

Abstract
Here we show that treatment of the HPV16‐positive tonsillar cancer cell line HN26 with DNA alkylating cancer drug melphalan‐induced p53 and activated apoptosis. Melphalan reduced the levels of RNA polymerase II and cellular transcription factor Sp1 that were associated with HPV16 DNA. The resulting inhibition of transcription caused a rapid loss of the HPV16 early mRNAs encoding E6 and E7 as a result of their inherent instability. As a consequence of HPV16 E6 and E7 down‐regulation, the DNA damage inflicted on the cells by melphalan caused induction of p53 and activation of apoptosis in the HN26 cells. The BARD1‐negative phenotype of the HN26 cells may have contributed to the failure to repair DNA damage caused by melphalan, as well as to the efficient apoptosis induction. Finally, nude mice carrying the HPV16 positive tonsillar cancer cells responded better to melphalan than to cisplatin, the chemotherapeutic drug of choice for tonsillar cancer. We concluded that the short half‐life of the HPV16 E6 and E7 mRNAs renders HPV16‐driven tonsillar cancer cells particularly sensitive to DNA damaging agents such as melphalan since melphalan both inhibits transcription and causes DNA damage.
Keywords: papillomavirus, tonsillar cancer, melphalan, apoptosis, p53
Short abstract
What's new?
In most tonsillar cancers involving human papillomavirus 16 (HPV16) infection, the p53 tumor suppressor is rendered nonfunctional by HPV16 E6 protein. As a result, tumor cells escape apoptosis and HPV replication proceeds unhindered. Here, the authors explored the ability of DNA‐damaging drugs to reverse these effects by liberating p53 in HPV‐positive tonsillar cancer cells. The data show that efficient reactivation of p53 and apoptosis requires degradation of HPV16 E6 and E7 mRNAs, as well as induction of the DNA damage response (DDR). The authors identify two drugs, melphalan and actinomycin D, that exert these dual effects in tonsillar cancer cells.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is a heterogeneous group of cancers that may be human papillomavirus (HPV) negative or positive.1, 2, 3 It is now appreciated that HPV negative and positive HNSCCs are two different entities and that the clinical outcome differs between the two.4, 5 More than 70% of all tonsillar cancers contain HPV DNA.6 Patients with HPV‐positive oropharyngeal SCC (OPSCC) have a better overall survival and respond better to treatment than patients with HPV‐negative OPSCC.7 It has become clear that the treatment strategies should differ between the groups and that new regimes should be developed specifically for HPV‐positive HNSCC.8 Despite the declining overall rate of HNSCC, the incidence of OPSCC is steadily increasing.9 This increase is attributed to increasing numbers of HPV‐associated OPSCC.10 The incidence of HPV‐associated OPSCC is now surpassing the incidence of cervical cancer in a number of developed countries.9, 11 The overwhelmingly predominant HPV type found in OPSCC is HPV16.6, 12
HPV‐positive cancers respond better to treatment than the HPV‐negative cancers as documented by a higher response rate to treatment and a higher survival rate.4, 5 One factor contributing to the better response to DNA damaging drugs of HPV‐positive cancer cells may be that p53 is mutated in 86% of HPV‐ negative cancers, but only in <3% in HPV‐positive tonsillar cancers.13 The p53 protein is incapacitated by the HPV16 E6 protein in HPV‐positive cancers. HPV16 E6 binds p53 and mediates efficient and continuous degradation of p53 in these cells.14 It is therefore of interest to investigate how p53 could be liberated from HPV16 E6 to induce apoptosis in the cancer cells.
The HPV16 life cycle is intimately linked to the differentiation pathway of the infected cell.15, 16, 17 Expression of the HPV16 genes is tightly controlled at the level of transcription18, 19, 20 and RNA processing.21, 22, 23 A plethora of alternatively spliced mRNAs24 are produced by HPV16, all encoding cis‐acting RNA elements that interact with cellular RNA‐binding proteins that control their relative expression levels.25, 26 The HPV16 DNA replicates with the help of the HPV16 early proteins E1 and E2,18, 19, 27 while the HPV16 E6 and E7 proteins interact with p53 and pRB to inhibit the functions of these proteins, Inactivation of p53 and pRB creates an intracellular environment that is optimal for HPV16 replication.14, 28 Once the HPV16 DNA has replicated, the HPV16 structural proteins L1 and L2 are produced and the HPV16 DNA is encapsidated to form virus particles.
We have previously established a HPV16‐positive OPSCC cell line as a tumor xenograft in nude mice,29 and more recently a cell line named LU‐HNSCC‐26. Only a small number of HPV16‐positive HNSCC cell lines are available.30 Our results show that the short half‐life of the HPV16 E6 and E7 mRNAs renders HPV16‐positive tonsillar cancer cells particularly vulnerable to DNA damaging drugs that both activate the cellular DNA damage response (DDR) and inhibit transcription.
Materials and Methods
Cell lines
The HN26 cell line (full name LU‐HNSCC‐26) was isolated from a 48‐year‐old nonsmoking man diagnosed with poorly differentiated, p16‐positive squamous cell tonsillar carcinoma stage T2N0M0 with HPV16 and has been previously described (Table 1).31 The C33A2 cell line is derived from the HPV‐negative and p53‐negative cervical cancer cell line C33A,32 whereas 3,310 cells were generated by transfection and immortalization of normal human primary keratinocytes with full‐length HPV16 genome plasmid pHPV16AN.32, 33
Table 1.
STR analysis
| LU‐HN‐26 | ||
|---|---|---|
| AMEL | X | Y |
| CSF1PO | 11 | 13 |
| D13S317 | 11 | 11 |
| D16S539 | 13 | 14 |
| D18S51 | 13 | 13 |
| D19S433 | 13 | 14 |
| D21S11 | 28 | 28 |
| D2S1338 | 16 | 16 |
| D3S1358 | 15 | 15 |
| D5S818 | 10 | 12 |
| D7S820 | 13 | 13 |
| D8S1179 | 14 | 15 |
| FGA | 20 | 22 |
| TH01 | 9 | 10 |
| TPOX | 8 | 11 |
| vWA | 19 | 21 |
Application of inhibitors and cancer drugs including melphalan to cultured cells
Cell culture medium was replaced with medium containing indicated concentrations of drugs for indicated time periods. All drugs were dissolved in DMSO, and DMSO in the absence of inhibitor was used as a control in all experiments. Substances in the initial experiments were from the “approved oncology drugs set IV” library obtained from the National Cancer Institute (NCI), USA (https://wiki.nci.nih.gov/display/NCIDTPdata/Compound+Sets). The after substances were purchased: Melphalan hydrochloride (Y0001158, Sigma), Etiposide (S1225, Selleckchem), Cisplatin (S1166, Selleckchem), actinomycin D (A9415, Sigma), alpha‐amanitin (A2263, Sigma) and PARP1 inhibitor A‐966492 (S2197, Selleckchem).
DNA fragmentation assay and MTT assay
Total DNA was prepared by lysis of cells in 10 mM Tris–HCl, pH 8.0, 100 mM NaCl, 25 mM EDTA, 0.5% SDS, 250 μg/mL proteinase K followed by phenol/chisam extraction and ethanol precipitation. Five micrograms of each DNA sample were analyzed on 1% agarose gels. MTT assay was performed according to the recommendations of the supplier (Sigma‐Aldrich).
RNA extraction and RT‐PCR
Total RNA was extracted using TRI Reagent and Direct‐zol RNA MiniPrep kit (ZYMO Research) according to the manufacturer's protocol. One micrograms of total RNA were reverse transcribed in a 20 μL reaction at 37 °C by using M‐MLV Reverse Transcriptase (Invitrogen) and random primers (Invitrogen) according to the protocol of the manufacturer. One microliter of cDNA was subjected to PCR amplification. E6, E6*IE7 and E6*IIE7 mRNAs were RT‐PCR‐amplified with primers 97S and 880A, E4 mRNAs spliced from SD880 to SA3358 were amplified with primers 773 s and E4as and E2 mRNAs with primers 773 s and E2as (Supporting Information Fig. 5A and B). RT‐PCR primers for BARD1‐, Bcl2‐, caspase‐3‐ and gapdh‐mRNAs are listed in Supporting Information Table T1 for all primer sequences.
