

Abstract
RGD‐cryptophycin and isoDGR‐cryptophycin conjugates were synthetized by combining peptidomimetic integrin ligands and cryptophycin, a highly potent tubulin‐binding antimitotic agent across lysosomally cleavable Val‐Ala or uncleavable linkers. The conjugates were able to effectively inhibit binding of biotinylated vitronectin to integrin αvβ3, showing a binding affinity in the same range as that of the free ligands. The antiproliferative activity of the novel conjugates was evaluated on human melanoma cells M21 and M21‐L with different expression levels of integrin αvβ3, showing nanomolar potency of all four compounds against both cell lines. Conjugates containing uncleavable linker show reduced activity compared to the corresponding cleavable conjugates, indicating efficient intracellular drug release in the case of cryptophycin‐based SMDCs. However, no significant correlation between the in vitro biological activity of the conjugates and the integrin αvβ3 expression level was observed, which is presumably due to a non‐integrin‐mediated uptake. This reveals the complexity of effective and selective αvβ3 integrin‐mediated drug delivery.
Keywords: antitumor agents, cancer, drug delivery, integrins, peptidomimetics
Successful delivery: Cryptophycin‐RGD/isoDGR conjugates with lysosomally cleavable or uncleavable linkers display high binding affinity towards integrin αvβ3 and show potent cytotoxicity in cell lines with different integrin expression levels.
1. Introduction
Chemotherapeutic agents used in traditional tumor therapy do not preferentially accumulate at the tumor site and are often associated with poor pharmacokinetics leading to inferior efficacy and severe side effects due to systemic toxicity. An emerging approach to minimize chemotherapy side effects and maximize tumor specificity, is the covalent conjugation of cytotoxic drugs to antibodies that provide site‐specific targeted delivery of the toxin.1, 2, 3 The resulting antibody‐drug conjugates (ADCs) have a definitive impact on anticancer drug development with currently four ADCs on the market (Adcetris®, Kadcyla®, Mylotarg®, and Besponsa®) and approximately 70 candidates in clinical trials.4 Although ADCs show great clinical benefit and success on the market, further development is required to address limitations like high manufacturing costs, inefficient tissue penetration and immunogenicity, in particular for the treatment of solid tumors.5 As an alternative, small‐molecule‐drug conjugates (SMDCs) have the potential to overcome drawbacks of ADCs. In this case, the cytotoxic agent is connected to a small synthetic ligand that selectively binds to an appropriate target overexpressed on the tumor cell surface.6, 7
Integrins, a family of 24 transmembrane heterodimers composed of 18α and 8β integrin subunits, are considerable targets in anticancer therapy due to their involvement in many steps of cancer progression.8 The cell adhesion protein, integrin αvβ3 is highly expressed on endothelial and tumor cells of various solid tumors such as glioma, gastric cancer, non‐small cell lung cancer, prostate, and pancreatic cancer.9, 10, 11 In addition, integrin αvβ3 plays a vital role in cancer progression, angiogenesis, and metastasis. Therefore, it has been widely considered as potential target antigen.8, 12, 13 Integrin αvβ3 binds endogenous ligands, like vitronectin, by recognizing the tripeptide sequence Arg−Gly−Asp (RGD) as the minimal binding motif.14 As a result, various RGD‐containing synthetic peptides and peptidomimetics have been developed and some of them demonstrate high binding affinity to different integrins.15, 16, 17 Several ligands with strong binding affinity and integrin subtype selectivity have been successfully conjugated to different cytotoxic agents, and have been investigated for tumor targeting applications.18, 19
Besides the RGD sequence, the isoaspartyl‐containing sequence isoAsp−Gly−Arg (isoDGR) was identified as an integrin ligand. Hence, there has been increasing interest in employing isoDGR peptides as RGD analogues for integrin targeting.20, 21 There is a growing body of evidence suggesting that this class of ligands are true integrin αvβ3 antagonists, unlike the RGD peptides, which induce integrin conformational changes upon binding, as well as receptor activation.22, 23 Based on these findings, a panel of cyclic RGD and isoDGR peptidomimetic integrin ligands containing a diketopiperazine (DKP) scaffold have been developed. Importantly, the cyclo[DKP‐RGD] 1 and cyclo[DKP‐isoDGR] 3 (Figure 1) are selective integrin αvβ3 ligands with low nanomolar affinity.24, 25 Functionalized analogues 2 and 4 have been conjugated to different anticancer drugs, such as paclitaxel,26, 27, 28, 29, 30, 31 camptothecin32 and α‐amanitin33 across different linkage systems including ester, ether, carbonate or carbamate bond, involving non‐cleavable, disulfide, elastase cleavable NPV, and lysosomally cleavable Val−Ala, Phe−Lys or GFLG linkers.
