

Abstract
NEPA is the first fixed‐combination antiemetic composed of the neurokinin‐1 receptor antagonist netupitant (netupitant; 300 mg) and the 5‐hydroxytryptamine‐3 receptor antagonist palonosetron (palonosetron; 0.50 mg). This study evaluated the pharmacokinetic profiles of netupitant and palonosetron. The pharmacokinetic profiles of both drugs were summarized using data from phase 1‐3 clinical trials. netupitant and palonosetron have high absolute bioavailability (63%‐87% and 97%, respectively). Their overall systemic exposures and maximum plasma concentrations are similar under fed and fasting conditions. netupitant binds to plasma proteins in a high degree (>99%), whereas palonosetron binds to a low extent (62%). Both drugs have large volumes of distribution (cancer patients: 1656‐2257 L and 483‐679 L, respectively). netupitant is metabolized by cytochrome P450 3A4 to 3 major pharmacologically active metabolites (M1, M2, and M3). palonosetron is metabolized by cytochrome P450 2D6 to 2 major substantially inactive metabolites (M4 and M9). Both drugs have similar intermediate‐to‐low systemic clearances and long half‐lives (cancer patients: netupitant, 19.5‐20.8 L/h and 56.0‐93.8 hours; palonosetron: 7.0‐11.3 L/h and 43.8‐65.7 hours, respectively). netupitant and its metabolites are eliminated via the hepatic/biliary route (87% of the administered dose), whereas palonosetron and its metabolites are mainly eliminated via the kidneys (85%‐93%). Altogether, these data explain the lack of pharmacokinetic interactions between netupitant and palonosetron at absorption, binding, metabolic, or excretory level, thus highlighting their compatibility as the oral fixed combination NEPA, with administration convenience that may reduce dosing mistakes and increase treatment compliance.
Keywords: complementary profiles, netupitant, palonosetron, pharmacokinetics
Chemotherapy‐induced nausea and vomiting (CINV) is a common side effect of cytotoxic chemotherapy that significantly impairs patients’ quality of life and may compromise adherence to anticancer treatment.1, 2 Depending on the time of onset, it is classified as acute (occurring 0 to 24 hours after chemotherapy administration) or delayed (occurring >24 hours after chemotherapy administration, and lasting up to 5 days).2
Cytotoxic chemotherapeutics trigger nausea and vomiting through a complex network of neuroanatomic centers and neurotransmitters.2 Two important neurotransmitters are serotonin and substance P, which act on the 5‐hydroxytryptamine‐3 (5‐HT3) and neurokinin‐1 (NK1) receptors, respectively. Serotonin acts via the peripheral pathway, binding to the 5‐HT3 receptors present on the terminal side of vagal nerve afferents from the gastrointestinal tract.3 This binding conducts information from chemical stimuli to the brain, which either initiates emesis (acute CINV) or sensitizes the vagal nerve to substance P. In its turn, substance P acts via the central pathway, binding mainly to the centrally located NK1 receptors, which mediate the induction of vomiting by transferring the signal, via the vagal afferent nerves, to the vomiting center.4 Peripherally released serotonin is thought to be the main mediator of CINV occurring within the first 8 to 12 hours following chemotherapy; thereafter, NK1‐dependent mechanisms are thought to become predominant.5 In this context, antagonists of the 5‐HT3 and NK1 receptors have been developed for CINV prevention. Relevant examples of first‐generation 5‐HT3 receptor antagonists, with similar efficacy and toxicities, include ondansetron, granisetron (administered orally as tablets, subcutaneously as extended‐release injection, or transdermally using patches), dolasetron, tropisetron, azasetron, and ramosetron.6 Considering that these agents’ efficacy in delayed CINV is suboptimal, second‐generation 5‐HT3 receptor antagonists with a longer half‐life and a higher receptor‐binding affinity, such as palonosetron (palonosetron), have been developed.7
International guideline committees consistently recommend combination antiemetic regimens targeting multiple emetic pathways as the standard of care for CINV prevention. For patients receiving highly emetogenic chemotherapy, current guidelines recommend the combined use of a 5‐HT3 receptor antagonist, an NK1 receptor antagonist, and a corticosteroid as antiemetic prophylaxis.8, 9, 10 Furthermore, the American Society of Clinical Oncology guidelines recommend adding olanzapine to antiemetic prophylaxis of patients receiving highly emetogenic chemotherapy because of its capacity for reducing the likelihood of nausea.11 For patients receiving moderately emetogenic chemotherapy, the general recommendation for antiemetic prophylaxis is treatment with a 5‐HT3 receptor antagonist plus dexamethasone; however, the Multinational Association of Supportive Care in Cancer/European Society for Medical Oncology guidelines advise that carboplatin‐receiving patients should be treated with a combination of an NK1 receptor antagonist, a 5‐HT3 receptor antagonist, and dexamethasone.8 A triplet NK1 receptor antagonist‐containing regimen is an option for moderately emetogenic chemotherapy‐receiving patients in the American Society of Clinical Oncology guidelines9 and in the National Comprehensive Cancer Network guidelines, if patients have high‐risk factors.10 However, increasing the number of antiemetics administered together with various chemotherapies leads to higher treatment complexity. Furthermore, this complexity is compounded when intravenously administering drugs such as rolapitant and fosaprepitant, which were reported to lead to injection‐site reactions (eg, pain, erythema, edema, and thrombophlebitis).12 This, in turn, increases the probability of nonadherence by patients, if the medication is not taken as prescribed.13 As such, development of a fixed‐combination antiemetic that targets dual antiemetic pathways with a single, straightforward administration, thus leading to a decrease in patient nonadherence to treatment, has been pursued.14 In developing such a combination antiemetic, the second‐generation 5‐HT3 receptor antagonist palonosetron was selected, because of its longer half‐life and higher receptor‐binding affinity, to be combined with the new, highly selective NK1 receptor antagonist netupitant (netupitant). One of the benefits of a fixed combination is the decrease in doses of antiemetic administered for CINV prophylaxis in patients receiving highly emetogenic chemotherapy, from 10 doses reported in the case of treatment with rolapitant regimens, to 8 doses with aprepitant regimens, and to 5 reported with the fixed combination of netupitant and palonosetron.15
NEPA is the first oral fixed‐combination antiemetic agent, composed of netupitant (300 mg), a new, highly selective NK1 receptor antagonist, and the second‐generation 5‐HT3 receptor antagonist palonosetron (0.50 mg). By combining these 2 agents in a single capsule, NEPA targets 2 critical pathways associated with CINV and provides control of acute and delayed CINV in a single administration (reviewed in Lorusso et al16). Previous in vitro studies with NG108‐15 cells, which express both the 5‐HT3 and NK1 receptors, have shown that netupitant and palonosetron exhibit synergistic effects in their capacity to inhibit substance P–mediated response17 and additive effects with respect to triggering NK1 receptor internalization.18 This pharmacologic synergy was demonstrated exclusively for palonosetron and netupitant and supports the concept of the 2 drugs being part of a fixed combination.
