

Abstract
Alternative splicing (AS) promotes transcriptome and proteome diversity during growth, development, and stress responses in eukaryotes. Genome-wide studies of AS in sugarcane (Saccharum spp.) are lacking, mainly due to the absence of a high-quality sequenced reference genome, sugarcane’s large, complex genome, and the variable chromosome numbers and polyploidy of sugarcane cultivars. Here, we analyzed changes in the sugarcane isoform-level transcriptome and AS landscape during infection with the smut fungus (Sporisorium scitamineum) using a hybrid approach involving Sorghum bicolor reference-based and Trinity de novo mapping tools. In total, this analysis detected 16,039 and 15,379 transcripts (≥2 FPKM) at 5 and 200 days after infection, respectively. A conservative estimate of isoform-level expression suggested that approximately 5,000 (14%) sugarcane genes undergo AS. Differential expression analysis of the alternatively spliced genes in healthy and smut-infected sugarcane revealed 896 AS events modulated at different stages of infection. Gene family and gene ontology functional enrichment analysis of the differentially spliced genes revealed overrepresentation of functional categories related to the cell wall, defense, and redox homeostasis pathways. Our study provides novel insight into the AS landscape of sugarcane during smut disease interactions.
Subject terms: Biotic, Plant molecular biology
Introduction
Sugarcane (Saccharum spp., Poaceae family) is a high-value C4 grass with a global estimated harvest yield of ~1.89 billion tons in 20161, contributing to ~75% of sugar and ~60% of ethanol production worldwide2,3. The biotrophic fungal pathogen Sporisorium scitamineum (Syd.) (previously known as Ustilago scitaminea4; causes wind- and soil-borne smut disease. Smut symptoms are characterized by powdery masses of teliospores, which form long black or gray whip-like structures emerging from the primary meristem of the sugarcane plant4–7. These whip-like structures contain a mixture of plant and pathogen cells with millions of teliospores. The teliospores can spread throughout a field, rapidly advancing the disease to new areas4,5,7. Smut is found worldwide and causes serious damage to sugarcane yield, juice quality, culms, and sucrose content5–8. Losses can range from 30–100% and, in extreme cases, can cause the demise of local varieties2,4,7,9. The severity of damage depends mostly on the smut race, sugarcane genotype, and neighboring environmental conditions7.
Development of smut-resistant sugarcane varieties is an arduous task due to 1) the complex sugarcane–smut pathosystem, 2) the many genes controlling resistance, which acts as a quantitative trait, and 3) the current poor understanding of gene-for-gene resistance7,8. Furthermore, considering annual increases in prevalence of sugarcane smut disease and lack of control strategies, development of smut-resistant sugarcane varieties has emerged as a key priority. Developing such varieties by manipulating the host system likely represents a safe, effective, economical, and environmentally friendly way of controlling smut disease2,7,8,10–13. Therefore, understanding sugarcane genetic responses to smut could provide key information for developing smut-resistant varieties.
We, and others, have reported genetic- and genome-level studies of sugarcane–smut interactions2,5,7,10,12,14. For instance10, studied the comparative response of reactive oxygen species (ROS) metabolism in smut-resistant and susceptible sugarcane genotypes. Transcriptome analysis of smut-infected sugarcane revealed modulation of several genes involved in diverse pathways, including lignin biosynthesis, providing clues for studying the progression of smut disease in sugarcane5,12. Another study showed differential expression of 13.5% of genes in the fungal genome at different developmental stages (5 and 200 DAI) of sugarcane–smut infection14. Moreover, integrative proteomics and transcriptomics analysis identified 273 and 341 differentially expressed proteins in smut-resistant and susceptible sugarcane genotypes, respectively13. These studies reveal significant information about the various transcription-level changes occurring during sugarcane–smut interactions. However, the extent of genome-wide changes in a key post-transcriptional process, alternative splicing (AS), in sugarcane has not yet been reported. This is mainly due to the lack of a sequenced reference genome and the daunting genome-level complexity of sugarcane hybrids with large polyploid genomes (~10 Gbp) and varying numbers of chromosomes15–17.
AS generates multiple transcripts (or isoforms) from a single precursor mRNA (pre-mRNA), increasing the diversity and complexity of the transcriptome and proteome18–22. AS can lead to truncated proteins and altered levels of protein activity22,23. Moreover, AS transcripts with varying sequences result in proteins with altered physical characteristics and molecular functions20,21, and it is estimated that 33 to 70%24 plant genes undergo AS25,26. In addition, AS regulates the level of functional transcripts by a mechanism called regulated unproductive splicing and translation (RUST)27. Splicing by the spliceosome complex composed of small nuclear riboproteins (snRNPs) occurs at exon-intron splice sites, usually GT-AG, which play a major role in forming alternative transcripts22. Differential splicing of multiexon genes results in four major types of AS: exon skipping (ES), intron retention (IR), alternative donor (AD), and alternative acceptor (AA) types20,26.
Several studies have shown that AS in plant genes can be modulated in response to biotic and abiotic stresses22,23,27. For example, the RPS4 disease resistance gene in Arabidopsis thaliana28 and the DREB transcription factor gene in wheat (Triticum aestivum L.)29 are alternatively spliced in response to biotic stress leading to various disease-induced isoforms. AS is also modulated during plant growth and development, photosynthesis, metabolic pathways, circadian clock function, and flowering22,26.
The advent of next-generation sequencing has enabled genome-wide RNA-sequencing studies to examine AS in several, primarily diploid, plants including Arabidopsis thaliana27, Brachypodium distachyon26, Zea mays30, and Physcomitrella patens19. AS events are often conserved among plant species. For instance, about 58% of AS events are conserved between rice (Oryza sativa) and Arabidopsis thaliana22. Also, AS events in an Arabidopsis splicing regulator gene (SCL33) are conserved in Brachypodium and other grasses26. In this study, we used RNA sequencing and a hybrid transcript mapping and assembly approach to decipher genome-wide expression changes at the isoform level and modulation of the AS landscapes following infection with Sporisorium scitamineum in a sugarcane hybrid showing intermediate smut resistance.
Results and Discussions
mRNA sequencing and Sorghum bicolor genome-based isoform calling
In the absence of a high-quality reference genome sequence for sugarcane, we leveraged the well-annotated, high-quality reference genome of the related grass Sorghum bicolor to establish AS events and isoform expression in sugarcane. During manuscript preparation, a draft monoploid sugarcane genome and Saccharum spontaneum genome sequences were released31,32. Unfortunately, neither of these published genomes included annotations for alternatively spliced transcripts. Moreover, the draft genomes also used Sorghum bicolor reference genome alignments as well as Trinity-based de novo transcript assemblies to annotate protein-coding genes. Sorghum bicolor and sugarcane share common ancestry, with extensive, genome-wide collinearity (80% with S. spontaneum L.) and few chromosomal rearrangements31–34. Comparative analysis of S. bicolor and sugarcane genome organization indicates that the diploid S. bicolor genome is a worthwhile resource for studying the highly complex polyploid sugarcane genome34.
Twelve samples representing control and smut-infected sugarcane at two stages of infection, i.e., early (5 DAI; before whip emergence) and late (200 DAI; after whip emergence), with three biological replicates each5 were subjected to paired-end Illumina HiScanSQ RNA-sequencing (Fig. 1). This produced 112 million raw reads (101 bp) and unambiguously aligned 107 million clean reads (Table 1) to the Sorghum bicolor (v3.1 release) genome35. The seemingly low overall alignment rate of less than 31% (Table 1) is due to the complex aneuploidy, heterozygous, and interspecific (hybrid) genome of sugarcane, when compared to the Sorghum genome. Genome-aligned sequence reads were further processed using Cufflinks, Cuffmerge, Cuffcompare, and Cuffdiff36 for discovery of reference and novel isoforms, and isoform-level differential expression analysis.
Figure 1.
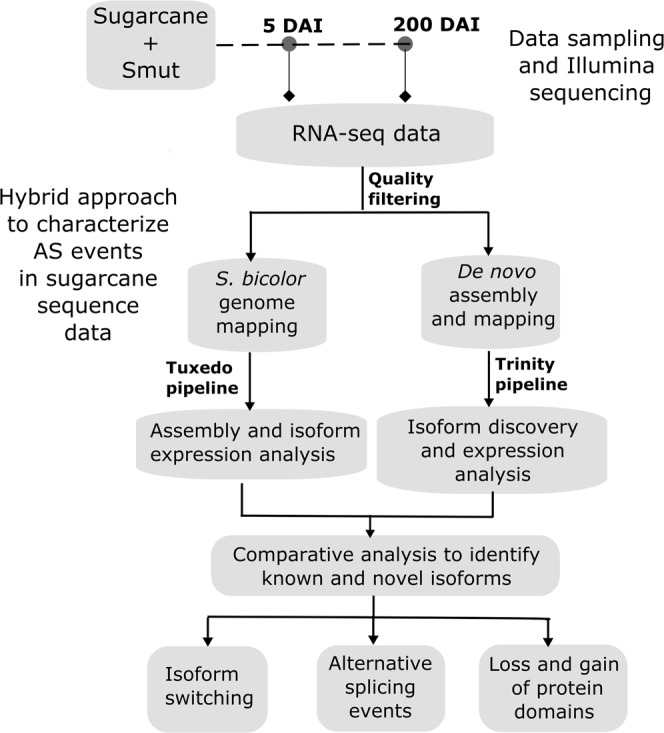
Experimental design and data analysis flowchart. Samples were collected at 5 and 200 DAI and subjected to RNA-seq analysis using three biological replicates for each sample. Twelve libraries were sequenced representing control and stress conditions at the two time points. Data were analyzed using a hybrid approach comprising reference genome (Sorghum bicolor)- and de novo (Trinity)-based mapping and assembly to identify alternative transcripts and isoform-level regulation.
Table 1.
Sequence read statistics for all RNA-seq libraries.
| Sample | Replicates | Total reads* | Total mapped readsa | Total uniquely mapped reads | Multiple mapped reads | Total reads mapped (%) |
|---|---|---|---|---|---|---|
| 5 DAI - Control | 3 | 27,783,727 (26,881,845) | 16,590,034 | 12,051,719 | 4,538,315 | 30.54 |
| 5 DAI - Infected | 3 | 28,179,752 (27,266,828) | 16,785,042 | 12,133,033 | 4,652,009 | 30.77 |
| 200 DAI - Control | 3 | 28,127,128 (26,228,856) | 11,459,114 | 10,187,726 | 1,271,388 | 21.84 |
| 200 DAI - Infected | 3 | 28,507,485 (26,628,131) | 13,302,600 | 8,138,565 | 5,164,035 | 24.97 |
*Paired-end RNA-seq reads; values in parentheses indicate cleaned, high-quality sequence reads.
aRNA-seq reads mapped to the Sorghum bicolor (v3.1 release) genome.
Expression analysis of samples collected at 200 DAI revealed that ~16,000 genes whose homologs are spread across all Sorghum bicolor chromosomes had transcriptional activity with log10 expression level > 1. To determine gene expression at the chromosome level, we plotted a Circos map of read density along the Sorghum bicolor chromosomes (Fig. 2). We observed a uniform distribution of sequence reads along the chromosomes, suggesting no systematic biases (Fig. 2). As expected, heterochromatic regions such as the centromeres showed little to no transcriptional activity. The Sorghum bicolor reference-based analysis identified ~52,567 sugarcane transcripts, of which ~4820 were novel isoforms.
Figure 2.

Global distribution of RNA-seq reads along Sorghum bicolor chromosomes. RNA-seq read density (log10 of absolute read count per gene) of the four sugarcane samples (5 DAI control, 5 DAI stress, 200 DAI control, and 200 DAI stress) across the 10 Sorghum bicolor chromosomes. Red dots indicate centromeric regions.
Trinity-based de novo isoform calling and annotation
To complement the alignments from Sorghum bicolor genome mapping, and to discover novel isoforms, we performed a de novo isoform assembly without any predefined genome annotations. We pooled sequence reads from all samples and performed de novo transcriptome assembly using Trinity37 to construct a consolidated reference transcriptome. Along with reporting assembled transcripts, Trinity resolves alternative isoforms of genes better than other transcriptome assembly tools37. Trinity assembled sequence reads into 322,205 transcripts constituting 212,023 unigenes. Among these unigenes, 36,925 had alternatively spliced isoforms (Supplementary Table S1). All unigenes and transcripts were annotated using public protein databases (see Materials and methods) and BLASTX38 with e-value cut-off < 1e-05. Priority for annotation was given to Sorghum bicolor protein databases35 followed by NCBI nr/nt39 and UniProtKB40 plant protein databases. Among 322,205 transcripts, 142,241 transcripts have shown significant similarity (e-value cut-off < 1e-05) to 28,613 (~60%) of sorghum proteins. Trinity-based de novo assembly with annotation using public protein databases allowed us to identify a substantial number of alternatively spliced transcripts.
Isoform-level expression changes in sugarcane–smut interactions
The Sorghum bicolor genome comprises 47,205 transcripts and 34,211 genes35. Of the total transcripts assembled by Cufflinks36, 15,379 (13,848 known and 1,531 novel) isoforms were expressed at FPKM ≥ 2 at 200 DAI. Similarly, 16,039 (14,423 known and 1,616 novel) isoforms were expressed at FPKM ≥ 2 at 5 DAI. Among these expressed transcripts, 394 and 324 transcripts at 5 and 200 DAI, respectively, did not have any biological annotation in Sorghum bicolor databases. The lower number of transcripts during later infection (200 DAI) compared to early infection (5 DAI) could be due to the low mapping rates of the 200 DAI samples (Table 1).
Differential expression analysis of the isoforms was performed using Cuffdiff36 to identify changes in AS under control and infected conditions. Low-abundance transcripts with FPKM < 2 were filtered out to preclude transcripts potentially resulting from incorrectly assembled transcripts or sequencing artifacts. We considered isoforms with log2 fold change ≥ 1 (21 absolute fold change) at P < 0.05 across control and infected conditions significantly differentially expressed in response to smut infection. Despite greater mapping rates, only 41 transcripts were differentially expressed at 5 DAI, while 855 isoforms were differentially regulated at 200 DAI (Fig. 3A,B; Supplementary Data Set 1). Among them, 530 and 11 isoforms were upregulated, and 325 and 30 isoforms were downregulated in response to smut infection at 200 and 5 DAI, respectively. The greater number of differentially expressed isoforms at 200 DAI compared with 5 DAI might result from enhanced plant stress and defense responses, related to the peak period of whip growth in sugarcane5. The 530 upregulated isoforms corresponded to 84 genes that are alternatively spliced or have more than one isoform.
Figure 3.

Isoform-level expression analysis in 5 and 200 DAI samples. (A,B) Volcano plots of differentially expressed isoforms at 5 and 200 DAI, respectively. Green and red dots indicate isoforms significantly upregulated (log2 ≥ 1 and P < 0.05) and downregulated (log2 ≤ -1 and P < 0.05), respectively, during smut infection. Hierarchical clustering and isoform-level expression dynamics of putative genes associated with (C) cell wall modification, (D) defense signaling, (E) transcription factors, and (F) ROS scavenging are represented by heatmaps (blue: upregulated isoforms; yellow: downregulated isoforms). List of the isoforms used in heatmap analysis are provided in Supplementary Data Set 2.
Of the 855 differentially regulated isoforms at 200 DAI, 676 isoforms were supported by the Trinity-assembled transcripts (Supplementary Data Set 1). To be conservative and rigorous in further AS analysis, we only explored differentially expressed isoforms identified by both Trinity and S. bicolor-based assembly.
Sugarcane genome-wide AS landscape affected by smut infection
Biotic and abiotic stresses modulate plant AS landscapes19,26,30. We determined AS events in sugarcane under control and smut-stress conditions based on S. bicolor mapped data. Using the S. bicolor annotation and spliced alignments, we categorized AS events into IR, ES, AD, AA, and other complex events.
We found 11,490, 10,699, 11,248, and 11,406 AS events in 200 DAI control, 200 DAI stress, 5 DAI control, and 5 DAI stress conditions, respectively. The distribution of individual IR, ES, AA, and AD events is shown in Fig. 4. AD and AA represented ~50% of the AS events followed by IR (~26%) and ES (~19%) (Fig. 4). We did not see any significant changes in the AS landscape between control and stress conditions, but the proportion of ES (19.57% vs 18.72%) and IR (26.87% vs 26.19%) events was higher under stress conditions than in controls at 200 DAI (Fig. 4).
Figure 4.

Frequency of AS events identified in RNA-seq data. Distribution of intron retention (IR), exon skipping (ES), alternative donor (AD), and alternative acceptor (AA) AS events under 5 and 200 DAI control (C) and infected (I) conditions.
Among the four major types of AS event, AD events appear to be predominant, while AA and ES were the least common at both 5 and 200 DAI (Fig. 4). The predominance of AD events contrasts with other plant AS landscapes where IR predominates26,41. This might be a unique feature of sugarcane, reflecting the complex genomic landscape of this polypoid and aneuploid species. Alternatively, the presence of multiple homeologs with polymorphisms could also result in an overestimation of AD or AA transcripts. Given the lack of a high-quality chromosome-level reference genome and annotation for the hybrid sugarcane, we interpret these results cautiously, and the possible scenarios need to be evaluated in the future.
Functional enrichment analysis of the alternatively spliced genes
To reveal the biological and molecular functions of the 855 isoforms (P < 0.05) that were differentially expressed in response to smut infection at 200 DAI, we performed functional GO and gene family (GenFam42; enrichment analyses. We characterized the enriched GO categories (Fisher exact test and FDR < 0.05) and visualized these as interaction networks based on parent–child relationships for ‘biological process,’ ‘molecular functions,’ and ‘cellular components’ (Fig. 5A). To avoid redundant functional analysis by GO categories, we also analyzed the data for gene family enrichment using GenFam. As expected, genes regulating cell wall biosynthesis and/or modifications, transcription factors, and biotic stress responses were highly enriched (Fig. 5B). Enriched GenFam and GO categories contained genes with alternatively spliced isoforms belonging to various functional categories such as cell wall fortification, defense signaling, and transcription factors.
Figure 5.

GO and gene family (GenFam) enrichment analysis of induced isoforms in response to smut infection at 200 DAI. (A) Enriched GO terms visualized using Cytoscape (see Materials and Methods). GO term interaction networks based on ancestor–child relationships for ‘biological process,’ ‘molecular function,’ and ‘cellular component’ categories. Node size indicates the number of genes in that GO term, and the color scale corresponds to Benjamini-Hochberg P-values. (B) Gene family enrichment analysis of induced isoforms in response to smut infection at 200 DAI using the GenFam tool.
GO terms enriched under the ‘biological process’ category were ‘response to stress’ (GO:0006950), ‘response to biotic stimulus’ (GO:0009607), and ‘transport’ (GO:0006810), while those enriched under ‘molecular functions’ were ‘transcription factor activity’ (GO:0003700), ‘hydrolase activity’ (GO:0016787), and ‘transporter activity’ (GO:0005215). Similarly, in the ‘cellular component’ category, cell wall related terms such as ‘cell wall’ (GO:0005618) and ‘plasma membrane’ (GO:0005886) were highly enriched (Fig. 5A). Comprehensive analysis of GO terms enriched under ‘molecular function,’ ‘biological process,’ and ‘cellular component’ were comparable to each other. Complementing the GO analysis, GenFam allowed identification of overrepresented gene families in biological processes such as cell wall modifications (expansins), transcription factors (HSF), transport (auxin permease), and defense signaling (calmodulin-binding and phosphatases genes) among the alternatively spliced genes (Fig. 5B).
Alternatively spliced genes perturbed during smut infection
A significant number of genes underwent AS in response to smut infection at 200 DAI, and some of these were related to defense responses. We categorized the alternatively spliced genes into different functional categories and found that several genes associated with cell wall modifications, transcription factors, ROS scavenging, and defense signaling were alternatively spliced during smut infection (Fig. 3; Supplementary Data Set 1 and 2). Characterization of AS among genes in these categories revealed multiple types of AS ranging from simple to complex isoform switching between control and smut-infected samples (Fig. 6; Supplementary Fig. S1).
Figure 6.

Gene isoform switching at 200 DAI in response to smut infection. Isoform expression changes in genes encoding A) xyloglucan endotransglucosylase/hydrolase (XTH) and D) lipoxygenase (LOX2) (200 C: 200 DAI Control; 200 S: 200 DAI infected). Isoform structures of B) XTH and E) LOX2 with known and novel isoforms, and their corresponding protein structures (C and E). Untranslated and intronic regions are represented by dotted and solid lines, respectively. Left- and right-facing arrows indicate transcript strand orientation.
The cell wall is a primary defense against fungal infection. Several sugarcane genes/isoforms involved in cell wall modification were significantly expressed and alternatively spliced during smut infection (Fig. 3A–C; Supplementary Data Set 1 and 2). For instance, a gene encoding a putative sugarcane xyloglucan endotransglucosylase/hydrolase (Sobic.002G324100, TRINITY_DN55793_c0_g1_i1, XTH, GO:0005618), which could function in cell wall strengthening43,44, was differentially regulated and alternatively spliced, with at least three isoforms expressed during smut infection. Increased activity of XTH has been linked with the pathogenic defense response by modifying cell wall structure44,45. The orthologous gene in sorghum, Sobic.002G324100, is annotated as producing two isoforms, with varying 5′ untranslated regions (UTRs). Sugarcane XTH has at least three isoforms—two corresponding to sorghum isoforms and a novel isoform predicted by cufflinks. Isoform 1 of XTH was the most abundant and could be the primary transcript (Fig. 6A). Isoforms 2 and 3 appeared to have alternative 5′ transcription start sites and exon/intron structures, compared to the primary transcript 1 (Fig. 6B). These XTH isoforms also mapped to a locus in the recently published monoploid sugarcane and Saccharum spontaneum draft genomes encoding a putative XTH gene, Sh02_t023560 and Sspon.002C0009350, respectively. We validated the presence of multiple XTH isoforms (numbered 1 and 2) by RT-PCR, and, as predicted, some isoforms were more common in smut-infected samples (Fig. 7). All three isoforms encoded Glyco_hydro_16 and XET_C domains (Fig. 6C), but had alterations in the N-terminal sequence. AS producing changes in the 5′ UTR of XTH could also affect transcript stability, translation efficiency and/or subcellular localization46,47.
Figure 7.

RT-PCR analysis of differentially expressed sugarcane gene isoforms. Alternative splicing in selected sugarcane genes was analyzed using RT-PCR. Molecular size (bp) of the isoforms is shown using a DNA ladder. Putative alternative transcripts/isoforms are labeled numerically. −: negative control/no template; *: primer-dimer. The top and bottom panels were cropped and grouped from different parts of the same gel for clarity. An uncropped image used to prepare this figure is presented in Supplementary Fig. S3.
We also identified differential splicing in a defense-related gene cluster (Fig. 3D). A lipoxygenase-encoding gene (Sobic.004G078600, TRINITY_DN62422_c1_g1_i1, LOX2, GO:0009607) was alternatively spliced, with two isoforms differentially expressed during smut infection (Fig. 6D). These isoforms had altered 5′ UTRs with different transcription start sites (Fig. 6E). Isoform 2 appeared to be highly induced in response to smut infection when compared to isoform 1. Both LOX2 isoforms encoded full-length PLAT and LOX domains with alterations in the N-terminal regions of the proteins (Fig. 6F). As discussed above for XTH, alteration of the 5′ UTR of LOX2 isoforms could affect transcript stability and relative abundance of isoforms encoding functional LOX2. RT-PCR analysis confirmed the presence of up to six LOX2 isoforms (Fig. 7). Several of these isoforms appeared to be differentially expressed in smut-infected and control samples (Fig. 7). In addition, we found splice variants for additional genes encoding stress-responsive transcription factors such as bZIP and HB by RT-PCR analysis (Fig. 7), providing some empirical evidence for our predictions and demonstrating the utility of the hybrid isoform calling approach (Fig. 1).
In addition to AS, stress induces several types of isoform-level changes in expression or switching48. In simple isoform switching, isoforms have varying expression levels but often antagonistic patterns, thus creating little or negligible change in overall gene-level expression (Mandadi and Scholthof, 2015b). In this study, a zinc finger family gene (Sobic.006G183000) showed simple isoform switching. Isoform 1 (Sobic.006G183000.1) was upregulated under control conditions, whereas isoform 2 (Sobic.006G183000.2) was downregulated under stress conditions (Supplementary Fig. S1). In contrast, both XTH and LOX2 genes demonstrated complex isoform switching patterns, with several isoforms showing various expression dynamics (Fig. 6).
To identify the effect of differential splicing on proteins, we determined if AS in multiexonic genes resulted in gain or loss of protein domains. We randomly selected two genes, one encoding tubulin alpha-5 (Sobic.002G350400; TRINITY_DN128043_c0_g1_i1) and the other encoding a floral homeotic protein (HUA1) (Sobic.001G132200; TRINITY_DN69225_c0_g1_i2) among the genes significantly induced in response to smut infection at 200 DAI. Tubulin alpha-5 has two known isoforms. In silico predictions suggested that isoform 1 has two tubulin domains, comprising the autoregulation signal and tubulin subunits, whereas isoform 2 lacks tubulin subunit (Supplementary Fig. S2B). During infection, only expression of isoform 1 (Sobic.002G350400.1) was significantly induced (Supplementary Fig. S2A). Tubulin proteins are major components of microtubules and are crucial for plant cell wall development49. AS-mediated changes in the tubulin alpha-5 protein structure could influence the overall levels and/or homeostasis of functional tubulin alpha-5 protein, having biological implications for sugarcane defense responses to smut. Similarly, smut infection induced expression of HUA1 isoform 1, while levels of isoform 2 were downregulated (Supplementary Fig. S2C). Isoform 1 was predicted to encode a protein with seven zinc finger domains, whereas isoform 2 lacked several zinc finger domains (Supplementary Fig. S2D). Zinc finger domains are required for RNA/DNA binding and play a crucial role in plant development50. In a manner similar to tubulin alpha-5, AS-mediated changes in HUA1 protein could have biological implications for sugarcane–smut interactions. Although we made in silico predictions and hypotheses regarding how AS could influence protein-level changes, further functional-genetic studies would be needed to test these hypotheses.
Conclusions
Analysis of AS and isoform-level expression changes in sugarcane has been hampered due to the complex polyploid genome, and lack of a high-quality sequenced reference genome in sugarcane. In this study, we used a hybrid (comparative and de novo) transcriptome mapping approach to determine AS patterns and alternatively spliced genes in sugarcane in response to infection with a biotrophic smut fungus. We identified several putative genes with splice variants/isoforms that are differentially expressed during smut infection. The alternatively spliced open reading frames encoded proteins with putative functions in cell wall modification, transcriptional regulation, ROS homeostasis, and defense hormone signaling. Often, AS resulted in truncated or altered protein domains, which could have implications for native protein function, localization and activity. Our study provides the first overview of the genome-wide AS landscape and posttranscriptional gene regulation in the complex sugarcane genome in response to smut infection.
Methods
Plant material, RNA extraction, and Illumina sequencing
Sugarcane (Saccharum spp.) cultivar ‘RB925345’ showing intermediate smut resistance was inoculated with teliospores of the biotrophic fungus Sporisorium scitamineum SSC39 using an artificial wounding protocol5,14. Plants were arranged in a randomized block design on greenhouse benches with three replicates of control and fungal treatments. Breaking buds and the culm region (up to 2 cm below culm) were collected and analyzed at an early stage, 5 days after inoculation (DAI), and a later stage after emergence of whips (200 DAI). Sporisorium scitamineum infection of sugarcane at 5 DAI was confirmed by rDNA ITS amplicon sequencing using Hs (AACACGGTTGCATCGGTTGGGTC) and Ha (GCTTCTTGCTCATCCTCACCACCAA) primers5,51. Total RNA was extracted from frozen tissues at each time point using the protocol described by14, and extracted RNA quality was verified using an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). Sequencing libraries were prepared using a TruSeq RNA Sample Prep v2 Low Throughput (LT) kit and as per the Illumina protocol (Illumina, CA, USA). The libraries were subjected to paired-end sequencing on an Illumina HiScanSQ system using Illumina TruSeq SBS reagents.
Reference-based transcript assembly and isoform calling
RNA-seq libraries from 12 sugarcane samples in response to smut disease (5 and 200 DAI) were filtered using in-house Python scripts to obtain high-quality reads that were mapped to the reference genome of Sorghum bicolor (v3.1 release) using the TopHat2 v2.1.152 spliced aligner using Bowtie253 alignment engine. Aligned Sorghum bicolor BAM files were further processed by Cufflinks v2.2.1 to assemble the aligned sequence reads into transcripts36, guided by the Sorghum bicolor annotation to predict novel genes and isoforms. Cufflinks was used to quantify transcript abundances using the fragments per kilobase of exon per million fragments mapped (FPKM) normalization. The Cuffmerge v2.2.136 script was used to create a high-quality merged assembly GTF file and filter out artifactual transfrags for all replicates under each experimental condition. The merged GTF and aligned BAM files among control and experimental conditions were processed by Cuffdiff v2.2.1 to identify significant gene and isoform changes.
De novo assembly, annotation, expression, and functional enrichment analysis
Sequence reads from all 12 RNA-seq libraries were assembled de novo using Trinity v2.1.137. The Ada cluster of the Texas A&M University High Performance Research Computing facility (http://hprc.tamu.edu/) with 256 GB memory and 20 core nodes was used to perform sequence assembly. The transcriptome was checked for redundancy using self-BLAST, and exact duplicates were removed. Trinity uses de Brujin graphs and has three core software modules, which first assemble the sequence data into unique contigs (Inchworm), cluster the contigs of a given gene and construct a de Brujin graph (Chrysalis), and lastly process the clusters and report the full-length alternatively spliced transcripts (Butterfly)54,55.
The whole transcriptome including alternatively spliced isoforms was annotated using BLASTX38 with e-value < 1e-05 against Sorghum bicolor proteins, NCBI nr/nt and UniProtKB reference databases. The alignment-based quantification method RSEM56 was used to quantify transcript abundance. RSEM uses the Bowtie53 aligner to map sequence reads to reference sequences. Differentially expressed transcripts were identified using the edgeR Bioconductor package57.
Absolute read counts were calculated using the HTSeq Python framework58 with aligned BAM files obtained from TopHat258. Global distribution of mapped RNA-seq reads on each chromosome were visualized using a Circos map59. The BiNGO60 functional enrichment tool was used for characterizing enriched isoforms with false discovery rate (FDR) < 0.05 for individual gene ontology (GO) terms. Results from BiNGO were visualized as GO networks using Cytoscape61. Gene family enrichment analysis was performed using the GenFam42 tool with Fisher exact test and Benjamini-Hochberg FDR method.
Reverse-transcription PCR (RT-PCR) validation of isoforms
Sugarcane ‘RB925345’ single-budded setts were surface disinfected and inoculated with Sporisorium scitamineum SSC39 spores (106 teliospores mL−1 in saline solution; NaCl2 0.85 M), previously tested for viability (Taniguti et al., 2015; Schaker et al., 2017). Mock-inoculated plants were prepared with only saline solution. Plants were placed on greenhouse benches in a completely randomized experimental design. After whip emission (120 DAI), meristems of infected and control plants (3 replicates each) were sampled and total RNA extracted using Trizol reagent (Invitrogen). Long cDNAs were obtained using a SMARTer PCR cDNA Synthesis kit (Clontech) and cDNA amplification using an Advantage 2 PCR kit (Clontech), according to the manufacturer’s instructions. cDNAs were diluted 20× and PCR-amplified using Trinity-based primers with a KAPA HiFi HotStart PCR kit (Kapa Biosystems). Reactions contained 1× KAPA HiFi buffer, 0.3 mM KAPA dNTP mix, 0.3 μM each primer, 1 µl diluted cDNA, 0.5 U KAPA HiFi HotStart DNA polymerase, and PCR-grade water to 25 µl. Cycling conditions were as follows: 95 °C for 3 min, 35 cycles of 98 °C for 20 s, 60 °C for 15 s, and 72 °C for 15 s. Results were analyzed in 1.5% agarose gels (0.5× TBE) using 100 bp (Sinapse) and 1 kb (Thermo Scientific) ladders and SYBR Green staining (Invitrogen).
Supplementary information
Acknowledgements
This study was supported in part by funds from USDA-NIFA-AFRI (2016-67013-24738) to K.K.M.; USDA-NIFA (HATCH TEX09621); Texas A&M AgriLife Research Bioenergy/Bioproducts Grant (124738-96210) to K.K.M and J.A.D, and (124738-92810) to J.A.D.; FAPESP (2017/13268-2) and CNPq (303965/2015-0) to C.B.M-V.; and FAPESP (2017/02434-9) to P.D.C.S.
Author Contributions
R.B., S.I., P.S., C.B.M.-V., J.A.D., and K.M. designed the experiments. R.B., S.I., and P.D.C.S. conducted the experiments, interpreted the data, and prepared the manuscript. S.I., C.B.M.-V., J.A.D., and K.M. supervised the study and edited the manuscript.
Data Availability
The raw RNA-seq data was deposited in the NCBI SRA database with BioProject accession PRJNA2918165. The assembled transcript sequences (excluding transcripts significantly matched to NCBI UniVec database) was deposited at DDBJ/ENA/GenBank database under accession GHKD00000000.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-45184-1.
References
- 1.FAO. (Latest update: 28 May, 2018. Accessed: 30 Nov, 2018. http://www.fao.org/faostat/en/#data/QC/visualize, 2018).
- 2.Que YX, Lin JW, Song XX, Xu LP, Chen RK. Differential gene expression in sugarcane in response to challenge by fungal pathogen Ustilago scitaminea revealed by cDNA-AFLP. J Biomed Biotechnol. 2011;2011:160934. doi: 10.1155/2011/160934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan NA, et al. Identification of cold-responsive genes in energycane for their use in genetic diversity analysis and future functional marker development. Plant Sci. 2013;211:122–131. doi: 10.1016/j.plantsci.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Que Y, et al. Genome sequencing of Sporisorium scitamineum provides insights into the pathogenic mechanisms of sugarcane smut. BMC Genomics. 2014;15:996. doi: 10.1186/1471-2164-15-996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaker PD, et al. RNAseq transcriptional profiling following whip development in sugarcane smut disease. PLoS One. 2016;11:e0162237. doi: 10.1371/journal.pone.0162237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh N, Somai BM, Pillay D. Smut disease assessment by PCR and microscopy in inoculated tissue cultured sugarcane cultivars. Plant Sci. 2004;167:987–994. doi: 10.1016/j.plantsci.2004.05.006. [DOI] [Google Scholar]
- 7.Carvalho G, et al. Sporisorium scitamineum colonisation of sugarcane genotypes susceptible and resistant to smut revealed by GFP‐tagged strains. Ann. Appl. Biol. 2016;169:329–341. doi: 10.1111/aab.12304. [DOI] [Google Scholar]
- 8.Sundar, A. R., Barnabas, E. L., Malathi, P. & Viswanathan, R. A mini-review on smut disease of sugarcane caused by Sporisorium scitamineum. (INTECH Open Access Publisher, 2012).
- 9.You-Xiong QUE, Zhi-Xia Y, Li-Ping XU, Ru-Kai C. Isolation and identification of differentially expressed genes in sugarcane infected by Ustilago scitaminea. Acta Agronomica Sinica. 2009;35:452–458. [Google Scholar]
- 10.Peters LP, et al. Functional analysis of oxidative burst in sugarcane smut-resistant and -susceptible genotypes. Planta. 2017;245:749–764. doi: 10.1007/s00425-016-2642-z. [DOI] [PubMed] [Google Scholar]
- 11.Bhuiyan SA, Croft BJ, Tucker GR, James R. Efficacy of flutriafol compared to other triazole fungicides for the control of sugarcane smut. Proceedings of Australian Society of Sugar Cane Technololgy. 2015;37:68–75. [Google Scholar]
- 12.Que Y, Su Y, Guo J, Wu Q, Xu L. A global view of transcriptome dynamics during Sporisorium scitamineum challenge in sugarcane by RNA-Seq. PLoS One. 2014;9:e106476. doi: 10.1371/journal.pone.0106476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su Y, et al. Comparative proteomics reveals that central metabolism changes are associated with resistance against Sporisorium scitamineum in sugarcane. BMC Genomics. 2016;17:800. doi: 10.1186/s12864-016-3146-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taniguti LM, et al. Complete Genome Sequence of Sporisorium scitamineum and Biotrophic Interaction Transcriptome with Sugarcane. PLoS One. 2015;10:e0129318. doi: 10.1371/journal.pone.0129318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grivet L, Arruda P. Sugarcane genomics: depicting the complex genome of an important tropical crop. Curr. Opin. Plant Biol. 2002;5:122–127. doi: 10.1016/S1369-5266(02)00234-0. [DOI] [PubMed] [Google Scholar]
- 16.Vermerris W. Survey of genomics approaches to improve bioenergy traits in maize, sorghum and sugarcane. J. Integr. Plant Biol. 2011;53:105–119. doi: 10.1111/j.1744-7909.2010.01020.x. [DOI] [PubMed] [Google Scholar]
- 17.de Setta N, et al. Building the sugarcane genome for biotechnology and identifying evolutionary trends. BMC Genomics. 2014;15:540. doi: 10.1186/1471-2164-15-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chamala S, Feng G, Chavarro C, Barbazuk WB. Genome-wide identification of evolutionarily conserved alternative splicing events in flowering plants. Front Bioeng Biotechnol. 2015;3:33. doi: 10.3389/fbioe.2015.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang CY, Lin WD, Tu SL. Genome-Wide Analysis of Heat-Sensitive Alternative Splicing in Physcomitrella patens. Plant Physiol. 2014;165:826–840. doi: 10.1104/pp.113.230540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satyawan D, Kim MY, Lee SH. Stochastic alternative splicing is prevalent in mungbean (Vigna radiata) Plant Biotechnol. J. 2017;15:174–182. doi: 10.1111/pbi.12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simpson CG, et al. Alternative splicing in plants. Biochem. Soc. Trans. 2008;36:508–510. doi: 10.1042/BST0360508. [DOI] [PubMed] [Google Scholar]
- 22.Barbazuk WB, Fu Y, McGinnis KM. Genome-wide analyses of alternative splicing in plants: opportunities and challenges. Genome Res. 2008;18:1381–1392. doi: 10.1101/gr.053678.106. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Dai C, Hu C, Liu Z, Kang C. Global identification of alternative splicing via comparative analysis of SMRT- and Illumina-based RNA-seq in strawberry. Plant J. 2017;90:164–176. doi: 10.1111/tpj.13462. [DOI] [PubMed] [Google Scholar]
- 24.Bedre, R., Irigoyen, S., Petrillo, E. & Mandadi, K. K. New Era in Plant Alternative Splicing Analysis Enabled by Advances in High-Throughput Sequencing (HTS) Technologies. Front Plant Sci10, (2019). [DOI] [PMC free article] [PubMed]
- 25.Shang, X., Cao, Y. & Ma, L. Alternative Splicing in Plant Genes: A Means of Regulating the Environmental Fitness of Plants. Int. J. Mol. Sci. 18, (2017). [DOI] [PMC free article] [PubMed]
- 26.Mandadi KK, Scholthof K-BG. Genome-wide analysis of alternative splicing landscapes modulated during plant-virus interactions in Brachypodium distachyon. Plant Cell. 2015;27:71–85. doi: 10.1105/tpc.114.133991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Filichkin SA, et al. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2010;20:45–58. doi: 10.1101/gr.093302.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X-C, Gassmann W. Alternative splicing and mRNA levels of the disease resistance gene RPS4 are induced during defense responses. Plant Physiol. 2007;145:1577–1587. doi: 10.1104/pp.107.108720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Egawa C, et al. Differential regulation of transcript accumulation and alternative splicing of a DREB2 homolog under abiotic stress conditions in common wheat. Genes Genet. Syst. 2006;81:77–91. doi: 10.1266/ggs.81.77. [DOI] [PubMed] [Google Scholar]
- 30.Thatcher SR, et al. Genome-Wide Analysis of Alternative Splicing during Development and Drought Stress in Maize. Plant Physiol. 2016;170:586–599. doi: 10.1104/pp.15.01267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018;50:1565–1573. doi: 10.1038/s41588-018-0237-2. [DOI] [PubMed] [Google Scholar]
- 32.Garsmeur O, et al. A mosaic monoploid reference sequence for the highly complex genome of sugarcane. Nat Commun. 2018;9:2638. doi: 10.1038/s41467-018-05051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Souza GM, et al. The sugarcane genome challenge: strategies for sequencing a highly complex genome. Trop. Plant Biol. 2011;4:145–156. doi: 10.1007/s12042-011-9079-0. [DOI] [Google Scholar]
- 34.Ming R, et al. Detailed alignment of saccharum and sorghum chromosomes: comparative organization of closely related diploid and polyploid genomes. Genetics. 1998;150:1663–1682. doi: 10.1093/genetics/150.4.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goodstein DM, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40:D1178–1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–U130. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coordinators NR. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2017;45:D12–D17. doi: 10.1093/nar/gkw1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boutet E, et al. UniProtKB/Swiss-Prot, the Manually Annotated Section of the UniProt KnowledgeBase: How to Use the Entry View. Methods Mol. Biol. 2016;1374:23–54. doi: 10.1007/978-1-4939-3167-5_2. [DOI] [PubMed] [Google Scholar]
- 41.Liu R, Loraine AE, Dickerson JA. Comparisons of computational methods for differential alternative splicing detection using RNA-seq in plant systems. BMC Bioinformatics. 2014;15:364. doi: 10.1186/s12859-014-0364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bedre, R. & Mandadi, K. GenFam: A new web application for gene family-based classification and functional enrichment analysis of plant genomes. bioRxiv (2018). [DOI] [PMC free article] [PubMed]
- 43.Sasidharan R, Voesenek LACJ, Pierik R. Cell Wall Modifying Proteins Mediate Plant Acclimatization to Biotic and Abiotic Stresses. Crit. Rev. Plant Sci. 2011;30:548–562. doi: 10.1080/07352689.2011.615706. [DOI] [Google Scholar]
- 44.Miedes E, Lorences EP. The implication of xyloglucan endotransglucosylase/hydrolase (XTHs) in tomato fruit infection by Penicillium expansum Link. A. J. Agric. Food Chem. 2007;55:9021–9026. doi: 10.1021/jf0718244. [DOI] [PubMed] [Google Scholar]
- 45.Bedre R, et al. Genome-wide transcriptome analysis of cotton (Gossypium hirsutum L.) identifies candidate gene signatures in response to aflatoxin producing fungus Aspergillus flavus. PLoS One. 2015;10:e0138025. doi: 10.1371/journal.pone.0138025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ozretic P, et al. Regulation of human PTCH1b expression by different 5’ untranslated region cis-regulatory elements. RNA Biol. 2015;12:290–304. doi: 10.1080/15476286.2015.1008929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee SK, et al. Identification of the ADP-glucose pyrophosphorylase isoforms essential for starch synthesis in the leaf and seed endosperm of rice (Oryza sativa L.) Plant Mol. Biol. 2007;65:531–546. doi: 10.1007/s11103-007-9153-z. [DOI] [PubMed] [Google Scholar]
- 48.Mandadi, K. K. & Scholthof, K.-B. G. Genomic architecture and functional relationships of intronless, constitutively- and alternatively-spliced genes in Brachypodium distachyon. Plant Signal Behav, (2015). [DOI] [PMC free article] [PubMed]
- 49.Ludwig SR, Oppenheimer DG, Silflow CD, Snustad DP. Characterization of the alpha-tubulin gene family of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA. 1987;84:5833–5837. doi: 10.1073/pnas.84.16.5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J, Jia D, Chen X. HUA1, a regulator of stamen and carpel identities in Arabidopsis, codes for a nuclear RNA binding protein. Plant Cell. 2001;13:2269–2281. doi: 10.1105/tpc.13.10.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bueno, C. R. N. C. Infection by Sporisorium scitamineum on sugarcane: influence of environmental variables and development of a method for early diagnosis Ph.D. thesis, University of São Paulo, (2011).
- 52.Khan NA, et al. Identification of cold-responsive genes in energycane for their use in genetic diversity analysis and future functional marker development. Plant Sci. 2013;211:122–131. doi: 10.1016/j.plantsci.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 53.Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol10, (2009). [DOI] [PMC free article] [PubMed]
- 54.Wang W, et al. Detection of alternative splice and gene duplication by RNA sequencing in Japanese flounder, Paralichthys olivaceus. G3 (Bethesda) 2014;4:2419–2424. doi: 10.1534/g3.114.012138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu B, Suo F, Lei W, Gu L. Comprehensive analysis of alternative splicing in Digitalis purpurea by strand-specific RNA-Seq. PLoS One. 2014;9:e106001. doi: 10.1371/journal.pone.0106001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 61.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw RNA-seq data was deposited in the NCBI SRA database with BioProject accession PRJNA2918165. The assembled transcript sequences (excluding transcripts significantly matched to NCBI UniVec database) was deposited at DDBJ/ENA/GenBank database under accession GHKD00000000.