Figure 1.
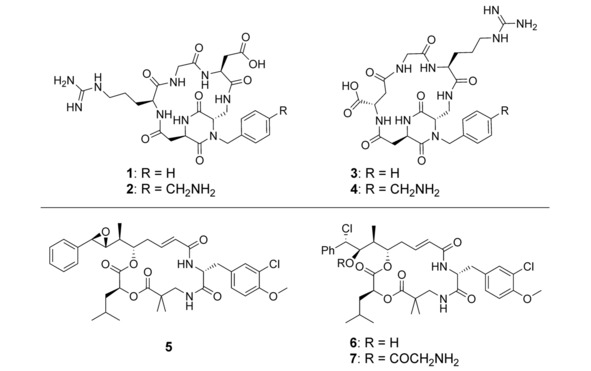
Chemical structure of ligands: cyclo[DKP‐RGD] (1), NH2CH2−cyclo[DKP‐RGD] (2), cyclo[DKP‐isoDGR] (3), NH2CH2−cyclo[DKP‐isoDGR] (4) and cytotoxic drugs: cryptophycin‐52 (5), cryptophycin‐55 (6) and cryptophycin‐55 glycinate (7).
Although the ligand plays a key role for efficient targeting, the high potency of the cytotoxic drug is also a crucial criterion for optimal SMDC design.2 Cryptophycins are compelling antimitotic payloads for tumor targeting applications, owing to their high activity as tubulin polymerization inhibitors, relative hydrophilicity and ability to overcome multidrug resistance.34 Initially the synthetic analogue cryptophycin‐52 (5, Figure 1) was developed and clinically investigated as an antitumor agent on its own. However, neurotoxic side effects and the lack of activity led to the failure of this drug candidate in clinical phase II.35, 36 Further research has been focused on the investigation of structural moieties that are essential for the biological activity. Consequently, a large variety of synthetic analogues have been designed and tested for structure‐activity relationship (SAR) studies.37, 38, 39 More recently, cryptophycin derivatives bearing a functional handle suitable for attachment to tumor targeting peptides or antibodies have come to the forefront.34, 40, 41, 42, 43 In this context, the unit A‐modified chlorohydrin derivative (6, Figure 1) and the glycinate of the chlorohydrin hydroxyl group (7, Figure 1) are highly potent in vitro and in vivo.44 The latter has been successfully conjugated to trastuzumab targeting HER2,45 acetazolamide targeting CAIX,46 and other RGD‐based integrin ligands.47
2. Results and Discussion
Here we focus on the conjugation of the cryptophycin‐55 glycinate derivative to the functionalized cyclo[DKP‐RGD] and cyclo[DKP‐isoDGR] peptidomimetics for integrin targeted delivery. The conjugates were designed to connect the integrin ligands to the cytotoxin across the lysosomally cleavable Val−Ala linker (10 and 11, Scheme 1). The cathepsin B sensitive peptide linker combined with the p‐aminobenzylcarbamate (PABC) self‐immolative spacer is widely used to ensure efficient release of the free drug upon endocytosis.48 Conjugates with uncleavable linker were also prepared (13 and 14, Scheme 1). Presumably the latter are not substrates of enzymatic cleavage; therefore, the drug should be released only after hydrolysis of ligand‐linker components resulting in decreased in vitro antitumor activity of the conjugate compared to the unconjugated drug and the conjugates with cleavable linker. To improve the water solubility of the conjugate and ensure flexibility of the construct, a PEG4 spacer was inserted between the ligand and the triazole ring, used as connection point with the linker.
Scheme 1.