Two pivotal studies with oral NEPA have shown superior CINV prevention in the delayed and overall phases and similar safety vs oral palonosetron 0.50 mg following highly emetogenic chemotherapy19 or anthracycline‐cyclophosphamide–based chemotherapy.14, 20 In addition, a third pivotal safety study has shown that oral NEPA is highly efficacious and well tolerated over multiple cycles of highly emetogenic chemotherapy (non‐anthracycline‐cyclophosphamide–based) or moderately emetogenic chemotherapy.21 On the basis of the results from these 3 trials, oral NEPA was approved by the US Food and Drug Administration in 2014 and by the European Medicines Agency in 2015 for the prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of cancer chemotherapy, including but not limited to highly emetogenic chemotherapy.22, 23 In 2015, the antiemetic guidelines from the Multinational Association of Supportive Care in Cancer/European Society for Medical Oncology, National Comprehensive Cancer Network, and American Society of Clinical Oncology included NEPA plus dexamethasone as a treatment option for patients receiving highly emetogenic chemotherapy or moderately emetogenic chemotherapy.8, 9, 10 An IV NEPA formulation has also been developed to further improve convenience and has just been approved by the Food and Drug Administration.22
In this publication, we aim to examine the pharmacokinetic (PK) profiles of netupitant and palonosetron on oral administration as a fixed combination (300 mg netupitant, 0.50 mg palonosetron) to offer further scientific and clinical reasons for these 2 drugs to be used in combination for prevention of CINV.
Methods
Study Design and Treatments
All the study protocols were approved by the ethical review committees for each center, subjects provided written informed consent, and investigators and site personnel followed the Good Clinical Practices, International Conference on Harmonization E6, Declaration of Helsinki (2008) ethical principles, and the local laws and regulations. The PK profiles of netupitant and palonosetron have been reviewed and summarized using data from unpublished and previously published phase 1‐3 clinical trials following administration of netupitant and palonosetron single agents, and of the oral NEPA combination in healthy volunteers and cancer patients. Table 1 lists all the clinical trials addressed in this manuscript; references are provided for the previously published trials (studies 2, 6, 7, and 8). The scope, design, and methods of the studies are illustrated in Table 1. For some, details have been reported previously, as indicated in the table.
Table 1.
Pharmacokinetic Studies of netupitant, palonosetron, and NEPA Included in This Analysisa
| Study | Study Design and Objective | Subjects Enrolled/Analyzed | Dose and Dosage Form |
|---|---|---|---|
| Single Agent in Healthy Volunteers | |||
| Study 122 | ADME study of [14C]‐palonosetron | 6 healthy volunteers (all M), age 30–54 yr | 0.75 mg [14C]‐palonosetron (oral solution), single administration (fasted state) |
| Study 228 | ADME study of [14C]‐netupitant | 6 healthy volunteers (all M), age 32–56 yr | 300 mg [14C]‐netupitant (oral suspension), single administration (fasted state) |
| NEPA Combination in Healthy Volunteers | |||
| Study 331 | PK drug interaction and safety study between netupitant and palonosetron | 18 healthy volunteers (9 M, 9 F), age 18–43 yr | netupitant 450‐mg capsules, single oral administration, alone or combined with palonosetron 0.75‐mg capsules |
| Study 4 | Pilot bioequivalence study of NEPA capsule vs a combination of netupitant 300 mg and palonosetron 0.50 mg | 8 healthy volunteers (all M), age 19–45 yr | NEPA capsules, netupitant capsules, palonosetron soft gel capsules, single oral administration (fasted state) |
| Study 5 | Bioequivalence study of NEPA capsule vs a combination of netupitant 300 mg and palonosetron 0.50 mg |
|
NEPA capsules, single oral administration (fasted state) |
| Study 642 | Crossover study to evaluate PK and safety of NEPA in combination with ethinylestradiol and levonorgestrel | 24 healthy volunteers (all F), age 19–40 yr | NEPA capsules + 2 tablets of ethinylestradiol and levonorgestrel, single oral administration (fasted state) vs 2 tablets of ethinylestradiol and levonorgestrel, single oral administration (fasted state) |
| Study 742 | Crossover study to evaluate the effect of concomitant administration of ketoconazole or rifampicin on the PK of netupitant and palonosetron | 36 healthy volunteers (21 M, 15 F); PK population: N = 35; Ketoconazole group: 17 subjects (6 F, 11 M), age 33–55 yr; rifampicin group: 18 subjects (8 F, 10 M), age 32–55 yr | NEPA capsules, single oral administration with/without coadministration of ketoconazole 400‐mg tablets (fasted state) × 12 consecutive days; NEPA, single oral administration with/without coadministration of rifampicin 600‐mg tablets (fasted state) × 17 consecutive days |
| Study 843 | Crossover study to investigate the effect of food (comparison fasted vs fed condition), with 1 parallel group of elderly subjects to investigate the effect of age (elderly vs younger subjects in the fasted group) on NEPA PK | 36 healthy volunteers (22 M, 14 F); PK population in crossover part: 22 healthy volunteers, age 22–45 yr; PK population in the parallel part: 12 healthy volunteers, age 66–79 yr | NEPA capsules, single oral administration (fasted or fed [standard high‐fat, high‐caloric breakfast] state) |
| Study 9 | Replicate crossover bioequivalence study between NEPA capsules produced by 2 different manufacturers: planned commercial product vs phase 3 and late phase 1 NEPA | PK population for netupitant: 82 healthy volunteers (65 M, 17 F); PK population for palonosetron: 79 healthy volunteers (63 M, 16 F) | NEPA capsules, single oral administration (fasted state) |
| Study 10 | Comparative bioavailability study of 3 different netupitant formulations | 24 healthy volunteers (all M), age 18–47 yr | NEPA capsules with standard dissolution, netupitant 300‐mg oral suspension, palonosetron 0.5‐mg soft gel capsules, single oral administration (fasted state) |
| NEPA Combination in Cancer Patients | |||
| Study 1144 | Population PK and pharmacodynamic modeling study of netupitant, its metabolites M1, M2, M3, and palonosetron, following NEPA administration in a phase 3 pivotal clinical trial | netupitant analysis: 117 cancer patients (4 M, 113 F), age 29–75 yr; palonosetron analysis: 118 cancer patients (5 M, 113 F), age 29–75 yr | NEPA tablets, single oral administration (fasted state) + dexamethasone 12 mg |
| Study 1222 | PK/safety and drug interaction study of NEPA with docetaxel, etoposide, or cyclophosphamide |
|
NEPA capsules, single oral administration (fasted state) + docetaxel 75 to 100 mg/m2 IV solution/etoposide 35 to 100 mg/m2 IV solution/cyclophosphamide 500 to 1000 mg/m2 IV solution |
ADME, absorption, distribution, metabolism, and excretion; F, female; IV, intravenous; M, male; NEPA, fixed combination of 300‐mg netupitant and 0.50‐mg palonosetron in a single capsule; PK, pharmacokinetic.
For the studies of NEPA, only PK values from the 300‐mg netupitant and 0.50‐mg palonosetron dose groups are included.
Assessments
Bioanalytical Methods
Blood samples for the determination of plasma concentrations of palonosetron and netupitant (and its metabolites M1, M2, and M3) were collected on day 1 at predose and at predefined times after study drug administration. Plasma samples were analyzed by 2 validated liquid chromatography–tandem mass spectrometry methods. Both methods proved to be specific, sensitive, accurate, and precise according to the acceptance criteria set forth in the relevant bioanalytic guidelines.24, 25 In the first method, netupitant and its metabolites M1, M2, and M3 were extracted from 50.0 μL of human K2–ethylenediaminetetraacetic acid plasma spiked with the respective deuterated internal standards by liquid‐liquid extraction. After the extraction, the organic layer was evaporated under a nitrogen stream. The extracts were reconstituted in the injection solvent, and the analytes were separated by reverse‐phase chromatography using a C18 column and isocratic elution. An API 4000 tandem mass spectrometer (AB SCIEX, Framingham, Massachusetts) equipped with a turbo ion spray (TIS) probe operating in multiple reaction monitoring in positive mode was used for analyte quantification. No interferences from endogenous plasma components, significant matrix effect, carryover, or effect of hemolyzed and hyperlipidemic plasma samples were observed on analyte quantification. Intra‐ and interrun precision and accuracy were estimated by replicate sample analysis at low, medium, and high concentrations. The validated calibration ranges were from 2 to 1000 ng/mL for netupitant and from 2 to 500 ng/mL for M1, M2, and M3. The lower limit of quantification was 2 ng/mL for the 4 analytes.