Real‐time quantitative PCR
The number of viral genomes per cell was quantified by carrying out two separate real‐time PCR tests, including appropriate standard curves, to amplify the HPV16 E7 gene and the human beta‐globin gene as described previously.29
Protein extraction and Western blotting
Western blotting was performed as described previously.34 For antibodies see Supporting Information Table T2. Secondary antibodies conjugated with horseradish peroxidase were used, and proteins were detected using the Clarity Western ECL Substrate (Bio Rad) or the Super Signal West Femto chemiluminescence substrate (Pierce).
DNA‐protein gel‐shift assay
Total cell extracts were prepared from subconfluent, untreated, DMSO‐treated or melphalan‐treated C33A2 cells or HN26 cells as previously described.35 Briefly, cells were lysed in lysis buffer (700 mM NaCl, 10 mM KCl, 2 mM MgCl2, 1 mM EGTA, 1 mM DTT, 0.1% triton X‐100, 1 mM sodium‐vanadate, 10 mM beta‐glycerol phosphate) at 4 °C for 30 min followed by centrifugation at 13,000g for 30 min. Supernatants were stored at −80 °C. A DNA probe representing the HPV16 p97 promoter was generated by PCR with primers 7860S and 160A followed by gel purification (Supporting Information Table T1). Seventy‐five nanograms of DNA probe was incubated with indicated concentrations of cell extract in binding buffer (10 mM Tris pH 7.8, 100 mM NaCl, 0.2 mM DTT, 0.1 mM EDTA, 5%glycerol) for 30 min at room temperature. DNA and DNA‐protein complexes were separated on 1% agarose gels. The DNA probe in the gel shift experiments was not radiolabeled but was detected by gel‐red staining.
Chromatin immunoprecipitation
Chromatin immunoprecipitations (Chlp) were performed using the SimpleChIPs Enzymatic Chromatin IP Kit (Cell Signaling) according to the manufacturer's instructions but with some adjustments (see Supporting Information for details).
Melphalan and cisplatin treatment of nude mice xenografted with HN26 cells
We used in‐house bred, athymic 5‐ to 8‐week‐old BALB/c nude (nu/nu) mice. The mice were given food and water ad libitum. Maximum tolerated dose (MTD) of melphalan and cisplatin was assessed in nontumor‐bearing nude mice. Melphalan (Aspen Pharma Trading, Dublin, Ireland), (15‐, 10‐ or 5‐mg/kg) or cisplatin (6‐, 4‐ or 2‐mg/kg) were injected intraperitoneally on day 0 (N = 5). Controls were injected with physiological NaCl. Body weight was measured for 16 days and related to the weight at day 0. Tumors were transplanted subcutaneously into the flank of the animals. Nude mice with growing xenografts of HN26 were treated with a single Intraperitoneal dose of melphalan (10 mg/kg body weight), cisplatin (4 mg/kg body weight) or physiological NaCl on day 0. Tumor size was measured with calipers three times a week for 27 days, and the relative tumor size (RTS) calculated in relation to the size at day 0. Body weight was measured three times a week for 18 days. The data points were fitted to a logarithmic equation using the GraphPad Prism (5.04) software package (GraphPad Software, La Jolla, CA). The experiment was repeated three times with similar results.
Results
Melphalan‐induced apoptosis in tonsillar cancer cell line HN26, but not in cervical cancer cell line C33A2
We wished to investigate how the HPV16‐positive tonsillar cancer cell line LU‐HNSCC‐26 (herein called HN26) responded to a series of cancer drugs, and to compare the effect of these drugs on HN26 cells with the effect of cisplatin on these cells. The HN26 tonsillar cancer cells have been shown previously to contain episomal HPV16 DNA and to produce HPV16 early mRNAs.31 The HN26 cells were incubated with 100uM each of the indicated cancer drugs from the “approved oncology drugs set IV” library obtained from the National Cancer Institute (NCI), USA (https://wiki.nci.nih.gov/display/NCIDTPdata/Compound+Sets) for 24 h followed by MTT assay to determine the number of viable cells. Cisplatin had a relatively modest effect on the viability of the HN26 tonsillar cancer cells compared to other DNA alkylating agents (melphalan and actinomycin D) (Fig. 1 a). Other cancer drugs such as DNA synthesis inhibitors (Nelarabine and flurouracil), topoisomerase inhibitors (teniposide and topotecan hydrochloride), tyrosine kinase inhibitor (lapatinib), androgen antagonist (megestrol acetate) and microtubule inhibitors (vinblastine sulfate and cabazitaxel) had a less pronounced effect on HN26 cells (Fig. 1 a). We decided to study further the effect of melphalan on the HN26 tonsillar cancer cells.
Figure 1.
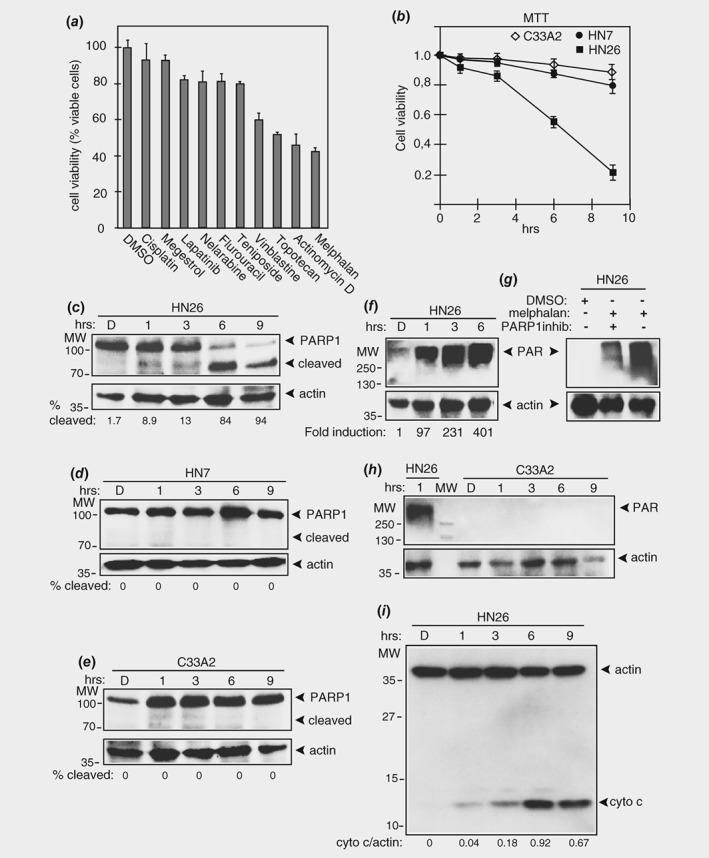
Melphalan reduces viability and induces apoptosis of HPV16 positive tonsillar cancer cells. (a) MTT assay on HN26 cells incubated for 24 h with 100 μM of the indicated cancer drugs or carrier substance DMSO. Mean values of triplicates are shown. (b) Viability of C33A2 cervical cancer cells, HN7 head and neck cancer cells or HN26 tonsillar cancer cells treated with DMSO or 100 μM melphalan for the indicated time periods was determined with an MTT assay as described in Materials and Methods and plotted against time in melphalan. (c–e) Western blots of full length and cleaved poly [ADP‐ribose] polymerase (PARP1) in extracts from HN26 cells, HN7 cells or1 C33A2 cells treated with 100 μM melphalan for the indicated time periods. Poly ADP‐ribosylation (parylation) was monitored by Western blotting with monospecific antibody to poly (ADP‐ribose) (PAR) in cell extracts from HN26 cells (f) or C33A2 cells (h) treated with 100 μM melphalan for the indicated time periods. (g) Western blotting with monospecific antibody to poly (ADP‐ribose) (PAR) in HN26 cells treated with 100 μM melphalan in the absence or presence of PARP1 inhibitor A‐966492. (i) Apoptosis‐mediated release of mitochondrial cytochrome c into the cytoplasmic space as a marker for apoptosis in HN26 cells treated with 100 μM melphalan for the indicated time points. Method is described in Supporting Information methods. Antibody is listed in Supporting Information Table T2. D, DMSO.