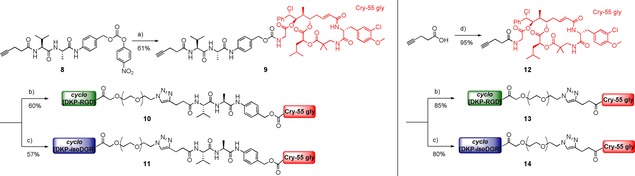
Synthesis of cyclo[DKP‐RGD]‐PEG4‐Val−Ala‐PABC‐Cry‐55gly (10), cyclo[DKP‐isoDGR]‐PEG4‐Val−Ala‐PABC‐Cry‐55gly (11), cyclo[DKP‐RGD]‐PEG4‐uncl‐Cry‐55gly (13), and cyclo[DKP‐isoDGR]‐PEG4‐uncl‐Cry‐55gly (14). Reagents and conditions: a) 7, DIPEA, DMF, RT, 4 h; b) cyclo[DKP‐RGD]‐PEG4‐N3, CuSO4 ⋅ 5H2O, sodium ascorbate, 1 : 1 DMF/H2O, 35 °C, 24 h; c) cyclo[DKP‐isoDGR]‐PEG4‐N3, CuSO4 ⋅ 5H2O, sodium ascorbate, 1 : 1 DMF/H2O, 35 °C, 24 h; d) 7, PyBOP, HOBt, DIPEA, DMF, RT, 4 h.
We evaluated the four conjugates for their ability to inhibit binding of biotinylated vitronectin to isolated integrin αvβ3 in comparison with the free ligands. We tested the in vitro cytotoxic activity of the conjugates against the human melanoma cells M21 and M21‐L with different integrin expression levels.
2.1. Synthesis
The RGD and isoDGR peptidomimetic‐cryptophycin conjugates 10, 11, 13, and 14 were synthetized as shown in Scheme 1.
The linker Fmoc−Val−Ala−PAB27 was deprotected by piperidine and the crude free amine was subsequently reacted with 4‐pentynoic acid. Then, the hydroxyl group was activated using bis(4‐nitrophenyl) carbonate (Scheme S1 in the Supporting Information). The resulting alkyne‐functionalized activated linker 8 was conjugated to cryptophycin‐55 glycinate46 by forming a carbamate to give compound 9. The PEG4‐containing azido‐functionalized cyclo[DKP‐RGD] and cyclo[DKP‐isoDGR] ligands were prepared as previously reported.29, 33 Final conjugation was achieved by copper(I)‐catalyzed azide−alkyne cycloaddition (CuAAC “click” reaction) between compound 9 and cyclo[DKP‐RGD]‐PEG4‐azide or cyclo[DKP‐isoDGR]‐PEG4‐azide, respectively.
For the synthesis of uncleavable conjugates 4‐pentynoic acid was coupled to cryptophycin‐55 glycinate (7) via amide bond to afford compound 12. This was subjected to CuAAC reaction with the respective ligands to give the final conjugates 13 and 14. Compounds 8–14 were characterized by NMR, HPLC‐MS and HRMS (see Supporting Information).
2.2. Competitive Integrin Binding Assays
Conjugates were evaluated in vitro for their binding affinity to human αvβ3 integrin in comparison with the free RGD and isoDGR ligands. Competitive binding assays were performed by incubation of immobilized αvβ3 integrin with increasing concentrations of test compounds (10−5–10−12 M) in the presence of the ECM protein, vitronectin (1 μg mL−1). The concentrations causing 50 % inhibition of biotinylated vitronectin (IC50) are listed in Table 1. As it is shown, free ligands as well as conjugates efficiently inhibit the biotinylated vitronectin binding to αvβ3 integrin with IC50 values in the low nanomolar range. Comparing the binding affinity of the conjugates and ligands we can conclude that conjugates retained high binding affinity. The linker type has only a minimal effect, conjugates with uncleavable linker displayed a slightly reduced affinity, presumably because of the shorter linker between the ligand and the drug. Similar results were reported for analogous conjugates of the cyclo[DKP‐RGD] and cyclo[DKP‐isoDGR] ligands and paclitaxel or α‐amanitin.29, 33 In addition, high selectivity for αvβ3 integrin was reported for cyclo[DKP‐RGD]‐paclitaxel conjugates involving various lysosomally cleavable Val−Ala or GFLG linkers.31 This indicates that conjugation of a drug to the respective ligands does not compromise specificity and we expect that the selectivity of the RGD‐ and isoDGR‐cryptophycin conjugates towards αvβ3 integrin subtype is maintained.
Table 1.