In the second method, palonosetron was extracted from 100.0 μL of human K2–ethylenediaminetetraacetic acid plasma spiked with deuterated palonosetron (internal standard) by solid‐phase extraction. The extract was taken to dryness under a nitrogen stream and reconstituted in the injection solvent. Separation was performed by reverse‐phase chromatography using a C18 column and gradient elution. An API 4000 tandem mass spectrometer equipped with a TIS probe operating in multiple reaction monitoring in positive mode was used for analyte quantification. No interferences from endogenous plasma components, significant matrix effect, carryover or effects of hemolyzed and hyperlipidemic plasma samples were observed on analyte quantification. The validated calibration range was from 0.05 to 2 ng/mL. The lower limit of quantification was 0.05 ng/mL.
Pharmacokinetic Data Analysis
Dense individual plasma concentration‐time profiles of netupitant, its metabolites M1, M2, and M3, and palonosetron were analyzed by noncompartmental methods using the WinNonlin software (version 6.3 and later, Certara Inc., Princeton, New Jersey). Main PK parameters estimated were the maximum plasma concentration (Cmax), time to reach maximum plasma concentration (tmax), bioavailability (F), area under the plasma concentration‐time curve from zero to infinity (AUCinf), volume of distribution in the postdistribution phase (Vz/F), systemic clearance (CL/F), and terminal half‐life (t1/2). Sparse population PK data were analyzed by the NONMEM® software (double precision, version VII, level 7.20, Icon plc, Dublin, Ireland), using a nonlinear mixed‐effects modeling (NM‐TRAN version III level 1.0, and PREDPP version IV level 1.0 or greater). The first‐order conditional estimation method with interaction was used for model development. Covariates were selected via exploratory methods, including graphic assessments and generalized additive model analysis. A stepwise addition (α = 0.01) and a backward elimination (α = 0.001) method was used to determine the covariates to be included in the final model. Goodness‐of‐fit plots and visual predictive check were used to determine if the model could adequately describe the netupitant and palonosetron concentrations. From the population PK data analysis, additional parameters such as typical values of volume of distribution of the central compartment, intercompartmental clearance, volume of the peripheral compartment, absorption rate constant, lag time, and model‐predicted individual Cmax, tmax, and AUCinf were estimated (Helsinn, data on file). In addition, other parameters such as the extent of excretion in urine and the unbound fraction in plasma (fu) were reported.
Statistical Analysis
The data are summarized by descriptive statistics. Arithmetic and geometric means, standard deviations, and the coefficient of variation (CV%) have been estimated.
Results
The PK profiles of netupitant, its active metabolites M1, M2, and M3, and palonosetron after oral administration of the fixed‐combination antiemetic NEPA (netupitant 300 mg and palonosetron 0.50 mg) are illustrated in Figure 1 (plasma concentration‐time profiles) and outlined in Tables 2 through 4 (PK parameters), which list data from previously unpublished as well as published clinical trials.
Figure 1.

Mean plasma concentration‐time (with standard deviation) curves of netupitant, its active metabolites M1, M2, and M3, and palonosetron after single administration of oral NEPA. NEPA, fixed combination of 300 mg netupitant and 0.50 mg palonosetron in a single capsule. Source: Study 8.
Table 2.
Pharmacokinetic Parameters for Netupitant and Palonosetron Following a Single Oral Administration of NEPA
| Study No. (Type) | Subjects (N) | Treatments (Dose and Route) | Cmax (ng/mL) | tmax (h) | AUCinf (ng ⋅ h/mL) | CL/F (L/h) | Vz/F (L) | t1/2 (h) |
|---|---|---|---|---|---|---|---|---|
| Netupitant Pharmacokinetic Parameters, Arithmetic Mean (CV%) or Range (Min‐Max) | ||||||||
| Study 4 (pilot bioequivalence study) | Healthy volunteers (8) | FDC (netupitant 300 mg palonosetron 0.50 mg orally) | 533.5 (45.8) | 5.38 (22.1) | 17 698 (42.2) | 20.9 (56.1) | 2783 (67.0) | 89.2 (43.24) |
| Study 5 (bioequivalence study) | Healthy volunteers (47) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) | 434.1 (55.8) | 5.07 (20.0) | 14 402 (50.7) | 26.3 (47.4) | 3314 (53.1) | 95.6 (61.54) |
| Study 6 (PK/safety DDI crossover study) | Healthy volunteers (24) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + oral contraceptives | 599.6 (61.7) | 4.36 (21.0) | 16 519 (42.2) | 25.1 (59.7) | 2755 (75.4) | 75.3 (44.8) |
| Study 7 (PK DDI crossover study) | Healthy volunteers, control group (17) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + ketoconazole (400 mg orally) | 546 (44.1) | 5.27 (15.3) | 17 971 (31.3) | 18.4 (33.5) | 2342 (45.8) | 86.6 (25.7) |
| Study 7 (PK DDI crossover study) | Healthy volunteers, control group (18) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + rifampicin (600 mg orally) | 498.1 (45.3) | 5.12 (10.4) | 16 944 (34.9) | 19.9 (38.2) | 2540 (76.4) | 87.6 (53.1) |
| Study 8 (PK food and age crossover study) | Healthy volunteers (22) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally), fasted | 596.4 (39.1) | 5.14 (17.3) | 20 039 (41.9) | 20.5 (52.7) | 2851 (57.3) | 101.2 (52.2) |
| Study 8 (PK food and age crossover study) | Healthy volunteers (22) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally), fed | 649.8 (21.8) | 5.66 (17.1) | 22 391 (38.6) | 16.3 (23.1) | 1984 (40.1) | 86.3 (46.2) |
| Study 9 (replicate crossover bioequivalence study) | Healthy volunteers (82) | FDC commercial product (netupitant 300 mg, palonosetron 0.50 mg orally) | 454.0 (52.4) | 5.01 (35.4) | 13 863 (41.6) | 26.2 (51.2) | 2737 (49.8) | 76.6 (37.6) |
| Study 10 (comparative bioavailability study) | Healthy volunteers (24) | FDC standard dissolution (netupitant 300 mg, palonosetron 0.50 mg orally) | 373.6 (37.1) | 5.15 (15.0) | 13 303 (34.5) | 25.7 (40.9) | 3162 (45.9) | 89.9 (39.7) |
| Healthy Volunteers, Min‐Max | 373.6–649.8 | 4.36–5.66 | 13 303–22 391 | 16.3–26.3 | 1984–3314 | 75.3–101.2 | ||