We compared the effect of melphalan on the HPV16‐positive and p53‐wild type HN26 tonsillar cancer cell line with the effect on the HPV‐negative head and neck cancer cell line HN7, and with the HPV‐negative and p53‐mutant cervical cancer cell line C33A2. We observed a striking difference in the effect of melphalan on these cell lines; while the HN7 cells and the C33A2 cells remained attached to the bottom of the plastic wells (Supporting Information Fig. S1A and S2), seemingly unaffected by melphalan, the HN26 cells were visibly affected and detached from the plastic wells (Supporting Information Fig. S1B). All experiments described in our study were repeated at least three times with similar results. An MTT assay revealed that the viability of the cells dropped in a time‐dependent manner, but more so for HN26 cells than for HN7 and C33A2 cells (Fig. 1 b).
Next, we investigated if melphalan‐induced apoptosis in the HN26 cells. Apoptosis should result in cleavage of PARP1 by caspase 3. Cleavage of PARP1 was observed in HN26 cells (Fig. 1 c), but not in HN7 cells or C33A2 cells (Figs. 1 d and 1 e). In addition, the PARP1 enzyme was highly activated in melphalan‐treated HN26 cells, resulting in high levels of PARylation in these cells (Fig. 1 f). Melphalan‐induced parylation in HN26 cells was strongly inhibited by addition of PARP1‐inhibitor A‐966492 (Fig. 1 g). In contrast, parylation was undetectable in C33A2 cells (Fig. 1 h). As an additional marker for apoptosis, we used cytochrome C release apoptosis assay kit to monitor apoptosis‐mediated cytochrome C release from mitochondria. As can be seen in Figure 1 i, cytoplasmic cytochrome c localization increased in HN26 cells over time after addition of melphalan (Fig. 1 i). Furthermore, initiator caspase 8 as well as executer caspase 3 were cleaved in HN26 cells treated with melphalan (Supporting Information Fig. S3A and 3B), while cleavage of these caspases in melphalan‐treated C33A2 cells was less efficient than in HN26 cells and could not be detected (Supporting Information Fig. S3A and 3B). Finally, we monitored cellular DNA fragmentation in C33A2 and HN26 cells. As can be seen in Supporting Information Figure 3C, fragmented DNA characteristic of apoptosis was observed in HN26 cells (Supporting Information Fig. S3C). Fragmented DNA was observed also in C33A2 cells, but to a lower extent (Supporting Information Fig. S3C). Taken together, our results demonstrated that melphalan‐induced apoptosis in HN26 cells.
Melphalan activated the DNA‐damage response in HPV16‐positive tonsillar cancer cell line HN26
Next we investigated if the DNA damage response was activated in the HN26 cell line. Induction of apoptosis occurred simultaneously with a melphalan‐induced DNA damage response in the HN26 cells as determined by the appearance of phosphorylated BRCA1 (Supporting Information Fig. S4A). BRCA1 was phosphorylated at both Ser988 and Ser1423, which occurred already after 1 h (Supporting Information Fig. S4B). Furthermore, also DDR factor Chk1 was phosphorylated in response to melphalan, but not by incubation of HN26 cells with etiposide (Supporting Information Fig. S4C). PARP1 was degraded in the same extracts as expected, whereas actin and core splicing factor SF3b were largely unaffected by melphalan (Supporting Information Fig. S4A). As further evidence of DDR induction in HN26 cells, we also performed immunofluorescence on melphalan‐treated HN26 cells with antibodies to phosphorylated Chk1 and to γH2AX. The results revealed the appearance of phosphorylated Chk1 in the cytoplasm of melphalan‐treated cells (Supporting Information Fig. S5A and C) and the appearance of nuclear staining of γH2AX in a dotted pattern (Supporting Information Fig. S5B and D). The γH2AX dots/cell increased over time in melphalan (Supporting Information Fig. S5E). Analysis of expression levels of BRCA1‐associated RING domain protein 1 (BARD1) revealed that this protein was undetectable in HN26 cells, but not in C33A2 cells (Supporting Information Fig. S4D). Furthermore, BARD1 mRNA was undetectable in HN26 cells, but readily detected in both C33A2 cells and HeLa cells (Supporting Information Fig. S4E and F). Since BARD1 is involved in DNA‐damage repair and has tumor suppressor functions, the lack of BARD1 in HN26 cells may contribute to the sensitivity of HN26 cells to melphalan. Inactivation of BARD1 could potentially be a significant feature of HPV16‐positive tonsillar cancer cells, but a previously published comprehensive genetic characterization of HPV positive and negative head and neck cancers revealed that BARD1 was not among significantly mutated genes, suggesting that BARD1 mutations are rare in HNSCC.36 We concluded that melphalan induced the DDR in HN26 cells.
Melphalan restored production of p53 in HPV16‐positive tonsillar cancer cell line HN26 but not in HPV16‐immortalized human keratinocytes
The p53 protein is a key player in apoptosis and is mutated and inactive in C33A2 cells, but not in HN26 cells.31 p53 is inactivated by HPV16 E6‐mediated degradation and is therefore undetectable in HN26 cells (Fig. 2 a). 293T cells constitutively produce high levels of mutant p53 protein and was used as positive control for p53 in Western blots (Fig. 2 a). High levels of mutant p53 were also present in C33A2 cells (Fig. 2 b) but was unaffected by melphalan (Fig. 2 b). In sharp contrast, the levels of p53 protein increased from undetectable, or barely detectable levels in HN26 cells, to high levels over time in melphalan (Figs. 2 c and 2 d). Figures 2 c and 2 d shows two different concentrations of melphalan. To investigate if melphalan could also induce apoptosis in HPV16‐immortalized, but nontransformed cells, we added melphalan to the previously described HPV16 immortalized human keratinocyte cell line 331033 and monitored apoptosis markers PARP1 and caspase 3, as well as p53 levels. In contrast to the effect of melphalan on the HN26 tonsillar cancer cell line, melphalan did not induce apoptosis in 3310 cells as determined by the absence of cleaved PARP1 and caspase 3 products (Fig. 2 e). Furthermore, p53 production was not induced by the addition of melphalan to 3,310 cells (Fig. 2 e). However, melphalan did induce the DDR in 3310 cells as evidenced by the appearance of phosphorylated ATM and Chk1 (Fig. 2 f). Our results indicated that melphalan induced p53 and apoptosis in HPV16‐positive HN26 tonsillar cancer cells, but not in HPV16‐immortalized human keratinocytes.
Figure 2.
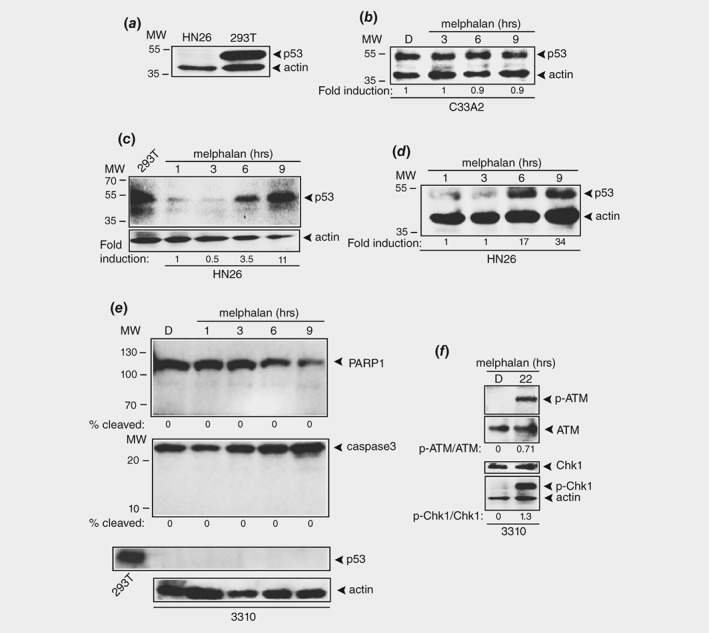
Melphalan activates p53 in HPV16 positive tonsillar cancer cells. (a) Western blot with monospecific antibody to p53 or actin on extracts from HN26 or 293T cells. Extract from 293T cells serves as a positive control for p53 protein. (b) Western blot with monospecific antibody to p53 or actin on extracts from C33A2 cells treated with DMSO or 100 μM melphalan for the indicated time points. (c, d) Western blot with monospecific antibody to p53 or actin on extracts from HN26 cells treated with DMSO or melphalan for the indicated time points. Two different concentrations of melphalan were used in c (100 μM) and d (50 μM). (e) Western blot with monospecific antibody to PARP1, caspase 3, p53 or actin on extracts from HPV16‐immortalized human keratinocyte cell line 3,310 treated with DMSO or 100 μM melphalan for the indicated time points. (f) Western blots with monospecific antibodies to phosphorylated ATM (p‐ATM), ATM, phosphorylated Chk1 (p‐Chk1), Chk1 or actin on extracts from 3,310 cells treated with DMSO or 100 μM melphalan for the indicated time points.