Inhibition of biotinylated vitronectin binding to human integrin αvβ3. IC50 values were determined as the compound concentration necessary for 50 % inhibition of biotinylated vitronectin binding as calculated using the software GraphPad Prism. All values are the average±standard deviation of duplicate measurements.
|
Cmpd |
Structure |
IC50 [nM] αvβ3 |
|---|---|---|
|
1 |
cyclo[DKP‐RGD] |
4.5±1.1 |
|
3 |
cyclo[DKP‐isoDGR] |
9.2±1.1 |
|
10 |
cyclo[DKP‐RGD]‐VA‐Cry‐55gly |
7.2±1.5 |
|
11 |
cyclo[DKP‐isoDGR]‐VA‐Cry‐55gly |
5.5±2.8 |
|
13 |
cyclo[DKP‐RGD]‐Cry‐55gly |
24.1±1.0 |
|
14 |
cyclo[DKP‐isoDGR]‐Cry‐55gly |
10.2±0.4 |
2.3. Cell Viability Assays
The cytotoxicity of the unconjugated drug (7) and cryptophycin‐conjugates (10, 11, 13, 14) was assessed for two cell lines with different expression levels of the integrin. The M21 human melanoma (αvβ3 positive) and M21‐L (αvβ3 negative) model was used to characterize in vitro the tumor‐targeting ability of the novel conjugates.49 The presence of the integrin subunit αv and the integrin heterodimer αvβ3 on the cell membrane was confirmed by flow cytometry analysis (Figure S1 in the Supporting Information). The cells were treated with increasing concentrations of the free drug and conjugates 10, 11, 13, 14, respectively, for 2 hours, then the medium was replaced with fresh medium and the incubation was continued for additional 70 hours. The short treatment time aimed to minimalize altered antiproliferative activity caused by incidental extracellular linker cleavage and undesired drug release, as well as intended to resemble more closely the in vivo experimental conditions. The cell viability was measured in an MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) assay (Figure 2, Table 2).
Figure 2.
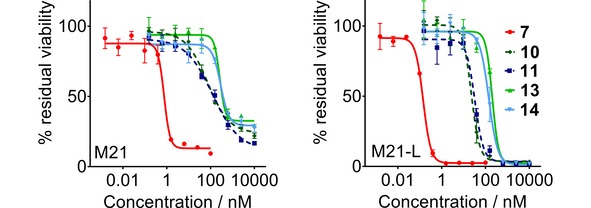
Cell viability determined in an MTT assay with M21 (left), M21‐L (right) cells after treatment with Cry‐55gly (7), cyclo[DKP‐RGD]‐cryptophycin and cyclo[DKP‐isoDGR]‐cryptophycin conjugates. Curves were obtained by non‐linear regression, error bars represent the standard deviation of triplicate measurements, the measurements were repeated twice.
Table 2.
Cytotoxic potencies of free cryptophycin‐55 glycinate and cryptophycin‐conjugates against M21 and M21‐L cell lines upon 2 h treatment and 70 h additional incubation.
|
Structure |
IC50 [nM] M21 (αvβ3+) |
IC50 [nM] M21‐L (αv−, αvβ3−) |
|---|---|---|
|
Cry‐55gly (7) |
0.75±0.11 |
0.14±0.08 |
|
cyclo[DKP‐RGD]‐VA‐Cry‐55gly (10) |
72.7±11.2 |
22.3±2.0 |
|
cyclo[DKP‐isoDGR]‐VA‐Cry‐55gly (11) |
122.6±18.6 |
32.6±3.0 |
|
cyclo[DKP‐RGD]‐Cry‐55gly (13) |
261.3±45.8 |
203.0±27.7 |
|
cyclo[DKP‐isoDGR]‐Cry‐55gly (14) |
287.5±35.9 |
140.8±13.3 |
The cryptophycin conjugates display high in vitro potency with IC50 values between 22.3–287.5 nm, showing slightly reduced activity compared to cryptophycin‐55 glycinate. Moreover, conjugates bearing cleavable linker (10, 11) are more cytotoxic than the corresponding uncleavable conjugates (13, 14). This underlines that higher potency of the conjugates may be associated with the intracellular cathepsin‐mediated drug release, while lower cytotoxicity correlates with less specific linker degradation. Although the intracellular enzymatic cleavage of peptide‐linked ADCs and SMDCs is a well‐known process,50 this study is the first head‐to‐head comparison for cryptophycin‐based SMDCs.