| Study 11 (population PK/pharmacodynamic study) | Patients (117) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally), PK population | 567.3 (38.2) | 4.02 (59.1) | 17 284 (57.0) | 20.5 (%RSE 16.3) | V0 = 486 (%RSE = 8.02) V1 = 1170 (%RSE = 45.0) | 92.8 (32.8) |
| Study 12 (PK/safety and DDI study) | Patients (8) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + docetaxel (75–100 mg/m2 IV) | 486.3 (51.2) | 4.56 (18.0) | 16 130 (30.7) | 19.5 (30.7) | 1859 (79.0) | 93.8 (47.7) |
| Study 12 (PK/safety and DDI study) | Patients (12) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + etoposide (35–100 mg/m2 IV) | 518.8 (50.7) | 4.54 (37.7) | 18 160 (45.7) | 20.8 (55.9) | 1856 (52.9) | 69.1 (30.9) |
| Study 12 (PK/safety and DDI study) | Patients (10) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + cyclophosphamide (500–1000 mg/m2 IV) | 477.3 (48.5) | 4.16 (23.7) | 16 440 (29.8) | 19.7 (31.4) | 2257 (30.5) | 88.2 (28.2) |
| Patients, Min‐Max | 477.3–567.3 | 4.02–4.56 | 16 130–18 160 | 19.5–20.8 | 1656–2257 | 56.0–93.8 | ||
| Palonosetron Pharmacokinetic Parameters, Arithmetic Mean (CV%) or Range (Min‐Max) | ||||||||
| Study 4 (pilot bioequivalence study) | Healthy volunteers (8) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) | 1.180 (20.6) | 4.19 (30.6) | 50.210 (29.0) | 11.5 (23.8) | 658 (18.7) | 42.1 (29.8) |
| Study 5 (bioequivalence study) | Healthy volunteers (47) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) | 1.530 (25.6) | 4.68 (51.2) | 56.710 (32.8) | 9.6 (28.3) | 586 (33.1) | 44.2 (34.3) |
| Study 6 (PK/safety DDI crossover study) | Healthy volunteers (24) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + oral contraceptives | 0.909 (17.6) | 3.92 (33.6) | 40.079 (22.5) | 14.5 (25.1) | 842 (27.4) | 41.4 (32.1) |
| Study 7 (PK DDI crossover study) | Healthy volunteers, control group (17) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + ketoconazole (400 mg orally) | 0.775 (23.9) | 4.83 (23.9) | 37.524 (25.5) | 14.2 (27.0) | 923 (33.0) | 48.3 (46.7) |
| Study 7 (PK DDI crossover study) | Healthy volunteers, control group (18) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + rifampicin (600 mg orally) | 0.772 (26.7) | 4.23 (24.8) | 35.714 (37.7) | 15.5 (29.2) | 799 (24.7) | 37.1 (20.4) |
| Study 8 (PK food and age crossover study) | Healthy volunteers (22) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally), fasted | 0.786 (28.4) | 4.34 (19.6) | 33.645 (26.7) | 17.9 (34.1) | 905 (20.7) | 36.9 (23.6) |
| Study 8 (PK food and age crossover study) | Healthy volunteers (22) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally), fed | 0.768 (20.7) | 4.96 (24.3) | 33.199 (20.9) | 17.8 (28.0) | 962 (24.5) | 38.9 (29.1) |
| Study 9 (replicate crossover bioequivalence study) | Healthy volunteers (82) | FDC commercial product (netupitant 300 mg, palonosetron 0.50 mg orally) | 1.271 (25.8) | 3.76 (40.6) | 48.165 (26.4) | 11.1 (27.6) | 575 (28.5) | 37.2 (29.1) |
| Healthy volunteers, Min‐Max | 0.768–1.530 | 3.76–4.96 | 33.199–56.710 | 9.6–17.9 | 575–962 | 36.9–48.3 | ||
| Study 11 (population PK/pharmacodynamic study) | Patients (117) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally), PK population | 1.378 (28.3) | 3.01 (66.4) | 68.611 (33.4) | 7.6 (%RSE = 3.64) | V0 = 367 (%RSE = 4.44) V1 = 116 (%RSE = 8.97) | NA |
| Study 12 (PK/safety and DDI study) | Patients (8) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + docetaxel (75–100 mg/m2 IV) | 1.158 (32.9) | 5.44 (57.8) | 85.580 (50.9) | 7.0 (39.8) | 633 (33.9) | 65.7 (20.5) |
| Study 12 (PK/safety and DDI study) | Patients (12) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + etoposide (35–100 mg/m2 IV) | 0.898 (38.4) | 5.33 (51.9) | 49.260 (25.9) | 10.8 (27.2) | 679 (22.7) | 45.7 (32.7) |
| Study 12 (PK/safety and DDI study) | Patients (10) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + cyclophosphamide (500–1000 mg/m2 IV) | 0.850 (22.3) | 4.41 (40.1) | 48.120 (32.4) | 11.3 (29.8) | 668 (18.6) | 43.9 (26.1) |
| Patients, Min‐Max | 0.850–1.378 | 3.01–5.33 | 48.120–85.580 | 7.0–11.3 | 483–679 | 43.8–65.7 | ||
%RSE: percent relative standard error; AUCinf indicates area under the plasma concentration‐time curve from time zero to infinity; CL/F, systemic clearance; Cmax, maximum plasma concentration; CV%, coefficient of variation; DDI, drug‐drug interaction; FDC, fixed‐dose combination; IV, intravenous; NA, not available; NEPA, fixed combination of 300 mg netupitant and 0.50 mg palonosetron in a single capsule; PK, pharmacokinetic; t1/2, terminal half‐life; tmax, time to maximum plasma concentration; V0, volume of central compartment; V1, volume of peripheral compartment; Vz/F, volume of distribution in the postdistribution phase.
Table 4.
Pharmacokinetic Parameters for netupitant, Its Active Metabolites M1, M2, and M3, and palonosetron Following Oral Administration of netupitant or palonosetron Alone, or netupitant and palonosetron Combined (Single Dose)a
| Treatment (Study 3) | Analyte | Cmax (ng/mL) | tmax (h) | AUCinf (ng ⋅ h/mL) | t1/2 (h) |
|---|---|---|---|---|---|
| netupitant 450 mg (N = 18) | netupitant | 650 (39.6) | 5.98 (82.8) | 25 927 (39.2) | 90.4 (59.1) |
| M1 | 60.7 (20.6) | 24.2 (68.1) | 8185 (31.1) | 69.6 (40.3) | |
| M2 | 228 (49.9) | 4.47 (9.9) | 2916 (47.5) | 67.2 (55.4) | |
| M3 | 93.0 (26.2) | 19.7 (80.5) | 8090 (33.2) | 67.9 (45.5) | |
| palonosetron 0.75 mg (N = 17) | palonosetron | 1.638 (25.4) | 5.50 (25.5) | 70.813 (28.8) | 37.1 (31.8) |
| netupitant 450 mg + palonosetron 0.75 mg (N = 18) | netupitant | 660 (49.4) | 5.61 (82.4) | 26 241 (50.4) | 88.1 (41.9) |
| palonosetron | 1.863 (26.1) | 4.69 (16.0) | 77.254 (32.9) | 38.5 (28.1) | |
| M1 | 59.1 (29.5) | 19.7 (64.8) | 8073 (45.2) | 73.8 (43.6) | |
| M2 | 219 (49.3) | 4.39 (8.2) | 2761 (58.3) | 68.6 (56.5) | |
| M3 | 97.7 (37.2) | 11.4 (74.3) | 7927 (50.5) | 78.8 (45.4) |
AUCinf, area under the plasma concentration‐time curve from time zero to infinity; Cmax, maximum plasma concentration; CV%, coefficient of variation; t1/2, terminal half‐life; tmax, time to maximum plasma concentration.
Arithmetic mean (CV%) values are indicated.
