

Abstract
Folic acid is a necessary micronutrient for normal human growth and development. Benzo(a)pyrene (BaP) is a ubiquitously distributed environmental pollutant and its metabolite, benzo(a)pyrene-diol-epoxide, is known to exert a strong teratogenic and carcinogenic effect on the body’s tissues and cells. The aim of this study was to investigate the mechanism by which folic acid can inhibit the toxic effects of BaP both in vivo and in vitro. We measured changes in 16HBE cell activity affected by the intervention of folic acid on BaP using the cell counting kit-8 assay and that of cell cycle distribution by flow cytometry. At the same time, we assessed the xeroderma pigmentosum group A, xeroderma pigmentosum group C, excision repair cross complementation group 1, cyclinD1, and CKD4 mRNAs, and their related protein expression both in mouse lung tissue and in 16HBE cells. In conclusion, the mechanisms by which this effect is mediated were not entirely elucidated by our study, possibly because folic acid antagonizes the toxic effects of BaP by upregulating the levels of excision repair cross complementation group 1, xeroderma pigmentosum group A, and xeroderma pigmentosum group C gene expression to improve the rate of DNA repair, in turn accelerating the speed of repair for DNA damage caused by BaP. Meanwhile, folic acid could restrain BaP-induced cyclinD1 protein expression, which could help cells return to their normal cell cycle.
Keywords: benzo(a)pyrene, cell cycle, folic acid, interference effect
Introduction
Benzo(a)pyrene (BaP), a polycyclic aromatic hydrocarbon, is a strong mutagen and human carcinogen, as confirmed in both experimental and epidemiological studies (Jeng and Bocca, 2013; Nebert, 2013). BaP is extensively and principally metabolized by cytochrome P450 enzymes (CYP1A family) and other enzymes into many reactive intermediates, such as benzo(a)pyrene-diol-epoxide (BPDE), which binds to DNA at guanine residues. The resulting BPDE–DNA adducts subsequently cause oxidative DNA damage and genetic mutations, and may be responsible for tumor initiation (An et al., 2011).
The DNA repair system is important in the removal of damaged DNA and to protect genomic integrity. Nucleotide excision repair (NER) is the major repair system capable of eliminating the chemical adducts formed by carcinogens. Celotti et al. (1993) reported that BPDE–DNA adducts can be removed by NER in the mammalian system. NER involves more than 25 polypeptides in a multistep process, including an initial damage recognition step, an incision at either side of the lesion, the removal of the damaged oligonucleotide, DNA repair synthesis, and the final resealing of the strand by a DNA ligase (Wood, 1996). Xeroderma pigmentosum group A (XPA), xeroderma pigmentosum group C(XPC), and excision repair cross complementation group 1 (ERCC1) are the main proteins in this pathway. ERCC1 is one of the critical genes within NER (Park et al., 1995) and may be responsible for the incision at the site of DNA damage (Duin et al., 1987). Chang et al. (1999) showed that increased ERCC1 expression in WI38 cells could result in significant promotion of NER capability. XPA and XPC are responsible for the modulation of damage recognition, and are involved in both global genome and transcription-coupled repair pathways (Volker et al., 2001). XPA has a high affinity for damaged DNA, and interacts with many repair factors, playing a crucial role in the correct positioning of the repair machinery around DNA lesions. Hamouchene et al. (2011) reported that following BaP exposure, genes that were found to have altered expression included those involved in xenobiotic metabolism, apoptosis, cell cycle regulation, and DNA repair. The effect of BaP on DNA repair efficiency is highly variable, but in general, the efficiency of DNA repair is correlated with the risk of cancer development.
One of the cellular responses to DNA damage is the activation of G1/S and G2/M checkpoints. BPDE–DNA adducts induce cell cycle arrest and alter cell cycle progression, causing cell damage. BaP can induce genetic mutations or alter the expression of cell cycle modulation-related genes, destroying cell cycle order and fidelity, and generating malignant transformation and cancer. Cyclin A and cyclin-dependent kinases (CDK4) are key regulators in the G1-S phase, the critical checkpoint in cell cycle progression. Jeffy et al. (2000) reported that BaP could promote cell growth and expression of cyclinD1 and CDK4, and cause the transition from the G1 to the S phase. The antioncogene p53 is a transcription factor, and modulates cell cycle progression by inducing p21 and inhibiting the gene expression of cyclinA, cyclinE, CDK2, and CDK4. Bjelogrlic et al. (1994) reported that the expression of p53 could be upregulated after stimulation with the chemical carcinogen BaP, inducing Gl arrest to repair the damaged DNA. However, p53 could not antagonize the effect of BaP at high concentrations and prolonged stimulus duration, where BaP could promote cell proliferation by upregulating the expression of cyclinD1, CDK2, and CDK4, eliminating the cell cycle modulation of p53. It is this property that is generally believed to account for the mutagenic and genotoxic activity of BaP. BaP can be easily formed in foods during baking, frying, and smoking processes in which combustion products come into direct contact with the food substance (Chen et al., 2012). In addition, BaP is a key contaminant in air pollution, and has attracted increasing attention for its impact on public health in developing countries (Zhang and Samet, 2015). Therefore, it is necessary and urgent to explore strategies for the prevention of BaP-mediated damage.
Folic acid (also called pterolyglutamic acid) is a micronutrient essential for normal human growth and development. Folates are involved in the transfer of one carbon unit in the methylation cycle and in DNA metabolism. Research suggests that dietary intake and blood levels of folate appear to be correlated negatively to the risk of tumorigenesis in several organs, such as the breast, colon, and lung (Le Marchand et al., 1989; Zhang, 1999; Van Guelpen 2006). Folic acid is involved in DNA repair and is necessary for the synthesis of DNA at appropriate doses (Vergote et al., 2015), which might explain its antimutagenic effect. This is consistent with Blount’s conclusion that folate deficiency causes uracil misincorporation into human DNA, leading to chromosome breakage, which in turn might induce cancer and neuronal damage (Blount et al., 1997). Our previous studies showed that folic acid significantly inhibited the reverse mutation of salmonella typhimurium strains TA98 and TA100 caused by BaP, and protected the viability of human liver cells against BaP (Zhang et al., 2015). However, the mechanism of action of folic acid in protecting against the toxic effects of BaP has still not been determined. Therefore, in the present study, we used human bronchial epithelial cells (16HBE), which are capable of activating parent BaP to reactive metabolites, to better understand the mechanism by which folic acid prevents the toxic effects of BaP. We measured alterations in 16HBE cell activity following the interaction of folic acid with BaP by the cell counting kit-8 (CCK-8) assay and cell cycle distribution by flow cytometry. We simultaneously assessed the XPA, XPC, ERCC1, cyclinD1, and CKD4 mRNA and protein expressions by RT-PCR and western blotting. Furthermore, the effects of folic acid at different doses on the mRNA transcription of NER pathway-associated factors induced by BaP, including ERCC1, XPA, and XPC, in mouse lung tissue were investigated in vivo.