All four conjugates were found to exhibit similar cytotoxic activity on the antigen‐positive and antigen‐negative cell line, indicating that there is no substantial correlation between the expression level of integrin and the observed biological activity. In this regard, it has to be noted that the αvβ3‐negative M21‐L cell line seems to be more sensitive to cryptophycin and cryptophycin conjugates. It was previously reported that the cytotoxicity of cyclo[DKP‐isoDGR]‐α‐amanitin conjugates was not integrin‐specific when tested on human glioblastoma U‐87 (αvβ3+), breast adenocarcinoma MDA‐MB‐468 (αvβ3−), and it was hypothesized that the internalization process was mediated by integrins different from αvβ3 (e. g. αvβ5).33 This observation prompted us to select the isogenic M21 and M21‐L cell lines for the in vitro evaluation of cryptophycin conjugates. The M21‐L cell line was considered a valid negative control, as these cells completely lack the integrin αv subunit. The deficient expression of the αv at the mRNA level leads to the absence of not only of integrin αvβ3, but also the other αv integrin heterodimers (e. g. αvβ5, αvβ6).51 Nevertheless, it is known that the RGD‐motif is not exclusively recognized by the αv‐integrin subfamily but also by other integrins, namely the αIIbβ3, α5β1, and α8β1.52 For this reason, targeting one distinct integrin subtype using small RGD ligand‐based SMDCs is considered to be challenging. Flow cytometry analysis shows similar expression level of integrin α5β1 in the M21 and M21‐L cells, that is comparable with the αvβ3 integrin expression level in the antigen positive cell line.53, 54, 55 Although cyclic RGD and isoDGR peptidomimetic ligands have been shown to bind to α5β1 100‐fold less effective than to αvβ3 (1: α5β1 IC50=532 nm,56 3: α5β1 IC50=1066 nm, determined according to ref. 56) the presence of this receptor might play a role in the recognition and internalization of RGD‐ and isoDGR‐cryptophycin conjugates, resulting in similar activity against both cell lines.
On the other hand, it has been shown that the cyclo‐RGDfK‐carboxyfluorescein peptide targeting αvβ3 integrin was taken up by antigen‐positive and antigen‐negative cells to a similar extent, suggesting that the endocytosis of cyclo‐RGD peptides is integrin‐independent, and proceeds by a fluid‐phase pathway.57 Furthermore, it has been reported that increased hydrophobicity of conjugates caused by the hydrophobic character of the cytotoxic drug may facilitate unwanted nonspecific interactions with the cell surface and can result in passive conjugate uptake into the antigen‐negative cell.58 Indeed, cryptophycins are highly hydrophobic and the cellular uptake of 3H‐cryptophycin‐52 by THP‐1 and H‐125 cells was reported to be fast and irreversible.59 The nonspecific intracellular uptake of the RGD‐ and isoDGR‐cryptophycin conjugates, promoted by the hydrophobic drug or the overall hydrophobicity of the conjugates, may be an additional factor implicated in the observed in vitro potencies. Further increasing the linker hydrophilicity may reduce these undesired interactions and can result in enhanced selectivity.
3. Conclusions
In summary, we have investigated the synthesis and biological activity of novel cyclo[DKP‐RGD]‐cryptophycin and cyclo[DKP‐isoDGR]‐cryptophycin conjugates aiming at integrin‐mediated drug delivery. The cytotoxic payload is connected to the respective peptidomimetic ligand via the protease cleavable Val−Ala linker in combination with PABC self‐immolative moiety or a non‐cleavable spacer. All four conjugates retain high binding affinity towards isolated αvβ3 integrin, similarly to the free ligands. In vitro, the cryptophycin‐conjugates were found to be highly potent against both human melanoma cells M21 and M21‐L with IC50 values in the nanomolar range. Conjugates containing uncleavable linker demonstrated moderately decreased activity in comparison with analogous cleavable conjugates. Although RGD‐ and isoDGR‐ligands show high specificity for αvβ3 integrin, no correlation could be found between the in vitro antitumor activity of the conjugates and the αvβ3 integrin expression level, revealing the complexity of effective and selective αvβ3 integrin‐mediated drug delivery. We assume that our SMDCs may internalize by an αv‐integrin independent pathway: endocytosis could either be mediated by integrin α5β1, or by nonspecific, fluid‐phase uptake, in which the high overall hydrophobicity of the conjugates could also play a role. Nonetheless, the high activity of cryptophycin conjugates encourages us to further investigate and improve the selectivity in tumor‐targeted drug delivery.