Absorption
According to the Biopharmaceutics Classification System, palonosetron and netupitant are class 1 and 2, respectively. palonosetron is freely soluble in water26 and has good permeability properties; its apical‐to‐basolateral apparent coefficient of permeability [Papp A/B] determined in a Madin‐Darby canine kidney strain II monolayer cell system ranged from 16*10−6 cm/sec (at 0.5 μM palonosetron) to 65*10−6 cm/sec (at 50 μM palonosetron) (Helsinn, data on file). netupitant is very slightly soluble in water22 and has high permeability properties; its Papp A/B, determined in a Caco‐2 monolayer cell system, is >20*10−6 cm/sec in the concentration range 1 to 100 μM netupitant (Helsinn, data on file). Permeability data were consistent with the octanol‐water partition coefficients (logP), which are 2.72 for palonosetron (calculated logP)27 and 5.1 for netupitant (experimental logP; Helsinn, data on file). palonosetron is a substrate, but not an inhibitor, of the P‐glycoprotein (P‐gp) efflux transporter. Differently, netupitant is a P‐gp inhibitor but not a P‐gp substrate.22 Clinical studies showed that the extent of oral bioavailability of palonosetron, close to 100% (Table 2), is not affected by its affinity for P‐gp and by netupitant coadministration (Table 4). Despite the significant presence in the gut wall and liver of cytochrome P450 3A4 (CYP3A4), the major CYP450 isoform involved in netupitant metabolism,22 and CYP2D6, the major isoform responsible for palonosetron metabolism22, the rate and extent of intestinal absorption of netupitant and palonosetron are not affected by a potential first‐pass effect. Altogether, the combination of these factors results in prompt, extended, and complete absorption of both drugs. netupitant and palonosetron were detected early in the systemic circulation after oral NEPA administration. Plasma concentrations were already measurable within 1 hour after drug administration, and, at 3 hours, plasma concentrations of netupitant and palonosetron represented approximately 80% of their mean Cmax. Population PK modeling in patients indicated absorption lag times of 0.77 hour and 0.79 hour for netupitant and palonosetron, respectively, with absorption rate constants of 0.956 h−1 and 2.66 h−1 for netupitant and palonosetron, respectively. netupitant and palonosetron have an extended absorption phase, with Cmax reached on average 4 to 5 hours after administration (Table 2). Drug concentrations fluctuating around the Cmax were observed between 3 and 8 hours after administration (Figure 1). The absolute bioavailability was estimated to reach 63% to 87% in the case of netupitant (Helsinn, data on file) and 97% for palonosetron.26 Food did not affect the rate and extent of absorption of both drugs. The overall systemic exposure (AUCinf) and Cmax were similar for both netupitant and palonosetron when they were administered under fed (breakfast with high fat content) or fasting conditions in an open, randomized, 2‐way crossover study in healthy volunteers (Table 2).
Distribution
Both netupitant and palonosetron distribute widely throughout the body, with a large apparent volume of distribution. In healthy subjects, mean Vz/F (CV%) ranged between 1984 (40.1) and 3314 (53.1) L for netupitant, and between 575 (28.5) and 962 (24.5) L for palonosetron (Table 2). Population PK modeling in patients indicated volumes of central and peripheral compartments (relative standard error) of 486 (8.02) L and 1170 (45.0) L, respectively, for netupitant, and 367 (4.44) L and 116 (8.97) L, respectively, for palonosetron. Other studies in cancer patients showed similar mean Vz/F (CV%) values for both netupitant, 1856 (52.9) to 2257 (30.5) L, and palonosetron, 633 (33.9) to 679 (22.7) L. netupitant shows a high degree of binding to plasma proteins, with fu <1%. The 3 main active metabolites (M1, M2, and M3) also show high protein binding, with fu <3%. Conversely, palonosetron shows a relatively low degree of binding to plasma proteins (fu = 38%).
Metabolism
Netupitant is largely metabolized to 3 main metabolites (M1, M2, and M3) and a minor metabolite (M4) (Figure 2). Metabolites are, respectively, the N‐demethyl derivative (M1), the N‐oxide derivative (M2), the hydroxymethyl derivative (M3), and the N‐oxide N‐demethyl derivative (M4) of netupitant.22 Metabolite‐to‐netupitant exposure ratios for the main metabolites were 29% (M1), 14% (M2), and 33% (M3).28 Exposure to metabolite M4 in the systemic circulation was lower (metabolite‐to‐netupitant exposure ratio 3%). Metabolites were all shown to be pharmacologically active in the gerbil foot‐tapping NK1 assay, where M3 was as potent as netupitant, and M1 and M2 were less potent. A binding assay was performed with M4 and, as in the case of M1 to M3, M4 showed high NK1 receptor‐binding activity (Helsinn, data on file).
Figure 2.
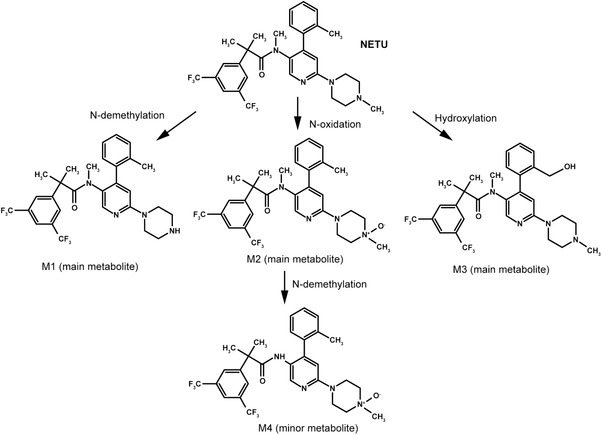
Proposed metabolic scheme for netupitant.
In humans, the 2 main metabolites of palonosetron are M4 (6‐S‐hydroxy‐palonosetron) and M9 (N‐oxide‐palonosetron).29 In the systemic circulation, AUCinf to M9 was 6% to 14% of that of palonosetron after both oral and intravenous administration. Mean exposure to M4 ranged from 9% to 16% of the parent. These metabolites are inactive, each having <1% of the 5‐HT3 receptor antagonist activity of palonosetron (Helsinn, data on file).
netupitant metabolism is mediated primarily by CYP3A4 and to a lesser extent by CYP2C9 and CYP2D6.22
The percent contribution of CYP3A4 to netupitant metabolism was estimated to be 95%, according to the following relationship30:
where CLint and CLint(I) are the intrinsic clearance values of netupitant in human liver microsomes, expressed as μL/min/mg protein, in the absence and presence of a selective CYP3A4 inhibitor, respectively (Helsinn, data on file). netupitant exposure has been shown to increase upon coadministration with strong CYP3A4 inhibitors (eg, ketoconazole) and to decrease upon coadministration with strong CYP3A4 inducers (eg, rifampicin). After administration of a single dose of NEPA, alone or in combination with 400‐mg ketoconazole for 12 days, netupitant Cmax and AUCinf increased 1.2‐fold and 2.4‐fold, respectively. Following single‐dose NEPA combined with a daily administration of 600‐mg rifampicin for 17 days, netupitant Cmax and AUCinf decreased 2.2‐fold and 4.9‐fold, respectively.31
In vitro, netupitant proved to be a moderate inhibitor of CYP3A4.32 In vivo, the exposure to the CYP3A4 substrates dexamethasone, erythromycin, and midazolam has been shown to increase upon coadministration with netupitant, whereas the exposure to the contraceptives ethinylestradiol and levonorgestrel was not affected by NEPA coadministration.31 Considering the likelihood of coadministration of netupitant and dexamethasone and the significant dose‐dependent increase in dexamethasone exposure following coadministration with netupitant, it is recommended that oral doses of dexamethasone should be reduced by approximately 50% when given in combination with netupitant.31
The potential for netupitant autoinhibition of CYP3A4 can be excluded because, in the clinical practice, NEPA is administered as a single‐dose treatment per chemotherapy cycle. Absence of netupitant autoinhibition was also confirmed by a multiple‐dose PK study in healthy subjects performed in the frame of a development program for an indication different than CINV. In this study, healthy volunteers received daily oral doses of 100, 300, and 450 mg netupitant (8 subjects per dose) for 7 days (Helsinn, data on file). netupitant exposure increased approximately 3‐fold after 7 days of dosing. The observed accumulation factor Rav, estimated as the ratio of the daily AUC on day 7 and day 1 after 300 mg NEPA, was 2.74, in keeping with predictions of netupitant accumulation at steady state (Rav = 2.80) obtained by 2‐compartment PK modeling, based on the assumption of time‐independent pharmacokinetics.
Palonosetron is primarily metabolized by CYP2D6 and to a lesser extent by CYP3A4 and CYP1A2 enzymes. Genetic polymorphism of CYP2D6 showed no effect on the pharmacokinetics of palonosetron. The plasma concentrations and PK parameters of palonosetron and M9 were similar in subjects with extensive or poor metabolizer CYP2D6 status.26 Coadministration of palonosetron with metoclopramide, a mechanism‐based inhibitor of CYP2D6, or aprepitant, a moderate CYP3A4 inhibitor, did not alter palonosetron pharmacokinetics. No differences between palonosetron plasma concentrations were observed after administration of palonosetron alone or combined with the above‐mentioned drugs.26
Elimination
Both netupitant and palonosetron have an intermediate‐to‐low CL/F. The CL/F of netupitant and palonosetron, accounting for metabolic and nonmetabolic elimination, is similar for both drugs, with mean (CV%) values ranging from 16.3 (23.1) to 26.3 (47.4) L/h (netupitant) and from 9.6 (28.3) to 17.9 (34.1) L/h (palonosetron) in healthy subjects. In cancer patients, mean CL/F varied, similarly to healthy subjects, from 19.5 (30.7) to 20.8 (55.9) L/h (netupitant) and 7.0 (39.8) to 11.3 (29.8) L/h (palonosetron) (Table 2). As a result of the high Vz/F of netupitant and palonosetron, the intermediate‐to‐low CL/F, and the high degree of binding to plasma proteins of netupitant, both netupitant and palonosetron have a long t1/2 (Table 2). For both compounds, t1/2 values are similar in healthy subjects (netupitant mean [CV%] t1/2 was 75.3 [44.8] to 101.2 [52.2] hours, palonosetron mean t1/2 was 36.9 [23.6] to 48.3 [46.7] hours) and in patients (netupitant mean t1/2 [CV%] was 69.1 [30.9] to 93.8 [47.7] h and palonosetron mean [CV%] t1/2 was 43.9 [26.1] to 65.7 [20.5] hours) (Table 2).
The major elimination route for netupitant and its metabolites is hepatic/biliary, with 86.5% of the administered dose excreted through the feces, along with a minor percentage via the renal route.28 The mass balance recovery of the total radioactivity of [14C]‐netupitant and its metabolites M1, M2, and M3 was estimated to be 4.75% in urine.28 Elimination of approximately 90% of netupitant is estimated at day 29 after administration. Following a single oral 0.75 mg dose of [14C]‐palonosetron, palonosetron and its metabolites are primarily excreted via the kidney (85% to 93%), with only 5% to 8% of the dose eliminated with feces.22 In urine, palonosetron represented approximately 40% of the administered dose after both IV and oral administration, whereas M9 accounted for approximately 13% of the IV and oral palonosetron dose, and M4 for 11.5% after IV and 17.2% after oral administrations.22, 29
The potential for netupitant, its metabolites, and palonosetron to act as substrates for transporters has been evaluated in vitro.22 netupitant, M1, M2, and M3 are not substrates for the uptake transporters organic‐anion‐transport polypeptides 1B1 and 1B3, the organic‐cation‐transport proteins 1 and 2, the organic‐anion‐transport proteins 1 and 3, and the efflux transporters multidrug resistance protein 1/P‐glycoprotein (MDR1/P‐gp; with the only exception of M2), breast cancer resistance protein (BCRP), and bile salt export pump. Thus, the elimination of netupitant and its metabolites, primarily occurring via the hepatobiliary route, is unlikely mediated by hepatic transporters.
Palonosetron is a substrate for MDR1/P‐gp but not for other transporters. At the renal level, P‐gp may behave as a luminal efflux transporter for palonosetron elimination in urine. palonosetron renal clearance is, in fact, approximately 2‐fold greater than that expected for a drug with an unbound fraction in plasma of 0.38, such as palonosetron, undergoing glomerular filtration only; hence, P‐gp–mediated palonosetron active secretion may concur with palonosetron renal clearance.
Pharmacokinetic Profile of Active netupitant Metabolites
Metabolite M2 reached maximum concentrations at approximately 5 hours after NEPA administration, whereas metabolites M1 and M3 peaked later, showing mean tmax between 10 and 17 hours (Table 3). The concentration of M2 decreased more rapidly after the peak as compared with M1 and M3, and entered an apparent terminal elimination phase at approximately 48 hours. Metabolites M1 and M3 showed rather flat concentrations up to 24 (M3) or 72 hours (M1), subsequently entering the terminal elimination phase. The extent of metabolite formation is higher for M1 and M3 as compared with M2, as deduced from the metabolite‐to‐parent AUCinf ratio (Table 3). Plasma concentrations of M2 dropped below the lower limit of quantification of the bioanalytical method 4 days after dosing, whereas M1 and M3 were measurable, like their parent drug netupitant, up to the last observation time, that is, up to 10 days after NEPA administration. The elimination of metabolites appears to be formation‐rate limited, as shown by the parallel decline of the plasma curves of netupitant and its metabolites on the semilogarithmic scale (Figure 1) and similar t1/2 values (Table 3).
Table 3.
Pharmacokinetic Parameters for the Active Metabolites of netupitant M1, M2, and M3 Following a Single Oral Administration of NEPA
| Study No. (Type) | Subjects (N) | Treatments | Cmax (ng/mL) | tmax (h) | AUCinf (ng ⋅ h/mL) | t1/2 (h) |
|---|---|---|---|---|---|---|
| Metabolite M1 Pharmacokinetic Parameters, Arithmetic Mean (CV%) or Range (Min‐Max) | ||||||
| Study 7 (PK DDI crossover study) | Healthy volunteers, control group (17) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + ketoconazole (400 mg orally) | 39.2 (26.1) | 14.6 (72.5) | 5307 (24.7) | 81.4 (38.5) |
| Study 7 (PK DDI crossover study) | Healthy volunteers, control group (18) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + rifampicin (600 mg orally) | 40.0 (26.9) | 11.7 (53.2) | 4944 (34.8) | 73.3 (35.4) |
| Study 8 (PK food and age crossover study) | Healthy volunteers (22) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally), fasted | 43.7 (28.4) | 13.8 (54.3) | 5886 (38.0) | 82.2 (45.0) |
| Study 8 (PK food and age crossover study) | Healthy volunteers (22) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally), fed | 46.8 (13.7) | 15.3 (65.4) | 6700 (28.7) | 82.9 (32.8) |
| Study 10 (comparative bioavailability study) | Healthy volunteers (24) | FDC standard dissolution (netupitant 300 mg, palonosetron 0.50 mg orally) | 44.1 (31.5) | 9.85 (92.0) | 5165 (34.2) | 73.6 (35.2) |
| Healthy Volunteers, Min‐Max | 39.2–46.8 | 9.85–15.3 | 4944–6700 | 73.3–82.9 | ||
| Study 12 (PK/safety and DDI study) | Patients (8) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + docetaxel (75–100 mg/m2 IV) | 36.0 (32.4) | 12.0 (46.5) | AUClast, 4356 (40.8) | – |
| Study 12 (PK/safety and DDI study) | Patients (12) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + etoposide (35–100 mg/m2 IV) | 40.6 (33.8) | 16.9 (44.7) | 4203 (44.2) | 81.7 (28.4) |
| Study 12 (PK/safety and DDI study) | Patients (10) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + cyclophosphamide (500–1000 mg/m2 IV) | 39.8 (32.3) | 13.0 (105.5) | 5993 (18.3) | 91.4 (40.5) |
| Patients, Min‐Max | 36.0–40.6 | 12.0–16.9 | 4203–5993 | 81.7–91.4 | ||
| Metabolite M2 PK Parameters, Arithmetic Mean (CV%) or Range (Min‐Max) | ||||||
| Study 7 (PK DDI crossover study) | Healthy volunteers, control group (17) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + ketoconazole (400 mg orally) | 195.1 (47.4) | 5.06 (10.3) | 2161 (48.7) | 46.4 (75.3) |
| Study 7 (PK DDI crossover study) | Healthy volunteers, control group (18) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + rifampicin (600 mg orally) | 174.4 (37.8) | 4.73 (17.8) | 1854 (42.5) | 33.5 (57.0) |
| Study 8 (PK food and age crossover study) | Healthy volunteers (22) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally), fasted | 202.2 (48.1) | 4.64 (16.4) | 2254 (41.9) | 48.9 (93.4) |
| Study 8 (PK food and age crossover study) | Healthy volunteers (22) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally), fed | 147.4 (31.7) | 5.57 (12.8) | 1951 (32.9) | 43.0 (69.5) |
| Study 10 (comparative bioavailability study) | Healthy volunteers (24) | FDC standard dissolution (netupitant 300 mg, palonosetron 0.50 mg orally) | 165.6 (32.1) | 4.67 (19.4) | 1707 (41.4) | 55.9 (97.9) |
| Healthy volunteers, Min‐Max | 147.4–202.2 | 4.64–5.57 | 1707–2254 | 33.5–55.9 | ||
| Study 12 (PK/safety and DDI study) | Patients (8) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + docetaxel (75–100 mg/m2 IV) | 361.0 (56.9) | 4.13 (23.1) | 8527 (10.3) | 67.6 (30.9) |
| Study 12 (PK/safety and DDI study) | Patients (12) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + etoposide (35–100 mg/m2 IV) | 219.2 (55.2) | 3.88 (40.8) | 3719 (40.6) | 63.1 (49.0) |
| Study 12 (PK/safety and DDI study) | Patients (10) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + cyclophosphamide | 214.9 (27.9) | 4.45 (18.7) | 3061 (30.1) | 56.2 (35.6) |
| Patients, Min‐Max | 214.9–361.0 | 3.88–4.45 | 3061–8527 | 56.2–67.6 | ||
| Metabolite M3 PK Parameters, Arithmetic Mean (CV%) or Range (Min‐Max) | ||||||
| Study 7 (PK DDI crossover study) | Healthy volunteers, control group (17) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + ketoconazole (400 mg orally) | 74.5 (35.3) | 12.7 (64.2) | 4851 (29.8) | 64.0 (40.8) |
| Study 7 (PK DDI crossover study) | Healthy volunteers, control group (18) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + rifampicin (600 mg orally) | 66.4 (26.9) | 10.5 (54.1) | 4491 (42.3) | 48.8 (27.6) |
| Study 8 (PK food and age crossover study) | Healthy volunteers (22) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally), fasted | 81.8 (46.3) | 13.9 (58.9) | 5841 (45.4) | 65.6 (44.4) |
| Study 8 (PK food and age crossover study) | Healthy volunteers (22) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally), fed | 73.1 (24.6) | 16.7 (42.6) | 5747 (33.5) | 61.9 (33.1) |
| Study 10 (comparative bioavailability study) | Healthy volunteers (24) | FDC standard dissolution (netupitant 300 mg, palonosetron 0.50 mg orally) | 60.1 (30.5) | 11.0 (67.3) | 4054 (32.1) | 67.8 (30.5) |
| Healthy volunteers, Min‐Max | 60.1–81.8 | 10.5–16.7 | 4054–5841 | 48.8–67.8 | ||
| Study 12 (PK/safety and DDI study) | Patients (8) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + docetaxel (75–100 mg/m2 IV) | 64.4 (30.4) | 12.3 (42.9) | 5946 (34.2) | 80.3 (35.1) |
| Study 12 (PK/safety and DDI study) | Patients (12) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + etoposide (35–100 mg/m2 IV) | 74.3 (43.6) | 15.1 (55.2) | 5294 (31.9) | 65.4 (41.5) |
| Study 12 (PK/safety and DDI study) | Patients (10) | FDC (netupitant 300 mg, palonosetron 0.50 mg orally) + cyclophosphamide (500–1000 mg/m2 IV) | 68.2 (57.6) | 16.0 (45.6) | 5821 (32.9) | 72.8 (31.0) |
| Patients, Min‐Max | 64.4–74.3 | 12.3–16.0 | 5294–5946 | 65.4–80.3 | ||
AUCinf, area under the plasma concentration‐time curve from time zero to infinity; AUClast, area under the plasma concentration‐time curve from time zero to time of last quantifiable concentration; Cmax, maximum plasma concentration; CV%, coefficient of variation; DDI, drug‐drug interaction; FDC, fixed‐dose combination; IV, intravenous; NEPA, fixed combination of 300 mg netupitant and 0.50 mg palonosetron in a single capsule; t1/2, terminal half‐life; tmax, time to maximum plasma concentration.
In vitro, the inhibition properties of netupitant metabolites were investigated toward the main CYP450 isoforms (Helsinn, data on file). Metabolite M1 showed a CYP3A4 inhibition activity comparable to that of the parent drug and also moderate inhibition of CYP2D6, CYP2C8, and CYP2B6. M2 showed weak or no inhibition toward all CYP450 isoforms. M3 showed a moderate inhibition of CYP3A4 and weak or no inhibition toward the other CYP450 isoforms. With the exception of M1, which may contribute to the inhibitory activity of CYP3A4 observed in vivo after netupitant administration, the in vitro interactions of all metabolites toward the main CYP450 isoforms are devoid of clinical relevance considering that the ratio between the metabolites’ Cmax after administration of 300 mg netupitant and the respective in vitro concentrations required to achieve 50% of the CYP450 isoforms inhibition (IC50) were lower than 0.1. Metabolites M1, M2, and M3 did not induce CYP450 isoforms in human hepatocytes at in vivo relevant concentrations (Helsinn, data on file).
M2 only is a P‐gp substrate. netupitant metabolites M1, M2, and M3 are P‐gp and BCRP inhibitors in vitro (Helsinn, data on file). Considering that the metabolites’ Cmax after administration of 300 mg oral netupitant to the respective P‐gp and BCRP IC50 ratios are lower than 0.1, no interaction is expected in vivo between netupitant metabolites and P‐gp or BCRP substrates, similarly to what has been concluded for the parent drug.
Dose Proportionality
Dose‐proportional increases in systemic exposure were observed after single oral doses of netupitant from 300 to 450 mg (1‐ and 1.5‐fold of the recommended dose in oral NEPA capsules), and palonosetron from 0.25 to 6.8 mg (0.5‐ to 13.6‐fold of the recommended dose in oral NEPA capsules).22
Lack of Drug Interaction Between netupitant and palonosetron
netupitant and palonosetron show no relevant PK interactions at absorption, binding, metabolic, or excretory level. No change of the PK profiles of netupitant and palonosetron occurs after coadministration of the combined drugs as compared with the pharmacokinetics of single agents. In a study performed in healthy volunteers investigating possible PK interactions between netupitant (450 mg orally) and palonosetron (0.75 mg orally), mean (CV%) Cmax values of netupitant and palonosetron administered alone were 650 (39.6) ng/mL and 1.638 (25.4) ng/mL, respectively. These were similar to the values observed upon combined administration of netupitant and palonosetron (660 [49.4] ng/mL and 1.863 [26.1] ng/mL, respectively; Table 4). Additionally, values of AUCinf and t1/2 were similar across all study subjects, when netupitant or palonosetron was administered alone, as compared with combined netupitant and palonosetron administration31 (Table 4).