Melphalan caused rapid downregulation of HPV16 E6 and E7 mRNAs in HPV16‐positive tonsillar cancer cell line HN26
To investigate if melphalan affected the HPV16 E6 and E7 mRNAs, we monitored the levels of these HPV16 mRNAs as well as the E2 and E4 mRNAs. E6*I and E6*II are alternative forms of the E6 protein produced by alternatively spliced HPV16 mRNAs shown schematically in Supporting Information Figure S6A and B. The levels of HPV16 E2 and E4 mRNAs dropped rapidly over time in HN26 cells incubated with melphalan (Figs. 3 a and 3 b). More importantly, so did the levels of the E6 and E7 mRNAs (Figs. 3 a and 3 b). Quantitation of the E6 and E7 mRNAs revealed a half‐life of approximately 3 h in melphalan‐treated HN26 cells, whereas gapdh mRNAs remained stable over time (Figs. 3 a and 3 b). The mRNAs encoding the pro‐apoptotic caspase 3 protein increased in response to melphalan treatment of the HN26 cells, as expected (Figs. 3 a and 3 c), whereas the levels of the anti‐apoptotic Bcl‐2 protein quickly declined, as expected (Figs. 3 a and 3 c).
Figure 3.
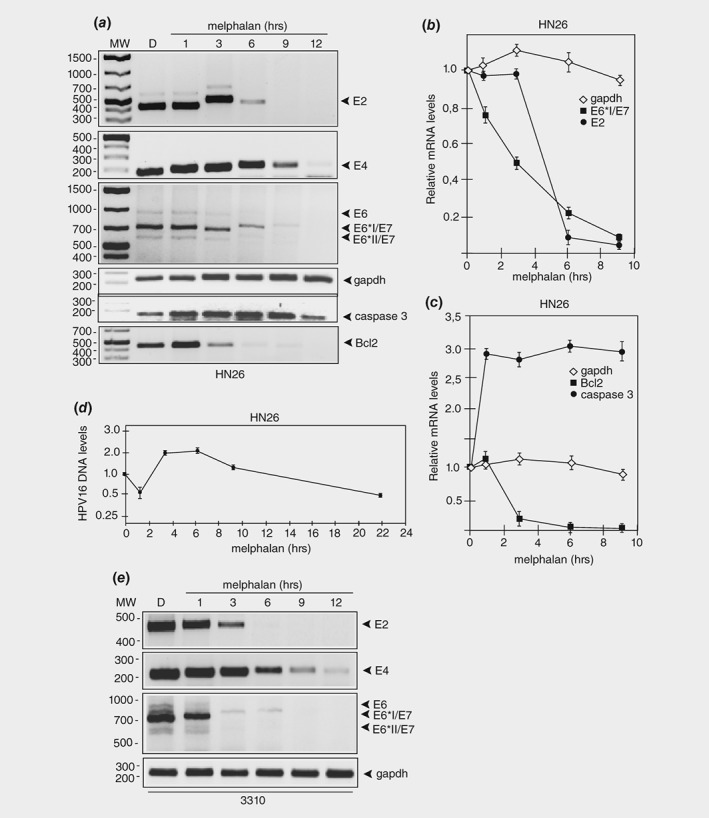
Melphalan causes rapid degradation of HPV16 E6 and E7 oncogene mRNAs in HPV16 positive tonsillar cancer cells. (a) RT‐PCR on total RNA extracted from HN26 cells treated with DMSO alone or 100 μM of melphalan for the indicated time periods. HPV16 E2, E4, E6, E6*I/E7 and E6*II/E7 mRNAs were monitored as well as spliced cellular gapdh, caspase 3 and Bcl2 mRNAs. (b, c) The RT‐PCR bands representing HPV16 mRNAs (b) or cellular mRNAs (c) were quantified and plotted against hours of melphalan treatment of the HN26 cells. (d) Quantitation of the HPV16 DNA genome with TaqMan PCR on Hirt DNA extracted at different time points from HN26 cells incubated in 100 μM melphalan. (e) RT‐PCR on total RNA extracted from HPV16‐immortalized human keratinocyte cell line 3,310 treated with DMSO alone or 100 μM of melphalan for the indicated time periods. HPV16 E2, E4, E6, E6*I/E7 and E6*II/E7 mRNAs were monitored as well as spliced cellular gapdh mRNAs. The location of the RT‐PCR primers in the HPV16 genome is shown in Supporting Information Figure 5B and the sequences of the RT‐PCR primers are listed in Supporting Information Table T1.
We reasoned that melphalan induced a drop in HPV16 E6 and E7 mRNA levels either by inhibiting HPV16 DNA replication, or by reducing the synthesis of the E6 and E7 mRNAs directly. To distinguish between the two possibilities, we first monitored the effects of melphalan on the HPV16 DNA levels over time in HN26 cells using a previously described quantitative TaqMan PCR protocol.29 Although we did observe some changes in HPV16 DNA levels, they did not decrease over time in the melphalan‐treated HN26 cells (Fig. 3 d). These results suggested that HPV16 mRNA synthesis was inhibited.
Next we investigated if melphalan reduced HPV16 early mRNA levels also in noncancerous cell line 3,310 in which melphalan activated the DDR but did not induce apoptosis. The results revealed that HPV16 early mRNAs were quickly degraded in response to melphalan (Fig. 3 e), whereas cellular gapdh mRNAs were largely unaffected by melphalan (Fig. 3 e). Since melphalan induced p53 and apoptosis in HN26 cells, but not in 3310 cells, our results suggested that induction of p53 in HN26 cells required both inhibition of E6 expression and irreparable DNA damage. Either the HPV16 positive tonsillar cancer cell line was particularly sensitive to melphalan, or HPV‐driven cancer cells were more sensitive to melphalan than HPV16 immortalized cells. Treatment of HPV18‐driven HeLa, cervical cancer cell line with melphalan, reduced HPV18 E6 and E7 mRNA levels (Supporting Information Fig. S7A, left panel) and activated the DDR (appearance of γH2AX) (Supporting Information Fig. S7B, left panel), but did not induce detectable levels of apoptosis. We did not detect caspase 3 cleavage or PARP1 cleavage in melphalan‐treated HeLa cells (Supporting Information Fig. 7C, left panel, and Supporting Information Fig. 7D). We did not detect p53 in melphalan‐treated HeLa cells either (not shown). Addition of melphalan to the HPV16‐driven SiHa cervical cancer cell line did not affect HPV16 E6 and E7 mRNA levels (Supporting Information Fig. S7A, right panel), but it did activate the DDR (Supporting Information Fig. S7B, right panel), but not apoptosis (Supporting Information Fig. S7C, right panel). We also analyzed E6 and E7 mRNA levels, caspase cleavage and DDR at a later time point (22 h) in SiHa cells, but we did not observe any effects on E6 and E7 mRNAs or apoptosis, only the DDR was induced (Supporting Information Fig. S7A, B, C, right panel). We concluded that the HPV16 positive tonsillar cancer cell line HN26 was particularly sensitive to melphalan. Due to the absence of BARD1 in HN26 cells, these cells may be less able to repair DNA damages than the 3,310 cells, HeLa or SiHa cells. Alternatively, HeLa and SiHa cells may have acquired additional mutations that render them less sensitive to melphalan than HN26 cells. The effects of melphalan on the various cell lines have been summarized in Supporting Information Table T3. We concluded that the HPV16 positive tonsillar cancer cell line HN26 was particularly sensitive to DNA alkylating agent melphalan.