Conflict of interest
A.M. is an employee of Exiris, P.G. is a shareholder of Exiris. C.S. is a shareholder of Exiris.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska‐Curie grant agreement No 642004 (ETN MAGICBULLET). We acknowledge the financial support of the German Research Foundation (DFG) and the Open Access Publication Fund of Bielefeld University for the article processing charge. The authors want to acknowledge Georg Falck for flow cytometry analysis, Marco Wißbrock, Anke Nieß, and Carmela Michalek for technical support. Human melanoma cells (M21 and M21‐L) were kindly provided by Dr. David Cheresh and The Scripps Research Institute (La Jolla, CA, USA).
A. Borbély, E. Figueras, A. Martins, L. Bodero, A. Raposo Moreira Dias, P. López Rivas, A. Pina, D. Arosio, P. Gallinari, M. Frese, C. Steinkühler, C. Gennari, U. Piarulli, N. Sewald, ChemistryOpen 2019, 8, 737.
References
- 1. Chari R. V. J., Miller M. L., Widdison W. C., Angew. Chem. Int. Ed. 2014, 53, 3796–3827; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3872–3904. [Google Scholar]
- 2. Beck A., Goetsch L., Dumontet C., Corvaïa N., Nat. Rev. Drug Discovery 2017, 16, 315–337. [DOI] [PubMed] [Google Scholar]
- 3. Lambert J. M., Morris C. Q., Adv. Ther. 2017, 34, 1015–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lu Z.-R., Qiao P., Mol. Pharm. 2018, 15, 3603–3616. [DOI] [PubMed] [Google Scholar]
- 5. Krall N., Scheuermann J., Neri D., Angew. Chem. Int. Ed. 2013, 52, 1384–1402; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1424–1443. [Google Scholar]
- 6. Kue C. S., Kamkaew A., Burgess K., Kiew L. V., Chung L. Y., Lee H. B., Med. Res. Rev. 2016, 36, 494–575. [DOI] [PubMed] [Google Scholar]
- 7. Srinivasarao M., Low P. S., Chem. Rev. 2017, 117, 12133–12164. [DOI] [PubMed] [Google Scholar]
- 8. Hamidi H., Ivaska J., Nat. Rev. Cancer 2018, 18, 533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nieberler M., Reuning U., Reichart F., Notni J., Wester H.-J., Schwaiger M., Weinmüller M., Räder A., Steiger K., Kessler H., Cancers 2017, 9, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Desgrosellier J. S., Cheresh D. A., Nat. Rev. Cancer 2010, 10, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Danhier F., Le Breton A., Préat V., Mol. Pharm. 2012, 9, 2961–2973. [DOI] [PubMed] [Google Scholar]
- 12. Avraamides C. J., Garmy-Susini B., Varner J. A., Nat. Rev. Cancer 2008, 8, 604–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jin H., Varner J., Br. J. Cancer 2004, 90, 561–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ruoslahti E., Pierschbacher M., Science 1987, 238, 491–497. [DOI] [PubMed] [Google Scholar]
- 15. Kapp T. G., Rechenmacher F., Neubauer S., Maltsev O. V., Cavalcanti-Adam E. A., Zarka R., Reuning U., Notni J., Wester H.-J., Mas-Moruno C., Spatz J., Geiger B., Kessler H., Sci. Rep. 2017, 7, 39805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Auzzas L., Zanardi F., Battistini L., Burreddu P., Carta P., Rassu G., Curti C., Casiraghi G., Curr. Med. Chem. 2010, 17, 1255–1299. [DOI] [PubMed] [Google Scholar]
- 17. Gottschalk K., Kessler H., Angew. Chem. Int. Ed. 2002, 41, 3767–3774; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 3919–3927. [Google Scholar]
- 18. Katsamakas S., Chatzisideri T., Thysiadis S., Sarli V., Future Med. Chem. 2017, 9, 579–604. [DOI] [PubMed] [Google Scholar]
- 19. Chatzisideri T., Leonidis G., Sarli V., Future Med. Chem. 2018, 10, 2201–2226. [DOI] [PubMed] [Google Scholar]
- 20. Corti A., Curnis F., J. Cell Sci. 2011, 124, 515–522. [DOI] [PubMed] [Google Scholar]