Materials and methods
Chemicals
Folic acid (97% purity) and BaP (96% purity) were purchased from Sigma Chemical Co. (St Louis, Missouri, USA). Cell Counting Kit-8, Dulbecco’s modified Eagle’s medium (DMEM), penicillin–streptomycin, trypsin, fetal bovine serum, and PBS were obtained from Gibco (Grand Island, New York, USA). The Cell Cycle Kit was acquired from Nanjing KeyGEN Biotech. Co. (Nanjing, China). SYBR Premix Ex Taq II (Tli RNaseH Plus) and PrimeScript RT reagent Kit (Perfect Real Time) were acquired from Takara (Dalian, China). TRIzol, DEPC water, RIPA lysis buffer, BCA Protein Assay Kit, SDS-PAGE Gel Preparation Kit, 5×SDS-PAGE Sample Loading Buffer, and Bovine Serum Albumin were procured from Beyotime (Wuhan, China). Anti-GAPDH (loading control), horseradish peroxidase-conjugated goat anti-rabbit IgG and goat anti-mice IgG, anti-cyclinD1, anti-CDK4, and anti-ERCC1 were obtained from Abcam (Cambridge, UK). Chemiluminescence reagent was purchased from Pierce (Rockford, Illinois, USA).
Cell culture
Human bronchial epithelial cell line (16HBE) was obtained from the Shenzhen Center for Disease Control and Prevention (Shenzhen, Guangdong, China) and maintained in DEME containing 10% fetal bovine serum, supplemented with 50 U/ml penicillin, and 50 μg/ml streptomycin at 37°C in a humidified atmosphere with 5% CO2. The cell density was adjusted to less than 5×104 cells/ml. All experiments were conducted with cells in the logarithmic phase of growth.
Preparation of liver postmitochondrial supernatant (S9) and the S9 mixture
The liver S9 fraction was prepared according to the procedure of Maron and Ames (1983). The S9 microsome fraction was prepared from the livers of rats treated with Aroclor 1254 (Supelco; Bel- lefonte, Pennsylvania, USA). The S9 mix was composed of 8 mmol/l MgCl2, 32.5 mmol/l KCl, 5 mmol/l glucose-6-phosphate, 4 mmol/l nicotinamide-adenine dinucleotide phosphate, and 0.1 mol/l sodium phosphate buffer (pH 7.4), and the S9 fraction was at a concentration of 0.04 ml/ml (S9 mix). The mixture was prepared immediately before each treatment and maintained at 4°C.
Preparation of test compounds
The stock solution of BaP dissolved in dimethyl sulfoxide was maintained at −20°C in the dark. It was diluted into different concentrations using the culture medium (DMEM) and the S9 mixture (3% of the final solution).
The stock solution of folic acid dissolved in ultrapure water was stored in the Amber Laboratory Bottle. It was diluted into different concentrations using the culture medium (DMEM).
Treatment of cells
Before each experiment, cell density was adjusted to less than 5×104 cells/ml. 16HBE cells were seeded in triplicate in six-well tissue culture plates. The cells were treated with folic acid at concentrations of 50, 100, 250, and 500 nmol/l at 2 h before exposure to 32 μmol/l BaP for 24 h. All experiments included negative (DMEM with a 3% liver S9 fraction) and positive controls (32 μmol/l BaP with 3% liver S9 fraction), and were conducted independently three times.
Cell viability
Cell viability was detected using the CCK-8. Briefly, the 16HBE cells were seeded in a 96-well plate (200 μl per well) and treated with (a) 4, 8, 16, 32, 64 μmol/l BaP, (b) 50, 100, 250, and 500 nmol/l folic acid, (c) 50, 100, 250, and 500 nmol/l folic acid+32 μmol/l BaP for 12, 24, and 48 h. Negative and positive controls were prepared, and five parallel wells were used for each concentration. Following treatment, 10 μl of the CCK-8 solution was added to each well, the 96-well plate was incubated continuously at 37°C for 1 h, and then the OD value for each well was read on a microplate reader (PerkinElmer; Connecticut, USA) at 450 nm wavelength to determine cell viability. The assay was repeated three times. The cell viability (%) was calculated according to the following equation:
 |
Flow cytometric analysis of cell cycle distribution
After the treatments, the 16HBE cells were harvested with trypsin and fixed in 70% ethanol at −20C. Before analysis, the cells were incubated with RNase and stained with propidium iodide (70 μmol/l in PBS). Samples were analyzed immediately by flow cytometry using a FACscan (Becton-Dickinson, Franklin Lakes, New Jersey, USA), and cell fractions in the sub-G1, G1, S, and G2/M phases were quantified in histograms using a CELLQuest program (Becton Dickinson and Company; New Jersey, USA).