Melphalan downregulated RNA polymerase II and reduced association of RNA polymerase II with HPV16 DNA in HN26 cells
To determine how the HPV16 early E6/E7 mRNA levels were reduced in HN26 cells in response to melphalan, we monitored the levels of RNA polymerase II. RNA polymerase levels dropped in both C33A2 cells and HN26 cells treated with melphalan (Figs. 4 a and 4 b). This reduction in RNA polymerase II levels occurred in a time‐dependent manner (Figs. 4 a and 4 b) and indicated that loss of HPV16 E6/E7 mRNAs could be explained by reduced RNA polymerase activity in the melphalan‐treated cells. To determine if less RNA polymerase II was associated with the HPV16 DNA in the presence of melphalan, a ChIP analysis on RNA polymerase II was performed. The ChIP results confirmed that the amount of RNA polymerase II associated with HPV16 DNA decreased over time in melphalan (Fig. 4 c). The reduction in association of RNA polymerase II with HPV16 DNA in the presence of melphalan is bigger than the drop in overall levels of RNA polymerase II, indicating that melphalan affects both total levels of RNA polymerase and the association of RNA polymerase II with HPV16 DNA. In contrast, the association of RNA polymerase II with the cellular PHB2 gene was largely unaffected by melphalan (Fig. 4 d).
Figure 4.
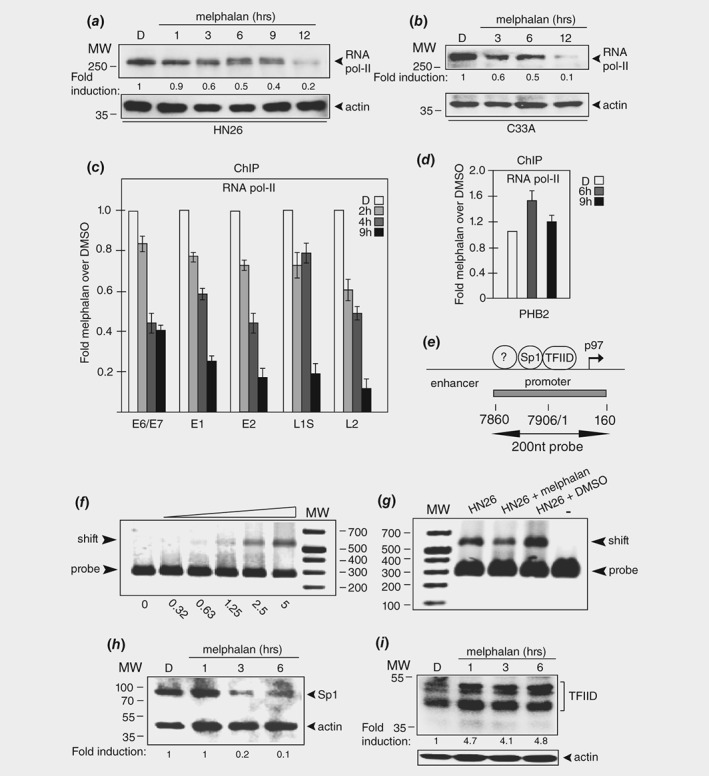
Melphalan inhibits transcription of the HPV16 E6 and E7 oncogenes. (a, b) Western blot with monospecific antibody to cellular RNA polymerase II in extracts from HN26 cells (a) or C33A2 cells (b) treated with DMSO or 100 μM melphalan for the indicated time points. (c) ChIP analyses on nucleosomes prepared from HN26 cells with monospecific antibody to RNA polymerase II and quantitative PCR (qPCR) of the indicated HPV16 amplicons. Hundred micromolar of melphalan was used. Primers are listed in Supporting Information Table T1. Mean values with standard deviations of the amount of immunoprecipitated DNA compared to input DNA are displayed. The q‐PCR values obtained for each primer pair with DNA extracted from DMSO‐treated HN26 cells were set to 1 to correct for differences between different ChIP extracts. Chip extracts were prepared from HN26 cells treated with melphalan for the indicated time‐periods. All samples were analyzed in two independent ChIP assays and all qPCR reactions were performed in triplicates. (d) ChIP analysis with monospecific antibody to RNA polymerase II of the cellular PHB2 gene. (e) Schematic representation of the enhancer/promoter at the HPV16 early promoter p97. The 200 nt dsDNA gel shift probe is indicated. Numbers refer to genomic positions in the HPV16R genome. (f) DNA‐protein gel‐shift assay with the HPV16 probe shown in Figure 7E and a two‐fold serial dilution of 5 μg of extract from HN26 cells. (g) DNA‐protein gel‐shift assay with the HPV16 probe shown in Figure 7E and extracts from untreated HN26 cells (HN26) or HN26 cells treated with DMSO or 100 μM melphalan. (h) Western blot on extracts from HN26 cells incubated with DMSO (D) or 100 μM melphalan for the indicated time‐periods with monospecific antibody to transcription factor Sp1 and actin. (i) Western blot on extracts from HN26 cells incubated with DMSO (D) or 100 μM melphalan for the indicated time‐periods with monospecific antibody to transcription factor TFIID and actin.
We speculated that reduced association of cellular transcription factors with the HPV16 p97 early promoter could also contribute to reduced transcription. To investigate this further, cell extracts were prepared from untreated, DMSO‐ or melphalan‐treated HN26 cells. A Western blot on these extracts revealed that actin protein levels were similar in DMSO‐ and melphalan‐treated HN26 cells, indicating that both extracts had similar quality (Supporting Information Fig. S8). Incubation of the HPV16 promoter‐DNA probe (Fig. 4 e) with total cell extract from untreated cells resulted in a concentration‐dependent shift of the probe as expected (Fig. 4 f). Gel‐shift experiments with cell extracts from DMSO‐ or melphalan‐treated HN26 Cells exhibited reduced binding to the HPV16 p97 promoter DNA probe in extracts from melphalan‐treated cells (Fig. 4 g). These experiments were repeated three times, all with similar results. We were unable to identify individual proteins binding to the HPV16 DNA using antibodies to transcription factors. However, the levels of transcription factor Sp1, that binds the HPV16 early p97 promoter,37 dropped in HN26 cells treated with melphalan (Fig. 4 h). The levels of the general transcription factor TFIID were not reduced but appeared to increase 3–4‐fold (Fig. 4 i). Taken together, our results demonstrated that melphalan reduced the levels of RNA polymerase II and transcription factor Sp1 and reduced the number of RNA polymerase II molecules associated with HPV16 DNA. These results suggested that reduced transcription of the HPV16 early genes in the presence of melphalan, reduced HPV16 E6 levels and restored p53.
Actinomycin D inhibited HPV16 early mRNA transcription, induced p53 and activated apoptosis in HN26 cells
To further substantiate the idea that reduced HPV16 transcription restored p53 within hours, we treated HN26 cells with actinomycin D, a substance that is a well‐characterized inhibitor of transcription and that is known to cause DNA damage through its DNA alkylating activity. The levels of the HPV16 E6 and E7 mRNAs, as well as E2 and E4 mRNAs, were quickly reduced after addition of actinomycin D. These results confirmed a short half‐life of these mRNAs (Figs. 5 a and 5 b), while gapdh mRNA levels were unaffected (Figs. 5 a and 5 b). The effects of actinomycin D on HPV16 E4, E6 and E7 mRNAs mimicked the effect of melphalan on the same mRNAs (compare Figs. 3 b and 5 b). Quantitation of the RT‐PCR levels in actinomycin D‐treated HN26 cells suggested a t1/2 of approximately 3 h for the E6 and E7 mRNAs (monitored here as the half‐life of the E6*I mRNA). Taken together, our results indicated that melphalan inhibited transcription to the same extent as actinomycin D, and that this inhibition quickly reduced the levels of the early HPV16 mRNAs as a result of their inherent short half‐lives. These results predicted that actinomycin D should also induce p53 and apoptosis in HN26 cells. Indeed, the p53 protein re‐appeared in a time dependent manner after addition of actinomycin D to HN26 cells (Fig. 5 c). Furthermore, cleavage of PARP1 in the presence of actinomycin D indicated that apoptosis was activated (Fig. 5 d). Actinomycin D also caused rapid degradation of HPV18 E6 and E7 mRNAs (Supporting Information Fig. S9A), similarly to melphalan (Supporting Information Fig. S7A). In contrast, the half‐life of HPV16 E6 and E7 mRNAs in SiHa cells appeared largely unaffected by actinomycin D (Supporting Information Fig. S9B). These results mimicked the results of melphalan (Supporting Information Fig. S7A), suggesting that SiHa cells were relatively insensitive to both melphalan and actinomycin D.