- 21. Spitaleri A., Mari S., Curnis F., Traversari C., Longhi R., Bordignon C., Corti A., Rizzardi G.-P., Musco G., J. Biol. Chem. 2008, 283, 19757–19768. [DOI] [PubMed] [Google Scholar]
- 22. Ghitti M., Spitaleri A., Valentinis B., Mari S., Asperti C., Traversari C., Rizzardi G. P., Musco G., Angew. Chem. Int. Ed. 2012, 51, 7702–7705; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7822–7825. [Google Scholar]
- 23. Nardelli F., Paissoni C., Quilici G., Gori A., Traversari C., Valentinis B., Sacchi A., Corti A., Curnis F., Ghitti M., Ghitti M., J. Med. Chem. 2018, 61, 7474–7485. [DOI] [PubMed] [Google Scholar]
- 24. da Ressurreição A. S. M., Vidu A., Civera M., Belvisi L., Potenza D., Manzoni L., Ongeri S., Gennari C., Piarulli U., Chem. Eur. J. 2009, 15, 12184–12188. [DOI] [PubMed] [Google Scholar]
- 25. Mingozzi M., Dal Corso A., Marchini M., Guzzetti I., Civera M., Piarulli U., Arosio D., Belvisi L., Potenza D., Pignataro L., Gennari C., Chem. Eur. J. 2013, 19, 3563–3567. [DOI] [PubMed] [Google Scholar]
- 26. Colombo R., Mingozzi M., Belvisi L., Arosio D., Piarulli U., Carenini N., Perego P., Zaffaroni N., De Cesare M., Castiglioni V., Scanziani E., Gennari C., J. Med. Chem. 2012, 55, 10460–10474. [DOI] [PubMed] [Google Scholar]
- 27. Dal Corso A., Caruso M., Belvisi L., Arosio D., Piarulli U., Albanese C., Gasparri F., Marsiglio A., Sola F., Troiani S., Valsasina B., Pignataro L., Donati D., Gennari C., Chem. Eur. J. 2015, 21, 6921–6929. [DOI] [PubMed] [Google Scholar]
- 28. Zanella S., Angerani S., Pina A., López Rivas P., Giannini C., Panzeri S., Arosio D., Caruso M., Gasparri F., Fraietta I., Albanese C., Marsiglio A., Pignataro L., Belvisi L., Piarulli U., Gennari C., Chem. Eur. J. 2017, 23, 7910–7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Raposo Moreira Dias A., Pina A., Dal Corso A., Arosio D., Belvisi L., Pignataro L., Caruso M., Gennari C., Chem. Eur. J. 2017, 23, 14410–14415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Raposo Moreira Dias A., Pina A., Dean A., Lerchen H.-G., Caruso M., Gasparri F., Fraietta I., Troiani S., Arosio D., Belvisi L., Pignataro L., Dal Corso A., Gennari C., Chem. Eur. J. 2019, 25, 1696–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. López Rivas P., Ranđelović I., Raposo Moreira Dias A., Pina A., Arosio D., Tóvári J., Mező G., Dal Corso A., Pignataro L., Gennari C., Eur. J. Org. Chem. 2018, 2018, 2902–2909. [Google Scholar]
- 32. Pina A., Dal Corso A., Caruso M., Belvisi L., Arosio D., Zanella S., Gasparri F., Albanese C., Cucchi U., Fraietta I., Marsiglio A., Pignataro L., Donati D., Gennari C., ChemistrySelect 2017, 2, 4759–4766. [Google Scholar]
- 33. Bodero L., López Rivas P., Korsak B., Hechler T., Pahl A., Müller C., Arosio D., Pignataro L., Gennari C., Piarulli U., Beilstein J. Org. Chem. 2018, 14, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weiss C., Figueras E., Borbely A. N., Sewald N., J. Pept. Sci. 2017, 23, 514–531. [DOI] [PubMed] [Google Scholar]
- 35. D'Agostino G., del Campo J., Mellado B., Izquierdo M. A., Minarik T., Cirri L., Marini L., Perez-Gracia J. L., Scambia G., Int. J. Gynecol. cancer 2006, 16, 71–76. [DOI] [PubMed] [Google Scholar]
- 36. Edelman M. J., Gandara D. R., Hausner P., Israel V., Thornton D., DeSanto J., Doyle L. A., Lung cancer 2003, 39, 197–199. [DOI] [PubMed] [Google Scholar]
- 37. Sammet B., Bogner T., Nahrwold M., Weiss C., Sewald N., J. Org. Chem. 2010, 75, 6953–6960. [DOI] [PubMed] [Google Scholar]
