

Summary
Asthma is a chronic inflammatory disease that involves a variety of cytokines and cells. Interleukin‐16 (IL‐16) is highly expressed during allergic airway inflammation and is involved in its development. However, its specific mechanism of action remains unclear. In the present study, we used an animal model of ovalbumin (OVA)‐induced allergic asthma with mice harboring an IL‐16 gene deletion to investigate the role of this cytokine in asthma, in addition to its underlying mechanism. Increased IL‐16 expression was observed during OVA‐induced asthma in C57BL/6J mice. However, when OVA was used to induce asthma in IL‐16−/− mice, a diminished inflammatory reaction, decreased bronchoalveolar lavage fluid (BALF) eosinophil numbers, and the suppression of OVA‐specific IgE levels in the serum and BALF were observed. The results also demonstrated decreased levels of T helper type 2 (Th2) and Th17 cytokines upon OVA‐induced asthma in IL‐16−/− mice. Hence, we confirmed that IL‐16 enhances the lung allergic inflammatory response and suggest a mechanism possibly associated with the up‐regulation of IgE and the promotion of Th2 and Th17 cytokine production. This work explored the mechanism underlying the regulation of IL‐16 in asthma and provides a new target for the clinical treatment of asthma.
Keywords: IgE, interleukin‐16, ovalbumin‐induced asthma, T helper type 2 cytokines, T helper type 17 cytokines
Abbreviations
- AHR
airway hyper‐responsiveness
- BALF
bronchoalveolar lavage fluid
- Con
control group
- ELISA
enzyme‐linked immunosorbent assay
- H&E
hematoxylin and eosin
- IFN‐γ
interferon‐γ
- IL‐4
interleukin‐4
- OVA sIgE
OVA‐specific IgE
- OVA
ovalbumin
- PAS
Periodic Acid‐Schiff stain
- PBS
phosphate‐buffered saline
- qPCR
quantitative real‐time polymerase chain reaction
- Th
helper T
- WT
wild‐type
Introduction
Bronchial asthma, a chronic airway inflammatory disease that seriously endangers human health, is associated with a high global incidence.1 Because its course can last for several decades, it has resulted in a heavy economic burden for patients and society. The acute challenge mouse models reproduce many key features of clinical asthma, for example airway inflammation, elevated levels of IgE, goblet cell hyperplasia, epithelial hypertrophy and airway hyper‐responsiveness (AHR) to specific stimuli.2 Hence, mouse models of the acute allergic response to inhaled allergens have been widely used to clarify the mechanisms underlying the immunological and inflammatory responses in asthma.3, 4, 5
It has been shown that the pathogenesis of asthma is closely related to the dysfunction of helper T (Th) cell subsets, especially Th2 cells. Both clinical and animal experiments have confirmed the production of Th2 cytokines in asthma, such as interleukin‐4 (IL‐4), IL‐5 and IL‐13, which can induce the activation of initial effector cells such as eosinophils and mast cells, and subsequently activate many inflammatory mediators, resulting in chronic airway inflammation.6, 7, 8 In addition, the applications of immunoregulatory therapies that induce a transformation from Th2 to Th1 responses have also been explored.9
Further, as a newly discovered CD4+ T‐cell subset, Th17 cells have been reported to be involved in the pathogenesis of asthma.10 Th17 cells secrete several cytokines including IL‐17 (also known as IL‐17A), IL‐22, IL‐6 and tumor necrosis factor α. As effective inducers of inflammation, Th17 and IL‐17 are significantly associated with asthma cells and have been considered the definitive factors of severe asthma.11, 12, 13, 14 Moreover, Th17 cytokines induce the release of pro‐fibrotic cytokines from human eosinophils in asthmatics.15
Interleukin‐16 is a pro‐inflammatory cytokine that is produced by many cells such as the lymphocytes, monocytes, eosinophils, mast cells and airway epithelial cells in individuals with asthma.16 Interleukin‐16 was shown to use the CD4 molecule as its receptor and mediate the chemotactic activity of CD4+ T lymphocytes, eosinophils and monocytes. Because IL‐16 acts specifically on CD4+ cells, it is not surprising that it plays an important role in allergic inflammation, allergic asthma, rheumatoid arthritis and multiple sclerosis.17, 18, 19 Previous studies have shown that serum levels of IL‐16 in patients with asthma are significantly higher than those in healthy individuals and that IL‐16 expression in airway epithelial cells is increased.20, 21 In an ovalbumin (OVA)‐induced mouse model of asthma, the expression of IL‐16 in serum and bronchoalveolar lavage fluid (BALF) was increased, and an IL‐16 neutralizing antibody was found to down‐regulate OVA‐specific IgE (OVA sIgE) and reduce AHR.22 Further, intra‐airway administration of an IL‐16‐blocking peptide largely inhibited the development of antigen‐induced AHR.23 These studies indicate that IL‐16 plays a pro‐inflammatory role during the progression of asthma. However, other studies showed that IL‐16 might exert an anti‐inflammatory effect on asthma as treatment with this cytokine can inhibit antigen‐induced airway hyper‐reactivity, in addition to decreasing the number of eosinophils in the BALF and Th2‐type cytokine production in mice.24, 25 We suggested that the better way to investigate the immunomodulatory effect of IL‐16 during asthma was to use knockout mice.
In the present study, we identified increased IL‐16 activity in the lung, serum and BALF of C57BL/6J mice upon OVA‐induced allergic inflammation. We used an IL‐16‐deletion mouse model to then research the role of this cytokine in OVA‐induced acute allergic inflammation. We discovered that IL‐16−/− mice are less susceptible to OVA‐induced allergic asthma than wild‐type (WT) mice. In addition, we found that IL‐16 deletion results in a significant decrease in the proportion of Th2 and Th17 cells in the spleen, and an obvious reduction in IL‐4 and IL‐17A levels. Accordingly, we speculate that IL‐16 perhaps affects the extent of the inflammatory reaction during OVA‐induced allergic asthma in mice by regulating Th2 and Th17 cells. Although for years it has been known that IL‐16 is up‐regulated during asthma, this is the first report to determine the pro‐inflammatory effect of IL‐16 using specific IL‐16−/− mice.
Materials and methods
Mice
C57BL/6J WT mice were obtained from Shandong Experimental Animal Center. IL‐16 gene‐knockout (IL‐16−/−) mice were generated by CRISPR/Cas9‐mediated genome engineering at the Nanjing Biomedical Research Institute of Nanjing University.26 Mice were viable and fertile, and no abnormalities were found in thymus, spleen and lymph node. All mice were maintained under standard conditions of temperature and humidity with a 12/12‐hr light/dark cycle and specific pathogen‐free conditions at Jining Medical University. For all experiments, 6‐ to 8‐week‐old female mice were used. All animals were cared for according to the guidelines of the Jining Medical University Animal Care and Use Committee.