RT-PCR
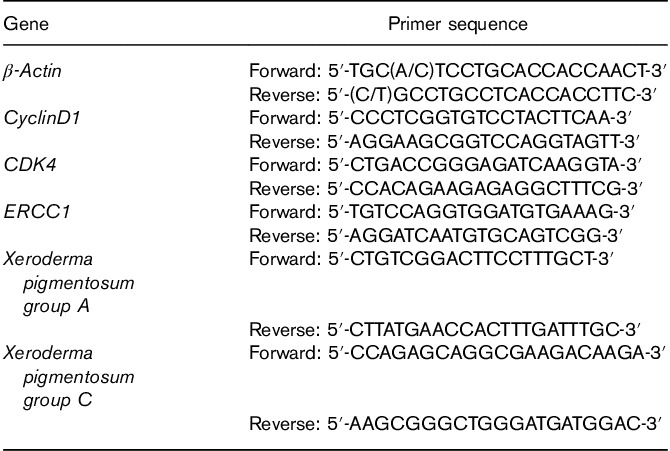
Total RNA was isolated from 16HBE cells with TRIzol (Beyotime Biotechnology, Wuhan, China) according to the manufacturer’s instruction. The concentration and purity of RNA were determined using the Ultramicro Nucleic Acid Protein Tester (Bio-Rad Laboratories, California, USA). Reverse transcription (RT) was performed using the PrimeScript RT reagent Kit (TaKaRa Biotechnology; Dalian, China) according to the instructions provided. Briefly, 500 ng of total RNA was combined with 2 μl of 5×PrimeScript RT buffer, 0.5 μl of PrimeScript RT Enzyme Mix I, 0.5 μl of Oligo dT Primer (50 μmol/l), and 0.5 μl of Random 6-mers primer (100 μmol/l), and RNase-free water was added to a total volume of 10 μl. The mixture was incubated at 37°C for 15 min, and the reaction was stopped by heating at 85°C for 5 s. The product was stored at −20°C after reverse transcription until used for PCR. Primers were designed (Table 1) and synthesized by Sangon (Sangon, Shanghai, China). All of the primers were designed based on BLAST searches. Real-time quantitative PCR was carried out using the SYBR Premix Ex Taq kit (TaKaRa Biotechnology) in an RT-PCR System (Bio-Rad Laboratories). The composition of PCR mix was as follows: 12.5 μl SYBR Premix Ex Taq II, 1 μl of each primer, 2 μl cDNA, and 8.5 μl dH2O in a final volume of 25 μl. The PCR program consisted of the initial denaturation at 95°C for 30 s, followed by 40 PCR cycles: 95°C for 10 s and 62°C for 30 s. Finally, the relative mRNA expression levels were analyzed using the  method with β-actin used as a reference gene.
method with β-actin used as a reference gene.
Table 1.
Real-time RT-PCR primer sequence of target genes
Western blot analysis
After treatments, 16HBE cells were washed twice with cold PBS and harvested on ice in RIPA lysis buffer (Beyotime; Biotechnology, Wuhan, China). The suspension was incubated in an ice box for 30 min. Cellular debris was then pelleted by centrifugation for 30 min at 12 000 rpm at 4°C, the supernatant was collected, and the concentration of protein was determined using the BCA Protein Assay Kit (Beyotime; Biotechnology). Cell extracts were normalized to the protein content and the concentration of protein was determined using the BCA Protein Assay Kit (Beyotime; Biotechnology). Then, proteins were separated using the gradient SDS-polyacrylamide gel electrophoresis kit (Beyotime; Biotechnology) according to the instructions provided. The same amount of loading buffer was added and mixed again. The samples were then denatured at 95°C for 10 min. About 200 μg total protein was loaded in each well and separated on 10% SDS-polyacrylamide electrophoresis gels. The separated proteins were transferred onto nitrocellulose membranes. The membranes were saturated and blocked with 5% fat-free milk at 37°C for 1 h, and were incubated with the first antibody (Rabbit anti-GAPDH, rabbit anti-cyclinD1, mouse anti-CDK4, and rabbit anti-ERCC1) for 2 h at 37°C. After extensive washing, the second antibody (horseradish peroxidase-conjugated goat anti-rabbit and rabbit anti-goat IgGs) was added, respectively, and the membranes were incubated for 45 min, followed by extensive washes (1–2 h). The immunocomplexes were detected by enhanced chemiluminescence. Specific antibody–antigen complexes were detected using the ECL western blot detection kit. Graphs of blots were obtained using an image analysis system (ChemiDoc MP; Bio-Rad Laboratories) in the linear range of detection, and were quantified for the level of specific induction by scanning laser densitometry. All experiments were conducted independently three times, and the averages were used for comparison.
Treatment of animals
Specified pathogen-free Kunming male and female mice in the weight range of 20–30 g were purchased from the Hubei Provincial Center for Disease Control and Prevention (number: 42000600001415). The animals were housed in polypropylene cages under hygienic conditions in departmental animal housing in strict accordance with the guidelines outlined by the institutional ethics committee.
Animals were divided into five treatment groups (five males and five females per group). Animals in group I served as normal controls and were administered ultrapure water intragastrically. Animals in group II through V were administered a single intraperitoneal injection of BaP at a dose level of 100 mg/kg body weight dissolved in corn oil. Groups III–V were administered folic acid in intragastrically at a dose level of 0.014, 0.07, and 0.35 mg/kg body weight once every 48 h. Folic acid treatment of animals from groups III–V was started 2 weeks before BaP injection. All the animals had free access to food and water, and the total duration of treatments was 60 days. At the end of the study, the animals were anesthetized using mild ether anesthesia and were then killed. The lungs were removed immediately and washed with ice-chilled saline, frozen in liquid nitrogen, and stored at −80°C.
Statistical analysis
GraphPad prism software (GraphPad Software Inc., San Diego, California, USA) was used for the statistical analysis of the experimental data and for graphing the results. All measurements were collected from three replicates and are represented as mean±SD values. The presence of statistical differences among groups was determined by one-way analysis of variance. The method of least significant difference was used to compare the differences among the folic acid pretreatment groups and the negative or positive control groups. Significant differences were defined as P value less than 0.05.
Results
The 16HBE cell activity affected by the interaction of folic acid with benzo(a)pyrene
Human bronchial cells (16HBE) were exposed to a single BaP dose at different concentrations and time intervals. Cell viability was detected using the CCK-8 assay, where cell viability was decreased in response to increased doses of BaP, indicating a clear dose–response relationship (Fig. 1) in the BaP-treated groups. Specifically, the viability of cells exposed to BaP for 24 h was significantly lower than those for 12 and 48 h.
Fig. 1.
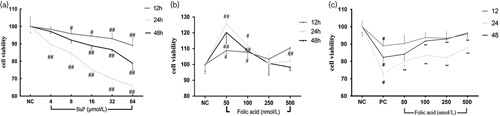
(a) Cell viability in human bronchial epithelia cells treated with 4, 8, 16, 32, and 64 μmol/l benzo(a)pyrene (BaP) for 12, 24, and 48 h. (b) Cell viability in human bronchial epithelia cells treated with 0, 50, 100, 250, and 500 nmol/l folic acid for 12, 24, and 48 h. (c) Cell viability in human bronchial epithelia cells pretreated with 50, 100, 250, and 500 nmol/l folic acid and exposed to 32 μmol/l BaP for 12, 24, and 48 h. Data represent the mean±standard error values of three independent experiments. Compared with the positive control group, and **P<0.01, compared with the negative control group, #P<0.05 and ##P<0.01.