- 38. Weiss C., Sammet B., Sewald N., Nat. Prod. Rep. 2013, 30, 924–40. [DOI] [PubMed] [Google Scholar]
- 39. Eißler S., Stoncius A., Nahrwold M., Sewald N., Synthesis 2006, 2006, 3747–3789. [Google Scholar]
- 40. Nahrwold M., Weiß C., Bogner T., Mertink F., Conradi J., Sammet B., Palmisano R., Royo Gracia S., Preuße T., Sewald N., J. Med. Chem. 2013, 56, 1853–1864. [DOI] [PubMed] [Google Scholar]
- 41. Verma V. A., Pillow T. H., DePalatis L., Li G., Phillips G. L., Polson A. G., Raab H. E., Spencer S., Zheng B., Bioorg. Med. Chem. Lett. 2015, 25, 864–868. [DOI] [PubMed] [Google Scholar]
- 42. Chen H., Lin Z., Arnst K. E., Miller D. D., Li W., Molecules 2017, 22, 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Figueras E., Borbély A., Ismail M., Frese M., Sewald N., Beilstein J. Org. Chem. 2018, 14, 1281–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liang J., Moore R. E., Moher E. D., Munroe J. E., Al-awar R. S., Hay D. A., Varie D. L., Zhang T. Y., Aikins J. A., Martinelli M. J., Shih C., Ray J. E., Gibson L. L., Vasudevan V., Polin L., White K., Kushner J., Simpson C., Pugh S., Corbett T. H., Invest. New Drugs 2005, 23, 213–224. [DOI] [PubMed] [Google Scholar]
- 45.C. Steinkühler, P. Gallinari, B. Osswald, N. Sewald, M. Ritzefeld, M. Frese, E. Figueras, L. Pethö, 2016, WO2016/146638 A1.
- 46. Cazzamalli S., Figueras E., Pethő L., Borbély A., Steinkühler C., Neri D., Sewald N., ACS Omega 2018, 3, 14726–14731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Borbély A., Figueras E., Martins A., Esposito S., Auciello G., Monteagudo E., Di Marco A., Summa V., Cordella P., Perego R., Kemker I., Frese M., Gallinari P., Steinkühler C., Sewald N., Pharmaceuticals 2019, 11, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dal Corso A., Pignataro L., Belvisi L., Gennari C., Curr. Top. Med. Chem. 2016, 16, 314–329. [DOI] [PubMed] [Google Scholar]
- 49. Cheresh D. A., Spiro R. C., J. Biol. Chem. 1987, 262, 17703–17711. [PubMed] [Google Scholar]
- 50. Jain N., Smith S. W., Ghone S., Tomczuk B., Pharm. Res. 2015, 32, 3526–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goodman S. L., Grote H. J., Wilm C., Biol. Open 2012, 1, 329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hatley R. J. D., Macdonald S. J. F., Slack R. J., Le J., Ludbrook S. B., Lukey P. T., Angew. Chem. Int. Ed. 2018, 57, 3298–3321; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3354–3379. [Google Scholar]
- 53. Felding-Habermann B., Mueller B. M., Romerdahl C. A., Cheresh D. A., J. Clin. Invest. 1992, 89, 2018–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Felding-Habermann B., Fransvea E., O'Toole T. E., Manzuk L., Faha B., Hensler M., Clin. Exp. Metastasis 2002, 19, 427–436. [DOI] [PubMed] [Google Scholar]
- 55. Lange J. R., Goldmann W. H., Alonso J. L., Biochem. Biophys. Res. Commun. 2016, 478, 1280–1285. [DOI] [PubMed] [Google Scholar]
- 56. Guzzetti I., Civera M., Vasile F., Arosio D., Tringali C., Piarulli U., Gennari C., Pignataro L., Belvisi L., Potenza D., ChemistryOpen 2017, 6, 128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Castel S., Pagan R., Mitjans F., Piulats J., Goodman S., Jonczyk A., Huber F., Vilaró S., Reina M., Lab. Invest. 2001, 81, 1615–1626. [DOI] [PubMed] [Google Scholar]
- 58. Srinivasarao M., Galliford C. V., Low P. S., Nat. Rev. Drug Discovery 2015, 14, 203–219. [DOI] [PubMed] [Google Scholar]
- 59. Chen B. D. C., Nakeff A. N., Valeriote F. V., Int. J. Cancer 1998, 77, 869–873. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary