

Summary
Overcoming the immunosuppressive tumour microenvironment is the major challenge impeding cancer immunotherapy today. Regulatory T‐cells (Tregs) are prevalent in nearly all cancers and, as immunosuppressive regulators of immune responses, they are the principal opponents of cancer immunotherapy. However, disabling Tregs systemically causes severe autoimmune toxicity, hastening the need for more selective methods to target intratumoural Tregs. In this review, we discuss a burgeoning new modality to specifically target tumour‐infiltrating Tregs (TI‐Tregs) by reprogramming their functionality from immunosuppressive to immune stimulatory within tumours. As the basis for therapeutic selectivity of TI‐Tregs, we will focus on the defining features of Tregs within cancer: their highly activated state controlled by the engagement of key surface receptors, their distinct metabolic programme, and their unique transcriptional programme. By identifying proteins and pathways that distinguish TI‐Tregs from other Tregs in the body, as well as from the beneficial antitumour effector T‐cells within tumours, we highlight mechanisms to selectively reprogramme TI‐Tregs for the treatment of cancer.
Keywords: cancer, Treg, tumour immunology
Introduction
The recent success of multiple forms of immune‐based cancer therapies that mobilize cytotoxic T‐cells to attack cancer cells has revolutionized the treatment of this disease.1 However, it is already apparent that these immune‐boosting drugs do not benefit all patients, highlighting the need for therapies that do more than ramp up the immune response. Two key hurdles remain to be cleared for the broadest impact of cancer immunotherapies. First, treatments are needed that can overcome the immunosuppressive tumour microenvironment (TME). Second, and perhaps more important, strategies for enhancing tumour‐specific anti‐tumour immunity locally within tumours are necessary to reduce the severe autoimmune side‐effects in non‐cancerous tissues that are associated with all current cancer immunotherapies.2
Regulatory T‐cells (Tregs), a subset of immunosuppressive CD4+ T‐cells defined by their expression of the Foxp3 transcription factor, may be the perfect target to achieve both of these goals.3 In cancer, Tregs infiltrate tumours where they dampen anti‐tumour immune responses and are commonly associated with poorer prognoses.4 However, Tregs play an essential role in preventing autoimmunity and, while Treg depletion can mobilize anti‐tumour immune responses in some instances, it has always come with the significant cost of severe autoimmunity.5
In this review, we will describe a newly evolving approach that aims to selectively reprogramme the functionality of Tregs within tumours by discovering and targeting the unique characteristics of tumour‐infiltrating Tregs (TI‐Tregs). We will focus on three defining aspects of TI‐Tregs that, when disrupted, can reprogramme TI‐Treg function: (i) their activation state via stimulatory cell surface receptors; (ii) their metabolic state; and (iii) their transcriptional state as controlled by critical transcription factors and chromatin regulators (Fig. 1). We define Treg reprogramming in this review broadly.6 It encompasses the loss of immunosuppressive activity, which may include loss of Foxp3 expression (often described as Foxp3 or Treg instability), but does not necessitate it, and/or the acquisition of pro‐inflammatory or immune stimulatory activities.7 By reprogramming the functionality of Tregs from an immunosuppressive to pro‐inflammatory state specifically within the TME, reprogramming therapies will not only circumvent immune regulation in tumours, by removing immunosuppressive cells, but also provide heightened precision for cancers, by converting immunosuppressive Tregs into immunostimulatory cells specifically within cancer. Indeed, as discussed in this review, many drugs are already approved, or are in development, which have the potential to reprogramme TI‐Tregs in support of cancer immunotherapies (Table 1).
Figure 1.
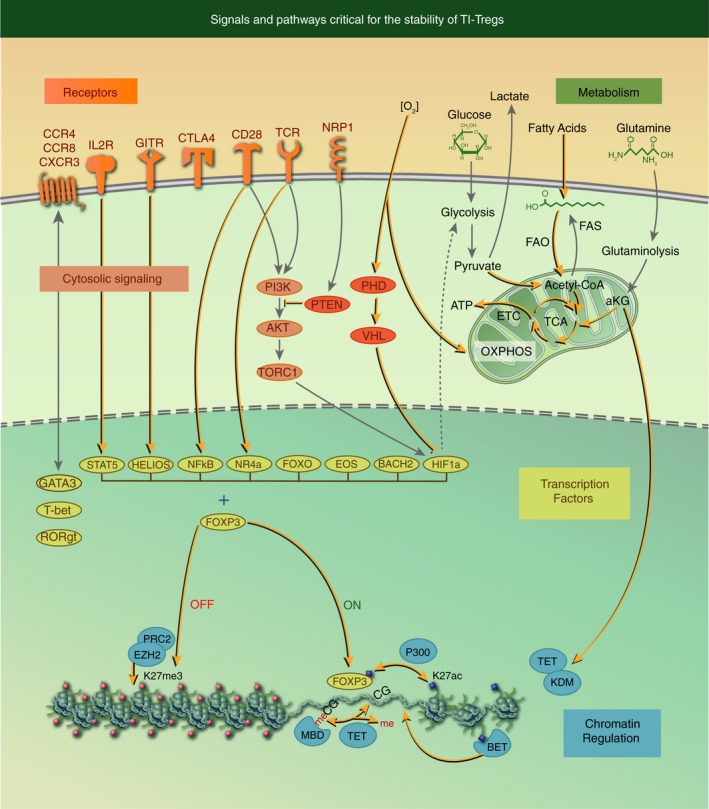
Signals and pathways critical for the stability of tumour‐infiltrating regulatory T‐cells (TI‐Tregs). Here we demarcate five interconnected nodes (receptors, cytosolic signalling, metabolism, transcription factors and chromatin regulation) that together programme Treg immunosuppression. Reprogramming of TI‐Tregs from immunosuppressive to immune stimulatory activities is achieved by the disruption of these critical pathways (see highlighted lines and arrows in the figure).
Table 1.
Druggable mechanisms to reprogramme TI‐Tregs
| Target | Agent | Mechanism of action | Study reference |
|---|---|---|---|
| CD28 | Pentoxifylline (PTXF), CPI‐1205 | c‐REL inhibition, EZH2 inhibition | 103, 117 |
| CTLA4 | Anti‐CTLA4 antibodies | Selectively deplete TI‐Tregs | 19 |
| CD25 (IL2R) | Anti‐CD25 antibody, ONTAK | Reprogramme, deplete Tregs | 29 |
| GITR | Anti‐GITR antibody | Reprogramme TI‐Tregs, reduce expression of HELIOS | 35 |
| NRP‐1 | Anti‐NRP1 antibody, Sema4a‐Ig | Reprogramme TI‐Tregs, block PTEN recruitment | 30, 31, 32 |
| CCR4/CCR8 | Anti‐CCR4, anti‐CCR8 antibodies | Selectively deplete/reprogramme Th2‐like Tregs | 9, 10, 40, 41 |
| CXCR3 | AMG487 | CXCR3 inhibitor | 45 |
| LCK | Imatinib, Dasatinib | Inhibit LCK kinase | 8, 20 |
| PI3K | CAL‐101 | Inhibit PI3K kinase subunit p110δ | 25 |
| PTEN | VO‐OHpic | Inhibit PTEN and increase Akt phosphorylation | 23 |
| AKT | SC79 | AKT activating chemical | 54 |
| Glycolysis | Dichloroacetate (DCA), 2‐DG | Inhibit glucose uptake and aerobic glycolysis | 52, 62 |
| Lipid metabolism | 5‐(tetradecyloxy)‐2‐Furoic acid (TOFA); Etomoxir | Inhibit FAO and FAS and destabilize Treg | 52, 53 |
| Glutaminolysis | dimethyl αKG (DMK) | Activate mTOR | 63 |
| FOXP3 | C646, CPI703, CPI644 | P300 (HAT) inhibitor destabilize Foxp3 protein | 86, 87 |
| CBP/EP300 | CPI703, CPI644 | CBP/EP300 inhibitors alter epigenome of Tregs | 87 |
| NF‐kB | Pentoxifylline (PTXF) | Inhibits c‐REL expression and TI‐Treg stability | 8 |
| EOS | TLR9 agonist | TLR9‐mediated inflammation downregulates EOS | 103 |
| FOXOs | SC79 | AKT activating, FOXO inhibiting | 54 |
| EZH2 | CPI‐1205, EPZ011989 | EZH2 inhibitors reprogramme TI‐Tregs, synergize with CBI | 117 |
| BET proteins | JQ1 | Selectively deplete TI‐Tregs, synergize with CBI | 88 |
Activation and differentiation of TI‐Tregs
A defining feature of TI‐Tregs is their highly activated phenotype.8, 9, 10 Treg activation within the TME leads to the induction of a repertoire of suppressive molecules, such as CTLA‐4, GITR, PD‐1 and LAG‐3, which control antitumour immune responses. However, the activation of Tregs can also drive the destabilization of Foxp3 expression and induce the production of pro‐inflammatory cytokines,11, 12, 13 revealing that maintenance of Treg identity is tenuous during activation. Treg activation within the context of the TME also imparts a unique transcriptional programme that distinguishes TI‐Tregs from Tregs present in normal tissues, indicating that Treg differentiation may adapt within cancers.9, 10, 14 Therefore, approaches that target the highly activated state of TI‐Tregs or their unique differentiation state within cancer may prove to be powerful mechanisms for specifically disrupting Tregs within tumours and reprogramming their functions. We will begin our review by discussing the critical surface receptors that control Treg activation or contribute to their differentiated state in tumours.