When 16HBE cells were exposed to a single folic acid dose at different concentrations and time intervals, folic acid promoted cell activity at different concentrations and exposure times (Fig. 2), but without a linear relationship.
Fig. 2.
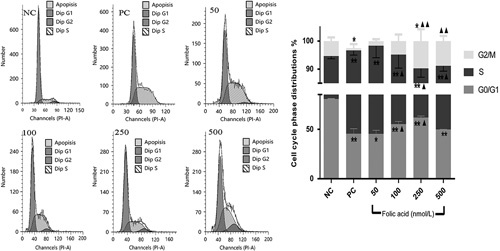
Cell cycle phase distributions and analysis after cells were pretreated with 50, 100, 250, and 500 nmol/l folic acid and exposed to 32 μmol/l benzo(a)pyrene (BaP) for 24 h. Cell cycle profiles were analyzed by flow cytometry. Bars represent the percentages of cells in the G0/G1, S, and G2/M phases, respectively. Data represent the mean±standard error values of three independent experiments. Compared with the positive control group, ▲P<0.05 and ▲▲P<0.01, Compared with the negative control group, *P<0.05 and **P<0.01.
The 16HBE cells were treated with folic acid at concentrations of 50, 100, 250, and 500 nmol/l for 2 h before exposure to BaP for 12, 24, and 48 h. As shown in Fig. 3, compared with the negative control group, cell activity in the positive control group decreased significantly (P<0.01) for 12, 24, and 48 h. Experimental results showed that folic acid exerted an inhibitory effect at doses of 50, 100, 250, and 500 nmol/l on 32 μmol/l Bap-induced 16HBE cell activity between 12 and 48 h. In addition, at the high dose of folic acid (500 nmol/l), cell viability reached 95.66, 87.84, and 96.40, during 12, 24, and 48 h, respectively, compared with the control group. There was a significant trend toward increased cell viability with increased doses of folic acid treatment, showing a clear dose–response relationship.
Fig. 3.
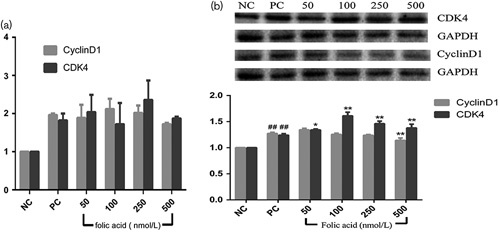
(a) mRNA expression levels of cyclinD1 and CDK4 genes in human bronchial epithelia cells pretreated with 50, 100, 250, and 500 nmol/l folic acid and exposed to 32 μmol/l benzo(a)pyrene (BaP) for 24 h. Data represent the mean±standard error values of three independent experiments. (b) CyclinD1 and CDK4 protein expression levels in human bronchial epithelia cells pretreated with 50, 100, 250, and 500 nmol/l folic acid and exposed to 32 μmol/l BaP for 24 h. Data represent the mean±standard error values of three independent experiments. Compared with the positive control group, *P<0.05 and **P<0.01, compared with the negative control group, and ##P<0.01.
The 16HBE cell cycle distribution and related genes and protein expression affected by folic acid intervention with benzo(a)pyrene toxicity
The present study reports on a method for detecting cell cycle using flow cytometry, which can be used to determine the interference effect of folic acid on advanced apoptosis and changes in the cell cycle of 16HBE induced by BaP. As shown in Fig. 3, the test results showed that the proportion of 16HBE cells in the G0/G1 phase decreased rapidly by 35.13% when exposed to 32 μmol/l Bap. However, the proportion of cells in the S-phase increased by 37.16% compared with the negative control, which suggests that BaP-induced the S-phase arrest of the 16HBE cell cycle. Compared with the positive control group, different doses of folic acid (50, 100, 250, 500 nmol/l), interacting with 32 μmol/l BaP, increased the proportion of G0/G1-phase cells by −0.14, 9.70, 16.41, and 4.33%, respectively, and decreased the proportion of S-phase cells by −1.78, 10.18, 22.79, and 9.84%, respectively, as shown in Fig. 2. In addition, compared with the positive control, the 16HBE cells pretreated with 100, 250, and 500 nmol/l folic acid could significantly remit (P<0.05) the S-phase arrest caused by BaP. Moreover, the 16HBE cells pretreated with 250 and 500 nmol/l folic acid significantly increased (P<0.01) the proportion of G2/M-phase cells compared with the positive control. Thus, folic acid at the doses of 100, 250, and 500 nmol/l played a significant role (P<0.05) in regulating BaP-induced S-phase arrest of 16HBE cell cycle progression. None of the experimental groups showed any obvious signs of advanced apoptosis.
RT-PCR was used to detect the transcription level of the cyclinD1 and CDK4 genes of 16HBE cells affected by the folic acid interference in BaP action. As shown in Fig. 3a, the expression of cyclinD1 and CDK4 increased by 1.96- and 1.91-fold, respectively, after exposure to 32 μmol/l BaP compared with the negative group. However, the results showed that there were no significant inhibitory effects on the overtranscription of cyclinD1 and CDK4 genes induced by BaP within a certain dose range of folic acid intervention on 16HBE cells. In contrast, 500 nmol/l folic acid pretreated cells could slightly decrease the mRNA transcription of cyclinD1 by 0.25-fold compared with the positive control group. For all doses of folic acid-pretreated groups, the mRNA transcription levels of cyclinD1 and CDK4 were elevated compared with the negative control group.
Protein expression was measured by western blot. Figure 3b shows the protein expression of cyclinD1 and CDK4 in cells pretreated with folic acid for 2 h before BaP induction, and then the relative levels of cyclinD1 and CDK4 for each concentration of folic acid in the pretreated groups to the control group. The results show that 32 μmol/l BaP could increase the levels of cyclinD1 and CDK4 compared with the negative control group. However, in the folic acid-pretreated groups (50, 100, 250, 500 nmol/l), the levels of cyclinD1 decreased, except in the 50 nmol/l folic acid group, and peaked in cells pretreated with 500 nmol/l folic acid compared with the positive control group (P<0.01). Moreover, folic acid did not reduce the levels of CDK4, but significantly improved the levels.
The 16HBE cell nucleotide excision repair-related genes and protein expression affected by intervention of folic acid on benzo(a)pyrene
RT-PCR was used to detect the transcription levels of ERCC1, XPA, and XPC genes in 16HBE cells treated with folic acid intervention with BaP. As shown in Fig. 4a, 32 μmol/l BaP could inhibit the transcription levels of ERCC1, XPA, and XPC mRNA in 16HBE cells, decreasing them by 0.91-, 0.50-, and 0.76-fold, respectively, compared with the negative control group. However, there was a significant trend where ERCC1, XPA, and XPC mRNA levels were enhanced with increased doses of folic acid treatment (P<0.05). In addition, compared with the positive control, the levels of ERCC1, XPA, and XPC increased significantly, reaching their peak in cells pretreated with 250 nmol/l folic acid.
Fig. 4.
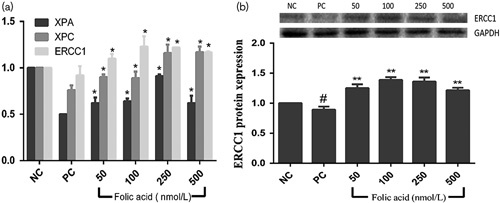
(a) Excision repair cross complementation group 1 (ERCC1), xeroderma pigmentosum group A (XPA), and xeroderma pigmentosum group C (XPC) mRNA expression levels in human bronchial epithelial cells pretreated with 50, 100, 250, and 500 nmol/l folic acid and exposed to 32 μmol/l benzo(a)pyrene (BaP)for 12, 24, and 48 h. (b) ERCC1 protein expression levels in human bronchial epithelia cells pretreated with 50, 100, 250, and 500 nmol/l folic acid and exposed to 32 μmol/l BaP for 24 h. Data represent the mean±standard error values of three independent experiments. Compared with the positive control, *P<0.05 and **P<0.01. Compared with the negative control, #P<0.05.
Protein expression was detected by western blot. Figure 4b shows the protein expression levels of ERCC1 in cells pretreated with folic acid for 2 h before BaP induction, and the relative ratios of ERCC1 for each concentration of folic acid pretreatment compared with the control group. The ratio of ERCC1 was the most significant, and increased by 1.40-, 1.55-, 1.52-, and 1.36-fold, compared with the positive control group, in cells pretreated by 50, 100, 250, and 500 nmol/l folic acid, respectively. The results of all the western blot experiments indicate that folic acid significantly restrains BaP-induced ERCC1 protein expression (P<0.01).
Mouse lung tissue nucleotide excision repair-related gene expression affected by intervention of folic acid on benzo(a)pyrene
mRNA transcription of NER repair pathways in-vivo was investigated using RT-PCR. The results of these experiments showed that the administration of folic acid to BaP-treated animals induced ERCC1, XPA, and XPC mRNA transcription in mouse lung tissue, as shown in Fig. 5, with the following doses of folic acid: high-dose group (0.35 mg/kg/day), intermediate-dose group (0.07 mg/kg/day), and low-dose group (0.014 mg/kg/day). BaP treatment significantly decreased the levels of ERCC1, XPA, and XPC gene expression in mouse lung tissue. However, groups treated with folic acid showed significantly increased levels of ERCC1, XPA, and XPC mRNA transcription in mouse lung tissue compared with the negative control. In addition, the results show that all three folic acid treatment groups (low-dose, intermediate-dose, high-dose groups) could strongly antagonize the low transcription of ERCC1 mRNA in mouse lung tissue induced by BaP, increasing them by 2.34-, 3.44-, and 3.01-fold, respectively, compared with the negative control group. Moreover, all the folic acid treatment groups (low-dose, intermediate-dose, high-dose group) could reverse the inhibitory effects of BaP on XPA and XPC mRNA transcription, and increased their levels 1.06-, 1.06-, and 1.05-fold and 1.04-, 1.18-, and 1.15-fold, respectively, compared with the positive control group. These results show that folic acid can significantly restrain the inhibitory effect of BaP on mRNA transcription of ERCC1, XPC, and XPA in mouse lung, reaching its peak in the intermediate-dose group, and increasing the ERCC1, XPA, and XPC mRNA levels to 3.44-, 0.93-, and 1.03-fold that of the negative control group, respectively.
Fig. 5.
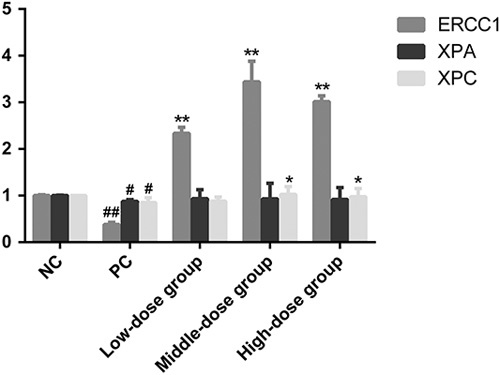
ERCC1, xeroderma pigmentosum group A (XPA), and xeroderma pigmentosum group C (XPC) mRNA expression levels in mouse lung tissue pretreated with 50, 100, 250, and 500 nmol/l folic acid and exposed to 32 μmol/l BaP for 24 h. Data represent the mean±standard error values of three independent experiments. Compared with the positive control, *P<0.05 and **P<0.01. Compared with the negative control, #P<0.05 and ##P<0.01.
