

Abstract
Macrophage activation plays a central role in neoatherosclerosis and in‐stent restenosis after percutaneous coronary intervention (PCI). Galectin‐3, mainly expressed on macrophages, is an important regulator of inflammation. This study aimed to investigate the effects of berberine (BBR) on oxidized low‐density lipoprotein (ox‐LDL)‐induced macrophage activation and galectin‐3 expression and their underlying mechanisms. THP‐1‐derived macrophages were pretreated with BBR prior to stimulation with ox‐LDL. Galectin‐3 expression was measured by real‐time PCR, Western blotting, and confocal microscopy. Macrophage activation was assessed by lipid accumulation, expression of inflammatory cytokines, and CD11b and CD86. Plasma galectin‐3 levels were measured in patients undergoing PCI at baseline and after BBR treatment for 3 months. BBR suppressed ox‐LDL‐induced upregulation of galectin‐3 and macrophage activation. Overexpression of galectin‐3 intervened the inhibitory effect of BBR on macrophage activation. BBR activated phospho‐AMPK and inhibited phospho‐NF‐κB p65 nuclear translocation. AMPK inhibition and NF‐κB activation abolished the inhibitory effects of BBR on galectin‐3 expression and macrophage activation. Combination of BBR and rosuvastatin exerted greater effects than BBR or rosuvastatin alone. However, BBR treatment did not further reduce plasma galectin‐3 after PCI in patients receiving standard therapy. In conclusion, BBR alleviates ox‐LDL‐induced macrophage activation by downregulating galectin‐3 via the NF‐κB and AMPK signaling pathways.
Keywords: berberine, galectin‐3, macrophage activation, rosuvastatin
1. INTRODUCTION
Despite years of effort in developing newer generation drug‐eluting stents (Didier et al., 2017; Giustino et al., 2017), intensive lipid management (Fujisue & Tsujita, 2017; Vinten‐Johansen et al., 2005; B. Zhang, Pei, et al., 2017), and prolonged antiplatelet therapy (Colombo et al., 2014; Didier et al., 2017; Mauri et al., 2014), the incidence of neoatherosclerosis (NA) and in‐stent restenosis (ISR) after percutaneous coronary intervention (PCI) still remains a concern. The mechanisms underlying the development of NA and ISR are not fully understood. However, it has been shown that endothelial injury, platelet/fibrin aggression, and leukocyte infiltration are the distinctive characteristics of immediate inflammatory response to stent injury, whereas macrophage infiltration is the main manifestation of delayed in‐stent neointimal formation (Libby, Schwartz, Brogi, Tanaka, & Clinton, 1992; Welt & Rogers, 2002; M. Zhang et al., 2014). Clinical studies have demonstrated that in‐stent NA and ISR are associated with accumulation of lipid‐laden foamy macrophages within the neointima (Jinnouchi et al., 2017; Nakazawa et al., 2011). Classically activated foamy macrophage clusters have been a promising target in the progression of NA and ISR (Nakazawa, Ladich, Finn, & Virmani, 2008; Otsuka et al., 2015).
Galectin‐3, a β‐galactoside‐binding lectin, is abundantly expressed and secreted by macrophages and foam cells (Bekkers, Yazdani, Virmani, & Waltenberger, 2010; van der Veer et al., 2007). Levels of galectin‐3 are increased in atherosclerotic lesions (Ito, 2006), and galectin‐3 amplifies inflammation through macrophage activation in atherosclerotic progression (Funaro et al., 2011; Hamirani, Wong, Kramer, & Salerno, 2014; Ito, 2006; Papaspyridonos et al., 2008; Taylor et al., 2004). Clinical studies have also shown that plasma galectin‐3 is an independent predictor of all‐cause mortality, cardiovascular death, reinfarction, and occurrence of heart failure following acute coronary syndrome (ACS; Dall'Armellina et al., 2011; Ganame et al., 2009; Harrison et al., 2013; Ito, 2006; Kloner, 2011).
Berberine (BBR) has been used as part of the traditional Chinese medicine to treat diarrhea and gastrointestinal disorders for centuries in China and Korea (Choi et al., 2003), and it has also been shown to inhibit carcinogenesis (Tsang et al., 2013) and exert antibacterial properties (Roser, Grundemann, Engels, & Huber, 2016). In recent decades, studies have shown that BBR has various beneficial effects on the cardiovascular system, including improvement of insulin resistance (Ye et al., 2016) and inhibition of inflammation (Fan et al., 2015) and atherosclerosis (Chen et al., 2014). However, the exact mechanisms of BBR on macrophage activation deserve further research. In the present study, we investigated the effects of BBR on macrophage activation and galectin‐3 expression compared with rosuvastatin and their underlying molecular mechanisms. In addition, we investigated whether additive BBR treatment for 3 months further reduced plasma galectin‐3 levels in ACS patients following PCI on top of standard therapy including statin.
2. MATERIALS AND METHODS
2.1. Participants
The population for the study comprised patients who successfully underwent primary or elective PCI for ACS at Xinhua Hospital affiliated to Shanghai Jiaotong University School of Medicine, Shanghai, China, between July 1, 2016 and June 30, 2017. Patients with an angiographically visible thrombus within target lesions, cardiogenic shock, New York Heart Association Class III/IV heart failure, abnormal liver or renal function, various inflammatory diseases, or infectious diseases were excluded from the study. A total of 45 ACS patients were included in this prospective study. Patients were single‐blind and divided into two cohorts after PCI according to 2:1 randomization ratio: 30 patients received 300‐mg BBR hydrochloride (Shanghai Sine Pharmaceutical Ltd., Shanghai, China) t.i.d., a common clinical dose, in addition to standard therapy, whereas 15 patients received standard therapy alone. As for standard therapy, all patients received clopidogrel (300‐mg loading dose and then 75‐mg/day maintenance dose), aspirin (300 mg loading dose and then 100 mg/day maintenance dose), and rosuvastatin (20 mg/day); the use of angiotensin‐converting enzyme inhibitors or angiotensin receptor blockers, calcium channel blockers, beta‐blockers, and/or antidiabetic therapy (including insulin or oral medication) was decided on an individual basis by the attending physician (Table 1). The duration of BBR hydrochloride treatment was 3 months. BBR hydrochloride was stopped if patients complained of any discomfort.
Table 1.
Baseline characteristics of study subjects
| BBR (n = 30) | Control (n = 15) | p value | |
|---|---|---|---|
| Demographics | |||
| Age (years) | 64.7 ± 7.1 | 68.4 ± 11.7 | 0.067 |
| Male, n (%) | 81.5 | 55.6 | 0.094 |
| BMI (kg/m2) | 25.3 ± 5.0 | 24.3 ± 2.7 | 0.602 |
| Medical history, n (%) | |||
| Family history of CAD | 81.5 | 50 | 0.047* |
| Hypertension | 74.1 | 61.1 | 0.512 |
| Diabetes | 25.9 | 27.8 | 1.000 |
| Smoking | 51.9 | 22.2 | 0.065 |
| History of PCI/CABG | 22.2 | 5.6 | 0.215 |
| Baseline medications, n (%) | |||
| Aspirin | 48.1 | 38.9 | 0.760 |
| Beta‐blocker | 33.3 | 22.2 | 0.514 |
| Calcium‐channel blocker | 40.7 | 22.2 | 0.333 |
| ACE inhibitors/ARBs | 48.1 | 33.3 | 0.371 |
| Statin | 44.4 | 22.2 | 0.204 |
| Hypoglycemic agent/insulin | 29.6 | 27.8 | 1.000 |
| Biological test | |||
| TC (mmol/L) | 3.95 ± 0.98 | 4.32 ± 1.06 | 0.228 |
| TG (mmol/L) | 1.68 ± 0.81 | 1.98 ± 1.39 | 0.763 |
| LDL (mg/dl) | 2.12 ± 0.69 | 2.30 ± 0.70 | 0.330 |
| HDL (mg/dl) | 1.04 ± 0.22 | 1.11 ± 0.24 | 0.411 |
| Glucose (mmol/L) | 5.62 ± 0.97 | 6.03 ± 1.61 | 0.643 |
| hsCRP (mg/L) | 5.70 ± 15.85 | 5.67 ± 13.52 | 0.233 |
| ALT (U/L) | 21.15 ± 12.52 | 23.81 ± 14.11 | 0.190 |
| AST (U/L) | 22.78 ± 14.82 | 26.17 ± 9.73 | 0.060 |
| BUN (mg/dl) | 5.32 ± 1.38 | 5.63 ± 1.88 | 0.754 |
| Cr (mmol/L) | 74.92 ± 16.95 | 67.82 ± 15.98 | 0.539 |
| eGFR (60 ml/min/1.73 m2) | 93.08 ± 23.37 | 90.72 ± 22.01 | 0.963 |
| Uric acid (mmol/24 hr) | 385.31 ± 80.88 | 351.52 ± 86.47 | 0.183 |
| TnT (ng/ml) | 1.63 ± 6.48 | 0.64 ± 2.42 | 0.770 |
| proBNP (ng/L) | 335.95 ± 1054.60 | 504.25 ± 678.97 | 0.336 |
| CAG details | |||
| Gensini score | 42.70 ± 28.76 | 33.28 ± 20.34 | 0.261 |
| Number of stents | 1.52 ± 0.64 | 1.39 ± 0.61 | 0.469 |
Note. Continuous variables were reported as mean ± SD. Dichotomous variables were reported as percentages. Continuous variables were compared using the Wilcoxon signed‐rank test (two‐tailed). Dichotomous variables were compared with the χ2 test (two‐sided) or Fisher's exact test.
ACE: angiotensin‐converting enzyme inhibitors; ALT: alanine aminotransferase; ARB: angiotensin receptor blockers; AST: aminotransferase; BBR: berberine; BMI: body mass index; BUN: blood urea nitrogen; CABG: coronary artery bypass grafting; CAD: coronary artery disease; CAG: coronary angiography; Cr: creatinine; eGFR: estimated glomerular filtration rate; HDL: high‐density lipoprotein; hsCRP: hypersensitive C‐reactive protein; LDL: low‐density lipoprotein; PCI: percutaneous coronary intervention; proBNP: pro‐B‐type natriuretic peptide; TC: total cholesterol; TG: triglycerides; TnT: troponin T.
The present study complied with the Declaration of Helsinki and was registered in the Chinese Clinical Trial Registry (no. ChiCTR‐TRC‐10000868; http://www.chictr.org, accessed May 24, 2010) and approved by the Ethics Committee of Experimental Research, Shanghai Jiaotong University. All patients provided written informed consent.
2.2. Biochemical analysis
Blood was taken on the morning before coronary angiography for baseline measurements and again after treatment for 3 months. Serum samples were obtained by centrifuging the blood at 1,600 g for 15 min at room temperature within 30 min of venipuncture, and aliquots were stored immediately at −80°C for future analysis. Total cholesterol (TC) were determined by 3‐methoxy‐5‐methylaniline methods (cholesterol multipurpose liquid reagent), low‐density lipoprotein cholesterol (LDL‐C) by direct method (LDL‐C kit), high‐density lipoprotein cholesterol (HDL‐C) by antibody hindrance homogenous method (highly density lipoprotein cholesterol kit), and triglycerides (TG) by 3‐methoxy‐5‐methylaniline methods (free glycerol determination kit) on the day of blood collection in the laboratories of Xinhua Hospital. Fasting plasma glucose (FPG) was measured by the hexokinase method (Glucose assay kit). All the above reagents were from Wako Pure Chemical Industries, Ltd., Odakyu Sharyo Kogyo, Japan, and assays were performed on Hitachi 008As (Hitachi, Tokyo, Japan) on the day of blood collection in the laboratories of Xinhua Hospital. Westgard multi‐rule quality control method was used as the decision rules of quality control of our laboratory in determining both plasma lipid profiles and FPG. The reference materials of lipid profile (Bio‐Rad, Foster City, California, USA) and FPG (Beckman Coulter, Carlsbad, California, USA) were distributed by Shanghai Clinical Laboratory Quality Control Center. Serum hypersensitive C‐reactive protein (hsCRP) was detected by the particle‐enhanced turbidimetric immunoassay. Serum aspartate aminotransferase, alanine aminotransferase, blood urea nitrogen, urea acid, and creatinine were detected using routine biochemical methods in the Central Clinical Laboratory of Xinhua Hospital. All assays were performed in a blinded manner.
2.3. Quantification of plasma galectin‐3 levels by enzyme‐linked immunosorbent assay
Plasma galectin‐3 was measured by enzyme‐linked immunosorbent assat kits (eBioscience, San Diego, CA, USA) according to the manufacturer's instructions. All samples were assayed in duplicate, and values were analyzed according to standard curves. The lower detection limit for this assay is 0.005 ng/ml. Blood samples used for this analysis were restricted to a single freeze–thaw cycle.
2.4. Cell culture
THP‐1 cells (ATCC, Rockville, Maryland, USA) were maintained in RPMI‐1640 medium (Gibco, Grand Island, New York, USA) containing 10% fetal bovine serum (Gibco), 0.05‐mM 2‐mercaptoethanol, 10‐mM HEPES, 1‐mM sodium pyruvate, 4.5‐g/L glucose, and 1.5‐g/L bicarbonate in a humidified atmosphere of 5% CO2 and 95% air at 37°C. The differentiation of THP‐1 monocytes into macrophages was induced by exposure to 100‐nM phorbol 12‐myristate 13‐acetate (PMA; Sigma‐Aldrich, St. Louis, MO, USA) for 48 hr. The differentiated THP‐1 macrophages were washed in phosphate‐buffered saline (PBS) before being used in the experiments. Macrophages were pretreated with different concentrations of BBR hydrochloride (Sigma‐Aldrich) or rosuvastatin (Sigma‐Aldrich) for 1 hr before stimulation with oxidized low‐density lipoprotein (ox‐LDL; 100 μg/ml; Sigma‐Aldrich) for 24 hr.
2.5. Galectin‐3 short hairpin RNA, lentiviral vector construction, and infection
Short hairpin RNA (ShRNA)‐galectin‐3, lentivirus‐galectin‐3, and their relative negative control (NC) were designed and synthesized by Shanghai GenePharma Co., Ltd, Shanghai, China. For shRNA‐galectin‐3 construction, one RNA interference vector pGLV3/H1/GFP&Puro and three specific shRNAs for galectin‐3 (shRNA‐Gal‐3‐599, shRNA‐Gal‐3‐651, and shRNA‐Gal‐3‐683) were designed and synthesized on the basis of the human galectin‐3 target sequence (NCBI Gene ID: 3958). An NC was produced according to the same design principle for shRNA. The above three shRNAs and the shRNA‐NC were transfected into THP‐1 cell line using X‐tremeGENE HP DNA transfection reagent (Roche, Basel, Switzerland) following the manufacturer's protocol. After 24 hr, the optimal RNA interference vector was identified by qPCR and shRNA‐Gal‐3‐651: 5′‐GCCACTGATTGTGCCTTATAA‐3′ was selected.
The PCR products of galectin‐3, with the forward primer 5′‐GCCTACCCATCTTCTGGACA and reverse primer 3′‐CCAGGCAAAGGCAGGTTAT, were cloned into the lentiviral expression plasmid pLV5/SmaI/GFP&Puro for lentivirus‐galectin‐3 construction. The construct of galectin‐3 was confirmed by sequencing. The plasmid DNA was co‐transfected into HEK293 T cells with pCDH‐galectin‐3, psPAX2, pMD packaging construct using RNAi‐Mate (GenePharma, Shanghai, China) according to the manufacturer's protocol. An NC was also produced according to the manufacturer's protocol.
Medium was refreshed after 6 hr, and lentiviral supernatant was collected 48 hr later. THP‐1 cells were seeded into 6‐well plates (5 × 106 cells/well) and transfected with different lentiviral vectors at a multiplicity of infection of 100. The culture medium was changed every 2 days. Selection was performed for about 2 weeks. Galectin‐3 expression was examined by real‐time PCR and Western blotting after transfection.
2.6. Protein isolation and Western blotting analysis
Total cellular protein was extracted in RIPA lysis buffer (Beyotime, Shanghai, China) supplemented with 1% phenylmethanesulfonyl fluoride (Beyotime, Shanghai, China) and 1% phosphatase inhibitor (Beyotime, Shanghai, China). For the analysis of nuclear NF‐κB p65, and phospho‐NF‐κB p65 (p‐p65) (Ser536), nuclear and cytoplasmic extraction reagents (Beyotime, Shanghai, China) were used to separate cytoplasmic and nuclear fractions according to the manufacturer's instructions. Equal amounts of protein (30 μg) were separated through a 10% or 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a poly(vinylidene difluoride) membrane. Membranes were first probed with primary antibodies for NF‐κB p65, p‐p65 (Ser536), AMPK, phospho‐AMPK (Thr172), and GAPDH (all the above five antibodies: 1:1000; from Cell Signaling Technology, Inc., Danvers, MA, USA), β‐actin and histone H3 (these two antibodies: 1:1000; from Abcam, Cambridge, MA, USA), and then incubated with anti‐rabbit or anti‐mouse secondary antibodies (1:1000; Beyotime, Shanghai, China). All signals were detected by the Molecular Imager ChemiDocTM XRS+ System (BIO‐RAD, Hercules, CA, USA). Galectin‐3 was normalized by GAPDH levels. Phospho‐AMPK (Thr172) was normalized by total AMPK. p‐p65 (Ser536) was normalized by NF‐κB p65. GAPDH was the indicator for cytoplasm protein. Histone H3 was the indicator for nuclear protein. β‐Actin was the indicator for both cytoplasm and nuclear protein.
2.7. RNA isolation and real‐time PCR
Total RNA was isolated from cells using Trizol (Invitrogen, Carlsbad, CA, USA). Real‐time PCR was performed to determine gene expression of galectin‐3, interleukin‐6 (IL‐6), tumor necrosis factor‐α (TNF‐α), and interleukin‐1β (IL‐1β). The primer sequences are shown in Table 2. The PCR reaction was performed as follows: Stage 1, 94°C for 2 min and Stage 2, 94°C for 20 s and 60°C for 34 s. Stage 2 was repeated for 40 cycles. Real‐time PCR was performed using SYBR&ROX PCR master mix (Takara, Japan) with Applied Biosystems ABI7500 Real‐time PCR System (Applied Biosystems, Foster City, California, USA). GAPDH was used as an endogenous control. All samples were normalized to internal controls, and the relative expression level was calculated using the 2−ΔΔCt analysis method.
Table 2.
PCR primer used for cDNA
| Genes | Primers |
|---|---|
| Galectin‐3 | F: 5′‐TATAAGATCTGAGGATAGGTGGGTTCCCGAGAACT‐3′ |
| R: 5′‐ATATGAATTCTCTCAGGGCTATGCCGCCTAAGTAC‐3′ | |
| IL‐6 | F: 5′‐GAGTAGTGAGGAACAAGCCAGAG‐3′ |
| R: 5′‐GCAGGCTGGCATTTGTGGTT‐3′ | |
| TNF‐α | F: 5′‐AGCCCATGTTGTAGCAAACC‐3′ |
| R: 5′‐TGAGGTACAGGCCCTCTGAT‐3′ | |
| IL‐1β | F: 5′‐CCTGAGCTCGCCAGTGAAAT‐3′ |
| R: 5′‐GGTGGTCGGAGATTCGTAGC‐3′ | |
| GAPDH | F: 5′‐GACAGTCAGCCGCATCTTCT‐3 |
| R: 5′‐TTAAAAGCAGCCCTGGTGAG‐3′ |
2.8. Confocal microscopy
For confocal microscopy, different groups of THP‐1‐derived macrophages were blocked with 1% bovine serum albumin (Sigma‐Aldrich)/PBS for 1 hr at room temperature, and cells were then incubated for 1 hr at room temperature with a rabbit anti‐human galectin‐3 antibody (1:100; Abcam). After being washed with PBS containing 0.1% Tween‐20, samples were incubated with a secondary antibody (Alexa Fluor 647 mouse‐anti‐rabbit IgG at 1:200; Invitrogen) for 2 hr at room temperature. Following fixation, the cell nucleus was stained with 4,6‐diamino‐2‐phenylindole (eBioscience). Cells were then examined on a LSM 510 confocal laser scanning microscope (Carl Zeiss Inc., Maple Grove, Minnesota, USA).
2.9. Flow cytometry
The expression of CD86 and CD11b on the surface of macrophages was determined by flow cytometry. After the removal of medium from the wells, cells were collected, incubated with FcR blocking reagent and stained directly with (APC)‐conjugated anti‐human‐CD86 antibody (BioLegend, San Diego, CA, USA) and APC‐cy7‐ conjugated anti‐human‐CD11b antibody (BioLegend) according to the manufacturer's instruction. An isotype control (BioLegend) was included in each experiment to identify background staining. Samples were analyzed on a BD FACS Calibur flow cytometer (Becton Dickinson, Franklin Lakes, New Jersey, USA).
2.10. Oil red O staining
To examine lipid accumulation in macrophages, oil red O staining was performed. Cells were washed with PBS and then fixed by 4% paraformaldehyde. Then, cells were stained for 30 min in the oil red O working solution (Sigma‐Aldrich), followed by isopropyl alcohol decolorization for 30 s. Lastly, cells were washed with PBS, and cells then examined on a Leica microscope (Leica Microsystems Inc., Buffalo Grove, IL, USA).
2.11. Statistical analysis
Continuous variables were reported as mean ± SD, and dichotomous variables were reported as percentage. Baseline demographics of patients between two groups were compared with the χ2 test (two‐sided) or Fisher's exact test for dichotomous variables and Wilcoxon signed‐rank test for continuous variables. Differences in galectin‐3, hsCRP, lipid profiles, and liver and renal function in the two groups were compared using the Wilcoxon matched‐pairs signed‐rank test (two‐tailed). Each cell experiment was performed at least three times. Data were compared using 2‐tailed Student's t test for two independent samples or one‐way analysis of variance followed by Kruskal–Wallis H test for more than two groups. p < 0.05 was considered statistically significant. All statistical analyses were performed with IBM SPSS statistics for Windows 24.0 (SPSS, Inc., Chicago, IL, USA).
3. RESULTS
3.1. Baseline clinical demographics
Baseline characteristics were presented in Table 1. There were no significant differences in age, gender, body mass index, hypertension, diabetes, smoking, history of PCI/coronary artery bypass grafting, and medications between the BBR‐treated and the control groups. The difference of the frequency of family history of coronary artery disease between the BBR‐treated and the control groups was significant (p < 0.05; Table 1). Baseline TC, TG, HDL‐C, LDL‐C, glucose, and hsCRP were not significantly different between the two groups. There were no significant differences in plasma galectin‐3, liver and renal function, and coronary angiography details between the two groups.
3.2. Plasma galectin‐3 levels, serum lipids, and inflammatory markers of ACS patients after 3 months
Plasma levels of galectin‐3 after 3 months were significantly reduced in both the control group who received standard therapy alone and in additive BBR‐treated group compared with baseline levels (both p < 0.05; Figure 1a). The decrease of hsCRP in additive BBR‐treated group was significant (p < 0.05), whereas the decrease in hsCRP in the control group did not reach significance (p > 0.05; Figure 1b). Compared with baseline values, serum levels of TC and LDL after 3 months were significantly reduced in additive BBR‐treated group (p < 0.001; Figure 1c,e), and in the control group who received standard therapy alone (p < 0.01; Figure 1c,e). Serum levels of TG after 3 months were significantly reduced in additive BBR‐treated group (p < 0.001; Figure 1d) but not in the control group. Mild elevation in HDL‐C was observed in both groups (p < 0.05; Figure 1f).
Figure 1.
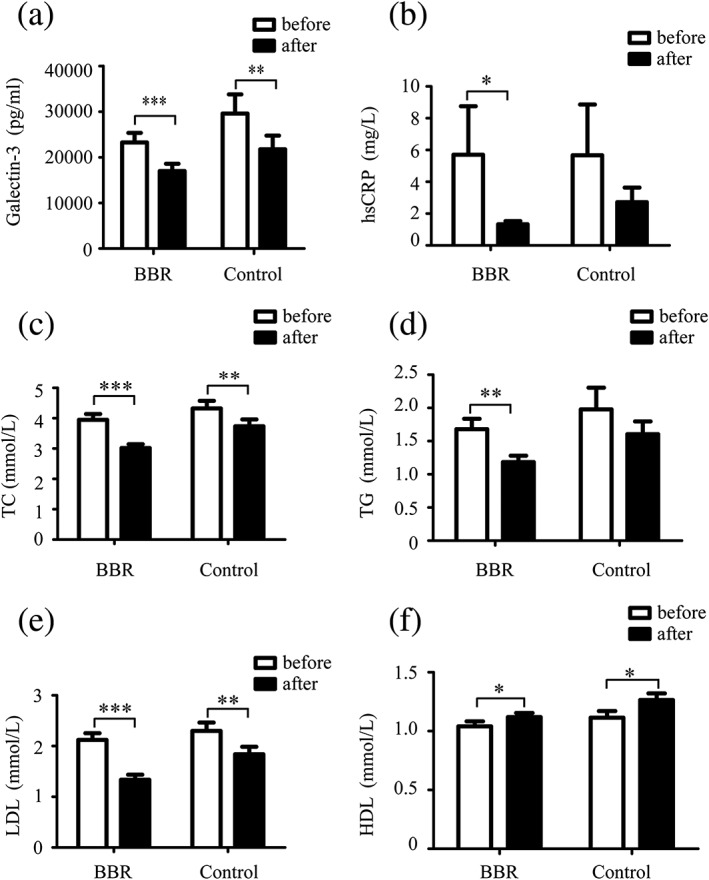
Effects of berberine (BBR), rosuvastatin, combination of BBR and rosuvastatin on plasma galectin‐3 level and serum lipids. (a) Plasma galectin‐3 level, (b) hypersensitive C‐reactive protein (hsCRP), and (c–f) lipid profile were examined in additive BBR‐treated group who received BBR, 300 mg, t.i.d., for 3 months in addition to standard therapy (n = 30) and in the control group who received standard therapy alone (n = 15). Data are shown in mean ± SD. * p < 0.05, ** p < 0.01, ***p < 0.001. The standard therapy contains clopidogrel (300 mg loading dose and then 75 mg/day maintenance dose), aspirin (300 mg loading dose and then 100 mg/day maintenance dose), and rosuvastatin (20 mg/day). HDL: high‐density lipoprotein; LDL: low‐density lipoprotein; TC: total cholesterol; TG: triglycerides
3.3. Safety of BBR
Compared with baseline values, alanine aminotransferase, aspartate aminotransferase, blood urea nitrogen, creatinine, urea acid, and estimated glomerular filtration rate after 3 months were not changed in either BBR group or the control group (p > 0.05; Figure S1), suggesting that BBR has no adverse effect on liver and renal function. No patients from the BBR‐treated group complained of any side effects. Overall, BBR at the dose administered in the present study is very safe in ACS patients.
3.4. BBR decreased galectin‐3 expression in THP‐1‐derived macrophages
Next, we investigated the effect of BBR on galectin‐3 expression on THP‐1‐derived macrophages in vitro. Macrophages were pretreated with BBR (5, 10, and 25 μM), rosuvastatin (5, 10, and 25 μM), and combination of BBR and rosuvastatin for 1 hr before stimulation with ox‐LDL for 24 hr. Galectin‐3 protein and mRNA were upregulated by ox‐LDL stimulation. BBR and rosuvastatin at 25 μM (but not at 5 or 10 μM; data not shown) abolished the effect of ox‐LDL on galectin‐3 gene and protein expression, whereas combination of rosuvastatin and BBR (25 μM) further decreased galectin‐3 expression compared with BBR or rosuvastatin alone (Figure 2a,b). Laser scanning confocal microscopy also confirmed that BBR and rosuvastatin (25 μM) decreased galectin‐3 expression induced by ox‐LDL on the cell surface of macrophages; combination of rosuvastatin and BBR exerted the greatest effect (Figure 2c). For the following experiments, 25 μM of BBR and 25 μM of rosuvastatin were used.
Figure 2.
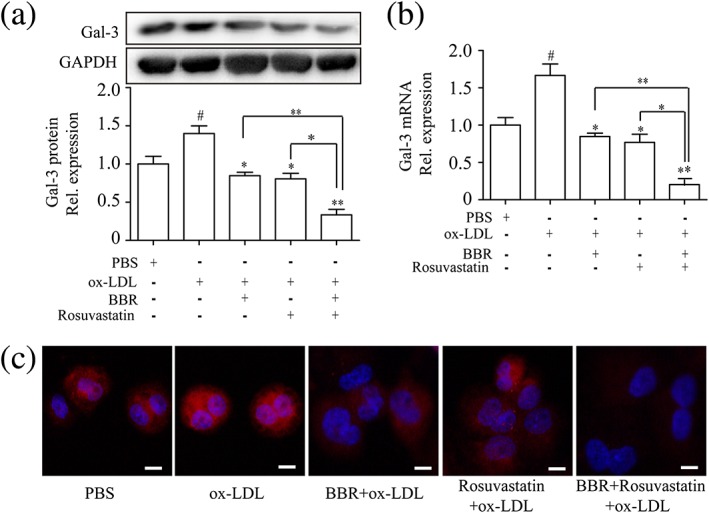
The effect of berberine (BBR), rosuvastatin, combination of BBR and rosuvastatin on galectin‐3 expression on THP‐1‐derived macrophages. THP‐1‐derived macrophages were pretreated with phosphate‐buffered saline (PBS), BBR (25 μM), rosuvastatin (25 μM), and BBR (25 μM) and rosuvastatin (25 μM) for 1 hr and then stimulated by 100‐μg/ml oxidized low‐density lipoprotein (ox‐LDL). (a) Western blots show the protein expression of galectin‐3 at 24 hr after stimulation by ox‐LDL. (b) Relative gene expression of galectin‐3 in THP‐1‐derived macrophages was determined by real‐time PCR at 12 hr after stimulation by ox‐LDL. (c) Laser scanning confocal microscopy was used to confirm galectin‐3 expression on the cell membrane of THP‐1‐derived macrophages in different groups. Data are represented as mean ± SD. n ≥ 3. * p < 0.05, ** p < 0.01 versus ox‐LDL group; #p < 0.05 versus PBS group. Scale bar = 100 μm [Colour figure can be viewed at wileyonlinelibrary.com]
3.5. BBR inhibited ox‐LDL‐induced lipid accumulation and expression of inflammatory markers
We then examined the effects of BBR on macrophage activation as compared with rosuvastatin. Macrophages were pretreated with BBR, rosuvastatin, and combination of rosuvastatin and BBR for 1 hr before stimulation with ox‐LDL for 24 hr. Lipid accumulation in macrophages induced by ox‐LDL was suppressed by BBR, rosuvastatin, and combination of BBR and rosuvastatin (Figure 3a). Both BBR and rosuvastatin inhibited expression of inflammatory markers (TNF‐α, IL‐6, and IL‐1β; Figure 3b) and CD11b and CD86 (Figure 3c,d) induced by ox‐LDL, and BBR exerted additional benefit on top of rosuvastatin.
Figure 3.
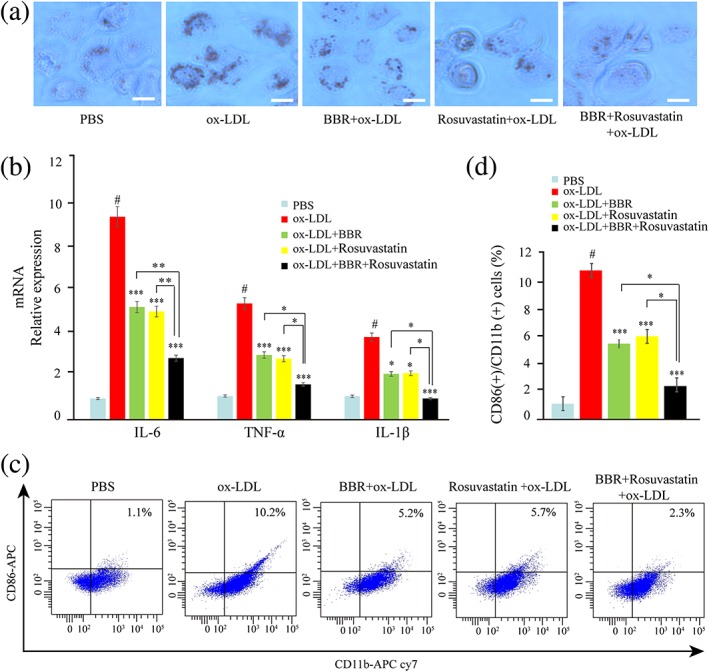
The effect of berberine (BBR), rosuvastatin, and combination of BBR and rosuvastatin on THP‐1‐derived macrophage activation induced by oxidized low‐density lipoprotein (ox‐LDL). THP‐1‐derived macrophages were pretreated with phosphate‐buffered saline (PBS), BBR (25 μM), rosuvastatin (25 μM), and combination of BBR (25 μM) and rosuvastatin (25 μM) for 1 hr and then stimulated 24 hr by 100 μg/ml ox‐LDL. (a) The lipid accumulation was assessed by oil red O stain. (b) IL‐6, TNF‐α, and IL‐1β expression in macrophages was evaluated by real‐time PCR. (c, d) CD11b and CD86 expression on macrophages was measured by flow cytometry. Data are represented as mean ± SD. n ≥ 3. * p < 0.05, ** p < 0.01, and *** p < 0.001 versus ox‐LDL group; #p < 0.05 versus PBS group. Scale bar = 100 μm [Colour figure can be viewed at wileyonlinelibrary.com]
3.6. Inhibition of galectin‐3 abrogated ox‐LDL‐induced macrophage activation
Next, we examined the effect of galectin‐3 on macrophage activation by manipulating galectin‐3 expression. We first confirmed that inhibition of galectin‐3 by shRNA‐Gal‐3 (p < 0.005; Figure 4a,b) downregulated, whereas overexpression of galectin‐3 by lentivirus‐Gal‐3 (p < 0.05; Figure 4a,b) upregulated protein expression of galectin‐3, measured by Western blotting. We then found that inhibition of galectin‐3 abrogated the effects of ox‐LDL on lipid accumulation (Figure 4c) and induction of inflammatory markers (Figure 4d) and CD11b and CD86 expression (Figure 4e,f), whereas overexpression of galectin‐3 enhanced these effects of ox‐LDL.
Figure 4.
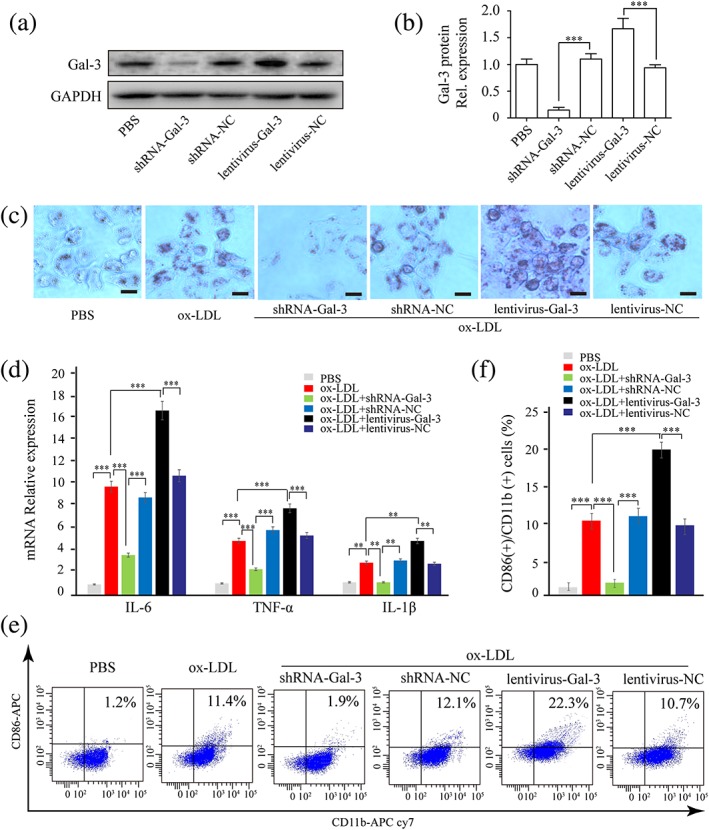
Galectin‐3 promoted THP‐1‐derived macrophage activation. THP‐1 cells were infected by short hairpin RNA (shRNA)‐Gal‐3, shRNA‐negative control (NC), lentivirus‐Gal‐3, lentivirus‐NC, then induced to differentiate into macrophage by phorbol 12‐myristate 13‐acetate. (a, b) Galectin‐3 protein was examined by Western blotting after infection. (c−f) Macrophages were stimulated with oxidized low‐density lipoprotein (ox‐LDL) for 24 hr. (c) Lipid accumulation was assessed by oil red O stain. (d) IL‐6, TNF‐α, and IL‐1β expression in macrophage was evaluated by real‐time PCR. (e, f) CD11b and CD86 expression on macrophage was measured by flow cytometry. Data are represented as mean ± SD. n ≥ 3. * p < 0.05, ** p < 0.01, and *** p < 0.001 versus respective control group. Scale bar = 100 μm. PBS: phosphate‐buffered saline [Colour figure can be viewed at wileyonlinelibrary.com]
3.7. Overexpression of galectin‐3 intervened the inhibitory effect of BBR on the macrophage activation
We then investigated whether manipulation on the gene expression of galectin‐3 could intervene the inhibitory effects of BBR on the ox‐LDL‐induced macrophage activation. THP‐1 cells were infected by lentivirus‐Gal‐3 or lentivirus‐NC and then induced to differentiate into macrophage by PMA. THP‐1‐derived macrophages were pretreated with PBS and BBR (25 μM) for 1 hr before being induced by 100 μg/ml ox‐LDL for 24 hr. We found that overexpression of galectin‐3 abrogated the inhibitory effects of BBR on lipid accumulation (Figure 5a) and induction of inflammatory markers (Figure 5b) and CD11b and CD86 expression (Figure 5c,d) induced by ox‐LDL.
Figure 5.
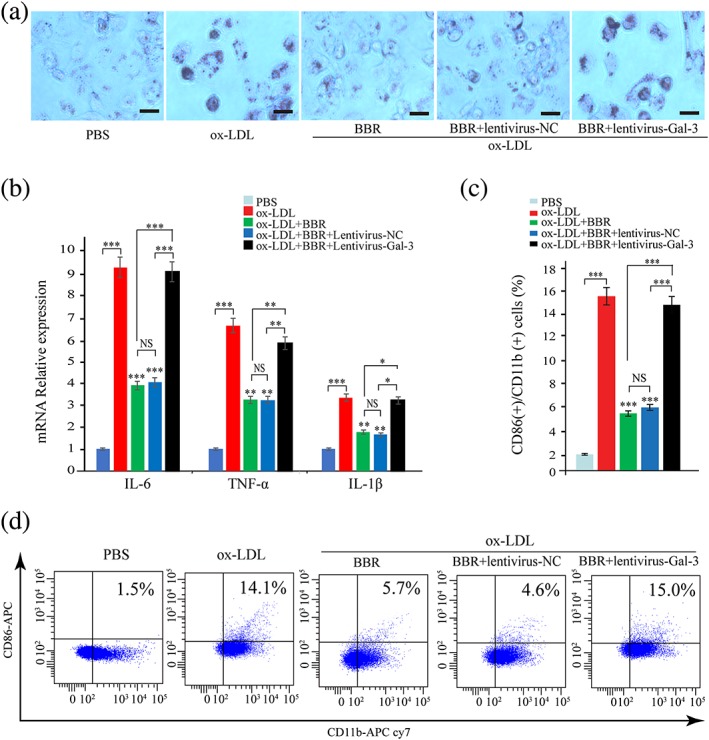
Overexpression of galectin‐3 intervened the inhibitory effect of berberine (BBR) on the macrophage activation. THP‐1 cells were infected by lentivirus‐Gal‐3 or lentivirus‐negative control (NC) and then induced to differentiate into macrophage by phorbol 12‐myristate 13‐acetate. THP‐1‐derived macrophages were pretreated with phosphate‐buffered saline (PBS) and BBR (25 μM) for 1 hr and then induced by 100 μg/ml oxidized low‐density lipoprotein (ox‐LDL) for 24 hr. (a) The lipid accumulation was confirmed by oil red O stain. (b) The IL‐6, TNF‐α, IL‐1β expression in macrophages was evaluated by real‐time PCR. (c, d) CD11b and CD86 on macrophages were measured by flow cytometry. Data are represented as mean ± SD. n ≥ 3. * p < 0.05, ** p < 0.01, and *** p < 0.001 versus ox‐LDL group. Scale bar = 100 μm. NS: non‐significant [Colour figure can be viewed at wileyonlinelibrary.com]
3.8. Role of the AMPK and NF‐κB signaling pathways in BBR mediated decrease in galectin‐3 expression and macrophage activation
We later examined the involvement of signaling pathways in regulating galectin‐3 and macrophage activation by BBR. First, we examined the regulation of signaling pathways in macrophages by BBR. THP‐1‐derived macrophages were pretreated with BBR, rosuvastatin, and combination with BBR and rosuvastatin for 1 hr, and signaling pathway proteins (p‐AMPK, AMPK, NF‐κB p65, and p‐p65) were examined by Western blotting at 30 min after the induction by ox‐LDL (Figure 6a,b). We found that p‐AMPK expression was upregulated by BBR, combination of BBR and rosuvastatin, but not by rosuvastatin (Figure 6a). Intranuclear p‐p65 was downregulated in BBR and rosuvastatin group, compared with PBS group, whereas combination group further downregulated p‐p65 expression compared with BBR or rosuvastatin group (Figure 6b). Combination of BBR and rosuvastatin but not BBR or rosuvastatin alone significantly decreased cytoplasma p‐p65. These results indicate that both BBR and rosuvastatin mainly inhibited p‐p65 nuclear translocation. Then, we examined the effect of blocking AMPK signaling pathway with a specific inhibitor, compound C (Selleck Chemicals, Houston, TX, USA), or activating NF‐κB signaling pathway with an activator, prostratin (Sigma‐Aldrich), on galectin‐3 expression. THP‐1‐derived macrophages were pretreated with BBR, rosuvastatin, and BBR and rosuvastatin, in the presence or absence of compound C (10 μg/ml) or prostratin (10 μg/ml) for 1 hr before being induced by 100 μg/ml ox‐LDL. Compound C abolished the inhibitory effect of BBR on galectin‐3 protein and mRNA expression but had no influence on the effect of rosuvastatin on galectin‐3 expression (Figure 6c,d). Prostratin abolished the inhibitory effects of both BBR and rosuvastatin on galectin‐3 protein and mRNA expression (Figure 6e,f). Compound C also counteracted the inhibitory effects of BBR, but not of rosuvastatin, on lipid accumulation (Figure 7a) and expression of inflammatory cytokines (Figure 7b) and CD11b and CD86 induced by ox‐LDL (Figure 7c,d). Prostratin counteracted the inhibitory effects of both BBR and rosuvastatin on lipid accumulation (Figure 8a) and expression of inflammatory cytokines (Figure 8b) and CD11b and CD86 (Figure 8c,d) induced by ox‐LDL.
Figure 6.
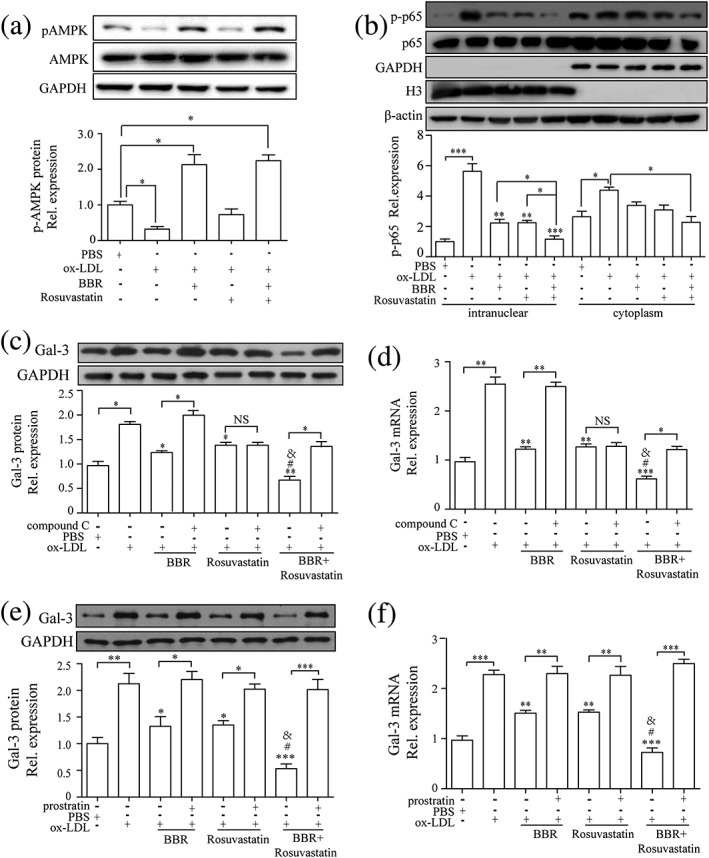
Berberine (BBR) downregulated galectin‐3 expression by inhibiting NF‐κB and activating AMPK signaling pathway. (a–d) Macrophages were pretreated with phosphate‐buffered saline (PBS), BBR (25 μM), rosuvastatin (25 μM), and combination of BBR (25 μM) and rosuvastatin (25 μM) for 1 hr and then stimulated by 100 μg/ml oxidized low‐density lipoprotein (ox‐LDL) for 30 min. Western blots and quantification of (a) AMPK and phospho‐AMPK (p‐AMPK) and (b) NF‐κB p65 and phospho‐NF‐κB p65 (p‐p65). Protein and mRNA expression of galectin‐3 measured by Western blotting and real‐time PCR on THP‐1‐derived macrophages pretreated with PBS, BBR, rosuvastatin, and BBR and rosuvastatin, in the presence or absence of (c and d) compound C (10 μg/ml) or (e and f) prostratin (10 μg/ml) for 1 hr before the induction by 100 μg/ml ox‐LDL for 24 hr. Galectin‐3 was normalized by GAPDH levels. Phospho‐AMPK (Thr172) was normalized by total AMPK. p‐p65 (Ser536) was normalized by NF‐κB p65. GAPDH was the indicator for cytoplasm protein. Histone H3 was the indicator for nuclear protein. β‐Actin was the indicator for both cytoplasm protein and nuclear protein. Data are represented as mean ± SD. n ≥ 3. * p < 0.05, ** p < 0.01, *** p < 0.01 versus ox‐LDL group; # p < 0.05 versus ox‐LDL + BBR group; & p < 0.05 versus ox‐LDL + rosuvastatin group. NS: non‐significant
Figure 7.
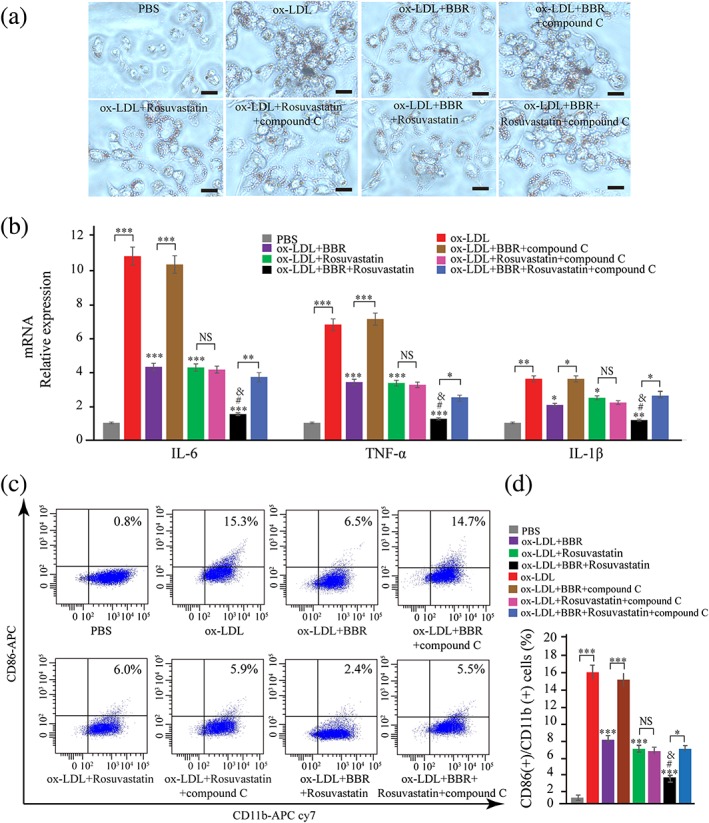
Compound C alleviates oxidized low‐density lipoprotein (ox‐LDL)‐induced classical macrophage activation. THP‐1‐derived macrophages were pretreated with PBS, BBR (25 μM), rosuvastatin (25 μM), and BBR (25 μM) and rosuvastatin (25 μM), in the presence or absence of compound C (10 μg/ml) for 1 hr and then induced for 24 hr by 100 μg/ml ox‐LDL. (a) The lipid accumulation was confirmed by oil red O stain. (b) The IL‐6, TNF‐α, and IL‐1β expression in macrophages was evaluated by real‐time PCR. (c, d) CD11b and CD86 on macrophages were measured by flow cytometry. Data are represented as mean ± SD. n ≥ 3. * p < 0.05, ** p < 0.01, and *** p < 0.001 versus ox‐LDL group; versus ox‐LDL group; # p < 0.05 versus ox‐LDL + BBR group; & p < 0.05 versus ox‐LDL + rosuvastatin group. Scale bar = 100 μm. PBS: phosphate‐buffered saline. NS: non‐significant [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 8.
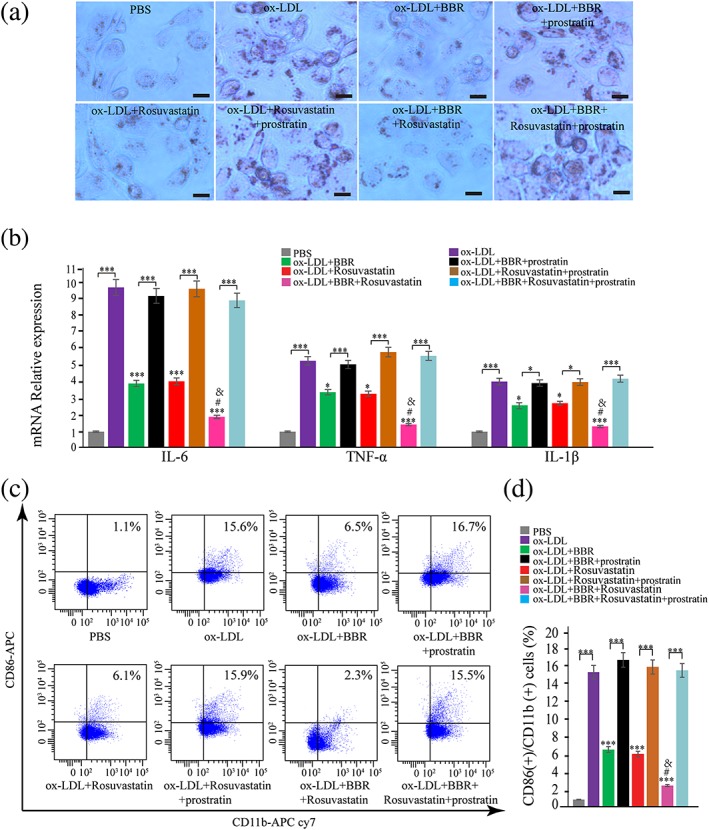
Prostratin intervened the inhibitory effect of berberine (BBR) on oxidized low‐density lipoprotein (ox‐LDL)‐induced macrophage activation. THP‐1‐derived macrophages were pretreated with phosphate‐buffered saline (PBS), BBR (25 μM), rosuvastatin (25 μM), and BBR (25 μM) and rosuvastatin (25 μM), in the presence or absence of prostratin (10 μg/ml) for 1 hr and then induced for 24 hr by 100 μg/ml ox‐LDL. (a) The lipid accumulation was confirmed by oil red O stain. (b) The IL‐6, TNF‐α, and IL‐1β expression in macrophages was evaluated by real‐time PCR. (c, d) CD11b and CD86 on macrophages were measured by flow cytometry. Data are represented as mean ± SD. n ≥ 3. ** p < 0.01 and *** p < 0.001 versus ox‐LDL group; versus ox‐LDL group; # p < 0.05 versus ox‐LDL + BBR group; & p < 0.05 versus ox‐LDL + rosuvastatin group. Scale bar = 100 μm [Colour figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
In the present study, we examined the mechanisms underlying the effect of BBR on macrophage activation and made some important findings. First, we demonstrated that BBR suppressed macrophage activation and galectin‐3 expression on macrophages induced by ox‐LDL, and inhibition of galectin‐3 abrogated macrophage activation. Overexpression of galectin‐3 intervened the inhibitory effect of BBR on macrophage activation. Second, BBR activated p‐AMPK and inhibited p‐p65 nuclear translocation, and a specific inhibitor of p‐AMPK, compound C, and an activator of NF‐κB, prostratin, abolished the effects of BBR on inhibiting galectin‐3 expression and macrophage activation. Third, in all the above experiments, we compared BBR with rosuvastatin and compared combined treatment of BBR and rosuvastatin with BBR or rosuvastatin alone. BBR and rosuvastatin had similar effects except that rosuvastatin failed to activate p‐AMPK, and compound C failed to abolish the effects of rosuvastatin on galetin‐3 and macrophage activation. Combination of BBR and rosuvastatin exerted greater effects than BBR or rosuvastatin alone. However, in our clinical study, additive BBR treatment on top of standard therapy did not further reduce plasma levels of galectin‐3 in ACS patients undergoing PCI. Taken together, our results suggest that BBR alleviates ox‐LDL‐induced macrophage activation by downregulating galectin‐3 via the NF‐κB and AMPK signaling pathways on top of rosuvastatin.
Studies of optical coherence tomography proved that NA and ISR lesion was associated with the presence of activated lipid‐laden foamy macrophages (Jinnouchi et al., 2017; Nakazawa et al., 2011). Thus, inhibiting macrophage infiltration and foam cell formation may limit NA and ISR. Over the last few years, our team has made great efforts to investigate the effects of BBR on macrophage activation and atherosclerosis. We first demonstrated that BBR markedly inhibited matrix metalloproteinase 9 in PMA‐induced macrophages (Huang et al., 2011). We then found that BBR treatment for 30 days further decreased circulating levels of matrix metalloproteinase 9, intercellular adhesion molecule‐1, and vascular cell adhesion molecule‐1 in ACS patients after PCI compared with the standard therapy alone (Meng et al., 2012). In ApoE(−/−) mice, we found that BBR derivatives (dhBER and Di‐MeBER) inhibited inflammatory response and reduced plaque size and vulnerability (Chen et al., 2014). In the present study, we further examined the effect of BBR on ox‐LDL‐induced activation of THP‐1‐derived macrophages and demonstrated that BBR suppressed ox‐LDL‐induced macrophage activation as indicated by reduced lipid accumulation and decreased expression of inflammatory cytokine (TNF‐α, IL‐1β, and IL‐6) and CD11b and CD86 on macrophages. Consistently, a very recent paper found that BBR suppressed inflammatory responses in RAW 264.7 macrophages (H. Zhang, Shan, et al., 2017).
Galectin‐3, abundantly expressed on macrophages and lipid‐laden foam cells (Bekkers et al., 2010; van der Veer et al., 2007), contributes to atherosclerotic progression via enhancing the recruitment of monocytes and macrophages and amplifying inflammatory state through macrophage activation in atherosclerotic lesions (Papaspyridonos et al., 2008; Taylor et al., 2004). It has been shown that both genetic and pharmacological inhibition of galectin‐3 reduces atherosclerotic lesions and slows atherosclerotic plaque‐progression in ApoE knockout mice (Funaro et al., 2011; Hamirani et al., 2014). Plasma galectin‐3 has also been reported as a major predictor of cardiovascular mortality in ACS patients following PCI (Ito, 2006; Kloner, 2011; Kumar et al., 2011). In the present study, we demonstrated that BBR downregulated ox‐LDL‐induced galectin‐3 expression on macrophages, and knockdown of galectin‐3 abrogated, whereas overexpression of galectin‐3 enhanced the effects of ox‐LDL on macrophage activation. Overexpression of galectin‐3 intervened the inhibitory effect of BBR on macrophage activation. These results suggest that galectin‐3 mediates the effect of BBR on macrophage activation. However, we did not find that additive BBR treatment for 3 months further reduced plasma galectin‐3 levels in ACS patients following PCI on top of standard therapy including statin. Such discrepancy between in vitro experiments and the clinical study deserves some comments. First, plasma galectin‐3 derives from different cell types and tissues (Payne et al., 2011). Second, THP‐1‐derived macrophages were pretreated with BBR before stimulation with ox‐LDL, but BBR was given to patients with established coronary artery disease and after PCI. Notably, the sample size of our patients was quite small.
AMPK activation improves macrophage cholesterol homeostasis in mice (Fullerton et al., 2015), inhibits PMA‐induced monocyte‐to‐macrophage differentiation (Vasamsetti et al., 2015), and attenuates atherosclerosis by enhancing the anti‐atherogenic effects of high‐density lipoproteins (Ma, Wang, Yang, An, & Zhu, 2017) and reducing atheroma‐inducing macrophages formation in ApoE (−/−) mice (Wang, Ma, Zhao, & Zhu, 2017). In the present study, phosphate AMPK protein was downregulated on ox‐LDL‐induced macrophages, which was rescued by BBR. Pretreated with compound C (a specific inhibitor of AMPK) abolished the effect of BBR on galectin‐3 protein and macrophage activation. NF‐κB signaling pathway was commonly regarded as a key pathological mechanism of atherosclerosis and other inflammation diseases (Pamukcu, Lip, & Shantsila, 2011; Yu, Zheng, & Tang, 2015). In the present study, BBR also suppressed ox‐LDL‐induced p‐p65 protein nuclear translocation. Activating NF‐κB signaling pathway by prostratin counteracted the inhibitory effect of BBR on galectin‐3 expression and macrophage activation. Dumic, Lauc, and Flogel (2000) found that NF‐κB p65 was involved in the regulation of galectin‐3 expression in a transcriptional manner. Taken together, the effects of BBR on galectin‐3 and macrophage activation are mediated by the AMPK and NF‐κB signaling pathways.
Statins not only lower lipid effectively but also have beneficial cardiovascular pleiotropic effects including inhibiting inflammatory responses, protecting blood vessel endothelium, and stabilizing atherosclerotic plaques. Statins attenuate plaque vulnerability via reducing the number and activity of macrophages on blood vessels intima and downregulating inflammatory and thrombotic signals in the endothelium. Statins appear to be safe for use in the majority of patients, but still a proportion of patients are at risk of side effects such as myopathy, hepatic damage, and diabetes from long‐term statin use (Patel, Martin, & Banach, 2016). BBR is a safe Chinese medicine without adverse effects on liver and kidney and can also improve insulin resistance (Ye et al., 2016). In the present study, we found that BBR and rosuvastatin had similar effects on downregulating galectin‐3 and inhibiting macrophage activation. However, although both BBR and rosuvastatin inhibited p‐p65 nuclear translocation, BBR, but not rosuvastatin, activated phospholation of AMPK. Activating NF‐κB signaling pathway counteracted the inhibitory effects of both BBR and rosuvastatin on galectin‐3 expression and macrophage activation. However, blocking AMPK only abolished the effect of BBR, but not of rosuvastatin, on inhibiting galectin‐3 expression and macrophage activation. So the underlying mechanisms for the actions of BBR and statins may be a bit different. Moreover, we proved that combined treatment of BBR and rosuvastatin exerted greater effect than BBR or rosuvastatin alone. So our results suggest that BBR could be an effective alternative drug for those who cannot tolerate statins, and BBR may provide additional benefits in preventing ISR or NA for ACS patients who undergo PCI procedure and receive standard therapy.
There are several limitations of this study. First, as mentioned above, the sample size of our patients was small. It is reasonable to call for more information at a larger patient base to further study the issue. Second, we did not collect follow‐up data to examine whether BBR had a preventive effect on NA or ISR, which warrants further investigation. In addition, because BBR was used as a pretreatment before the stimulation of ox‐LDL, the effects of BBR on macrophage activation and galectin‐3 expression are protective rather than therapeutic. Whether BBR has therapeutic effects on macrophage activation and galectin‐3 expression is yet to be explored.
In conclusion, BBR alleviates ox‐LDL‐induced macrophage activation by downregulating galectin‐3 via suppressing NF‐κB and activating AMPK signaling pathway, whereas combination of BBR and rosuvastatin exerts greater effects than rosuvastatin alone.
CONFLICT OF INTEREST
The authors have declared that there are no conflicts of interest.
Supporting information
Figure S1.
Safety of the BBR Renal (A and B) and live function (C, D, E, F) in BBR‐treated patients who received 300 mg BBR, tid, for 3 months in addition to standard therapy (n = 30) and control patients who received standard therapy alone (n = 15) at baseline time and after 3 months of treatment. Data are shown in mean ± SD.
ACKNOWLEDGMENTS
This project was supported by the National Natural Science Foundation of China (to S. M., Grant 81270207) and the Science and Technology Commission of Shanghai Municipality (to S. M., Grant 16401972000).
Pei CZ, Zhang Y, Wang P, et al. Berberine alleviates oxidized low‐density lipoprotein‐induced macrophage activation by downregulating galectin‐3 via the NF‐κB and AMPK signaling pathways. Phytotherapy Research. 2019;33:294–308. 10.1002/ptr.6217
REFERENCES
- Bekkers, S. C. , Yazdani, S. K. , Virmani, R. , & Waltenberger, J. (2010). Microvascular obstruction: Underlying pathophysiology and clinical diagnosis. Journal of the American College of Cardiology, 55(16), 1649–1660. 10.1016/j.jacc.2009.12.037 [DOI] [PubMed] [Google Scholar]
- Chen, J. , Cao, J. , Fang, L. , Liu, B. , Zhou, Q. , Sun, Y. , … Meng, S. (2014). Berberine derivatives reduce atherosclerotic plaque size and vulnerability in ApoE(−/−) mice. Journal of Translational Medicine, 12, 326 10.1186/s12967-014-0326-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, S. H. , Cho, S. K. , Kang, S. S. , Bae, C. S. , Bai, Y. H. , Lee, S. H. , & Pak, S. C. (2003). Effect of apitherapy in piglets with preweaning diarrhea. The American Journal of Chinese Medicine, 31(2), 321–326. 10.1142/S0192415X03001004 [DOI] [PubMed] [Google Scholar]
- Colombo, A. , Chieffo, A. , Frasheri, A. , Garbo, R. , Masotti‐Centol, M. , Salvatella, N. , … Sardella, G. (2014). Second‐generation drug‐eluting stent implantation followed by 6‐ versus 12‐month dual antiplatelet therapy: The SECURITY randomized clinical trial. Journal of the American College of Cardiology, 64(20), 2086–2097. 10.1016/j.jacc.2014.09.008 [DOI] [PubMed] [Google Scholar]
- Dall'Armellina, E. , Karia, N. , Lindsay, A. C. , Karamitsos, T. D. , Ferreira, V. , Robson, M. D. , … Choudhury, R. P. (2011). Dynamic changes of edema and late gadolinium enhancement after acute myocardial infarction and their relationship to functional recovery and salvage index. Circulation. Cardiovascular Imaging, 4(3), 228–236. 10.1161/CIRCIMAGING.111.963421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didier, R. , Morice, M. C. , Barragan, P. , Noryani, A. A. L. , Noor, H. A. , Majwal, T. , … Gilard, M. (2017). 6‐ versus 24‐month dual antiplatelet therapy after implantation of drug‐eluting stents in patients nonresistant to aspirin: Final results of the ITALIC trial (Is There a Life for DES After Discontinuation of Clopidogrel). JACC. Cardiovascular Interventions, 10(12), 1202–1210. 10.1016/j.jcin.2017.03.049 [DOI] [PubMed] [Google Scholar]
- Dumic, J. , Lauc, G. , & Flogel, M. (2000). Expression of galectin‐3 in cells exposed to stress‐roles of jun and NF‐kappaB. Cellular Physiology and Biochemistry: International Journal of Experimental Cellular Physiology, Biochemistry, and Pharmacology, 10(3), 149–158. [DOI] [PubMed] [Google Scholar]
- Fan, X. , Wang, J. , Hou, J. , Lin, C. , Bensoussan, A. , Chang, D. , … Wang, B. (2015). Berberine alleviates ox‐LDL induced inflammatory factors by up‐regulation of autophagy via AMPK/mTOR signaling pathway. Journal of Translational Medicine, 13, 92 10.1186/s12967-015-0450-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisue, K. , & Tsujita, K. (2017). Current status of lipid management in acute coronary syndrome. Journal of Cardiology, 70(2), 101–106. 10.1016/j.jjcc.2017.02.004 [DOI] [PubMed] [Google Scholar]
- Fullerton, M. D. , Ford, R. J. , McGregor, C. P. , LeBlond, N. D. , Snider, S. A. , Stypa, S. A. , … Kemp, B. E. (2015). Salicylate improves macrophage cholesterol homeostasis via activation of Ampk. Journal of Lipid Research, 56(5), 1025–1033. 10.1194/jlr.M058875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funaro, S. , Galiuto, L. , Boccalini, F. , Cimino, S. , Canali, E. , Evangelio, F. , … Agati, L. (2011). Determinants of microvascular damage recovery after acute myocardial infarction: Results from the acute Myocardial Infarction Contrast Imaging (AMICI) multi‐centre study. European Journal of Echocardiography: The Journal of the Working Group on Echocardiography of the European Society of Cardiology, 12(4), 306–312. [DOI] [PubMed] [Google Scholar]
- Ganame, J. , Messalli, G. , Dymarkowski, S. , Rademakers, F. E. , Desmet, W. , Van de Werf, F. , & Bogaert, J. (2009). Impact of myocardial haemorrhage on left ventricular function and remodelling in patients with reperfused acute myocardial infarction. European Heart Journal, 30(12), 1440–1449. 10.1093/eurheartj/ehp093 [DOI] [PubMed] [Google Scholar]
- Giustino, G. , Harari, R. , Baber, U. , Sartori, S. , Stone, G. W. , Leon, M. B. , … Mehran, R. (2017). Long‐term safety and efficacy of new‐generation drug‐eluting stents in women with acute myocardial infarction: From the Women in Innovation and Drug‐Eluting Stents (WIN‐DES) collaboration. JAMA Cardiology, 2(8), 855–862. 10.1001/jamacardio.2017.1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamirani, Y. S. , Wong, A. , Kramer, C. M. , & Salerno, M. (2014). Effect of microvascular obstruction and intramyocardial hemorrhage by CMR on LV remodeling and outcomes after myocardial infarction: A systematic review and meta‐analysis. JACC. Cardiovascular Imaging, 7(9), 940–952. 10.1016/j.jcmg.2014.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, R. W. , Aggarwal, A. , Ou, F. S. , Klein, L. W. , Rumsfeld, J. S. , Roe, M. T. , … American College of Cardiology National Cardiovascular Data Registry (2013). Incidence and outcomes of no‐reflow phenomenon during percutaneous coronary intervention among patients with acute myocardial infarction. The American Journal of Cardiology, 111(2), 178–184. 10.1016/j.amjcard.2012.09.015 [DOI] [PubMed] [Google Scholar]
- Huang, Z. , Wang, L. , Meng, S. , Wang, Y. , Chen, T. , & Wang, C. (2011). Berberine reduces both MMP‐9 and EMMPRIN expression through prevention of p38 pathway activation in PMA‐induced macrophages. International Journal of Cardiology, 146(2), 153–158. 10.1016/j.ijcard.2009.06.023 [DOI] [PubMed] [Google Scholar]
- Ito, H. (2006). No‐reflow phenomenon and prognosis in patients with acute myocardial infarction. Nature Clinical Practice. Cardiovascular Medicine, 3(9), 499–506. 10.1038/ncpcardio0632 [DOI] [PubMed] [Google Scholar]
- Jinnouchi, H. , Kuramitsu, S. , Shinozaki, T. , Tomoi, Y. , Hiromasa, T. , Kobayashi, Y. , … Ando, K. (2017). Difference of tissue characteristics between early and late restenosis after second‐generation drug‐eluting stents implantation—An optical coherence tomography study. Circulation Journal: Official Journal of the Japanese Circulation Society, 81(4), 450–457. [DOI] [PubMed] [Google Scholar]
- Kloner, R. A. (2011). No‐reflow phenomenon: Maintaining vascular integrity. Journal of Cardiovascular Pharmacology and Therapeutics, 16(3–4), 244–250. 10.1177/1074248411405990 [DOI] [PubMed] [Google Scholar]
- Kumar, A. , Green, J. D. , Sykes, J. M. , Ephrat, P. , Carson, J. J. , Mitchell, A. J. , … Friedrich, M. G. (2011). Detection and quantification of myocardial reperfusion hemorrhage using T2*‐weighted CMR. JACC. Cardiovascular Imaging, 4(12), 1274–1283. 10.1016/j.jcmg.2011.08.016 [DOI] [PubMed] [Google Scholar]
- Libby, P. , Schwartz, D. , Brogi, E. , Tanaka, H. , & Clinton, S. K. (1992). A cascade model for restenosis. A special case of atherosclerosis progression. Circulation, 86(6 Suppl), III47–III52. [PubMed] [Google Scholar]
- Ma, A. , Wang, J. , Yang, L. , An, Y. , & Zhu, H. (2017). AMPK activation enhances the anti‐atherogenic effects of high density lipoproteins in ApoE−/− mice. Journal of Lipid Research, 58(8), 1536–1547. 10.1194/jlr.M073270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauri, L. , Kereiakes, D. J. , Yeh, R. W. , Driscoll‐Shempp, P. , Cutlip, D. E. , Steg, P. G. , … DAPT Study Investigators (2014). Twelve or 30 months of dual antiplatelet therapy after drug‐eluting stents. The New England Journal of Medicine, 371(23), 2155–2166. 10.1056/NEJMoa1409312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, S. , Wang, L. S. , Huang, Z. Q. , Zhou, Q. , Sun, Y. G. , Cao, J. T. , … Wang, C. Q. (2012). Berberine ameliorates inflammation in patients with acute coronary syndrome following percutaneous coronary intervention. Clinical and Experimental Pharmacology & Physiology, 39(5), 406–411. 10.1111/j.1440-1681.2012.05670.x [DOI] [PubMed] [Google Scholar]
- Nakazawa, G. , Ladich, E. , Finn, A. V. , & Virmani, R. (2008). Pathophysiology of vascular healing and stent mediated arterial injury. EuroIntervention: Journal OF EuroPCR in Collaboration with the Working Group on Interventional Cardiology of the European Society of Cardiology, 4(Suppl C), C7–C10. [PubMed] [Google Scholar]
- Nakazawa, G. , Otsuka, F. , Nakano, M. , Vorpahl, M. , Yazdani, S. K. , Ladich, E. , … Virmani, R. (2011). The pathology of neoatherosclerosis in human coronary implants bare‐metal and drug‐eluting stents. Journal of the American College of Cardiology, 57(11), 1314–1322. 10.1016/j.jacc.2011.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka, F. , Byrne, R. A. , Yahagi, K. , Mori, H. , Ladich, E. , Fowler, D. R. , … Joner, M. (2015). Neoatherosclerosis: overview of histopathologic findings and implications for intravascular imaging assessment. European Heart Journal, 36(32), 2147–2159. 10.1093/eurheartj/ehv205 [DOI] [PubMed] [Google Scholar]
- Pamukcu, B. , Lip, G. Y. , & Shantsila, E. (2011). The nuclear factor‐‐kappa B pathway in atherosclerosis: A potential therapeutic target for atherothrombotic vascular disease. Thrombosis Research, 128(2), 117–123. 10.1016/j.thromres.2011.03.025 [DOI] [PubMed] [Google Scholar]
- Papaspyridonos, M. , McNeill, E. , de Bono, J. P. , Smith, A. , Burnand, K. G. , Channon, K. M. , & Greaves, D. R. (2008). Galectin‐3 is an amplifier of inflammation in atherosclerotic plaque progression through macrophage activation and monocyte chemoattraction. Arteriosclerosis, Thrombosis, and Vascular Biology, 28(3), 433–440. 10.1161/ATVBAHA.107.159160 [DOI] [PubMed] [Google Scholar]
- Patel, J. , Martin, S. S. , & Banach, M. (2016). Expert opinion: The therapeutic challenges faced by statin intolerance. Expert Opinion on Pharmacotherapy, 17(11), 1497–1507. 10.1080/14656566.2016.1197202 [DOI] [PubMed] [Google Scholar]
- Payne, A. R. , Berry, C. , Kellman, P. , Anderson, R. , Hsu, L. Y. , Chen, M. Y. , … Arai, A. E. (2011). Bright‐blood T(2)‐weighted MRI has high diagnostic accuracy for myocardial hemorrhage in myocardial infarction: A preclinical validation study in swine. Circulation. Cardiovascular Imaging, 4(6), 738–745. 10.1161/CIRCIMAGING.111.965095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roser, E. , Grundemann, C. , Engels, I. , & Huber, R. (2016). Antibacterial in vitro effects of preparations from anthroposophical medicine. BMC Complementary and Alternative Medicine, 16(1), 372 10.1186/s12906-016-1350-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, A. J. , Al‐Saadi, N. , Abdel‐Aty, H. , Schulz‐Menger, J. , Messroghli, D. R. , & Friedrich, M. G. (2004). Detection of acutely impaired microvascular reperfusion after infarct angioplasty with magnetic resonance imaging. Circulation, 109(17), 2080–2085. 10.1161/01.CIR.0000127812.62277.50 [DOI] [PubMed] [Google Scholar]
- Tsang, C. M. , Cheung, Y. C. , Lui, V. W. , Yip, Y. L. , Zhang, G. , Lin, V. W. , … Tsao, S. W. (2013). Berberine suppresses tumorigenicity and growth of nasopharyngeal carcinoma cells by inhibiting STAT3 activation induced by tumor associated fibroblasts. BMC Cancer, 13, 619 10.1186/1471-2407-13-619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veer, E. , Ho, C. , O'Neil, C. , Barbosa, N. , Scott, R. , Cregan, S. P. , & Pickering, J. G. (2007). Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. The Journal of Biological Chemistry, 282(15), 10841–10845. 10.1074/jbc.C700018200 [DOI] [PubMed] [Google Scholar]
- Vasamsetti, S. B. , Karnewar, S. , Kanugula, A. K. , Thatipalli, A. R. , Kumar, J. M. , & Kotamraju, S. (2015). Metformin inhibits monocyte‐to‐macrophage differentiation via AMPK‐mediated inhibition of STAT3 activation: Potential role in atherosclerosis. Diabetes, 64(6), 2028–2041. 10.2337/db14-1225 [DOI] [PubMed] [Google Scholar]
- Vinten‐Johansen, J. , Zhao, Z. Q. , Zatta, A. J. , Kin, H. , Halkos, M. E. , & Kerendi, F. (2005). Postconditioning—A new link in nature's armor against myocardial ischemia‐reperfusion injury. Basic Research in Cardiology, 100(4), 295–310. 10.1007/s00395-005-0523-x [DOI] [PubMed] [Google Scholar]
- Wang, J. , Ma, A. , Zhao, M. , & Zhu, H. (2017). AMPK activation reduces the number of atheromata macrophages in ApoE deficient mice. Atherosclerosis, 258, 97–107. 10.1016/j.atherosclerosis.2017.01.036 [DOI] [PubMed] [Google Scholar]
- Welt, F. G. , & Rogers, C. (2002). Inflammation and restenosis in the stent era. Arteriosclerosis, Thrombosis, and Vascular Biology, 22(11), 1769–1776. 10.1161/01.ATV.0000037100.44766.5B [DOI] [PubMed] [Google Scholar]
- Ye, L. , Liang, S. , Guo, C. , Yu, X. , Zhao, J. , Zhang, H. , & Shang, W. (2016). Inhibition of M1 macrophage activation in adipose tissue by berberine improves insulin resistance. Life Sciences, 166, 82–91. 10.1016/j.lfs.2016.09.025 [DOI] [PubMed] [Google Scholar]
- Yu, X. H. , Zheng, X. L. , & Tang, C. K. (2015). Nuclear factor‐kB activation as a pathological mechanism of lipid metabolism and atherosclerosis. Advances in Clinical Chemistry, 70, 1–30. 10.1016/bs.acc.2015.03.004 [DOI] [PubMed] [Google Scholar]
- Zhang, B. , Pei, C. , Zhang, Y. , Sun, Y. , & Meng, S. (2017). High resting heart rate and high BMI predicted severe coronary atherosclerosis burden in patients with stable angina pectoris by SYNTAX score. Angiology, 3319717715579. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Shan, Y. , Wu, Y. , Xu, C. , Yu, X. , Zhao, J. , … Shang, W. (2017). Berberine suppresses LPS‐induced inflammation through modulating Sirt1/NF‐kB signaling pathway in RAW264.7 cells. International Immunopharmacology, 52, 93–100. 10.1016/j.intimp.2017.08.032 [DOI] [PubMed] [Google Scholar]
- Zhang, M. , Cresswell, N. , Tavora, F. , Mont, E. , Zhao, Z. , & Burke, A. (2014). In‐stent restenosis is associated with neointimal angiogenesis and macrophage infiltrates. Pathology, Research and Practice, 210(12), 1026–1030. 10.1016/j.prp.2014.04.004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Safety of the BBR Renal (A and B) and live function (C, D, E, F) in BBR‐treated patients who received 300 mg BBR, tid, for 3 months in addition to standard therapy (n = 30) and control patients who received standard therapy alone (n = 15) at baseline time and after 3 months of treatment. Data are shown in mean ± SD.