least in vitro, is dependent on opsonization. You should expect to observe that C. neoformans crosses in a time dependent manner, and that opsonization increases the number of transmigrated cells significantly. You should also notice that, using the number of cryptococcal cells suggested here, there are minimal changes to the TEER of the barrier. (This observation indicates that the main route used by C. neoformans is the transcellular route, whereby fungal cells get internalized on the apical side of the brain endothelial cells and then exit on the basolateral side. Paracellular crossing, where fungi crosses between endothelial cells by disrupting the cell-cell junctions, does not play a big role under this in vitro model.) This assay is easily adaptable to multiple experiments, for example comparing wild-type to mutant strains, or free fungal cells to internalized cells (Trojan horse route; (Santiago-Tirado et al., 2017)).
To analyze results stringently, appropriate statistical tests and careful analysis should be used. For example, a T-test can be used to determine whether the difference between any two conditions is significant; for three or more conditions (e.g. testing crossing of wild-type, mutant, and complemented strains) other appropriate tests (e.g. one-way ANOVA) should be applied. This is a complicated system with multiple variables, so every experiment must have multiple biological and technical replicates and results should be consistent across multiple repeated experiments before conclusions are drawn.
Time Considerations
Preliminary preparations (making solutions, media or pouring plates) can be done before starting the experiment and take 2 – 3 hr. The procedure for growth and subculture of hCMEC/D takes 1.5 – 2 hr every 3 – 4 days. If planned correctly, procedures from protocols 1 through 3 should not take more than 2 – 3 hr daily. Many of the procedures involve long incubations, like coating plastic culture surfaces or inserts with collagen, during which time other steps can be performed. Once the barriers are seeded, approximately five days will be required to achieve confluence. An additional day is required to induce differentiation and increase TEER values. During this day, start growing your C. neoformans cells and double check your reagents and solutions (e.g. have enough YPD plates for sampling timepoints). If measuring TEER daily, TEER measurements for 12-well insert plates, including sterilizing the electrodes between readings, take 30 min – 1 hr. On the day of the transmigration experiment, moving the inserts to a new plate, measuring the TEER, and plating for CFU should not take more than 1 hr for each time point if doing a simple experiment like that illustrated in Figure 1 (testing 2 conditions). Once plated, C. neoformans colonies take 2 – 3 days to be ideally sized to count. Overall, a transmigration experiment will take approximately 10 days if media and plates are prepared on Day 1, inserts seeded on day 2, transmigration experiment performed on Day 7, and colonies counted on Day 10.
Table 1:
Troubleshooting Guide
| Problem | Possible cause | Possible solutions |
|---|---|---|
| Barrier development is minimal (TEER < 10 ohms* cm2) | Contamination | Check under a microscope for the presence of bacteria or fungi. Remember to include 1% penicillin-streptomycin in your media. |
| Bad or expired culture media | Prepare fresh culture media. | |
| Wrong pore size inserts | For hCMEC/D3, TEER development is slow and minimal when using pore sizes of 3-micron or less. Use an 8-micron pore insert. | |
| hCMEC/D3 seeding density too low | Review your numbers for the initial hCMEC/D3 seeding density. If too low, cells won’t establish a monolayer. | |
| TEER drops suddenly | Contamination | Check under a microscope for the presence of bacteria or fungi. Remember to include 1% penicillin-streptomycin in your media. |
| During insert manipulation (media replacement, TEER measurement, etc.) the monolayer or membrane broke | Note which insert is the problem. If the monolayer was only disrupted, the TEER will rebound within hours, but if the membrane was broken, the TEER will remain low. If the latter discard that insert. | |
| Too high of an inoculum. | In 0.9 cm2 inserts, more than 107 C. neoformans cells will cause the barrier to break down. | |
| CFUs arising from control wells | Cross-contamination from insert manipulation | When replacing media, make sure you use sterile tips. When measuring TEER, clean the electrodes with ethanol and PBS between readings. Make sure the PBS or other media you use is sterile. |
| Using contaminated PBS for plating dilutions | Make sure the PBS (or another diluent) you are using is sterile. | |
| No CFUs obtained from any wells | Inoculum too low | Increase the C. neoformans inoculum. |
| Wrong agar plates | Make sure you are using YPD plates for CFU determination. | |
| Wrong size pore insert | Make sure you are using 8-micron (or larger) pore inserts. | |
| Absolute transmigration varies widely between replicates | Different ratios of serum used during opsonization | Make sure you use the same ratio of cryptococcal cells to 40% serum between your replicates. |
| OD600 too different and/or age of cryptococcal starter culture too old | When using different strains within an experiment, or the same strain between experiment, make sure to grow them to OD600 of 0.8 – 1.2 (log phase). Also, cells from old plates (> 4 weeks) behave heterogeneously, so make sure to start your culture from freshly streaked cells. | |
Significance Statement.
Cryptococcal meningitis (CM) is a leading cause of death among AIDS patients that also has significant impact on other immunocompromised populations; this disease also occurs, although less commonly, in immunocompetent individuals. This lethal infection is caused by the environmental yeast Cryptococcus neoformans, and although it is contracted via inhalation, the fatal pathology occurs in the brain. In order to reach the brain, the yeast must cross the endothelial cell barriers protecting this site. Understanding this traversal will increase understanding of fundamental biological mechanisms and may enable the development of novel, more effective strategies to prevent or treat CM.
ACKNOWLEDGEMENT
Development of these methods was supported by NIH grant AI114549 to TLD and RSK; NIH grants AI078795 and AI102882 to TLD. FHS’s training was supported in part by NIH T32 grant AI007172 and a Postdoctoral Enrichment Program Award from the Burroughs Wellcome Fund.
LITERATURE CITED
- Bongomin F, Gago S, Oladele RO, & Denning DW (2017). Global and multi-national prevalence of fungal diseases-estimate precision. Journal of Fungi (Basel, Switzerland), 3(4), 57. doi: 10.3390/jof3040057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YC, Stins MF, McCaffery MJ, Miller GF, Pare DR, Dam T, … Kwon-Chung KJ (2004). Cryptococcal yeast cells invade the central nervous system via transcellular penetration of the blood-brain barrier. Infection and Immunity, 72(9), 4985–4995. doi: 10.1128/IAI.72.9.4985-4995.2004 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Stins MF, Huang SH, Chen YH, Kwon-Chung KJ, Chang Y, … Jong AY (2003). Cryptococcus neoformans induces alterations in the cytoskeleton of human brain microvascular endothelial cells. Journal of Medical Microbiology, 52(Pt 11), 961–970. doi: 10.1099/jmm.0.05230-0 [doi] [DOI] [PubMed] [Google Scholar]
- Daneman R, & Prat A (2015). The blood-brain barrier. Cold Spring Harbor Perspectives in Biology, 7(1), a020412. doi: 10.1101/cshperspect.a020412 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazakas C, Wilhelm I, Nagyoszi P, Farkas AE, Hasko J, Molnar J, … Krizbai IA (2011). Transmigration of melanoma cells through the blood-brain barrier: Role of endothelial tight junctions and melanoma-released serine proteases. PloS One, 6(6), e20758. doi: 10.1371/journal.pone.0020758 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmesser M, Rivera J, Kress Y, Kozel TR, & Casadevall A (2000). Antibody interactions with the capsule of cryptococcus neoformans. Infection and Immunity, 68(6), 3642–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster C, Burek M, Romero IA, Weksler B, Couraud PO, & Drenckhahn D (2008). Differential effects of hydrocortisone and TNFalpha on tight junction proteins in an in vitro model of the human blood-brain barrier. The Journal of Physiology, 586(7), 1937–1949. doi: 10.1113/jphysiol.2007.146852 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms HC, Abbott NJ, Burek M, Cecchelli R, Couraud PO, Deli MA, … Brodin B (2016). In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. Journal of Cerebral Blood Flow and Metabolism : Official Journal of the International Society of Cerebral Blood Flow and Metabolism, 36(5), 862–890. doi: 10.1177/0271678X16630991 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RS, Garber C, & Howard N (2017). Infectious immunity in the central nervous system and brain function. Nature Immunology, 18(2), 132–141. doi: 10.1038/ni.3656 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon-Chung KJ, Edman JC, & Wickes BL (1992). Genetic association of mating types and virulence in cryptococcus neoformans. Infection and Immunity, 60(2), 602–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Lee SC, & Casadevall A (1995). Antibodies to cryptococcus neoformans glucuronoxylomannan enhance antifungal activity of murine macrophages. Infection and Immunity, 63(2), 573–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen K, Cox GM, Wang P, Toffaletti DL, Perfect JR, & Heitman J (2003). Sexual cycle of cryptococcus neoformans var. grubii and virulence of congenic a and alpha isolates. Infection and Immunity, 71(9), 4831–4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Halloran JA, Powderly WG, & Spec A (2017). Cryptococcosis today: It is not all about HIV infection. Current Clinical Microbiology Reports, 4(2), 88–95. doi: 10.1007/s40588-017-0064-8 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman NA, Rasil ANHM, Meyding-Lamade U, Craemer EM, Diah S, Tuah AA, & Muharram SH (2016). Immortalized endothelial cell lines for in vitro blood-brain barrier models: A systematic review. Brain Research, 1642, 532–545. doi:S0006-8993(16)30232-3 [pii] [DOI] [PubMed] [Google Scholar]
- Rajasingham R, Smith RM, Park BJ, Jarvis JN, Govender NP, Chiller TM, … Boulware DR (2017). Global burden of disease of HIV-associated cryptococcal meningitis: An updated analysis. The Lancet. Infectious Diseases, 17(8), 873–881. doi: 10.1016/S1473-3099(17)30243-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabiiti W, & May RC (2012). Capsule independent uptake of the fungal pathogen cryptococcus neoformans into brain microvascular endothelial cells. PloS One, 7(4), e35455. doi: 10.1371/journal.pone.0035455 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Tirado FH, Onken MD, Cooper JA, Klein RS, & Doering TL (2017). Trojan horse transit contributes to blood-brain barrier crossing of a eukaryotic pathogen. mBio, 8(1), 16. doi:e02183-16 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrell TC, Juillard PG, Djordjevic JT, Kaufman-Francis K, Dietmann A, Milonig A, … Grau GE (2016). Cryptococcal transmigration across a model brain blood-barrier: Evidence of the trojan horse mechanism and differences between cryptococcus neoformans var. grubii strain H99 and cryptococcus gattii strain R265. Microbes and Infection, 18(1), 57–67. doi: 10.1016/j.micinf.2015.08.017 [doi] [DOI] [PubMed] [Google Scholar]
- Srikanta D, Santiago-Tirado FH, & Doering TL (2014). Cryptococcus neoformans: Historical curiosity to modern pathogen. Yeast (Chichester, England), 31(2), 47–60. doi: 10.1002/yea.2997 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu K, Eigenheer RA, Phinney BS, & Gelli A (2013). Cryptococcus neoformans promotes its transmigration into the central nervous system by inducing molecular and cellular changes in brain endothelial cells. Infection and Immunity, 81(9), 3139–3147. doi: 10.1128/IAI.00554-13 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu K, Weksler B, Romero I, Couraud PO, & Gelli A (2009). Immortalized human brain endothelial cell line HCMEC/D3 as a model of the blood-brain barrier facilitates in vitro studies of central nervous system infection by cryptococcus neoformans. Eukaryotic Cell, 8(11), 1803–1807. doi: 10.1128/EC.00240-09 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weksler BB, Subileau EA, Perriere N, Charneau P, Holloway K, Leveque M, … Couraud PO (2005). Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology, 19(13), 1872–1874. doi:04-3458fje [pii] [DOI] [PubMed] [Google Scholar]