Discussion
BaP is a well-known ubiquitous carcinogen (Delft et al., 2012; Zhu et al., 2014), a large class of chemicals in the atmosphere, soil, waterways, and food chain (Issaq et al., 1979). BaP is a main member of polycyclic aromatic hydrocarbons, a potent carcinogen, as confirmed in several different animal models (Cui et al., 2016). BaP is metabolized by cytochrome P450 enzymes into mutagenic derivatives that can form DNA adducts (Uno and Makishima, 2009). The formation and persistence of carcinogen–DNA adducts are believed to be critical events for DNA damage and alteration of cell cycle progression. The DNA repair system of somatic cells plays a key role in the removal of damaged DNA for the protection and maintenance of genomic integrity. BaP adducts are removed by NER (Celotti et al., 1993). The effect of BaP on DNA repair efficiency is highly variable. For instance, Athas et al. (1991) have shown that BaP treatment reduces the repair efficiency of UV-damaged viral DNA plasmid reporter in lymphocytes of NER-deficient xeroderma pigmentosum patients. Folates include the family of B-group vitamins composed of an aromatic pteridine ring attached through a methylene group to p-aminobenzoic acid and a glutamate residue (Shane, 2001). Folate metabolism plays a vital role in nucleic acid synthesis, methionine regeneration, shuttling, and redox reaction of one carbon units required for normal metabolism and regulation (Nazki et al., 2014). Folate also plays an essential role in the de-novo synthesis of purines and thymidylate, which is required in DNA replication and repair (Kim, 2000). Thus, we hypothesized that folic acid could improve the rate of DNA repair to block the toxic effects of BaP. In this study, we used immortalized human bronchial epithelial cells (16HBE) to investigate the possible intervention mechanism of folic acid in preventing the toxic effects of BaP.
Our results showed that cellular activity decreased in a concentration-dependent manner in cells exposed to BaP at different time points, and that 32 μmol/l BaP inhibited the activity of cells significantly at all-time points. Folic acid exposure promoted cellular activity at different concentrations and exposure times, but without a linear relationship; on treatment with 50 nmol/l folic acid for 24 and 48 h, and 500 nmol/l for 12 h, cell viability increased significantly. We also observed that different concentrations of folic acid were correlated significantly with cell viability when treated with BaP for 24 h. There was a significant trend, with increased cell viability correlated with increased doses of folic acid treatment. This clear dose–effect relationship suggests that there are certain protective effects of folic acid for maintaining cell viability against BaP-induced toxicity.
Studies have shown that BaP causes DNA damage and increases lung cancer in both animals and humans (Lin et al., 2016). It is well known that genotoxic events leading to cell cycle arrest can decrease cell viability. Hamouchene et al. (2011) observed that following BaP exposure, cells evaded G1 arrest and accumulated in the S phase, which is consistent with the results of our study. Moreover, our results show that folates significantly relieved the cell cycle arrest induced by BaP. Cell cycle regulators include many cyclins, CDKs, and cyclin-dependent kinase inhibitors. Previous studies showed that cell cycle regulators are frequently mutated in many human tumors, resulting in overexpression of cyclins and CDKs, which promote cellular proliferation (Chu et al., 2005). CyclinD1 is a key regulator of cell proliferation, acting as a mitogen sensor, and linking extracellular signaling to the cell cycle machinery (Hizli et al., 2006). CDK4 is a master integrator that couples mitogenic and antimitogenic extracellular signals with the cell cycle, and is also crucial for many oncogenic transformational processes. To probe the mechanism of folic acid function against the toxic effects of BaP, the expression of cyclinD1 and CDK4 was determined. This study found that BaP increased both the mRNA and the protein expression of cyclinD1 and CDK4. The results showed that the cell cycle arrest mechanism of BaP appeared to be related to the upregulation of cyclinD1 and CDK4 expression. However, we did not observe the folates alleviating the situation, but rather, they promoted the mRNA levels of cyclinD1 and CDK4. This finding did not support our prediction that folic acid reduces protein expression by decreasing the mRNA levels of cyclinD1 and CDK4. In the western blot experiments, we observed that folic acid improved the protein expression of CDK4 while reducing cyclinD1 protein expression. Therefore, we speculated that cyclinD1 protein expression might be regulated by miRNA after transcription. miRNA, are small, noncoding RNA molecules (on average 22 nucleotides in length) that are involved in post-transcriptional gene silencing (Filipowicz et al., 2008). Card et al. (2008) reported that miR-302 translationally represses cyclinD1, which may be linked it to cell cycle regulation. Moreover, dietary folate has been found to modulate miRNA expression in different model systems, which may be related to its role in cancer prevention and risk (Ross and Davis, 2011). Therefore, in the present study, we hypothesized that folic acid may modulate miRNA expression to downregulate cyclinD1 protein expression, eventually enabling cells to recover their normal cell cycle.
BaP can be activated metabolically by CYP enzymes and epoxide hydrolase to form DNA adducts, thus exerting its mutagenic and carcinogenic effects (Harrigan et al., 2004). The formation and persistence of carcinogen–DNA adducts are believed to be critical events in DNA damage. In BaP-treated cells, BaP–DNA adducts are removed by NER (Celott et al., 1993). XPA, XPC, and ERCC1 are the main proteins in this pathway. ERCC1 is one of the critical genes within NER (Park et al., 1995) and may be responsible for the incision 5′ to the site of DNA damage. (Duin et al., 1987). Chang et al. (1999) showed that an increase in ERCC1 expression in WI38 cells could result in significant promotion of NER capability, suggesting a role for ERCC1 in the repair of modified nucleotides, specifically increased removal of DNA adducts (Li et al., 2000). The damage recognition complex, XPC-HR23B, has been shown to be essential for the recruitment of all subsequent NER factors in the preincision step (Venema et al., 1991). XPA is also responsible for the modulation of damage recognition, and is involved in both global genome-coupled and transcription-coupled repair pathways (Hamouchene et al., 2011). XPA has a high affinity for damaged DNA, and interacts with many repair factors, playing a vital role in correctly positioning the repair machinery around DNA lesions. Huang et al. (1999) showed that folate deficiency leads to cell apoptosis, coinciding with an accumulation of cells in the S phase. They believed that the perturbation of dNTP pools could act as a biochemical signal to trigger DNA fragmentation in different cell types. The dNTP pool imbalance may potentially cause the failure of DNA replication and repair (Bebenek et al., 1992). Folate deficiency has been shown to induce dNTP imbalance in lymphocytes (Courtemanche et al., 2004) and in Chinese hamster ovary cell lines (James et al., 1994). Folate also plays an essential role in the de-novo synthesis of purines and thymidylate, which are required for DNA replication and repair (Kim, 2000). Choi et al. (1998) observed a significantly decreased rate of DNA excision repair in folate-deficient colonocytes. Thus, we hypothesized that folic acid could antagonize the toxic effects of BaP by balancing the dNTP pool and increasing the expression levels of ERCC1, XPA, and XPC genes.