We performed genotyping using genomic DNA isolated from mouse tail biopsies, and the primers are listed as follows: IL‐16‐forward: TGGTACCCCAGTTAGGTGTCATCC, IL‐16‐reverse: GACCAGAAAATCGTCCTCCATCTT. PCR genotyping results are shown in the Supplementary material (Fig. S1).
OVA‐induced mouse model of airway allergic inflammation
The OVA‐induced mouse model of airway allergic inflammation was performed as described by Reddy et al.27 Mice were arbitrarily assigned to two or four groups, with each consisting of eight mice. Six‐week‐old female mice were sensitized on days 0 and 14 through an intraperitoneal injection of 20 μg of OVA (Sigma‐Aldrich, St. Louis, MO, grade V) adsorbed to 2 mg of an aluminum hydroxide gel (Inject alum; Thermo Fisher Scientific, Waltham, MA, USA) in 0·2 ml of phosphate‐buffered saline (PBS, pH 7·4). Then, mice were challenged with 1% aerosolized OVA (Sigma‐Aldrich, grade II) for 30 min each day for six consecutive days on days 21–26. Control animals received an intraperitoneal injection of 0·2 ml of PBS containing 0·05 ml of aluminum hydroxide gel and were challenged with PBS only. Twenty‐four hours after the last challenge, mice were killed to harvest the serum, BALF, lungs and spleen (Fig. 1).
Figure 1.
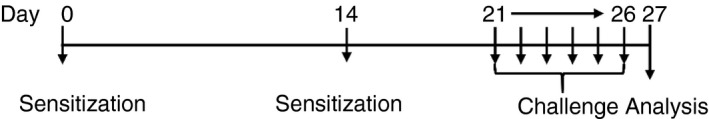
Protocol for asthma allergic mouse model. Experimental protocol of the study, n = 8 per group.
Airway hyperresponsiveness
Twenty‐four hours after the last challenge, AHR was determined using whole‐body plethysmography (Buxco, St. Paul, MN, USA). Briefly, mice were placed in the body and respiration was kept stead for 10 min. Different concentrations of methacholine (0, 6·25, 12·5, 25, 50 mg/ml; Sigma‐Aldrich) were given at intervals of more than 5 min per dose. The airway resistance was evaluated by enhancing respiratory intermission (enhanced pause, Penh) values in all groups of mice. Penh (%) = (T expiratory time/T relaxation time − 1) × peak respiratory flow/peak inspiratory flow × 100%.
Collection of blood and BALF
Twenty‐four hours after the last challenge, the blood of mice was collected via the retroorbital plexus and sera were obtained by centrifugation (1485 g, 10 min) and stored at −80° for enzyme‐linked immunosorbent assay (ELISA). BALF was collected by delivering 0·8 ml of cold PBS into the airway with a self‐made tracheal intubation and the liquid was gently pumped. This process was repeated three times to recover a total volume of 2–2·5 ml per mouse. The BALF was centrifuged at 209 g at 4° for 10 min and the supernatants were stored at −80° until analysis. The pellet was resuspended in 200 μl of PBS, centrifuged onto slides, and then stained with Wright–Giemsa; neutrophils, eosinophils, lymphocytes and macrophages were identified based on standard morphology by microscopy. The total number of cells was counted using a hemocytometer. Two different blinded investigators counted the cells with a microscope, and approximately 200 cells were counted in each of four different randomized positions.
ELISA
The concentrations of serum and BALF IL‐16, OVA‐specific IgE, IL‐4, IL‐17A and interferon‐γ (IFN‐γ) in mice were detected using respective mouse ELISA kits (Biolegend, San Diego, CA, USA) according to the manufacturer's protocol. Briefly, 100 μl of serum or BALF from each sample was added to the well and incubated at room temperature for 2 hr. After washing the plate with wash buffer four times, 100 μl of detection antibody was added to each well and samples were incubated at room temperature for 1 hr. Then, after washing the plate with wash buffer four times, 100 μl of avidin‐horseradish peroxidase was added to each well, which was incubated at room temperature for 30 min. After washing the plate with wash buffer five more times, 100 μl of substrate solution was added to each well and the plate was incubated in the dark until the desired color developed, at which time 100 μl of stop solution was added to each well and the absorbance was read at 450 nm with a Microplate reader (BioTek, Winooski, VT, USA). All measurements were performed in duplicate.
Quantitative real‐time polymerase chain reaction
Total RNA was extracted from the lung tissue of mice with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and converted to cDNA using a RevertAid First Stand cDNA Synthesis Kit based on the manufacturer's instructions (Thermo Fisher Scientific, Waltham, MA, USA). Quantitative real‐time polymerase chain reaction (qPCR) was performed using SYBR Green PCR Master Mix (Vazyme, Q111‐02, Jiangsu, China), and the 2−ΔΔCt method was used for gene expression analysis. All quantified data were normalized to mouse GAPDH levels. The primers used for real‐time were as follows: IL‐16, forward, 5′‐GCAAGACCAACTCGGTCACT‐3′, reverse, 5′‐GCCCTTCATCAGCACTATGTT‐3′; IL‐4, forward, 5′‐GGTCTCAACCCCCAGCTAGT‐3′, reverse, 5′‐GCCGATGATCTCTCTCAAGTGAT‐3′; IL‐17A, forward, 5′‐GAAGCTCAGTGCCGCCA‐3′ reverse, 5′‐TTCATGTGGTGGTCCAGCTTT‐3′; IFN‐γ, forward, 5′‐ATGAACGCTACACACTGCATC‐3′, reverse, 5′‐CCATCCTTTTGCCAGTTCCTC‐3′; GAPDH, forward 5′‐AGGTCGGTGTGAACGGATTTG‐3′, reverse, 5′‐TGTAGACCATGTAGTTGAGGTCA‐3′.
Histological assessment
The lungs from each mouse were fixed with 4% paraformaldehyde (Sigma) at 4°. After 24 hr, the fixed lung tissues were embedded in paraffin, and then cut into 4‐μm sections with a microtome and stained with hematoxylin and eosin (H&E) to analyze lung inflammation. Some sections were stained with Periodic Acid‐Schiff stain (PAS) to assess mucin. Lung inflammation and mucin production were assessed using a microscope in a blinded fashion as previously described.28, 29 Briefly, the severity of infiltration was assessed based on a five‐point inflammation scoring system as follows: 0, no inflammatory cells; 1, a few cells; 2, a ring of inflammatory cells one cell‐layer deep; 3, a ring of inflammatory cells two to four cell‐layers deep; 4, a ring of inflammatory cells more than four cell‐layers deep. The level of mucus secretion was evaluated based on the number of PAS‐positive mucus‐containing cells as follows: 0, < 0·5% PAS‐positive cells; 1, 0·5–25%; 2, 25–50%; 3, 50–75%; 4, > 75%. A mean score was obtained from three animals.
Immunohistochemistry
For this, 4‐μm‐thick, paraffin‐embedded lung‐tissue sections were heated for 1 hr at 65° and dehydrated with a gradient of ethanol solutions comprising different concentrations. After incubating with 0·3% H2O2 to inhibit endogenous peroxidase activity and blocking with normal goat serum for 30 min, sections were incubated with a mouse anti‐IL‐16‐specific antibody (1 : 200; Abcam; ab180792, Cambridge, UK) overnight at 4°. After washing with PBS, slides were incubated with biotinylated rabbit anti‐mouse IgG (1 : 1000 dilution; Streptavidin‐Peroxidase Immunohistochemical staining kit, SP‐0022) for 20 min at 37°. Then, after washing with PBS, sections were incubated with a streptavidin‐peroxidase reagent for another 20 min at 37°. After three additional PBS washes, sections were stained with diaminobenzidine under a microscope until intense staining could be observed. After hematoxylin staining and dehydration with a gradient of ethanol, sections were analyzed with a microscope (Olympus, Tokyo, Japan).
Flow cytometry
To determine the percentages of Th1, Th2 and Th17 subsets of CD4+ Th cells, single‐cell suspensions (1 × 106) from mouse spleens were prepared and cultured for 2 hr with ionomycin (BioLegend) and phorbol 12‐myristate 13‐acetate (BioLegend); then, 1 × brefeldin A (BioLegend) was used to block the samples for 4 hr. After staining with mouse fluorescein isothiocyanate‐conjugated (FITC‐) CD4 antibodies (BioLegend), single‐cell suspensions were stained with mouse Peridin chlorophyll protein 5·5‐conjugated (PerCP5·5‐) IFN‐γ, phycoerythrin‐conjugated (PE‐) IL‐4 and allophycocyanin‐conjugated (APC‐) IL‐17A (BioLegend) antibodies to differentiate Th1, Th2 and Th17 cells after fixation and permeabilization. All cells were analyzed by flow cytometry (Becton Dickinson, NJ, USA). An isotype control was used for each antibody.
Statistical analysis
All values are expressed as means ± SEM. All statistical analysis and plotting were performed with graphpad prism 6 software by performing a one‐way analysis of variance. P < 0·05 was considered statistically significant.
Results
Levels of IL‐16 in serum, BALF and lung tissue are elevated in C57BL/6J mice upon OVA‐induced allergic inflammation
To determine levels of IL‐16 during allergic asthma, wild‐type C57BL/6J mice were randomly divided into two groups, namely the control group (Con) and asthma group (OVA). ELISA analysis showed that the levels of IL‐16 in the serum and BALF of the asthma group were higher than those in the control group (Fig. 2a,b); this was also confirmed based on mRNA levels in lung tissue (Fig. 2c). Moreover, as shown in Fig. 2(d,e), IL‐16 was mainly localized to the epithelium in all mice; however, compared with those in the control group, IL‐16‐positive cells were not only increased in the epithelium but also identified in the cellular infiltrate surrounding the blood vessels and tracheas. These results suggested increased expression of IL‐16 during OVA‐induced allergic inflammation.
Figure 2.
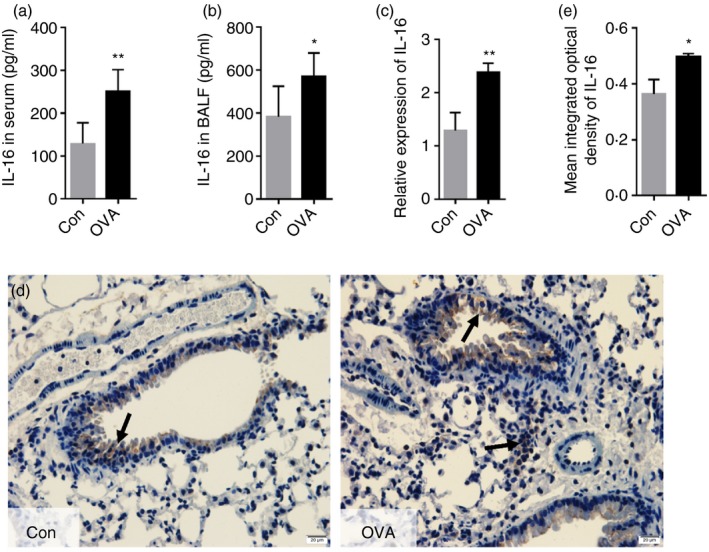
Levels of interleukin‐16 (IL‐16) in serum, bronchoalveolar lavage fluid (BALF), and lung tissue are elevated in C57BL/6 mice upon ovalbumin (OVA) ‐induced allergic inflammation. ELISA was used to detect the expression level of IL‐16 in serum (a) and BALF (b). The mRNA expression of IL‐16 in lung (c). IL‐16 protein expression in the paraffin section of lung was detected by Immunohistochemistry (Magnification: 100×) (d and e). The mean integrated optical density (IOD)was semiquantified using image‐proplus software. Date are expression as mean ± SEM (n ≥ 5). *P < 0·05 and **P < 0·01 versus control (Con).
There are no changes of spleen morphology and percentage of B cells, T cells, dendritic cells, macrophages, natural killer cells in the spleen between WT and IL‐16−/− mice
To test the effect of IL‐16 gene deficiency on the spleen, the morphology and some cells of the spleen were observed. In 8‐week‐old WT and IL‐16−/− mice, there was no significant difference in spleen morphology (Fig. 3a). Spleen cell suspensions from WT and IL‐16−/− mice were stained with mouse FITC‐B220, FITC‐CD11c, FITC‐F4/80, PE‐NK1·1, APC‐CD3, FITC‐CD4, and PE‐CD8 antibodies and flow cytometry was performed using a FACSCalibur. There were no changes in the percentage of B cells (Fig. 3b), dendritic cells (Fig. 3c), macrophages (Fig. 3d), natural killer (NK) cells, and T cells (Fig. 3e) between WT and IL‐16−/− mice. Results were similar in thymus and mesenteric lymph nodes (see Supplementary material, Figs S2 and S3).
Figure 3.
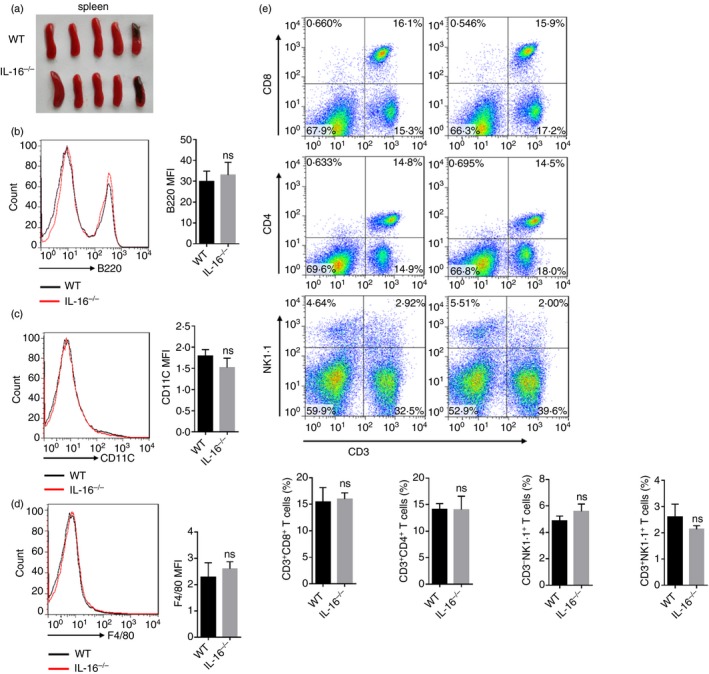
There was no difference in morphology and immune cell ratio of spleen between wild‐type (WT) and IL‐16 knockout (IL‐16−/−) mice. Morphology of spleen (a). Spleen cells stained successively with Anti‐B220 (FITC), Anti‐CD11c (FITC), Anti‐F4/80 (FITC), Anti‐NK1·1 (PE), Anti‐CD3 (APC), Anti‐CD4 (FITC), Anti‐CD8 (PE), percentages of B cells (b), DCs (c), macrophages (d), CD3+ CD8+ T cells, CD3+ CD4+ T cells, NKs and NKTs (e) were analyzed with flow cytometric analysis. The data are shown as the means ± SEM (n = 5 replicates/group) and ns denotes P > 0·05.
IL‐16 deletion effects on AHR and reduces OVA‐induced allergic inflammation in mice
The airway responsiveness to methacholine was significantly enhanced in OVA‐treated mice compared with PBS‐treated mice, as demonstrated by a significant increase in Penh values, an indirect parameter for airway function. In OVA‐treated mice, the level of AHR in IL‐16−/− mice tends to be reduced compared with that from WT mice; however, this was not significant (see Supplementary material, Fig. S4).
Next, we assessed changes in allergic airway inflammation after IL‐16 deletion. There was no difference in cell numbers in the BALF between PBS‐treated groups of both WT and IL‐16−/− mice; however, comparing OVA‐treated groups, the BALF from IL‐16−/− mice contained fewer eosinophils than that of WT mice (Fig. 4a). As shown in Fig. 4(b), compared with that in the PBS‐treated control group, the OVA group showed typical pathological features of allergic airway inflammation based on H&E staining, as well as inflammatory cell infiltration around the blood vessels and tracheas; interestingly, these cells were less abundant in IL‐16−/− mice compared with numbers in WT mice. As shown in Fig. 4(d), an obviously decreased inflammation score was noted in OVA‐treated IL‐16−/− mice compared with that in WT mice. In addition, many mucus‐containing epithelial cells were observed in OVA‐treated mice; however, these cells were relatively scarce in IL‐16−/− mice (Fig. 4c,e). Based on these data, we expounded that IL‐16 deletion in mice suppresses OVA‐induced allergic inflammation during asthma.
Figure 4.
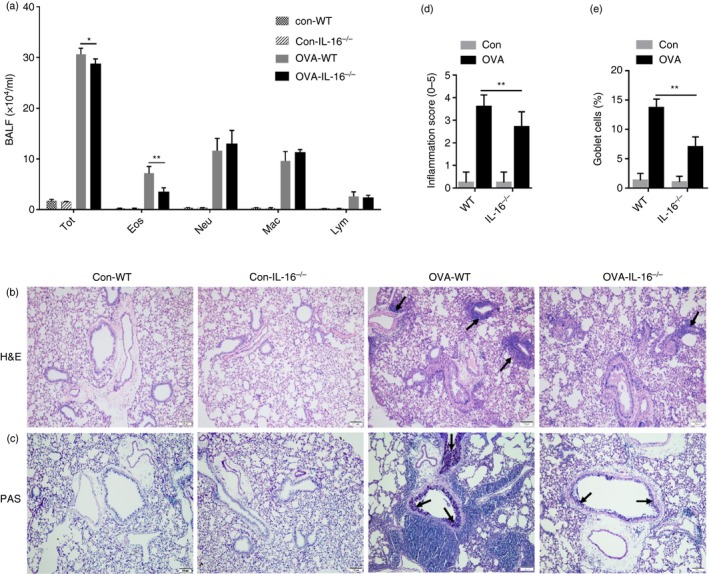
Interleukin‐16 (IL‐16) deletion reduces ovalbumin (OVA) ‐induced allergic inflammation in mice. The recruitment of total leukocytes and its differential counts in bronchoalveolar lavage fluid (BALF) (a). Lung tissue sections were stained with hematoxylin and eosin (H&E) (b). (Magnification: 100×). Periodic acid‐Schiff (PAS) staining of lung‐tissue sections (c). (Magnification: 100×). Histopathological changes in lung inflammation and mucus production were scored as described in the Materials and Methods (d and e). Date are expression as mean ± SEM (n ≥ 5). *P < 0·05 and **P < 0·01.
IL‐16 deletion down‐regulates OVA‐specific IgE in the serum and BALF
Ovalbumin‐specific IgE is an important indicator of asthma; therefore, we examined the effect of IL‐16 deletion on OVA‐specific IgE content. According to ELISA, these levels were increased in the serum and BALF of OVA‐treated groups, whereas this effect was diminished in IL‐16−/− mice (Fig. 5a,b). These data further reinforce that IL‐16 deletion in mice can indeed suppress OVA‐induced allergic inflammation during asthma.
Figure 5.
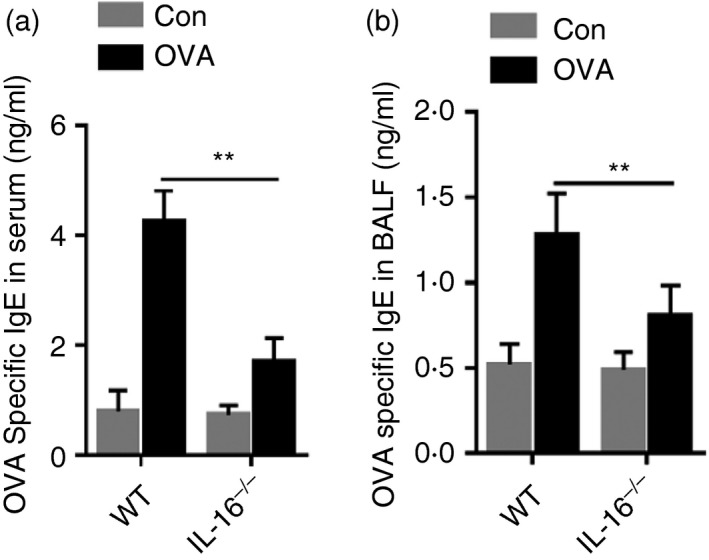
Interleukin‐16 (IL‐16) deletion down‐regulates ovalbumin (OVA) ‐specific IgE in serum and bronchoalveolar lavage fluid (BALF) of OVA‐challenged asthma mice. OVA‐specific IgE levels in serum of each group (a). OVA‐specific IgE levels in BALF of each group (b). The date are shown as mean ± SEM (n ≥ 5). **P < 0·01.
IL‐16 deletion inhibits the production and expression of Th2‐ and Th17‐specific cytokines in OVA‐challenged mice
Many cytokines secreted by Th1, Th2 and Th17 cells are involved in the pathophysiology of asthma. To investigate the effect of IL‐16 deletion on these cytokines, we detected their expression levels in BALF and serum samples from control and asthma group mice. ELISA results showed that levels of IL‐4 and IL‐17A were obviously increased in the BALF and serum of OVA‐treated animals compared with those in control groups. Furthermore, the levels of IL‐4 and IL‐17A in the serum (Fig. 6a,b) and BALF (Fig. 6d,e) of IL‐16−/− mice were lower than those in WT mice. Compared with those in control groups, levels of the Th1 cytokine IFN‐γ were significantly reduced in the BALF of OVA‐treated mice, and these levels were higher in IL‐16−/− mice than in WT mice (Fig. 6f). However, regarding serum levels of IFN‐γ, there were no differences between control and OVA‐treated groups or IL‐16−/− and WT mice (Fig. 6c). In addition, we tested the mRNA expression of genes encoding IL‐4, IL‐17A and IFN‐γ in lung tissues. As expected, qPCR results for IL4, IL17A and IFNg were consistent with ELISA results for the BALF (Fig. 6g–i).
Figure 6.
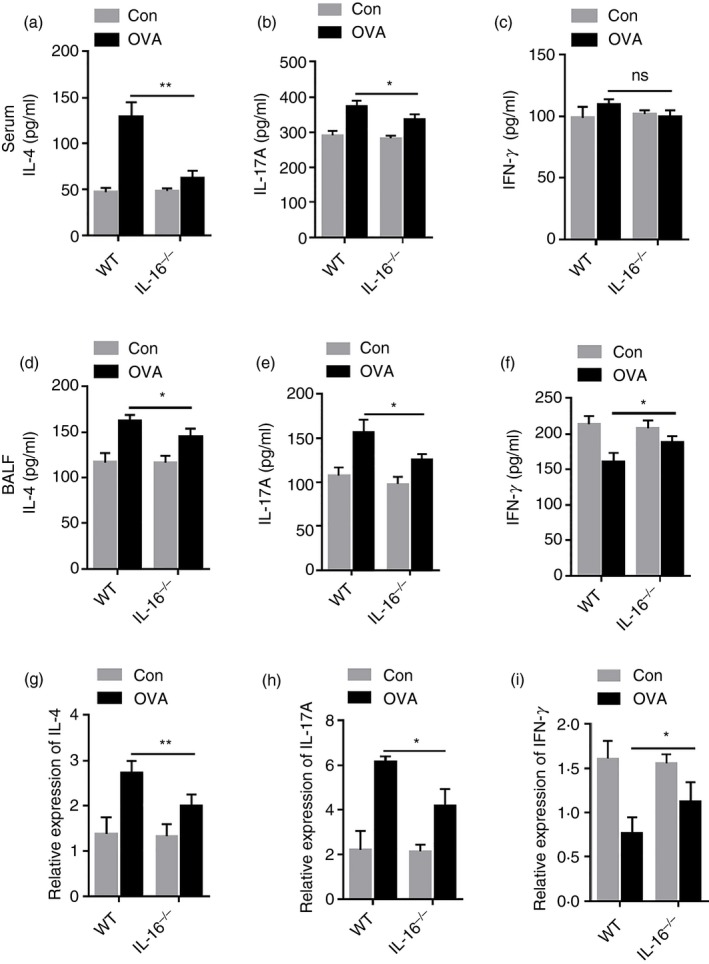
Interleukin‐16 (IL‐16) deletion inhibits the production and expression of T helper type 2 (Th2) ‐specific and Th17‐specific cytokines in ovalbumin (OVA) ‐challenged mice. The expression level of Th2 cytokines IL‐4 (a), Th17 cytokine IL‐17A (b) and Th1 cytokine IFN‐γ (c) in serum. The expression levels of Th2 cytokines IL‐4 (d), Th17 cytokine IL‐17A (e) and Th1 cytokine IFN‐γ (f) in BALF. The mRNA expression level of Th2 cytokines IL‐4 (g), Th17 cytokine IL‐17A (h) and Th1 cytokine IFN‐γ (i) in lung. The data are shown as mean ± SEM (n ≥ 5). *P < 0·05, **P < 0·01 and ns denotes P > 0·05.
IL‐16 deletion diminishes the Th2 and Th17 cell population in OVA‐challenged mice
Th1, Th2 and Th17 cells are involved in allergic inflammatory responses. The relative percentages of these cells in the spleen of WT and IL‐16−/− were detected by flow cytometry. As shown in Fig. 7(a–d), compared with those in the control group, the percentages of Th2 and Th17 cells were greatly increased and the percentage of Th1 cells was decreased in the asthma group. Moreover, in the asthma group, compared with those in WT mice, the percentages of Th2 and Th17 cells were obviously reduced, whereas the proportion of Th1 cells was increased in IL‐16−/− mice. Therefore, we concluded that IL‐16 deletion decreases the percentages of Th2 and Th17 cells and increases the proportion of Th1 cells. In addition, we also detected the percentages of CD4+ CD25+ Foxp3+ cells and Th9 cells in the spleen; however, there were no differences between IL‐16−/− mice and WT mice (see Supplementary material, Fig. S5a–c).
Figure 7.
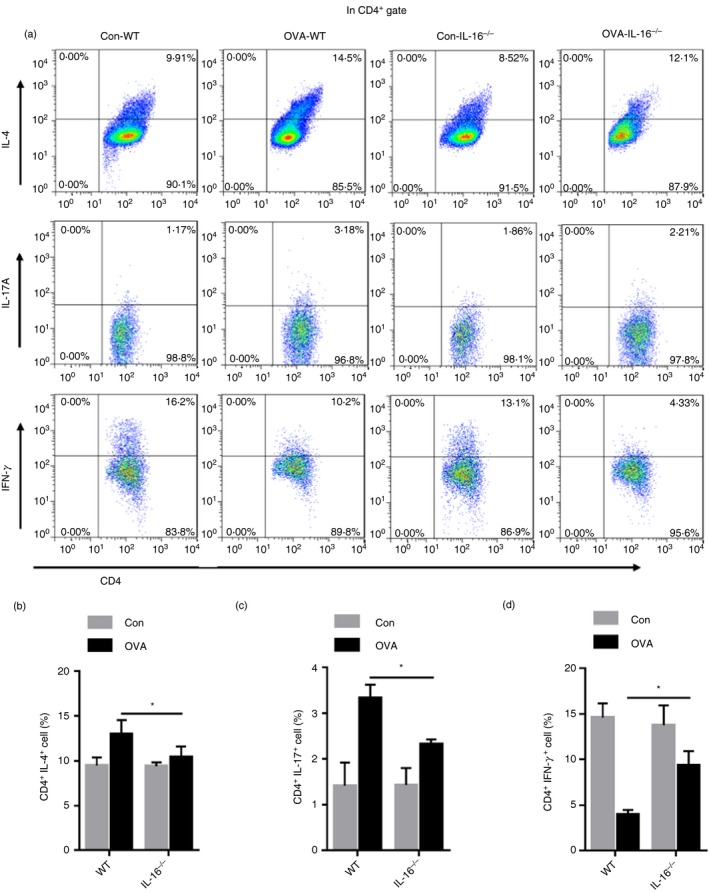
Interleukin‐16 (IL‐16) deletion diminishes the T helper type 2 (Th2) and Th17 cell population in ovalbumin (OVA) ‐challenged mice. Flow cytometric analysis of splenocytes stained successively with Anti‐CD4 (FITC), Anti‐IFN‐γ (PerCP5·5), Anti‐IL‐4 (PE), Anti‐IL‐17A (APC) (a). Bar plot of the average percentage of CD4+ IL‐4+ cells (b). Bar plot of the average percentage of CD4+ IL‐17A+ cells (c). Bar plot of the average percentage of CD4+ IFN‐γ + cells (d). The date are shown as mean ± SEM (n ≥ 5). *P < 0·05.
Discussion
In the present study, we assessed the effect of IL‐16 on OVA‐induced acute allergic asthma using IL‐16‐deletion mice. IL‐16−/− mice were less susceptible to OVA sensitization and challenge than WT mice. Interleukin‐16 deletion inhibited eosinophil accumulation and OVA‐specific IgE levels in serum and BALF, but did not affect AHR. The unfavorable effect of IL‐16 appears to be caused by potentiating the Th2 and Th17 response.
Many studies have shown that the increasing expression of IL‐16 has been demonstrated in a variety of diseases, including asthma.30 Interleukin‐16 was previously identified in the BALF 4 hr after antigen challenge in asthmatic subjects.31 Increased airway expression of IL‐16 was detected in bronchial biopsy samples obtained from allergic subjects with asthma 24 hr after allergen challenge.32 Levels of IL‐16 protein and mRNA were also readily detected and uniformly distributed in the airway epithelium and infiltrating CD4+ cells in biopsies from asthmatics.21 In our study, we identified increasing expression of IL‐16 in lung tissue, and specifically in both the serum and BALF of OVA‐induced allergic mice. Thus, IL‐16 is indeed highly expressed in asthma.
In this study, marked inflammatory cell infiltration into the lungs was observed in the airways of OVA‐induced allergic mice. The IL‐16 deletion diminished eosinophil infiltration inflammatory scores. Interleukin‐16 has been described as a potent human eosinophil chemoattractant in vitro.33, 34 Our data demonstrated that IL‐16 increased inflammation by chemotaxis of eosinophils in OVA‐induced allergic mice. The IL‐16 deletion decreased up‐regulation of OVA‐specific IgE in the serum and BALF in asthmatic mice, which agrees with findings that IL‐16 neutralizing antibody or blocking peptide could reduce OVA‐specific sIgE in a model of OVA‐induced asthma.22, 23 It may be related that IL‐16 binding to the CD4 molecule regulates B‐cell class switching to IgE through the contact between CD4+ T cells and B cells with lower levels of IL‐4. Hence, our data support that IL‐16 plays a pro‐inflammatory role in OVA‐induced asthma.
Interleukin‐16 neutralizing antibody or blocking peptide inhibited OVA‐induced AHR.22, 23 This sounds contradictory to later published papers, showing that AHR was diminished by intratracheal IL‐16 treatment and intraperitoneal administration of IL‐16.24, 25 However, in our study there was no significant correlation between IL‐16 and OVA‐induced AHR; AHR were not alleviated obviously after IL‐16 deletion in allergic asthma mice. Hence, the impact of IL‐16 on AHR remains controversial, further studies are needed.
It is established that Th2 cells produce IL‐4, IL‐5 etc., which are responsible for the development of asthma, Th1 cells and IFN‐γ have an opposite effect; Th1/Th2 cytokine levels comprise an important index to evaluate asthma.35 Furthermore, it was recently reported that Th17 cells and IL‐17A play important roles in asthma and allergic disease.36, 37, 38, 39, 40, 41 In vitro, recombinant human IL‐16 abrogates allergen‐mediated IL‐13 and IL‐5 production by blood mononuclear cells.42, 43 We have only limited knowledge on regulating Th1/Th2/Th17 cells and cytokines in asthmatic IL‐16−/− mice. We have shown here that deletion of IL‐16 down‐regulated Th2 and Th17 cells and inhibited IL‐4 and IL‐17A release in BALF, and up‐regulated Th1 and IFN‐γ in the BALF in OVA‐induced asthmatic mice. However, there was no significant change in serum IFN‐γ. It is consistent with our observations of the mRNA expression of IL4, IL17A and IFNg in lung tissues. Collectively, these data suggest that the deletion of IL‐16 could reduce allergic inflammation by down‐regulating the immune response of Th2 cells and Th17 cells. The increased levels of the Th1 cytokine IFN‐γ detected in IL‐16−/− asthmatic mice might have also contributed to the alleviation of inflammation. Accordingly, we have yet to determine the potential molecular mechanisms responsible for IL‐16 induction in T cells in response to OVA challenge. We hypothesize that this could be an effect related to the dendritic cell‐induced priming of T cells or that IL‐16 might directly stimulate CD4+ T cells to produce increased levels of Th2 or Th17 cytokines. Moreover, it is unknown if IL‐16 can alter the recruitment of distinct T‐cell subsets. These issues will be investigated further in our next study.
In conclusion, the present study has clarified the less inflammatory susceptibility of IL‐16 deletion in allergic asthma though decreasing OVA‐specific IgE and eosinophil recruitment. Furthermore, we also find that IL‐16 aggravates OVA‐induced allergic inflammation by enhancing Th2 and Th17 responses, which provides new insight into the mechanism of, and suggests a potential intervention target for, treatment of asthma.
Disclosures
These authors declare no conflict of interest.
Supporting information
Figure S1. PCR genotype results of mouse tail DNA.
Figure S2. There are no changes of thymus morphology, CD3+ CD8+ T cells and CD3+ CD4+ T cells in thymus between WT and IL‐16−/− mice.
Figure S3. There are no changes of mesenteric lymph node morphology, B cells, CD3+ CD8+ T cells and CD3+ CD4+ T cells in mesenteric lymph nodes between WT and IL‐16−/− mice.
Figure S4. There is no significant difference of airway hyper‐responsiveness assay between WT and IL‐16−/− mice sensitized and challenged with OVA.
Figure S5. IL‐16 deletion has no effect on CD4+ CD25+ Foxp3+ cell and Th9 cell population in OVA‐challenged mice.
Acknowledgements
HBX and CPS designed the research; CXL, QM, ZHL and FLY, performed most of the experiments and analyzed the data; CXL and GJD wrote the manuscript. JFZ and HZ provided technical support for the flow cytometric analysis of Th cells. JD provided logistical support and discussed the data. HS, BW, YZZ and XYY provided technical support and experimental assistance. This work was supported by grants from the National Natural Science Foundation of China (81671632, 81601426) and Supporting Fund for Teachers’ research of Jining Medical University (JYFC2018KJ029).
Contributor Information
Chuanping Si, Email: chpsi@mail.jnmc.edu.cn.
Huabao Xiong, Email: huabao.xiong@mssm.edu.
References
- 1. Pawankar R, Canonica GW, Holgate ST, Lockey RF. Allergic diseases and asthma: a major global health concern. Curr Opin Allergy Clin Immunol 2012; 12:39–41. [DOI] [PubMed] [Google Scholar]
- 2. Nials AT, Uddin S. Mouse models of allergic asthma: acute and chronic allergen challenge. Dis Model Mech 2008; 1:213–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tomkinson A, Duez C, Cieslewicz G, Pratt JC, Joetham A, Shanafelt MC et al A murine IL‐4 receptor antagonist that inhibits IL‐4‐ and IL‐13‐induced responses prevents antigen‐induced airway eosinophilia and airway hyperresponsiveness. J Immunol 2001; 166:5792–800. [DOI] [PubMed] [Google Scholar]
- 4. McMillan SJ, Bishop B, Townsend MJ, McKenzie AN, Lloyd CM. The absence of interleukin 9 does not affect the development of allergen‐induced pulmonary inflammation nor airway hyperreactivity. J Exp Med 2002; 195:51–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lloyd CM, Gonzalo JA, Nguyen T, Delaney T, Tian J, Oettgen H et al Resolution of bronchial hyperresponsiveness and pulmonary inflammation is associated with IL‐3 and tissue leukocyte apoptosis. J Immunol 2001; 166:2033–40. [DOI] [PubMed] [Google Scholar]
- 6. Mazzarella G, Bianco A, Catena E, De Palma R, Abbate GF. Th1/Th2 lymphocyte polarization in asthma. Allergy 2000; 55(Suppl 61):6–9. [DOI] [PubMed] [Google Scholar]
- 7. Shirai T, Suzuki K, Inui N, Suda T, Chida K, Nakamura H. Th1/Th2 profile in peripheral blood in atopic cough and atopic asthma. Clin Exp Allergy 2003; 33:84–9. [DOI] [PubMed] [Google Scholar]
- 8. Tang ML, Coleman J, Kemp AS. Interleukin‐4 and interferon‐γ production in atopic and non‐atopic children with asthma. Clin Exp Allergy 1995; 25:515–21. [DOI] [PubMed] [Google Scholar]
- 9. Thorburn AN, Hansbro PM. Harnessing regulatory T cells to suppress asthma: from potential to therapy. Am J Respir Cell Mol Biol 2010; 43:511–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alcorn JF, Crowe CR, Kolls JK. TH17 cells in asthma and COPD. Annu Rev Physiol 2010; 72:495–516. [DOI] [PubMed] [Google Scholar]
- 11. Halwani R, Sultana A, Vazquez‐Tello A, Jamhawi A, Al‐Masri AA, Al‐Muhsen S. Th‐17 regulatory cytokines IL‐21, IL‐23, and IL‐6 enhance neutrophil production of IL‐17 cytokines during asthma. J Asthma 2017; 54:893–904. [DOI] [PubMed] [Google Scholar]
- 12. Ji NF, Xie YC, Zhang MS, Zhao X, Cheng H, Wang H et al Ligustrazine corrects Th1/Th2 and Treg/Th17 imbalance in a mouse asthma model. Int Immunopharmacol 2014; 21:76–81. [DOI] [PubMed] [Google Scholar]
- 13. Kianmehr M, Haghmorad D, Nosratabadi R, Rezaei A, Alavinezhad A, Boskabady MH. The effect of Zataria multiflora on Th1/Th2 and Th17/T regulatory in a mouse model of allergic asthma. Front Pharmacol 2017; 8:458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tao B, Ruan G, Wang D, Li Y, Wang Z, Yin G. Imbalance of peripheral Th17 and regulatory T cells in children with allergic rhinitis and bronchial asthma. Iran J Allergy Asthma Immunol 2015; 14:273–9. [PubMed] [Google Scholar]
- 15. Al‐Muhsen S, Letuve S, Vazquez‐Tello A, Pureza MA, Al‐Jahdali H, Bahammam AS et al Th17 cytokines induce pro‐fibrotic cytokines release from human eosinophils. Respir Res 2013; 14:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Laberge S, Pinsonneault S, Ernst P, Olivenstein R, Ghaffar O, Center DM et al Phenotype of IL‐16‐producing cells in bronchial mucosa: evidence for the human eosinophil and mast cell as cellular sources of IL‐16 in asthma. Int Arch Allergy Immunol 1999; 119:120–5. [DOI] [PubMed] [Google Scholar]
- 17. Conti P, Kempuraj D, Boucher W, Letourneau R, Theoharides TC. Mechanism by which IL‐16 generated by mast cells mediates allergic inflammation. Int J Immunopathol Pharmacol 2001; 14:1–4. [DOI] [PubMed] [Google Scholar]
- 18. Majak P, Jerzynska J, Bojo M, Brzozowska A, Koczkowska M, Sielski P et al Cytokine profiling in exhaled breath condensate after exercise challenge in asthmatic children with post‐exercise symptoms. Arch Med Sci 2016; 12:778–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baumann R, Rabaszowski M, Stenin I, Tilgner L, Gaertner‐Akerboom M, Scheckenbach K et al Nasal levels of soluble IL‐33R ST2 and IL‐16 in allergic rhinitis: inverse correlation trends with disease severity. Clin Exp Allergy 2013; 43:1134–43. [DOI] [PubMed] [Google Scholar]
- 20. Afifi SS, ElArab AE, Mostafa SY. Interleukin 16 (IL‐16) in asthma and allergic rhinitis. A comparison between upper and lower airways. Egypt J Immunol 2004; 11:31–6. [PubMed] [Google Scholar]
- 21. Laberge S, Ernst P, Ghaffar O, Cruikshank WW, Kornfeld H, Center DM et al Increased expression of interleukin‐16 in bronchial mucosa of subjects with atopic asthma. Am J Respir Cell Mol Biol 1997; 17:193–202. [DOI] [PubMed] [Google Scholar]
- 22. Hessel EM, Cruikshank WW, Van Ark I, De Bie JJ, Van Esch B, Hofman G et al Involvement of IL‐16 in the induction of airway hyper‐responsiveness and up‐regulation of IgE in a murine model of allergic asthma. J Immunol 1998; 160:2998–3005. [PubMed] [Google Scholar]
- 23. de Bie JJ, Henricks PA, Cruikshank WW, Hofman G, Nijkamp FP, van Oosterhout AJ. Effect of interleukin‐16‐blocking peptide on parameters of allergic asthma in a murine model. Eur J Pharmacol 1999; 383:189–96. [DOI] [PubMed] [Google Scholar]
- 24. Little FF, de Bie J, van Oosterhout A, Kornfeld H, Center DM, Cruikshank WW. Immunomodulatory effect of interleukin‐16 on allergic airway inflammation. Chest 2003; 123:431S–2S. [DOI] [PubMed] [Google Scholar]
- 25. De Bie JJ, Jonker EH, Henricks PA, Hoevenaars J, Little FF, Cruikshank WW et al Exogenous interleukin‐16 inhibits antigen‐induced airway hyper‐reactivity, eosinophilia and Th2‐type cytokine production in mice. Clin Exp Allergy 2002; 32:1651–8. [DOI] [PubMed] [Google Scholar]
- 26. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE et al RNA‐guided human genome engineering via Cas9. Science 2013; 339:823–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reddy AT, Lakshmi SP, Reddy RC. Murine model of allergen induced asthma. J Vis Exp 2012:e3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kurakula K, Vos M, Logiantara A, Roelofs JJ, Nieuwenhuis MA, Koppelman GH et al Nuclear receptor Nur77 attenuates airway inflammation in mice by suppressing NF‐κB activity in lung epithelial cells. J Immunol 2015; 195:1388–98. [DOI] [PubMed] [Google Scholar]
- 29. Li K, Zhang Y, Liang KY, Xu S, Zhou XJ, Tan K et al Rheb1 deletion in myeloid cells aggravates OVA‐induced allergic inflammation in mice. Sci Rep 2017; 7:42655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoshimoto T, Wang CR, Yoneto T, Matsuzawa A, Cruikshank WW, Nariuchi H. Role of IL‐16 in delayed‐type hypersensitivity reaction. Blood 2000; 95:2869–74. [PubMed] [Google Scholar]
- 31. Cruikshank WW, Long A, Tarpy RE, Kornfeld H, Carroll MP, Teran L et al Early identification of interleukin‐16 (lymphocyte chemoattractant factor) and macrophage inflammatory protein 1 α (MIP1α) in bronchoalveolar lavage fluid of antigen‐challenged asthmatics. Am J Respir Cell Mol Biol 1995; 13:738–47. [DOI] [PubMed] [Google Scholar]
- 32. Laberge S, Pinsonneault S, Varga EM, Till SJ, Nouri‐Aria K, Jacobson M et al Increased expression of IL‐16 immunoreactivity in bronchial mucosa after segmental allergen challenge in patients with asthma. J Allergy Clin Immunol 2000; 106:293–301. [DOI] [PubMed] [Google Scholar]
- 33. Rand TH, Cruikshank WW, Center DM, Weller PF. CD4‐mediated stimulation of human eosinophils: lymphocyte chemoattractant factor and other CD4‐binding ligands elicit eosinophil migration. J Exp Med 1991; 173:1521–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mashikian MV, Tarpy RE, Saukkonen JJ, Lim KG, Fine GD, Cruikshank WW et al Identification of IL‐16 as the lymphocyte chemotactic activity in the bronchoalveolar lavage fluid of histamine‐challenged asthmatic patients. J Allergy Clin Immunol 1998; 101:786–92. [DOI] [PubMed] [Google Scholar]
- 35. Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest 2008; 118:3546–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chang Y, Al‐Alwan L, Risse PA, Halayko AJ, Martin JG, Baglole CJ et al Th17‐associated cytokines promote human airway smooth muscle cell proliferation. FASEB J 2012; 26:5152–60. [DOI] [PubMed] [Google Scholar]
- 37. Chesne J, Braza F, Mahay G, Brouard S, Aronica M, Magnan A. IL‐17 in severe asthma. Where do we stand? Am J Respir Crit Care Med 2014; 190:1094–101. [DOI] [PubMed] [Google Scholar]
- 38. Silva FMC, Oliveira EE, Gouveia ACC, Brugiolo ASS, Alves CC, Correa JOA et al Obesity promotes prolonged ovalbumin‐induced airway inflammation modulating T helper type 1 (Th1), Th2 and Th17 immune responses in BALB/c mice. Clin Exp Immunol 2017; 189:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma C, Ma Z, Fu Q, Ma S. Curcumin attenuates allergic airway inflammation by regulation of CD4+ CD25+ regulatory T cells (Tregs)/Th17 balance in ovalbumin‐sensitized mice. Fitoterapia 2013; 87:57–64. [DOI] [PubMed] [Google Scholar]
- 40. Wakashin H, Hirose K, Maezawa Y, Kagami S, Suto A, Watanabe N et al IL‐23 and Th17 cells enhance Th2‐cell‐mediated eosinophilic airway inflammation in mice. Am J Respir Crit Care Med 2008; 178:1023–32. [DOI] [PubMed] [Google Scholar]
- 41. Makihara S, Okano M, Fujiwara T, Kariya S, Noda Y, Higaki T et al Regulation and characterization of IL‐17A expression in patients with chronic rhinosinusitis and its relationship with eosinophilic inflammation. J Allergy Clin Immunol 2010; 126:397–400, e1–11. [DOI] [PubMed] [Google Scholar]
- 42. Pinsonneault S, El Bassam S, Mazer B, Cruikshank WW, Laberge S. IL‐16 inhibits IL‐5 production by antigen‐stimulated T cells in atopic subjects. J Allergy Clin Immunol 2001; 107:477–82. [DOI] [PubMed] [Google Scholar]
- 43. El Bassam S, Pinsonneault S, Kornfeld H, Ren F, Menezes J, Laberge S. Interleukin‐16 inhibits interleukin‐13 production by allergen‐stimulated blood mononuclear cells. Immunology 2006; 117:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. PCR genotype results of mouse tail DNA.
Figure S2. There are no changes of thymus morphology, CD3+ CD8+ T cells and CD3+ CD4+ T cells in thymus between WT and IL‐16−/− mice.
Figure S3. There are no changes of mesenteric lymph node morphology, B cells, CD3+ CD8+ T cells and CD3+ CD4+ T cells in mesenteric lymph nodes between WT and IL‐16−/− mice.
Figure S4. There is no significant difference of airway hyper‐responsiveness assay between WT and IL‐16−/− mice sensitized and challenged with OVA.
Figure S5. IL‐16 deletion has no effect on CD4+ CD25+ Foxp3+ cell and Th9 cell population in OVA‐challenged mice.