Figure 5.
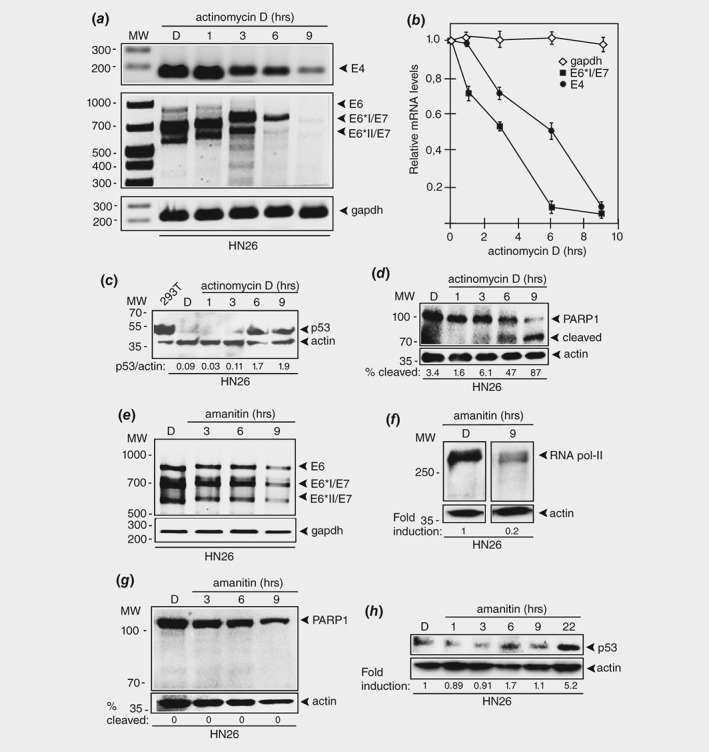
Actinomycin D causes rapid degradation of HPV16 E6 and E7 oncogene mRNAs in HPV16 positive tonsillar cancer cells. (a) RT‐PCR on total RNA extracted from HN26 cells treated with DMSO alone or 1.5 μM of actinomycin D for the indicated time periods. HPV16 E4, E6, E6*I/E7 and E6*II/E7 mRNAs were monitored as well as spliced cellular gapdh mRNA. The location of the RT‐PCR primers in the HPV16 genome is shown in Supporting Information Figure 5B. (b) The RT‐PCR bands representing HPV16 mRNAs were quantified and plotted against hours of 1.5 μM actinomycin D treatment of the HN26 cells. (c) Western blot with monospecific antibody to p53 on extracts from 293T cells or HN26 cells treated with DMSO (D) or actinomycin D for the indicated time points. (d) Western blot with monospecific antibody to PARP1 on extracts from HN26 cells treated with DMSO (D) or 1.5 μM actinomycin D for the indicated time points. (e) RT‐PCR on total RNA extracted from HN26 cells treated with DMSO alone or 3 μg/mL of alpha‐amanitin for the indicated time periods. HPV16 E6, E6*I/E7 and E6*II/E7 mRNAs were monitored as well as spliced cellular gapdh mRNA. (f) Western blot on extracts from HN26 cells incubated with DMSO (D) or 3 μg/mL of alpha‐amanitin for 9 h with monospecific antibody to RNA polymerase II and actin. (g) Western blot with monospecific antibody to PARP1 on extracts from HN26 cells treated with DMSO (D) or 3 μg/mL alpha‐amanitin for the indicated time points. (H) Western blot with monospecific antibody to p53 on extracts from 293T cells or HN26 cells treated with DMSO (D) or alpha‐amanitin (3 μg/mL) for the indicated time points.
To determine the effect on HN26 cells of a specific transcriptional inhibitor without DNA alkylating activity, we added RNA polymerase II inhibitor amanitin to HN26 cells. It reduced HPV16 E6 and E7 mRNA levels as expected (Fig. 5 e) and reduced the levels of RNA polymerase II (Fig. 5 f). However, we did not observe PARP1 cleavage as a sign of apoptosis (Fig. 5 g) nor was p53 induced (Fig. 5 h). Prolonging incubation of HN26 cells in amanitin to 22 h induced p53, but levels were lower than those with melphalan and actinomycin D and occurred several hours later (Fig. 5 h). Since amanitin did not restore p53 levels in HN26 cells as efficiently as melphalan and actinomycin D, we speculate that it is the dual ability of melphalan and actinomycin D to inhibit HPV16 E6 and E7 transcription and to cause DNA damage that efficiently activated p53 and induced apoptosis in the HN26 tonsillar cancer cells.
Melphalan inhibited growth of HPV16‐positive tonsillar cancer cells in mice
Our results indicated that HPV16‐driven cancers might be particularly vulnerable to DNA‐damaging drugs that also inhibit transcription of the short‐lived HPV16 early E6 and E7 mRNAs, such as melphalan. The reduced levels of E6 and E7 mRNAs would pave the way for induction of p53 and apoptosis in response to the DNA damage caused by melphalan. We wished to investigate if melphalan could inhibit growth of the HPV16‐positive HN26 tonsillar cancer cells in vivo, in nude mice. First the maximum tolerated dose of each drug was determined on nontumor bearing nude mice (Supporting Information Fig. S10A and B). Based on these results, nude mice xenotransplanted with HN26 cells were treated with a single dose of either melphalan (10 mg/kg body weight) or cisplatin (4 mg/kg body weight) or physiological NaCl on day 0. The size of the tumor was monitored three times per week and the relative tumor size was plotted against days after drug application. A picture of the xenograft is shown in Supporting Information Figure S11. As predicted by the documented therapeutic effect of cisplatin on tonsillar cancer, cisplatin affected tumor growth. Cisplatin delayed HN26 tumor growth in the mice (Fig. 6 a). As suggested by our results, melphalan inhibited the growth of the HPV16‐positive tonsillar cancer cells in the mice (Fig. 6 b). As a matter of fact, melphalan not only inhibited tumor growth, but reduced the size of the original tumor (Fig. 6 b). We concluded that melphalan inhibited the growth of HN26 tonsillar cancer cells in vitro and in vivo. These results suggested that melphalan, and possibly other substances that possess transcription inhibitory‐ as well as DNA damage inducing‐properties, may be particularly well suited for treatment of HPV‐driven tonsillar cancers.
Figure 6.
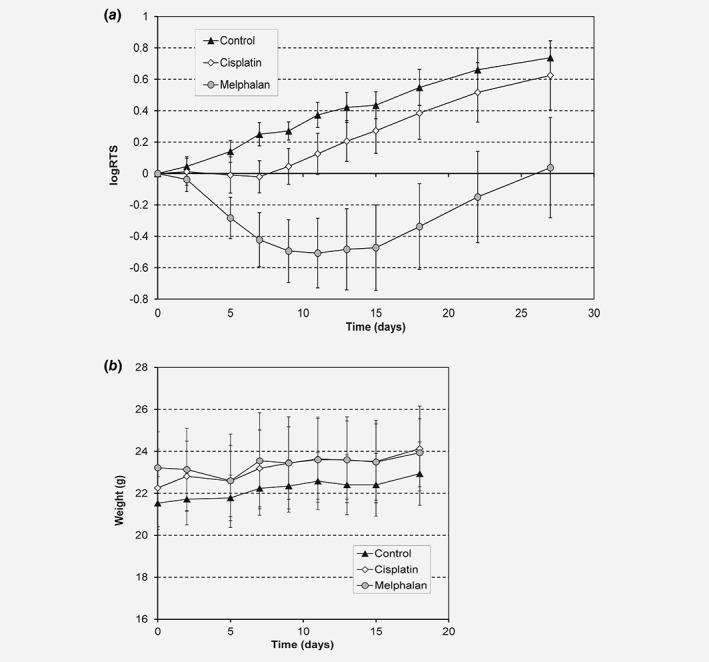
Melphalan reduces size of HPV16 positive tonsillar cancer in nude mice. (a) Nude mice with xenografts of LU‐HNSCC‐26 were treated with a single dose of melphalan, 10 mg/kg body weight, (circles), cisplatin, 4 mg/kg body weight (diamonds) or physiological NaCl (triangles) on day 0. Tumor size was measured three times a week for 27 days (melphalan, N = 21, cisplatin, N = 22, and control, N = 20), and the relative tumor size (RTS) calculated in relation to the size at day 0. (b) Body weight was measured three times a week for 18 days (melphalan, N = 11, cisplatin, N = 13, and control, N = 12). Error bars indicate standard deviation. The experiment was repeated three times with similar results.
Discussion
The short half‐life of the HPV16 early mRNAs encoding E6 and E7 oncogenes suggests that the HPV16 E6 and E7 proteins have short half‐lives as well, which has been shown previously.38, 39, 40 These earlier observations demonstrated that the E6 protein had a half‐life between 30 min and 4 h. Consequently, down regulation of the HPV16 early promoter will relatively rapidly deplete the cells of HPV16 E6 and E7 mRNAs and E6 and E7 proteins. Our results presented here suggest that the short half‐lives of the HPV16 oncogene mRNAs could potentially be exploited for therapeutic purposes. It is of interest to note that the various cancer cell lines and the HPV16‐immortalized 3,310 cell line used here respond differently to melphalan treatment (Supporting Information Table T3). Melphalan activated the DNA damage response in all cell lines, whereas apoptosis was readily detected only in tonsillar cancer cell line HN26. Apoptosis of HN26 cells correlated with the appearance of p53, and the decrease in HPV16 E6 and E7 mRNA levels, suggesting a causal relationship. However, low or no induction of p53 and apoptosis in the other cell lines may be the results of efficient DNA repair mechanisms or resistance toward melphalan caused by for example inefficient uptake or efficient export of melphalan. In support of these explanations, melphalan caused rapid degradation of HPV16 E6/E7 mRNAs in the immortalized cell line 3,310 and of HPV18 E6/E7 mRNAs in HeLa cells, yet apoptosis was not easily detected in 3310 cells or HeLa cells. It is reasonable to speculate that efficient DNA damage and/or inefficient DNA repair is a prerequisite for induction of apoptosis in response to inhibition of HPV E6/E7 expression. One may also speculate that HN26 cells may be particularly sensitive to DNA damaging substances due the inactive BARD1 gene. However, this remains to be confirmed. Furthermore, the relative resistance of the HPV16 E6/E7‐driven SiHa cancer cell line to melphalan, as well as the failure of melphalan to induce apoptosis of the HPV16‐immortalized keratinocyte cell line 3,310, would be of great interest to study further since these observations may have implications for treatment of cancer as well as HPV16 infections.
It has previously been shown that the AU‐rich HPV mRNAs are instable22 and that the HPV16 early untranslated region (UTR) mediates rapid mRNA degradation.41, 42 In cancers in which the HPV16 genome is found integrated, the HPV16 early UTR is replaced by the 3′‐end of a cellular mRNA.42 Such hybrid mRNAs may be more resistant to inhibition of transcription. However, it has been shown that treatment of HPV16 E7 transgenic mice with melphalan for 8 weeks affected HPV16 E7 biomarkers in the lung tissue of the mice.43 Other sequences on the HPV16 mRNAs may contribute to the short half‐life of the E6/E7 mRNAs.
Previous publications have shown that knock‐down of HPV16 or HPV18 E6 and E7 mRNAs either by overexpressing the papillomavirus E2 protein that inhibits transcription of HPV early genes, or by siRNAs or shRNAs directed to E6 and E7 mRNAs, induced p53 and pRb and activated apoptosis.44, 45, 46 Here we show that the two substances melphalan and actinomycin D that both inhibit transcription in the HPV16 positive tonsillar cancer cell line HN26‐induced p53 production and apoptosis. Inhibition of transcription only, was much less efficient in inducing p53 and apoptosis. Efficient induction of p53 and activation of apoptosis required a both inhibition of E6‐ and E7‐mRNA synthesis and damage of DNA that activated the DNA‐damage response. The successful induction of p53 and activation of apoptosis observed by other investigators who knocked down E6 and E7 expression by the overexpression of E2 may be the result of the dual role of E2 as an inducer of apoptosis47 inhibitor of HPV transcription. One may speculate that an efficient DNA damage repair counteracts the action of p53 in HPV‐infected cells, and that induction of apoptosis in these cells required both liberation of p53 and signals that activate apoptosis. Melphalan and actinomycin D could individually achieve both tasks.
The inherent short‐half‐life of the HPV16 early mRNAs and proteins may stem from the fact that the HPV16 life‐cycle is divided into an early and late stage.16, 48, 49 Shut‐down of the early genes encoding the cell‐growth promoting E6 and E7 proteins is an absolute necessity for cellular differentiation and entry into the late stage of the HPV life cycle. The HPV E2 protein inhibits transcription of the HPV early genes19 and inhibits the HPV16 early polyadenylation signal to pave the way for HPV late gene expression.34 A switch to the late gene expression program requires a rapid clearance of the HPV early mRNAs and proteins to allow for cell differentiation. Thus, it is unlikely that HPVs would encode early mRNAs with long half‐lives as it would avert a timely entry into the late, productive stage of the viral life cycle. The half‐life of the HPV late mRNAs is also short.22 In this case the short half‐life is believed to prevent premature expression of the HPV late genes.22 By extension, it is reasonable to speculate that other DNA viruses with life‐cycles characteristically divided into early and late stages, encode early mRNAs with short half‐lives.