CD28
In conjunction with T‐cell receptor (TCR) stimulation, the CD28 co‐stimulatory receptor represents the most potent secondary stimulus for Treg functionality. Blocking CD28 signalling to Tregs drastically inhibits their stability and function.15, 16 In the context of cancer, deleting CD28 in Tregs impairs their differentiation and function selectively within tumours, reducing their capacity to suppress anti‐tumour immune responses and promoting tumour control.17 CD28 binding is opposed by cytotoxic T lymphocyte‐associated protein‐4 (CTLA‐4), an inhibitory receptor that competes for binding to the same ligands, B7‐1 and B7‐2, on antigen‐presenting cells. CTLA‐4 expression is induced by CD28 activation as a negative feedback regulatory mechanism in effector T‐cells, and CTLA‐4 is a target for checkpoint blockade immunotherapy (CBI) for cancer.18 CLTA‐4 is also highly expressed on TI‐Tregs, most likely as a result of their highly activated state, and its expression on TI‐Tregs supports their immunosuppressive activity. Interestingly, a major mechanism of action of anti‐CTLA4 CBI may be to selectively deplete, or interfere with the function of, TI‐Tregs.18, 19
CD28 engagement activates several cytosolic signalling pathways, but activation of lymphocyte cell‐specific protein tyrosine kinase (LCK) seems to be particularly important for Tregs.20 LCK is central to the activation and nuclear localization of the transcription factor nuclear factor of kappa light polypeptide gene enhancer in B‐cells (NF‐κB), which, as will be discussed later, is essential for the maintenance of the immunosuppressive activity of Tregs in cancers.8, 20 Interestingly, the phosphatidylinositol 3‐kinase (PI3K)–AKT–mammalian target of rapamycin (mTOR) pathway, which is also activated in response to CD28, has not proven to be important in establishing and maintaining the immunosuppressive Treg state. In conventional CD4+ T‐cells, CD28 potently activates PI3K/AKT/TORC1 signalling, whereas in Tregs, AKT activation is significantly reduced in response to CD28 ligation. AKT activity is impaired in Tregs by the action of two phosphatases: phosphatase and tensin homologue (PTEN), a lipid phosphatase that counteracts PI3K‐mediated phosphorylation of phosphatidylinositol‐4,5‐bisphosphate (PIP2) to phosphatidylinositol‐3,4,5‐trisphosphate (PIP3); and the PH‐domain leucine‐rich‐repeat protein phosphatase (PHLPP), which directly dephosphorylates and inactivates AKT.21, 22 A critical role for suppressing PI3K/AKT/TORC1 signalling in TI‐Tregs was revealed by disrupting PTEN in Tregs, which led to increased expression of the pro‐inflammatory molecules IL‐2, CD40L and IL‐17 in TI‐Tregs and enhanced anti‐tumour immunity.23 Mechanistically, uncontrolled AKT activation may reprogramme Treg function by reducing their expression of CD25, inactivating FOXO transcription factors, or altering their metabolic state (see below).21, 24 However, some PI3K activity may be essential for Treg survival and function, as genetic deletion or pharmacological inhibition of the PI3K subunit p110δ selectively impairs TI‐Treg function and improves immune‐mediated control of murine tumours.25
Interleukin‐2 receptor
Interleukin‐2 (IL‐2) is the essential cytokine for the maintenance and function of Tregs.26 Binding of IL‐2 to the IL‐2 receptor leads to the phosphorylation and activation of signal transducer and activator of transcription 5 (STAT5), a critical transcription factor for programming immunosuppressive Tregs, largely through its direct regulation of Foxp3 expression.27 Disruption of STAT5 binding to the Foxp3 locus leads to Treg reprogramming, where Tregs switch from producing immunosuppressive cytokines to making pro‐inflammatory cytokines.27 The IL‐2 receptor is made up of three polypeptides. CD25, the IL‐2 receptor α chain, is highly upregulated in Tregs and distinguishes Tregs from other immune cells.28 Treatment of human Tregs in vitro with the FDA‐approved monoclonal antibody against the CD25 receptor, daclizumab, reduces CD25 expression on Tregs, decreasing expression of Foxp3 and increasing their secretion of IFN‐γ.29
Neuropilin 1
Neuropilin 1 (Nrp‐1) is a receptor that is highly expressed in thymic‐derived (natural) Tregs. In the context of several human cancers, Nrp‐1 is found to be highly induced in TI‐Tregs.30 In murine cancer models, deletion of Nrp‐1 specifically in Tregs leads to enhanced immunity to many transplantable tumours. Nrp‐1 deficiency in Tregs can selectively prevent their recruitment to cancers by disabling their capacity to sense vascular endothelial growth factor (VEGF), which is produced at high concentrations in many cancers, via Nrp‐1.31 More strikingly, Nrp‐1‐deficient Tregs that enter the TME produce large amounts of IFN‐γ, which acts in a paracrine fashion to bolster stronger CD8+ T‐cell responses within the TME and acts on other TI‐Tregs to promote their reprogramming to IFN‐γ‐producing cells.30 Mechanistically, Nrp‐1 recruits PTEN to the immunological synapse upon TCR engagement, blocking the potentially toxic activation of AKT, which can inhibit FOXO activity and drive glycolytic metabolism, thus preserving Treg stability and function.30, 32
GITR
Glucocorticoid‐induced tumour necrosis factor (TNF) receptor (GITR), a member of the TNF receptor (TNFR) superfamily, is highly upregulated upon T‐cell activation and is highly expressed on Tregs. Antibodies against GITR have long been used to selectively deplete Tregs and inhibit their suppressive capacity in vitro.33 However, recent evidence from murine tumour models suggests that anti‐GITR antibodies can also selectively reprogramme TI‐Tregs.34 Mechanistically, anti‐GITR causes downregulation of Foxp3, Helios and IL‐10, while increasing the levels of IFN‐γ production from TI‐Tregs.35 As will be discussed in the subsequent section, anti‐GITR antibodies may function via regulating Helios expression, as genetic deletion of Helios in Tregs phenocopies anti‐GITR treatment.36
Chemokine receptors
After activation, Tregs differentiate to suppress specific types of inflammation by expressing the same polarizing transcription factors, such as T‐bet, GATA3 or ROR‐γt, and the accompanying chemokine receptors to home to the same niche as the T‐cells they will control.37 The co‐expression of Foxp3 with transcription factors that control effector T‐cell functions may provide better precision for Treg immunosuppression but, also, competition between opposing T‐cell programmes can disrupt the immunosuppressive function of Tregs or the maintenance of Foxp3 expression itself.6, 13, 38 While it is not clear whether there is a common pattern of TI‐Treg differentiation in all cancers, selectively targeting specific subsets of TI‐Tregs based on chemokine receptor expression is an active area of investigation for the treatment of cancer.
Th2‐like Tregs, distinguished by the expression of the chemokine receptor CCR4, are increased in patients with melanoma and colorectal cancer, and exhibit a heightened capacity to suppress effector T‐cells.9, 39 Antibodies against CCR4 can reduce the number of TI‐Tregs in murine tumours,40 and a humanized anti‐CCR4 antibody, KW‐0761, is being tested in early clinical trials in patients.41 Interestingly, treating T‐cells with CCL17 (a CCR4 interacting chemokine) can directly inhibit the production of the Th1 cytokine IFN‐γ, thus implicating chemokines themselves as factors that can directly reprogramme the functionality of T‐cells.40 CCR8, another chemokine receptor associated with Th2 cells, may prove to be a superior target, as it was found highly expressed on TI‐Tregs in multiple solid tumours, but in a more restricted fashion than CCR4.9, 10 However, the type of TI‐Tregs may vary in different cancers and patients, as Th1‐like Tregs, defined by the expression of T‐bet, made up 40% of Foxp3+ cells in a murine model of lung carcinoma, and between 2% and 10% of Tregs from human non‐small cell lung carcinomas.42 CXCR3, the Th1‐defining chemokine receptor, was the most prevalent chemokine receptor on tumour‐infiltrating FOXP3+ Treg in human ovarian cancers.43 Because Th1‐like TI‐Tregs may best inhibit Th1 effector T‐cell responses that are associated with cytotoxic CD8+ T‐cells and IFN‐γ production, selectively eliminating these Tregs may have the greatest effect on potentiating the most effective type of anti‐tumour T‐cell responses.44, 45 A role for Th17‐like Treg reprogramming, defined by the transcription factor retinoic acid‐related orphan receptor‐γt (RORγt) and the chemokine receptor CCR6, in cancer is complicated. In inflammation‐driven cancers, such as colorectal cancer, pro‐inflammatory Th17‐like Tregs supported cancer development, while in other cancers, such as ovarian cancer that may not be driven by inflammation, Th17‐like Tregs have enhanced suppressive activity and support cancer development.46, 47
Metabolism in TI‐Tregs
The TME is characterized by a multitude of factors that impinge on the metabolism of infiltrating immune cells. Common TME characteristics such as hypoxia, low glucose and increased lactic acid may prohibit productive anti‐tumour immune responses by impeding the metabolic programmes required to support the tumour‐killing functions of effector T‐cells, while simultaneously favouring Treg metabolism.48 In this section of our review, we will highlight the capacity for Tregs to thrive in the TME by their use of a diversity of metabolic substrates, namely glucose, lipids and glutamine, to feed their metabolic requirements within the context of low oxygen tension. We will describe how disrupting the metabolic pathways used by TI‐Tregs may reprogramme their functionality selectively in cancers to drive more potent anti‐tumour immune responses.
Metabolism of glucose, lipids and glutamine
During the activation of T‐cells through their TCR, a switch in metabolism occurs from oxidative phosphorylation (OXPHOS), which maximizes ATP yield from glucose, to glycolysis, which supports cellular proliferation and effector functions, for example cytokine production.49 This switch is directed by CD28 co‐stimulation, which activates PI3K/AKT, inducing the expression of the glucose transporter Glut1, and increasing glucose uptake for glycolysis.50 However, Tregs exhibit a much less distinctive switch to glycolysis upon activation and instead utilize a more diverse set of energy‐generating pathways, including fatty acid oxidation (FAO) and glutaminolysis to support their preference for the OXPHOS pathway.51, 52, 53, 54 The importance of OXPHOS in Tregs is clear, as deletion of two important OXPHOS regulators, peroxisome proliferator‐activated receptor γ co‐activator 1a (Pgc1a) or sirtuin (Sirt) 3, impairs Tregs suppressive function both in vitro and in vivo.55
In the context of low glucose and high lactate, which is typical of most TMEs, Foxp3 expression in Tregs can increase OXPHOS, impede glycolysis by repressing the expression of Myc (a major transcriptional activator of glycolytic genes), and increase resistance to high concentrations of lactate.56 In multiple murine tumour models, TI‐Tregs have proven to be less vulnerable to glucose restriction than other effector T‐cells.52, 57, 58 However, in some settings, inhibition of glycolysis in TI‐Tregs reversed Treg suppressive function and promoted anti‐tumour immunity.59, 60, 61 This may be due to the capacity of Tregs to preferentially convert pyruvate into mitochondrial acetyl‐CoA, which would further support OXPHOS62 but, more likely, this reflects a requirement for some glycolytic activity in Tregs despite their increased utilization of OXPHOS compared with effector T‐cells.
Lipid and glutamine metabolism in Tregs versus effector T‐cells may provide a clearer metabolic distinction between these subsets. FAO also generates acetyl‐CoA to feed the citric acid cycle. FAO is favoured by Tregs compared with effector T‐cells in vitro,53 and inhibition of endogenous fatty acid synthesis (FAS) or FAO can attenuate Foxp3 expression and TI‐Treg function without affecting Th1 cell differentiation.52, 53 Cancers also exhibit high levels of glutaminolysis, wherein glutamine is diverted into metabolic intermediates to feed the citric acid cycle or provide a substrate for lipid biosynthesis. Similar to glucose deprivation in the TME, glutamine deprivation prevents Th1 differentiation but drives Treg conversion from naïve CD4+ T‐cells. This may be the result of glutamine conversion to α‐ketoglutarate (α‐KG), which can increase TORC1 activity and oppose Treg programming.63 However, systemic administration of glutamine to mice has also been reported to increase the frequency of Foxp3+ Tregs.64
PI3K/AKT/mTOR
As a central axis of metabolic control, the PI3K/AKT/mTOR signalling pathway provides the most direct route to reprogramming TI‐Tregs metabolically. As discussed in the previous section, during Treg activation, tight control of AKT activity is required for the suppressive phenotype of Tregs, and blocking the activity of the pathway supports the immunosuppressive Treg programme.65, 66 Furthermore, Th1 cells can be reprogrammed to Foxp3+ Tregs by reducing TCR/CD28‐mediated PI3K/AKT/mTOR activity, which causes a metabolic shift from glycolysis to OXPHOS.67 Conversely, pharmacological activation of AKT increases glucose uptake and glycolysis, destabilizing Tregs.54 As described earlier, PTEN deficiency in Tregs promotes Foxp3 instability and this is likely the result of elevated glycolytic metabolism and reduced OXPHOS.21, 23, 68, 69 However, inhibition of specific isoforms of PI3K or disruption of TORC1 can negatively impact the Treg programme, resulting in the loss of suppressive receptors, such as CTLA4, ICOS and PD‐1.70, 71 Nevertheless, interpreting these results is complicated, as regulatory feedback mechanisms, such as TORC1 inhibition of TORC2, can actually result in hyper‐activation of AKT in the absence of TORC1, as TORC2 phosphorylates and activates AKT.6
HIF1‐α
Due to poor vascularization of most solid tumours, TMEs are notoriously deprived of oxygen, or hypoxic. Respiratory hyperoxia has been shown to improve the anti‐tumour T‐cell response against metastatic melanoma in the lung, and this was associated with decreased Treg frequency, Foxp3 expression and suppressive function within tumours.72 Low oxygen tension, combined with TCR activation, can stabilize hypoxia‐inducible factor 1‐α (HIF1‐α), and this may promote Foxp3 expression in vitro.73, 74, 75 However, a number of studies also indicate that expression of HIF1‐α impairs Treg stability due to its transcriptional induction of glycolytic genes and its direct binding to Foxp3, which can drive Foxp3 degradation.76, 77, 78 Supporting the latter hypothesis, the oxygen‐sensing prolyl‐hydroxylase (PHD) proteins, which are suppressors of HIF1‐α, are required to induce Treg programming in metastatic niches.79 Similarly, targeted deletion of the HIF1‐α E3 ubiquitin ligase Von Hippel‐Lindau (VHL) in Tregs leads to elevated HIF1‐α that directly binds to the promoter of the Ifng gene and induces IFN‐γ expression in Tregs, resulting in their conversion into Th1‐like cells.80 This finding was also confirmed in TI‐Tregs, where increased HIF1‐α expression supported the production of IFN‐γ from Tregs, which led to the impairment of TI‐Treg function.30
Transcription in TI‐Treg
Changes in transcription strongly underlie the stability of the immunosuppressive Treg programme. Factors controlling Treg transcription, both transcription factors and the chromatin landscape, act in an independent and overlapping fashion to establish and maintain the Treg programme upon activation.81, 82 TI‐Tregs exhibit a distinctive transcriptional programme compared with Tregs in other sites of the body, opening up the possibility to specifically disrupt the TI‐Treg transcriptome as a mechanism to enhance antitumour immunity.14
Foxp3
Foremost in importance among transcription factors in Tregs is Foxp3, the lineage‐defining transcription factor of Tregs that is essential for their differentiation and function. Deficiency for Foxp3 leads to multi‐organ autoimmunity in mice and humans, and loss of Foxp3 in Tregs diminishes their immunosuppressive capacities, often leading to their acquisition of pro‐inflammatory activities.83, 84, 85 Several mechanisms have been discovered that regulate Foxp3 stability, either at the level of protein stability or at the level of transcription, and their disruption can selectively promote anti‐cancer immunity. Post‐transcriptional acetylation of Foxp3 by the histone acetyltransferase (HAT) EP300 enhances Foxp3 stability and activity. EP300 inhibition selectively reduces the frequency and suppressive function of Tregs within tumours by reducing acetylation of Foxp3 itself, as well as reducing histone acetylation (a stimulating transcriptional mark) at key Treg‐activated genes, leading to decreased expression of Foxp3, LAG‐3, CTLA‐4 and TIM‐3.86, 87 In addition, pharmacological inhibitors that block the interaction of other bromodomains with acetylated histones, such as JQ1, can selectively disrupt the function of Tregs in tumours while leaving antitumour effector T‐cells fully functional.86, 87, 88
Foxp3 transcription is also silenced epigenetically via DNA methylation of its locus at key conserved non‐coding sites. Importantly, evidence from animal models and humans indicates that these loci are stably unmethylated in TI‐Tregs, lending credence to the hypothesis that the TME naturally supports the immunosuppressive Treg programme.89, 90 However, DNA methylation is dynamic; thus, disruption of ten‐eleven translocation (TET) proteins that catalyse the first steps in DNA demethylation, or blocking their recruitment to the Foxp3 locus, can disrupt Foxp3 expression.91, 92, 93 In contrast, TET2 disruption in antitumour effector T‐cells may enhance their capacity to persist as memory cells and better regulate cancer, making targeting of TETs an attractive opportunity to selectively block immune suppression in the TME.94 2‐Hydroxyglutarate (2‐HG), a glutamine metabolite common in the TME, can negatively regulate TET proteins and other dioxygenases, such as lysine demethylases, thus linking glutamine deprivation (discussed in metabolism section) to an epigenetic mechanism of enhancing Treg stability in cancers.95
Cooperating transcription factors
Foxp3 drives the transcriptional programme of Tregs by interacting with a series of transcription factors, both through direct physical interactions within common complexes and by complementing Treg transcriptional programmes. Disruption of several of these transcription factors, discussed here, has been shown to reprogramme Tregs into pro‐inflammatory cells with anti‐cancer‐promoting activities.
NF‐kB
Activated upon TCR/CD28 stimulation, the NF‐kB subunits p65 and c‐Rel are required for the development of Tregs in the thymus. However, only disruption of c‐Rel, either genetically or pharmacologically, reprogrammes TI‐Tregs to enhance cancer immunity.8, 96 c‐Rel‐deficient TI‐Tregs exhibit reduced expression of genes associated with Treg activation – Itgae, Tigit, Klrg1, Il1r2, as well as downregulation of Foxp3, CD25 and Helios8 but, more importantly, these Tregs reprogramme into Th1‐like cells, expressing Eomes, Tbx21, Il2 and Ifng.8, 96
Helios
An Ikaros family transcription factor, Helios is co‐expressed in 70%–80% of mouse and human Foxp3+ Tregs, and its expression is a defining feature of thymic‐derived Tregs.97 While Helios appears dispensable in quiescent cells, upon Treg activation in the context of inflammation or cancer it is required for their stability.98, 99 Genetic disruption of Helios, or its down‐modulation by anti‐GITR antibodies, reprogrammes TI‐Tregs by decreasing Foxp3 and increasing an inflammatory Th programme (IFN‐γ, IL‐10, GATA3 and TBX21) and enhancing tumour immunity.35, 36, 99, 100
Eos
Also a member of the Ikaros family of transcription factors, Eos physically interacts with Foxp3 in a complex that represses genes necessary for the preservation of the Treg immunosuppressive programme.101 TI‐Tregs in murine models and from human patients with lung cancer are characterized by high levels of Eos.90, 102 Downregulation of Eos reprogrammes Tregs to gain immune‐stimulating capacity by increasing expression of CD40L, and this enhances CD8+ T‐cell responses against cancer.103, 104
Forkhead box proteins
Foxo1 and Foxo3 are essential transcription factors for Treg stability, binding directly within the Foxp3 locus to support its expression, while also acting at the Ifng locus to repress it.24, 105 Phosphorylation of Foxo proteins by AKT leads to their inactivation by nuclear exclusion, further establishing the toxicity of AKT activity for Treg identity. Paradoxically, by introducing mutations into Foxo proteins to prevent phosphorylation by AKT, the migration and function of Tregs into tumours was blocked and cancer immunity was enhanced.106 Therefore, it is clear that tight control of PI3K/AKT pathway signalling, both in the strength and timing of the signals, is paramount for optimizing the Treg programme.
Bach2
A member of the basic leucine zipper family of transcription factors, Bach2 polymorphisms are associated with multiple inflammatory diseases in humans.107 This is due to a direct role in Tregs, where Bach2 is required to suppress inflammatory transcriptional programmes. In the absence of Bach2, Tregs reprogramme to Th1‐ and Th2‐like cells and enhance CD8+ antitumour T‐cell responses, significantly impairing cancer progression.108, 109
Nr4a
Three orphan nuclear receptor 4A proteins (Nur77, Nurr1 and Nor1) act in a largely redundant fashion to induce and maintain Foxp3 expression.110 NR4A expression is increased as a direct result of the strength of TCR engagement.111 Therefore, NR4A proteins are found highly expressed in TI‐Tregs, where TCR‐mediated activation is high. Importantly, in preclinical models of cancer, genetic disruption of Nr4a1 and Nr4a2, or pharmacological inhibition of NR4As led to reductions in the levels of Foxp3, Il2ra, Ikzf4 and Ctla4 in TI‐Tregs and enhanced anti‐tumour immunity.112
Ezh2
In addition to activating genes upon TCR engagement, equally important for the maintenance of the Treg immunosuppressive state is the repression of genes that antagonize the Treg programme.113, 114 This is particularly important for preventing the production of pro‐inflammatory cytokines, as discussed with Eos and Bach2. However, beyond co‐opting transcription factors, Tregs also utilize histone modifications to alter chromatin structure and control gene expression. Mechanistic insight into how Foxp3 mediates gene repression came from the discovery that Foxp3 associates with Enhancer of Zeste Homolog 2 (Ezh2) after Treg activation, thereby guiding the Polycomb Repressive Complex 2 (PRC2) to deposit the repressive H3K27me3 chromatin mark to regions of the genome that should remain silent in Tregs.115 Deletion of Ezh2 in Tregs solidified the importance of the EZH2‐Foxp3 collaboration, with mice developing a late‐onset autoimmunity that was restricted to non‐lymphoid tissues.12 EZH2 is induced during Treg activation in a CD28‐dependent manner, likely via c‐Rel, which can directly bind to and activate Ezh2 transcription, thus connecting an extracellular cue to the epigenetic control of Treg transcription.116 Blocking the function of EZH2 in Tregs, either genetically or pharmacologically, leads to specific reprogramming of Treg function within cancers, with TI‐Tregs exhibiting reduced production of immunosuppressive IL‐10 and increased production of TNF‐α, IFN‐γ and IL‐2.117 Furthermore, anti‐CTLA4 checkpoint blockade dramatically increases EZH2 expression within the TME, presumably due to increased CD28 signalling, and may promote TI‐Treg suppressive activity. As a result, the combination of anti‐CTLA4 with EZH2 inhibition can act synergistically to engage potent anti‐cancer immunity in murine cancer models.118
Conclusion: Key challenges and next steps
Here we have highlighted a rapidly evolving paradigm for the immunotherapeutic treatment of cancer by reprogramming TI‐Treg function. We cite several pre‐clinical studies that have already revealed the power of TI‐Treg reprogramming. This approach is effective because it not only reduces Treg immunosuppression in tumours, but actually reverses it, by converting TI‐Tregs to pro‐inflammatory cells that stimulate the immune response within tumours. While the number of examples of TI‐Treg reprogramming per se is small, the idea of selectively targeting TI‐Tregs based on their unique properties within cancers has proven to be widely effective (e.g. anti‐CTLA4 or anti‐CCR4 antibodies). By pooling our knowledge of the state of the field, we simplified the characteristics of TI‐Tregs into three defining features: a high activation state; an altered metabolism favouring OXPHOS driven largely by lipid fuels; and a set of collaborating transcription factors and chromatin regulators that assist the Foxp3‐driven gene expression programme.
However, while the strategies highlighted here are predicted to selectively target TI‐Tregs versus other Treg populations in the body, for such basic discoveries to be applied to patients as therapies it is essential to consider the consequences of modulating these pathways in other types of T‐cells, other immune cells, and other cell types within the tumour and systemically. In several instances, there is already strong evidence to support the possibility that targeting these pathways in Tregs and other cell types would both benefit cancer immunotherapy. For instance, targeting co‐inhibitory receptors, such as CTLA‐4, PD‐1 or TIGIT, upregulated both on CD8+ T‐cells and Tregs in tumours can enhance anti‐tumour T‐cell functions while simultaneously disabling Tregs to potently enhance the cancer immune response.119 Similarly, antibodies against GITR, while capable of reprogramming Tregs, can also enhance the proliferation and cytokine production of intratumoural CD8+ T‐cells.120 Targeting Tregs through reprogramming metabolism is also promising, as the metabolic needs of TI‐Tregs appear to be unique and opposite of other types of T‐cells. For example, selectively blocking FAO or enhancing glycolysis may disrupt Treg stability while promoting anti‐tumour T‐cell function. Perhaps most remarkably, small molecule inhibitors of the chromatin modifier EZH2 may not only reprogramme TI‐Treg function, but also directly increase cytotoxic T‐cell activity and recruitment to the TME (by increasing Th1 chemokine production from tumour cells).117, 118, 121, 122 In other instances, targeting some of the pathways described here may have tempered effects due to blocking the function of beneficial immune cell populations attacking the cancer. For example, blocking CD28 can reprogramme Tregs, but CD28 is required for T‐cell activation, the acquisition of glycolytic metabolism necessary for effector functions, and the efficacy of PD‐1 checkpoint blockade therapy.50, 123 Thus, inhibiting CD28 may negate the benefits of Treg reprogramming. Similarly, blocking IL‐2 signalling may reprogramme Tregs, but IL‐2 signalling also stimulates natural killer cells and T‐cells; thus, its blockade may hinder the anti‐tumour activities of these cells. Efforts to direct these drugs exclusively to Tregs, such as with optimized antibodies that specifically deplete TI‐Tregs or engineered IL‐2 molecules that do not bind Tregs, may be critical to the success of targeting the IL‐2 pathway.124, 125 Further work identifying target pathways that act selectively in TI‐Tregs combined with new strategies for selectively delivering drugs to TI‐Tregs will be needed to expand the repertoire of therapeutic options to reprogramme TI‐Tregs.
Finally, the greatest challenge of cancer treatment is to slow or reverse metastatic disease, the most lethal form of cancer. Treg immunosuppression within primary tumours is thought to contribute to the correlations between Treg frequencies and increased incidence of metastatic disease in gastric,126 breast,127, 128 non–small cell lung,129 bladder130 and renal cell cancers.131 However, few studies have resolved whether Tregs play a more specific role in driving the process of metastasis. TI‐Tregs have been shown to promote the vascularization of ovarian cancers via their capacity to produce VEGF‐A, thus facilitating the establishment of a bloodstream to tumours that may be used by cancer cells to escape to distant sites in the body.132 TI‐Tregs were also shown to induce metastasis by their expression of RANKL, which interacts with the RANK receptor on mammary carcinoma cells and represses the expression of the metastasis inhibitor maspin.133 Furthermore, co‐administration of antibodies against RANKL and CTLA‐4 can exhibit synergistic effects, impairing tumour growth and metastasis in a murine melanoma model.134 Therefore, targeting TI‐Tregs may go beyond reversing immunosuppression in the TME to encompassing the prevention of metastases by blocking select pathways by which Tregs can promote the escape of tumour cells from their primary sites.
In this review, we have focused on revealing several commonalities in the mechanisms that control the immunosuppressive Treg state that can be targeted to reprogramme their function to become immune stimulatory, often with a high degree of intratumoural specificity. This has led to the identification of several specific and interconnected signalling nodes that may prove to be particularly potent targets for cancer immunotherapy: surface receptors (e.g. CD28‐NFkB), cytosolic signalling pathways (e.g. NRP1‐PTEN inhibition of PI3K/AKT/mTOR), metabolic pathways (e.g. FAO‐OXPHOS), transcription factors (e.g. GITR‐Helios) and chromatin regulators (e.g. Foxp3‐EZH2). In conclusion, the selective reprogramming of Tregs within cancers represents a singular methodology to overcome the last barriers that impede the broad success of cancer immunotherapy in patients by both overcoming immunosuppression in the TME and limiting autoimmune toxicity.
Disclosures
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by the University of California, Berkeley, and the St Baldrick's Foundation Hope with Hazel Scholar (to M.D.). M.D. generated the figures. M.D., M.M.A. and Z.L. conceived and wrote the manuscript.
References
- 1. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011; 480:480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med 2017; 23:540–7. [DOI] [PubMed] [Google Scholar]
- 3. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell 2008; 133:775–87. [DOI] [PubMed] [Google Scholar]
- 4. Liu C, Workman CJ, Vignali DA. Targeting regulatory T cells in tumors. FEBS J 2016; 283:2731–48. [DOI] [PubMed] [Google Scholar]
- 5. Joshi NS, Akama‐Garren EH, Lu Y, Lee DY, Chang GP, Li A et al Regulatory T cells in tumor‐associated tertiary lymphoid structures suppress anti‐tumor T cell responses. Immunity 2015; 43:579–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DuPage M, Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune‐mediated disease. Nat Rev Immunol 2016; 16:149–63. [DOI] [PubMed] [Google Scholar]
- 7. Munn DH, Sharma MD, Johnson TS. Treg destabilization and reprogramming: implications for cancer immunotherapy. Cancer Res 2018; 78:5191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grinberg‐Bleyer Y, Oh H, Desrichard A, Bhatt DM, Caron R, Chan TA et al NF‐kappaB c‐Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell 2017; 170:1096–108, e1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV et al Regulatory T cells exhibit distinct features in human breast cancer. Immunity 2016; 45:1122–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C et al Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor‐infiltrating T regulatory cells. Immunity 2016; 45:1135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bailey‐Bucktrout SL, Martinez‐Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H et al Self‐antigen‐driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity 2013; 39:949–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D et al The chromatin‐modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity 2015; 42:227–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Z, Zhang W, Guo J, Gu Q, Zhu X, Zhou X. Activation and functional specialization of regulatory T cells lead to the generation of Foxp3 instability. J Immunol 2017; 198:2612–25. [DOI] [PubMed] [Google Scholar]
- 14. Magnuson AM, Kiner E, Ergun A, Park JS, Asinovski N, Ortiz‐Lopez A et al Identification and validation of a tumor‐infiltrating Treg transcriptional signature conserved across species and tumor types. Proc Natl Acad Sci USA 2018; 115:E10672–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK et al Cutting edge: CD28 controls peripheral homeostasis of CD4 + CD25 + regulatory T cells. J Immunol 2003; 171:3348–52. [DOI] [PubMed] [Google Scholar]
- 16. Zhang R, Huynh A, Whitcher G, Chang J, Maltzman JS, Turka LA. An obligate cell‐intrinsic function for CD28 in Tregs. J Clin Invest 2013; 123:580–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marangoni F, Zhang R, Mani V, Thelen M, Ali Akbar NJ, Warner RD et al Tumor tolerance‐promoting function of regulatory T cells is optimized by CD28, but strictly dependent on Calcineurin. J Immunol 2018; 200:3647–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov 2018; 8:1069–86. [DOI] [PubMed] [Google Scholar]
- 19. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M et al Anti‐CTLA‐4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res 2013; 1:32–42. [DOI] [PubMed] [Google Scholar]
- 20. Vang KB, Yang J, Pagán AJ, Li LX, Wang J, Green JM et al Cutting edge: CD28 and c‐Rel‐dependent pathways initiate regulatory T cell development. J Immunol 2010; 184:4074–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM et al Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol 2015; 16:188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Patterson SJ, Han JM, Garcia R, Assi K, Gao T, O'Neill A et al Cutting edge: PHLPP regulates the development, function, and molecular signaling pathways of regulatory T cells. J Immunol 2011; 186:5533–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sharma MD, Shinde R, McGaha TL, Huang L, Holmgaard RB, Wolchok JD et al The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci Adv 2015; 1:e1500845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV et al Novel Foxo1‐dependent transcriptional programs control T(reg) cell function. Nature 2012; 491:554–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ahmad S, Abu‐Eid R, Shrimali R, Webb M, Verma V, Doroodchi A et al Differential PI3Kdelta signaling in CD4(+) T‐cell subsets enables selective targeting of T regulatory cells to enhance cancer immunotherapy. Cancer Res 2017; 77:1892–904. [DOI] [PubMed] [Google Scholar]
- 26. Abbas AK, Trotta E, Rsimeonov D, Marson A, Bluestone JA. Revisiting IL‐2: biology and therapeutic prospects. Sci Immunol 2018; 3:pii: eaat1482. [DOI] [PubMed] [Google Scholar]
- 27. Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 2014; 158:749–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self‐tolerance maintained by activated T cells expressing IL‐2 receptor alpha‐chains (CD25). Breakdown of a single mechanism of self‐tolerance causes various autoimmune diseases. J Immunol 1995; 155:1151–64. [PubMed] [Google Scholar]
- 29. Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr et al CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med 2012; 4:134ra162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Overacre‐Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G et al Interferon‐gamma drives Treg fragility to promote anti‐tumor immunity. Cell 2017; 169:1130–41. e1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hansen W, Hutzler M, Abel S, Alter C, Stockmann C, Kliche S et al Neuropilin 1 deficiency on CD4 + Foxp3 + regulatory T cells impairs mouse melanoma growth. J Exp Med 2012; 209:2001–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Delgoffe GM, Woo SR, Turnis ME, Gravano DM, Guy C, Overacre AE et al Stability and function of regulatory T cells is maintained by a neuropilin‐1‐semaphorin‐4a axis. Nature 2013; 501:252–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self‐tolerance. Nat Immunol 2002; 3:135–42. [DOI] [PubMed] [Google Scholar]
- 34. Cohen AD, Schaer DA, Liu C, Li Y, Hirschhorn‐Cymmerman D, Kim SC et al Agonist anti‐GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra‐tumor accumulation. PLoS ONE 2010; 5:e10436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schaer DA, Budhu S, Liu C, Bryson C, Malandro N, Cohen A et al GITR pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol Res 2013; 1:320–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakagawa H, Sido JM, Reyes EE, Kiers V, Cantor H, Kim HJ. Instability of Helios‐deficient Tregs is associated with conversion to a T‐effector phenotype and enhanced antitumor immunity. Proc Natl Acad Sci USA 2016; 113:6248–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3 + regulatory T cells. Nat Rev Immunol 2011; 11:119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu F, Sharma S, Edwards J, Feigenbaum L, Zhu J. Dynamic expression of transcription factors T‐bet and GATA‐3 by regulatory T cells maintains immunotolerance. Nat Immunol 2015; 16:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Halim L, Romano M, McGregor R, Correa I, Pavlidis P, Grageda N et al An atlas of human tegulatory T helper‐like cells reveals features of Th2‐like Tregs that support a tumorigenic environment. Cell Rep 2017; 20:757–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berlato C, Khan MN, Schioppa T, Thompson R, Maniati E, Montfort A et al A CCR4 antagonist reverses the tumor‐promoting microenvironment of renal cancer. J Clin Invest 2017; 127:801–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T et al Phase Ia study of FoxP3 + CD4 Treg depletion by infusion of a humanized anti‐CCR4 antibody, KW‐0761, in cancer patients. Clin Cancer Res 2015; 21:4327–36. [DOI] [PubMed] [Google Scholar]
- 42. Kachler K, Holzinger C, Trufa DI, Sirbu H, Finotto S. The role of Foxp3 and Tbet co‐expressing Treg cells in lung carcinoma. Oncoimmunology 2018; 7:e1456612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Redjimi N, Raffin C, Raimbaud I, Pignon P, Matsuzaki J, Odunsi K et al CXCR3 + T regulatory cells selectively accumulate in human ovarian carcinomas to limit type I immunity. Cancer Res 2012; 72:4351–60. [DOI] [PubMed] [Google Scholar]
- 44. Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M et al Stability and function of regulatory T cells expressing the transcription factor T‐bet. Nature 2017; 546:421–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Walser TC, Rifat S, Ma X, Kundu N, Ward C, Goloubeva O et al Antagonism of CXCR3 inhibits lung metastasis in a murine model of metastatic breast cancer. Cancer Res 2006; 66:7701–7. [DOI] [PubMed] [Google Scholar]
- 46. Keerthivasan S, Aghajani K, Dose M, Molinero L, Khan MW, Venkateswaran V et al Beta‐Catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Sci Transl Med 2014; 6:225ra228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Downs‐Canner S, Berkey S, Delgoffe GM, Edwards RP, Curiel T, Odunsi K et al Suppressive IL‐17A(+)Foxp3(+) and ex‐Th17 IL‐17A(neg)Foxp3(+) Treg cells are a source of tumour‐associated Treg cells. Nat Commun 2017; 8:14 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD et al Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015; 162:1229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science 2013; 342:1 242 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ et al Glucose uptake is limiting in T cell activation and requires CD28‐mediated Akt‐dependent and independent pathways. J Immunol 2008; 180:4476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Maj T, Wang W, Kryczek I, Zou W. Oxidative phosphorylation controls regulatory T cell suppressor activity in the tumor microenvironment. J Immunol 2016; 196. [Google Scholar]
- 52. Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D et al Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc Natl Acad Sci USA 2018; 115:E6546–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF et al Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4 + T cell subsets. J Immunol 2011; 186:3299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Basu S, Hubbard B, Shevach EM. Foxp3‐mediated inhibition of Akt inhibits Glut1 (glucose transporter 1) expression in human T regulatory cells. J Leukoc Biol 2015; 97:279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H et al Essential role of mitochondrial energy metabolism in Foxp3(+) T‐regulatory cell function and allograft survival. FASEB J 2015; 29:2315–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Angelin A, Gil‐de‐Gómez L, Dahiya S, Jiao J, Guo L, Levine MH et al Foxp3 reprograms T cell metabolism to function in low‐glucose, high‐lactate environments. Cell Metab 2017; 25:1282–93. e1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S et al Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD‐L1‐blockade resistance in tumor. Nat Immunol 2017; 18:1332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R et al Phosphoenolpyruvate Is a metabolic checkpoint of anti‐tumor T cell responses. Cell 2015; 162:1217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M et al The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity 2016; 44:712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C et al Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol 2015; 16:1174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li L, Liu X, Sanders KL, Edwards JL, Ye J, Si F et al TLR8‐mediated metabolic control of human Treg function: a mechanistic target for cancer immunotherapy. Cell Metab 2018; 29:103–123. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O et al Metabolic programming and PDHK1 control CD4 + T cell subsets and inflammation. J Clin Invest 2015; 125:194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L et al Glutamine‐dependent alpha‐ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal 2015; 8:ra97. [DOI] [PubMed] [Google Scholar]
- 64. Hsiung YC, Liu JJ, Hou YC, Yeh CL, Yeh SL. Effects of dietary glutamine on the homeostasis of CD4 + T cells in mice with dextran sulfate sodium‐induced acute colitis. PLoS ONE 2014; 9:e84410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Crellin NK, Garcia RV, Levings MK. Altered activation of AKT is required for the suppressive function of human CD4 + CD25 + T regulatory cells. Blood 2007; 109:2014–22. [DOI] [PubMed] [Google Scholar]
- 66. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M et al T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA 2008; 105:7797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kanamori M, Nakatsukasa H, Ito M, Chikuma S, Yoshimura A. Reprogramming of Th1 cells into regulatory T cells through rewiring of the metabolic status. Int Immunol 2018; 30:357–73. [DOI] [PubMed] [Google Scholar]
- 68. Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol 2015; 16:178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sharma MD, Rodriguez PC, Koehn BH, Baban B, Cui Y, Guo G et al Activation of p53 in immature myeloid precursor cells controls differentiation into Ly6c(+)CD103(+) monocytic antigen‐presenting cells in tumors. Immunity 2018; 48:91–106. e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)‐cell function. Nature 2013; 499:485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sun IH, Oh MH, Zhao L, Patel CH, Arwood ML, Xu W et al mTOR Complex 1 signaling regulates the generation and function of central and effector Foxp3(+) regulatory T cells. J Immunol 2018; 201:481–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R et al Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med 2015; 7:277ra230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Neildez‐Nguyen TMA, Bigot J, Da Rocha S, Corre G, Boisgerault F, Paldi A et al Hypoxic culture conditions enhance the generation of regulatory T cells. Immunology 2015; 144:431–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P et al Hypoxia‐inducible factor‐1 alpha‐dependent induction of FoxP3 drives regulatory T‐cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci USA 2012; 109:E2784–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ben‐Shoshan J, Maysel‐Auslender S, Mor A, Keren G, George J. Hypoxia controls CD4 + CD25 + regulatory T‐cell homeostasis via hypoxia‐inducible factor‐1alpha. Eur J Immunol 2008; 38:2412–8. [DOI] [PubMed] [Google Scholar]
- 76. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y et al Control of T(H)17/T(reg) balance by hypoxia‐inducible factor 1. Cell 2011; 146:772–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR et al HIF1alpha‐dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 2011; 208:1367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Feldhoff LM, Rueda CM, Moreno‐Fernandez ME, Sauer J, Jackson CM, Chougnet CA et al IL‐1beta induced HIF‐1alpha inhibits the differentiation of human FOXP3(+) T cells. Sci Rep 2017; 7:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Clever D, Roychoudhuri R, Constantinides MG, Askenase MH, Sukumar M, Klebanoff CA et al Oxygen sensing by T cells establishes an immunologically tolerant metastatic niche. Cell 2016; 166:1117–31. e1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee JH, Elly C, Park Y, Liu YC. E3 Ubiquitin ligase VHL regulates hypoxia‐inducible factor‐1alpha to maintain regulatory T cell stability and suppressive capacity. Immunity 2015; 42:1062–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Delacher M, Imbusch CD, Weichenhan D, Breiling A, Hotz‐Wagenblatt A, Träger U et al Genome‐wide DNA‐methylation landscape defines specialization of regulatory T cells in tissues. Nat Immunol 2017; 18:1160–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y et al T cell receptor stimulation‐induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 2012; 37:785–99. [DOI] [PubMed] [Google Scholar]
- 83. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L et al The immune dysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 2001; 27:20–1. [DOI] [PubMed] [Google Scholar]
- 84. Williams LM, Rudensky AY. Maintenance of the Foxp3‐dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol 2007; 8:277–84. [DOI] [PubMed] [Google Scholar]
- 85. Zhou X, Bailey‐Bucktrout SL, Jeker LT, Penaranda C, Martínez‐Llordella M, Ashby M et al Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo . Nat Immunol 2009; 10:1000–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Liu Y, Wang L, Predina J, Han R, Beier UH, Wang LC et al Inhibition of p300 impairs Foxp3(+) T regulatory cell function and promotes antitumor immunity. Nat Med 2013; 19:1173–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ghosh S, Taylor A, Chin M, Huang HR, Conery AR, Mertz JA et al Regulatory T cell modulation by CBP/EP300 bromodomain inhibition. J Biol Chem 2016; 291:13 014–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Adeegbe DO, Liu S, Hattersley MM, Bowden M, Zhou CW, Li S et al BET bromodomain inhibition cooperates with PD‐1 blockade to facilitate antitumor response in Kras‐mutant non‐small cell lung cancer. Cancer Immunol Res 2018; 6:1234–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ma H, Gao W, Sun X, Wang W. STAT5 and TET2 cooperate to regulate FOXP3‐TSDR demethylation in CD4(+) T cells of patients with colorectal cancer. J Immunol Res 2018; 2018:6 985 031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Waight JD, Takai S, Marelli B, Qin G, Hance KW, Zhang D et al Cutting edge: epigenetic regulation of Foxp3 defines a stable population of CD4 + regulatory T cells in tumors from mice and humans. J Immunol 2015; 194:878–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wang L, Liu Y, Han R, Beier UH, Thomas RM, Wells AD et al Mbd2 promotes foxp3 demethylation and T‐regulatory‐cell function. Mol Cell Biol 2013; 33:4106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yang R, Qu C, Zhou Y, Konkel JE, Shi S, Liu Y et al Hydrogen sulfide promotes Tet1‐ and Tet2‐mediated Foxp3 demethylation to drive regulatory T cell differentiation and maintain immune homeostasis. Immunity 2015; 43:251–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yue X, Trifari S, Äijö T, Tsagaratou A, Pastor WA, Zepeda‐Martínez JA et al Control of Foxp3 stability through modulation of TET activity. J Exp Med 2016; 213:377–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ et al Disruption of TET2 promotes the therapeutic efficacy of CD19‐targeted T cells. Nature 2018; 558:307–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK et al Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature 2017; 548:228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Oh H, Grinberg‐Bleyer Y, Liao W, Maloney D, Wang P, Wu Z et al An NF‐kappaB transcription‐factor‐dependent lineage‐specific transcriptional program promotes regulatory T cell identity and function. Immunity 2017; 47:450–65. e455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y et al Expression of Helios, an Ikaros transcription factor family member, differentiates thymic‐derived from peripherally induced Foxp3 + T regulatory cells. J Immunol 2010; 184:3433–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sebastian M, Lopez‐Ocasio M, Metidji A, Rieder SA, Shevach EM, Thornton AM. Helios controls a limited subset of regulatory T cell functions. J Immunol 2016; 196:144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kim HJ, Barnitz RA, Kreslavsky T, Brown FD, Moffett H, Lemieux ME et al Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science 2015; 350:334–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yates K, Bi K, Haining WN, Cantor H, Kim HJ. Comparative transcriptome analysis reveals distinct genetic modules associated with Helios expression in intratumoral regulatory T cells. Proc Natl Acad Sci USA 2018; 115:2162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Pan F, Yu H, Dang EV, Barbi J, Pan X, Grosso JF et al Eos mediates Foxp3‐dependent gene silencing in CD4 + regulatory T cells. Science 2009; 325:1142–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Akimova T, Zhang T, Negorev D, Singhal S, Stadanlick J, Rao A et al Human lung tumor FOXP3 + Tregs upregulate four “Treg‐locking” transcription factors. JCI Insight 2017; 2:pii: 94075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sharma MD, Huang L, Choi JH, Lee EJ, Wilson JM, Lemos H et al An inherently bifunctional subset of Foxp3 + T helper cells is controlled by the transcription factor eos. Immunity 2013; 38:998–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sharma MD, Hou DY, Baban B, Koni PA, He Y, Chandler PR et al Reprogrammed foxp3(+) regulatory T cells provide essential help to support cross‐presentation and CD8(+) T cell priming in naive mice. Immunity 2010; 33:942–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kerdiles YM, Stone EL, Beisner DR, McGargill MA, Ch'en IL, Stockmann C et al Foxo transcription factors control regulatory T cell development and function. Immunity 2010; 33:890–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Luo CT, Liao W, Dadi S, Toure A, Li MO. Graded Foxo1 activity in Treg cells differentiates tumour immunity from spontaneous autoimmunity. Nature 2016; 529:532–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Roychoudhuri R, Hirahara K, Mousavi K, Clever D, Klebanoff CA, Bonelli M et al BACH2 represses effector programs to stabilize T(reg)‐mediated immune homeostasis. Nature 2013; 498:506–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kim EH, Gasper DJ, Lee SH, Plisch EH, Svaren J, Suresh M. Bach2 regulates homeostasis of Foxp3 + regulatory T cells and protects against fatal lung disease in mice. J Immunol 2014; 192:985–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Roychoudhuri R, Eil RL, Clever D, Klebanoff CA, Sukumar M, Grant FM et al The transcription factor BACH2 promotes tumor immunosuppression. J Clin Invest 2016; 126:599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sekiya T, Kashiwagi I, Yoshida R, Fukaya T, Morita R, Kimura A et al Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis. Nat Immunol 2013; 14:230–7. [DOI] [PubMed] [Google Scholar]
- 111. Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J et al T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med 2011; 208:1279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hibino S, Chikuma S, Kondo T, Ito M, Nakatsukasa H, Omata‐Mise S et al Inhibition of Nr4a receptors enhances antitumor immunity by breaking Treg‐mediated immune tolerance. Cancer Res 2018; 78:3027–40. [DOI] [PubMed] [Google Scholar]
- 113. Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD et al Foxp3 occupancy and regulation of key target genes during T‐cell stimulation. Nature 2007; 445:931–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Morikawa H, Ohkura N, Vandenbon A, Itoh M, Nagao‐Sato S, Kawaji H et al Differential roles of epigenetic changes and Foxp3 expression in regulatory T cell‐specific transcriptional regulation. Proc Natl Acad Sci USA 2014; 111:5289–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Arvey A, van der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, Rudensky AY. Inflammation‐induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat Immunol 2014; 15:580–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Neo WH, Lim JF, Grumont R, Gerondakis S, Su IH. c‐Rel regulates Ezh2 expression in activated lymphocytes and malignant lymphoid cells. J Biol Chem 2014; 289:31 693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A et al Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep 2018; 23:3262–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Goswami S, Apostolou I, Zhang J, Skepner J, Anandhan S, Zhang X et al Modulation of EZH2 expression in T cells improves efficacy of anti‐CTLA‐4 therapy. J Clin Invest 2018; 128:3813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X et al Landscape of infiltrating T cells in liver cancer revealed by single‐cell sequencing. Cell 2017; 169:1342–56. e1316. [DOI] [PubMed] [Google Scholar]
- 120. Sabharwal SS, Rosen DB, Grein J, Tedesco D, Joyce‐Shaikh B, Ueda R et al GITR agonism enhances cellular metabolism to support CD8(+) T‐cell proliferation and effector cytokine production in a mouse tumor model. Cancer Immunol Res 2018; 6:1199–211. [DOI] [PubMed] [Google Scholar]
- 121. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W et al Epigenetic silencing of TH1‐type chemokines shapes tumour immunity and immunotherapy. Nature 2015; 527:249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Zingg D, Arenas‐Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J et al The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep 2017; 20:854–67. [DOI] [PubMed] [Google Scholar]
- 123. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL et al Rescue of exhausted CD8 T cells by PD‐1‐targeted therapies is CD28‐dependent. Science 2017; 355:1423–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Arenas‐Ramirez N, Zou C, Popp S, Zingg D, Brannetti B, Wirth E et al Improved cancer immunotherapy by a CD25‐mimobody conferring selectivity to human interleukin‐2. Sci Transl Med 2016; 8:367ra166. [DOI] [PubMed] [Google Scholar]
- 125. Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH et al Fc‐optimized anti‐CD25 depletes tumor‐infiltrating regulatory T cells and synergizes with PD‐1 blockade to eradicate established tumors. Immunity 2017; 46:577–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zhou S, Shen Z, Wang Y, Ma H, Xu S, Qin J et al CCR7 expression and intratumoral FOXP3 + regulatory T cells are correlated with overall survival and lymph node metastasis in gastric cancer. PLoS ONE 2013; 8:e74430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Ye J, Ma C, Wang F, Hsueh EC, Toth K, Huang Y et al Specific recruitment of gammadelta regulatory T cells in human breast cancer. Cancer Res 2013; 73:6137–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lal A, Chan L, Devries S, Chin K, Scott GK, Benz CC et al FOXP3‐positive regulatory T lymphocytes and epithelial FOXP3 expression in synchronous normal, ductal carcinoma in situ, and invasive cancer of the breast. Breast Cancer Res Treat 2013; 139:381–90. [DOI] [PubMed] [Google Scholar]
- 129. Lv M, Xu Y, Tang R, Ren J, Shen S, Chen Y et al miR141‐CXCL1‐CXCR2 signaling‐induced Treg recruitment regulates metastases and survival of non‐small cell lung cancer. Mol Cancer Ther 2014; 13:3152–62. [DOI] [PubMed] [Google Scholar]
- 130. Baras AS, Drake C, Liu JJ, Gandhi N, Kates M, Hoque MO et al The ratio of CD8 to Treg tumor‐infiltrating lymphocytes is associated with response to cisplatin‐based neoadjuvant chemotherapy in patients with muscle invasive urothelial carcinoma of the bladder. Oncoimmunology 2016; 5:e1134412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Giraldo NA, Becht E, Vano Y, Petitprez F, Lacroix L, Validire P et al Tumor‐infiltrating and peripheral blood t‐cell immunophenotypes predict early relapse in localized clear cell renal cell carcinoma. Clin Cancer Res 2017; 23:4416–28. [DOI] [PubMed] [Google Scholar]
- 132. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP et al Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011; 475:226–30. [DOI] [PubMed] [Google Scholar]
- 133. Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM et al Tumour‐infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL‐RANK signalling. Nature 2011; 470:548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ahern E, Harjunpää H, Barkauskas D, Allen S, Takeda K, Yagita H et al Co‐administration of RANKL and CTLA4 antibodies enhances lymphocyte‐mediated antitumor immunity in mice. Clin Cancer Res 2017; 23:5789–801. [DOI] [PubMed] [Google Scholar]