In our study, the levels of XPA, XPC, and ERCC1 mRNA transcription were detected by RT-PCR, and ERCC1 protein expression was evaluated by western blot. Our results showed a significant decrease in the mRNA expression of ERCC1, XPA, and XPC, as well as the ERCC1 protein in the BaP-treated cells, which was reversed with folic acid pretreatment as expected. Therefore, we showed that folates protect cells from DNA damage by improving the expression levels of ERCC1, XPA, and XPA, which accelerate the DNA repair process. In addition, these results were in line with our in-vivo gene expression levels obtained in mice. The results of the in-vivo experiments showed that different doses of folic acid could significantly inhibit the BaP-induced reduction of mRNA transcription of ERCC1, XPC, and XPA in mouse lung, and reached its peak in the intermediate-dose group. These results could provide a feasible reference point for determining the reasonable dosage of folic acid supplementation.
In summary, the present study assessed the mechanisms by which folic acid intervenes in BaP toxicity using in-vitro and in-vivo experiments. Although the details of how this effect is mediated have not been entirely revealed by our study, one possibility is that folic acid could antagonize the toxic effects of BaP by balancing the dNTP pool and increasing the levels of ERCC1, XPA, and XPC gene expression. This could improve the rate of DNA repair, by accelerating the repair of the DNA damage caused by BaP, and enabling the cells to complete DNA replication and mitosis. However, folic acid may be modulating miRNA expression to downregulate cyclinD1 protein expression, allowing cells to recover their normal cell cycle. The present study shows that folic acid plays an important role in inhibiting the toxicity induced by BaP both in vivo and in vitro. The study of vitamins with chemopreventive properties, and a better understanding of their health-related interactions could lead to improved dietary interventions to counteract the effects of environmental pollutants.
Acknowledgements
This project was supported by the National Natural Science Foundation of China (grant no. 21077082). The experiments conducted in this research comply with the current laws of the People’s Republic of China.
Zhiyong Gong and Yongning Wu conceived and designed the experiments. Lichun Wang, Yuwei Chen, Lei Wang, and Pengcheng Wang conducted the experiments. Pengcheng Wang and Lichun Wang analyzed the data. Zhiyong Gong, Yang Wu, and Yongning Wu contributed reagents, materials, and analysis tools. Pengcheng Wang, Zhiyong Gong, and Yang Wu wrote the manuscript. All authors have approved the final version of the manuscript.
Conflicts of interest
There are no conflicts of interest.
References
- An J, Yin L, Shang Y, Zhong Y, Zhang X. (2011). The combined effects of BDE47 and BaP on oxidatively generated DNA damage in L02 cells and the possible molecular mechanism. Mutat Res 721:192–198. [DOI] [PubMed] [Google Scholar]
- Athas WF, Hedayati MA, Matanoski GM, Farmer ER, Grossman L. (1991). Development and field-test validation of an assay for DNA repair in circulating human lymphocytes. Cancer Res 51:5786. [PubMed] [Google Scholar]
- Blount CB, Mark MM, Wehr M, MacGregor JT, Hiatt RA, Wang G, et al. (1997). Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: Implications for cancer and neuronal damage. Proc Natl Acad Sci USA 7:3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebenek K, Roberts JD, Kunkel TA. (1992). The effects of dNTP pool imbalances on frameshift fidelity during DNA replication. J Biol Chem 267:3589–3596. [PubMed] [Google Scholar]
- Bjelogrlic NM, Mäkinen M, Stenbäck F, Vähäkangas K. (1994). Benzo[a]pyrene-7,8-diol-9,10-epoxide-DNA adducts and increased p53 protein in mouse skin. Carcinogenesis 15:771–774. [DOI] [PubMed] [Google Scholar]
- Card DAG, Hebbar PB, Li L, Trotter KW, Komatsu Y, Mishina Y, Archer ATK. (2008). Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol Cell Biol 28:6426–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celotti L, Ferraro P, Furlan D, Zanesi N, Pavanello S. (1993). DNA repair in human lymphocytes treated in vitro with (+)-anti- and (+/−)-syn-benzo[a]pyrene diolepoxide. Mutat Res 2:117–126. [DOI] [PubMed] [Google Scholar]
- Chang LC, Sheu HM, Huang YS, Tsai TR, Kuo KW. (1999). A novel function of emodin: enhancement of the nucleotide excision repair of UV- and cisplatin-induced DNA damage in human cells. Biochem Pharmacol 58:49–57. [DOI] [PubMed] [Google Scholar]
- Chen YH, Xia EQ, Xu XR, Sha L, Ling WH, Shan W, et al. (2012). Evaluation of benzo[a]pyrene in food from china by high-performance liquid chromatography-fluorescence detection. Int J Environ Res Public Health 9:4159–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SW, Kim YI, Weitzel JN, Mason JB. (1998). Folate depletion impairs DNA excision repair in the colon of the rat. Gut 43:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu XJ, Lovey A, Depinto W, Bartkovitz D, So SS, Vu B, et al. (2005). The discovery of novel 2,4-diaminopyrimidines as potent and selective cyclin-dependent kinase (CDK) inhibitors with in vivo activity. Cancer Res 65. [Google Scholar]
- Courtemanche C, Elson-Schwab I, Mashiyama ST, Kerry N, Ames BN. (2004). Folate deficiency inhibits the proliferation of primary human CD8+ T lymphocytes in vitro. J Immunol 173:3186–3192. [DOI] [PubMed] [Google Scholar]
- Cui XY, Xiang P, He RW, Juhasz A, Ma LQ. (2016). Advances in in vitro methods to evaluate oral bioaccessibility of PAHs and PBDEs in environmental matrices. Chemosphere 150:378–389. [DOI] [PubMed] [Google Scholar]
- Delft JV, Gaj S, Lienhard M, Albrecht MW, Kirpiy A, Brauers K, et al. (2012). RNA-Seq provides new insights in the transcriptome responses induced by the carcinogen benzo[a]pyrene. Br J Dermatol 130:568–577. [DOI] [PubMed] [Google Scholar]