Melphalan and actinomycin D reduced HN26 cell viability to a greater extent than cisplatin as determined by MTT assay. It has previously been reported that cancer cells respond differently to melphalan and cisplatin.50 These results may have implications for treatment of HPV‐positive oropharyngeal carcinomas, which was suggested by the animal experiments presented here. Here we used only one dose of melphalan which caused the tumors in the mice to shrink, although they eventually gained momentum and started grew in size. It would therefore be of interest to combine the single dose of melphalan with either radiation or a second dose of melphalan to investigate if the HPV16‐containing tumor could be eradicated. In conclusion, our results suggest that the most efficient induction of apoptosis of the HPV16‐positive cancer cells was obtained with a substance that both inhibits transcription of the HPV16 E6 and E7 genes and induces apoptosis by causing irreparable DNA damage. These results are of interest in light of current suggestions of de‐escalated treatment of HPV16‐positive tonsillar carcinomas.51
Supporting information
Supporting Information
References
- 1. Zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2002;2:342–50. [DOI] [PubMed] [Google Scholar]
- 2. Schiffman M, Doorbar J, Wentzensen N, et al. Carcinogenic human papillomavirus infection. Nat Rev Dis Primers 2016;2:16086. [DOI] [PubMed] [Google Scholar]
- 3. Bouvard V, Baan R, Straif K, et al. A review of human carcinogens‐‐part B: biological agents. Lancet Oncol 2009;10:321–2. [DOI] [PubMed] [Google Scholar]
- 4. Fakhry C, Westra WH, Li S, et al. Improved survival of patients with human papillomavirus‐positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst 2008;100:261–9. [DOI] [PubMed] [Google Scholar]
- 5. Gillison ML, D'Souza G, Westra W, et al. Distinct risk factor profiles for human papillomavirus type 16‐positive and human papillomavirus type 16‐negative head and neck cancers. J Natl Cancer Inst 2008;100:407–20. [DOI] [PubMed] [Google Scholar]
- 6. Kreimer AR, Clifford GM, Boyle P, et al. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systematic review. Cancer Epidemiol Biomarkers Prev 2005;14:467–75. [DOI] [PubMed] [Google Scholar]
- 7. Dayyani F, Etzel CJ, Liu M, et al. Meta‐analysis of the impact of human papillomavirus (HPV) on cancer risk and overall survival in head and neck squamous cell carcinomas (HNSCC). Head Neck Oncol 2010;2:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang MB, Liu IY, Gornbein JA, et al. HPV‐positive oropharyngeal carcinoma: a systematic review of treatment and prognosis. Otolaryngol Head Neck Surg 2015;153:758–69. [DOI] [PubMed] [Google Scholar]
- 9. Chaturvedi AK, Engels EA, Pfeiffer RM, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol 2011;29:4294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. D'Souza G, Agrawal Y, Halpern J, et al. Oral sexual behaviors associated with prevalent oral human papillomavirus infection. J Infect Dis 2009;199:1263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chaturvedi AK, Anderson WF, Lortet‐Tieulent J, et al. Worldwide trends in incidence rates for oral cavity and oropharyngeal cancers. J Clin Oncol 2013;31:4550–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reinson T, Toots M, Kadaja M, et al. Engagement of the ATR‐dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J Virol 2013;87:951–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. TCGA network . Comprehensive genomic characterization of head and necksquamous cell carcinomas. Nature 2015;517:576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vande Pol SB, Klingelhutz AJ. Papillomavirus E6 oncoproteins. Virology 2013;445:115–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chow LT, Broker TR, Steinberg BM. The natural history of human papillomavirus infections of the mucosal epithelia. APMIS 2010;118:422–49. [DOI] [PubMed] [Google Scholar]
- 16. Doorbar J, Quint W, Banks L, et al. The biology and life‐cycle of human papillomaviruses. Vaccine 2012;30(Suppl 5):F55–70. [DOI] [PubMed] [Google Scholar]
- 17. Mighty KK, Laimins LA. The role of human papillomaviruses in oncogenesis. Recent Results Cancer Res 2014;193:135–48. [DOI] [PubMed] [Google Scholar]
- 18. McBride AA. The papillomavirus E2 proteins. Virology 2013;445:57–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thierry F. Transcriptional regulation of the papillomavirus oncogenes by cellular and viral transcription factors in cervical carcinoma. Virology 2009;384:375–9. [DOI] [PubMed] [Google Scholar]
- 20. Bernard HU. Regulatory elements in the viral genome. Virology 2013;445:197–204. [DOI] [PubMed] [Google Scholar]
- 21. Jia R, Zheng ZM. Regulation of bovine papillomavirus type 1 gene expression by RNA processing. Front Biosci 2009;14:1270–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johansson C, Schwartz S. Regulation of human papillomavirus gene expression by splicing and polyadenylation. Nature Rev Microbiol 2013;11:239–51. [DOI] [PubMed] [Google Scholar]
- 23. Graham SV, Faizo AA. Control of human papillomavirus gene expression by alternative splicing. Virus Res 2017;231:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schwartz S. Papillomavirus transcripts and posttranscriptional regulation. Virology 2013;445:187–96. [DOI] [PubMed] [Google Scholar]
- 25. Wu C, Kajitani N, Schwartz S. Splicing and polyadenylation of human papillomavirus type 16 mRNAs. Int J Mol Sci 2017;18, Article no. 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kajitani N, Glahder J, Wu C, et al. hnRNP L controls HPV16 RNA polyadenylation and splicing in an Akt‐kinase‐dependent manner. Nucleic Acids Res 2017;45:9654–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kadaja M, Silla T, Ustav E, et al. Papillomavirus DNA replication ‐ from initiation to genomic instability. Virology 2009;384:360–8. [DOI] [PubMed] [Google Scholar]
- 28. Roman A, Munger K. The papillomavirus E7 proteins. Virology 2013;445:138–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Letsolo BT, Faust H, Ekblad L, et al. Establishment and characterization of a human papillomavirus type 16‐positive tonsillar carcinoma xenograft in BALB/c nude mice. Head Neck 2016;38:417–25. [DOI] [PubMed] [Google Scholar]
- 30. Tang AL, Hauff SJ, Owen JH, et al. UM‐SCC‐104: a new human papillomavirus‐16‐positive cancer stem cell‐containing head and neck squamous cell carcinoma cell line. Head Neck 2012;34:1480–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Forslund O, Sugiyama N, Wu C, et al. A novel human in vitro papillomavirus type 16 positive tonsil cancer cell line with high sensitivity to radiation and cisplatin. in press. [DOI] [PMC free article] [PubMed]
- 32. Li X, Johansson C, Glahder J, et al. Suppression of HPV‐16 late L1 5′‐splice site SD3632 by binding of hnRNP D proteins and hnRNP A2/B1 to upstream AUAGUA RNA motifs. Nucleic Acids Res 2013;41:10488–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Johansson C, Jamal Fattah T, Yu H, et al. Acetylation of intragenic histones on HPV16 correlates with enhanced HPV16 gene expression. Virology 2015;482:244–59. [DOI] [PubMed] [Google Scholar]
- 34. Johansson C, Somberg M, Li X, et al. HPV‐16 E2 contributes to induction of HPV‐16 late gene expression by inhibiting early polyadenylation. EMBO J 2012;13:3212–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsai C, Smider V, Hwang BJ, et al. Electrophoretic mobility shift assays for protein‐DNA complexes involved in DNA repair. Methods Mol Biol 2012;920:53–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015;517:576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dong XP, Pfister H. Overlapping YY1‐ and aberrant SP1‐binding sites proximal to the early promoter of human papillomavirus type 16. J Gen Virol 1999;80(Pt 8):2097–101. [DOI] [PubMed] [Google Scholar]
- 38. Grossman SR, Laimins LA. E6 protein of human papillomavirus type 18 binds zinc. Oncogene 1989;4:1089–93. [PubMed] [Google Scholar]
- 39. Androphy EJ, Hubbert NL, Schiller JT, et al. Identification of the HPV‐16 E6 protein from transformed mouse cells and human cervical carcinoma cell lines. EMBO J 1987;6:989–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tomaic V, Pim D, Banks L. The stability of the human papillomavirus E6 oncoprotein is E6AP dependent. Virology 2009;393:7–10. [DOI] [PubMed] [Google Scholar]
- 41. Vinther J, Rosenstierne MW, Kristiansen K, et al. The 3′ region of human papillomavirus type 16 early mRNAs decrease expression. BMC Infect Dis 2005;5:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jeon S, Lambert PF. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc Natl Acad Sci USA 1995;92:1654–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim EJ, Kim H, Lee S, et al. Melphalan modulates the expression of E7‐specific biomarkers in E7‐Tg mice. Anticancer Res 2010;30:2773–83. [PubMed] [Google Scholar]
- 44. Sima N, Wang W, Kong D, et al. RNA interference against HPV16 E7 oncogene leads to viral E6 and E7 suppression in cervical cancer cells and apoptosis via upregulation of Rb and p53. Apoptosis 2008;13:273–81. [DOI] [PubMed] [Google Scholar]
- 45. Goodwin EC, Dimaio D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc Natl Acad Sci USA 2000;97:12513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rampias T, Sasaki C, Weinberger P, et al. E6 and e7 gene silencing and transformed phenotype of human papillomavirus 16‐positive oropharyngeal cancer cells. J Natl Cancer Inst 2009;101:412–23. [DOI] [PubMed] [Google Scholar]
- 47. Desaintes C, Demeret C, Goyat S, et al. Expression of the papillomavirus E2 protein in HeLa cells leads to apoptosis. EMBO J 1997;16:504–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kajitani N, Satsuka A, Kawate A, et al. Productive lifecycle of human papillomaviruses that depends upon squamous epithelial differentiation. Front Microbiol 2012;3:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hong S, Laimins LA. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Future Microbiol 2013;8:1547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Spanswick VJ, Lowe HL, Newton C, et al. Evidence for different mechanisms of 'unhooking' for melphalan and cisplatin‐induced DNA interstrand cross‐links in vitro and in clinical acquired resistant tumour samples. BMC Cancer 2012;12:436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yete S, D'Souza W, Saranath D. High‐risk human papillomavirus in Oral cancer: clinical implications. Oncology 2018;94:133–41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information