- Duin MV, Koken MHM, Tol JVD, Dijke PT, Odijk H, Westerveld A, et al. (1987). Genomic characterization of the human DNA excision repair gene ERCC-1. Nucleic Acids Res 15:9195–9213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. (2008). Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9:102. [DOI] [PubMed] [Google Scholar]
- Hamouchene H, Arlt VM, Giddings I, Phillips DH. (2011). Influence of cell cycle on responses of MCF-7 cells to benzo[a]pyrene. BMC Genomics 12:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrigan JA, Vezina CM, McGarrigle BP, Ersing N, Box HC, Maccubbin AE, Olson JR. (2004). DNA adduct formation in precision-cut rat liver and lung slices exposed to benzo(a)pyrene. Toxicol Sci 77:307–314. [DOI] [PubMed] [Google Scholar]
- Hizli AA, Black AR, Pysz MA, Black JD. (2006). Protein kinase C α signaling inhibits cyclin D1 translation in intestinal epithelial cells. J Biol Chem 281:14596–14603. [DOI] [PubMed] [Google Scholar]
- Huang RF, Ho YH, Lin HL, Wei JS, Liu TZ. (1999). Folate deficiency induces a cell cycle-specific apoptosis in HepG2 cells. J Nutr 129:25–31. [DOI] [PubMed] [Google Scholar]
- Issaq HJ, Andrews AW, Janini GM, Barr EW. (1979). Isolation of stable mutagenic photodecomposition products of benzo(a)pyrene by thin-layer chromatography. J Liq Chromatogr 2:319–325. [Google Scholar]
- James SJ, Basnakian AG, Miller BJ. (1994). In vitro folate deficiency induces deoxynucleotide pool imbalance, apoptosis, and mutagenesis in chinese hamster ovary cells. Cancer Res 54:5075–5080. [PubMed] [Google Scholar]
- Jeffy BD, Chen EJ, Gudas JM, Romagnolo DF. (2000). Disruption of cell cycle kinetics by benzo[a]pyrene: inverse expression patterns of BRCA-1 and p53 in MCF-7 cells arrested in S and G2. Neoplasia 5:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeng HA, Bocca SM. (2013). Influence of exposure to benzo[a]pyrene on mice testicular germ cells during spermatogenesis. J Toxicol 2013:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YI. (2000). Methylenetetrahydrofolate reductase polymorphisms, folate, and cancer risk: a paradigm of gene-nutrient interactions in carcinogenesis. Nutr Rev 58:205. [DOI] [PubMed] [Google Scholar]
- Le Marchand L, Yoshizawa CN, Kolonel LN, Hankin JH, Goodman MT. (1989). Vegetable consumption and lung cancer risk: a population-based case-control study in Hawaii. J Natl Cancer Inst 81:1158–1164. [DOI] [PubMed] [Google Scholar]
- Li Q, Yu JC, Yunmbam M, Slavsky D, Cross C, Bostick BF, Reed E. (2000). Association between the level of ERCC-1 expression and the repair of cisplatin-induced DNA damage in human ovarian cancer cells. Anticancer Res 20:645–652. [PubMed] [Google Scholar]
- Lin CS, Chiou WY, Lee KW, Chen TF, Lin YJ, Huang JL. (2016). Xeroderma pigmentosum, complementation group D expression in H1299 lung cancer cells following benzo[a]pyrene exposure as well as in head and neck cancer patients. J Toxicol Environ Health A 79:1. [DOI] [PubMed] [Google Scholar]
- Maron DM, Ames BN. (1983). Revised methods for the Salmonella mutagenicity test. Mutat Res 113:173–215. [DOI] [PubMed] [Google Scholar]
- Nazki FH, Sameer AS, Ganaie BA. (2014). Folate: metabolism, genes, polymorphisms and the associated diseases. Gene 533:11–20. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Shi Z, Gálvez-Peralta M, Uno S, Dragin N. (2013). Oral benzo[a]pyrene: understanding pharmacokinetics, detoxication, and consequences – Cyp1 knockout mouse lines as a paradigm. Mol Pharmacol 3:304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CH, Bessho T, Matsunaga T, Sancar A. (1995). Purification and characterization of the XPF-ERCC1 complex of human DNA repair excision nuclease. J Biol Chem 270:22657–22660. [DOI] [PubMed] [Google Scholar]
- Ross SA, Davis CD. (2011). MicroRNA, nutrition, and cancer prevention. Adv Nutr 2:472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shane B. (2001). Folate chemistry and metabolism. Clin Res Regul Aff 18:137–159. [Google Scholar]
- Uno S, Makishima M. (2009). Benzo[a]pyrene toxicity and inflammatory disease. Curr Rheumatol Rev 5:266–271. [Google Scholar]
- Van Guelpen B. (2006). Low folate levels may protect against colorectal cancer. Gut 55:1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venema J, Hoffen AV, Karcagi V, Natarajan AT, Zeeland AAV, Mullenders LH. (1991). Xeroderma pigmentosum complementation group C cells remove pyrimidine dimers selectively from the transcribed strand of active genes. Mol Cell Biol 11:4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergote IB, Marth C, Coleman RL. (2015). DNA adduct formation in precision-cut rat liver and lung slices exposed to benzo(a)pyrene. Cancer Metast Rev 34:41–52. [Google Scholar]
- Volker M, Moné MJ, Karmakar P, Hoffen VA, Schul W. (2001). Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell 8:213. [DOI] [PubMed] [Google Scholar]
- Wood RD. (1996). DNA repair in eukaryotes. Annu Rev Biochem 65:135–167. [DOI] [PubMed] [Google Scholar]
- Zhang S, Hunter DJ, Hankinson SE, Giovannucci EL, Rosner BA, Colditz GA, et al. (1999). A Prospective Study of Folate Intake and the Risk of Breast Cancer. Jama 281:1632–1637. [DOI] [PubMed] [Google Scholar]
- Zhang JJ, Samet JM. (2015). Chinese haze versus Western smog: lessons learned. J Thorac Dis 7:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Wu K, Zhan C, Liu X, Gong Z. (2016). Folic acid supplementation reduces the mutagenicity and genotoxicity caused by benzo(a)pyrene. J Nutr Sci Vitaminol (Tokyo) 62:26–31. [DOI] [PubMed] [Google Scholar]
- Zhu W, Cromie MM, Cai Q, Lv T, Singh K, Gao W. (2014). Curcumin and vitamin E protect against adverse effects of benzo[a]pyrene in lung epithelial cells. PLoS One 9:e92992. [DOI] [PMC free article] [PubMed] [Google Scholar]