Discussion
International guidelines for CINV management in patients with cancer recommend a multidrug prophylaxis regimen, which is characterized by considerable treatment complexity. Development of a fixed‐combination antiemetic that targets 2 critical CINV‐associated pathways with a single, convenient administration is desirable for avoiding errors in administration, as well as for enhancing patient compliance and adherence to guidelines. For this purpose, the second‐generation 5‐HT3 receptor antagonist palonosetron was selected because of its longer half‐life and higher receptor‐binding affinity. palonosetron was combined with the new, highly selective NK1 receptor antagonist netupitant into the first oral fixed‐combination antiemetic agent NEPA, thus allowing for a decrease in the doses of antiemetic administered.15 This article highlights the optimal fit of relevant PK properties of palonosetron and netupitant, which may account for the high efficacy of NEPA in preventing acute and delayed CINV, as well as lack of any drug‐drug interaction between the 2 drugs. Additionally, their complementary pharmacodynamic properties (ie, synergistic effects on NK1 receptor antagonism) also provide support for their combination.
The high bioavailability of both netupitant and palonosetron indicates that both drugs undergo limited or no presystemic clearance nor clinically relevant interactions with intestinal efflux transporters. Both drugs show a prolonged absorption phase with peak concentrations occurring, on average, 4 to 5 hours after administration. netupitant and palonosetron show different affinities to plasma proteins (ie, high for netupitant and relatively low for palonosetron). This difference excludes the risk for interaction between the 2 drugs at a protein‐binding level. Because of its poor affinity to plasma proteins, palonosetron is unlikely to displace protein‐bound netupitant, thus avoiding any possible transient increase of plasma concentration of unbound netupitant and the risk of netupitant‐related adverse events.
Netupitant and palonosetron metabolism is mediated by different CYP450 isoforms. While the principal CYP450 isoform responsible for the metabolism of netupitant is CYP3A4, that for palonosetron is CYP2D6. In addition, netupitant is not an inhibitor or inducer of CYP2D6, and palonosetron does not inhibit or induce CYP3A4. To a minor extent, CYP2D6 and CYP2C9 were shown in vitro to have a marginal role in netupitant metabolism,32 and CYP3A4 and CYP1A2 in palonosetron metabolism.26 Because of the involvement of different CYP450 enzymes in the metabolism of these 2 agents, no clinically relevant metabolism‐based drug‐drug interactions are expected between netupitant and palonosetron when administered in combination.
Furthermore, no interactions between netupitant and palonosetron are expected during their absorption through the gut mucosa and elimination through the liver and the kidneys. The P‐gp pump is a major efflux transporter involved in the drug absorption process through the gut mucosa and the drug elimination process via biliary and renal excretion.33 In vitro interaction studies of netupitant and palonosetron with uptake and efflux transporters indicated that palonosetron is a P‐gp substrate but not a P‐gp inhibitor, whereas netupitant proved to be a P‐gp inhibitor but not a P‐gp substrate (Helsinn, data on file).22 Concomitant administration of palonosetron with P‐gp inhibitors such as netupitant could, in principle, increase palonosetron bioavailability. However, palonosetron bioavailability cannot be further increased, because it is already complete when palonosetron is administered alone in fed and fasting conditions, as well as in combination with netupitant. Hence, potential P‐gp inhibition by netupitant or other P‐gp inhibitors should not affect the rate and extent of palonosetron absorption.
At the renal level, P‐gp acts as a luminal efflux transporter that mediates the active secretion of substrate drugs into the urine.33 P‐gp‐mediated renal active secretion of palonosetron may occur, as evidenced by the fact that mean palonosetron renal clearance (89.8 mL/min) is approximately 2‐fold greater than the expected renal clearance of a drug with an unbound fraction in plasma of 0.38 that simply undergoes passive glomerular filtration. In principle, netupitant P‐gp inhibition at the renal level might determine a reduction of palonosetron renal clearance and an increase in systemic exposure. However, in vivo P‐gp inhibition determined by netupitant at the renal level is likely negligible, if there is any, because the peak plasma concentration of unbound netupitant is approximately 5‐fold lower than the estimated inhibition constant (Ki) of netupitant for P‐gp (0.65 μM and 3 μM, respectively). Hence, netupitant P‐gp inhibition is not expected to remarkably affect palonosetron renal excretion and PK profile. Similarly, no clinically relevant interactions between netupitant and palonosetron at hepatic level are expected for P‐gp. This efflux transporter mediates the active secretion of substrate drugs into the bile. In the case of palonosetron, excretion in the bile is marginal, if it occurs at all, as determined by mass balance studies in healthy subjects dosed with [14C]‐palonosetron.22 palonosetron and/or palonosetron‐related species in feces accounted for 5% to 8% of the administered dose. Therefore, the role of P‐gp in the hepatic elimination of palonosetron appears to be negligible. Accordingly, hepatic P‐gp inhibition by a drug such as netupitant, administered concomitantly with palonosetron, is not expected to alter palonosetron hepatic elimination and PK profile. In addition, as mentioned for renal excretion, netupitant P‐gp inhibition is likely negligible because the peak plasma concentration of unbound netupitant is lower than the Ki of netupitant for P‐gp. Indeed, no interaction has been observed in vivo in humans after NEPA administration, when the PK profiles of netupitant and palonosetron have been studied after administration of the agents alone or in combination. Absence of drug‐drug interaction between netupitant and palonosetron minimizes the likelihood of adverse events, thereby accounting for a good NEPA safety profile.
Netupitant and palonosetron are characterized by intermediate‐to‐low CL/F and large volumes of distribution, which explains their prolonged presence in the systemic circulation (Figure 1) and their long t1/2 (Table 2). Compared with other NK1 receptor antagonists, the t1/2 of netupitant is higher than that of aprepitant (9 to 13 hours; reviewed in Navari34), and lower than that of rolapitant (approximately 180 hours).35 The t1/2 of palonosetron described in the present analysis is consistent with reported values ranging from 37.4 hours in healthy subjects29 to 49.8 hours in cancer patients36 following intravenous administration of a single dose, and higher than the t1/2 of other 5‐HT3 receptor antagonists (eg, 3 to 9 hours for ondansetron, granisetron, and dolasetron; reviewed in Navari34). The long t1/2 of netupitant allows long‐lasting NK1 receptor occupancy and antiemetic efficacy, as observed during both the acute and delayed postchemotherapy phases when a single NEPA dose is administered orally.37 Furthermore, the long t1/2 of both netupitant and palonosetron support a prolonged NK1 and 5‐HT3 receptor occupancy and, hence, a prolonged antiemetic effect when both compounds are administered as a fixed combination once per chemotherapy cycle.14, 19, 21 In contrast, use of an NK1 receptor antagonist with a shorter t1/2, such as aprepitant, requires daily administration to elicit and maintain the desired therapeutic effect. On the basis of the t1/2 of netupitant and palonosetron, modest or no accumulation of either compound is expected upon NEPA administration every 2 or 3 weeks in conjunction with chemotherapy cycles. Considering the t1/2 values of netupitant and palonosetron, it can be estimated that, in patients receiving chemotherapy every 3 weeks, approximately 99% of netupitant and 100% of palonosetron from the previous NEPA administration will have been eliminated at the time the next NEPA dose is received. Even in patients who undergo chemotherapy every 2 weeks at the time of starting a new chemotherapy cycle, approximately 95% of netupitant and more than 99% of palonosetron from the previous NEPA administration will have been eliminated, thus allowing for no clinically significant accumulation. In contrast, in case of an NK1 receptor antagonist with a longer t1/2, such as rolapitant (which has an average t1/2 of 180 hours35), administration every 2 or 3 weeks in conjunction with chemotherapy cycles would not allow a complete washout of rolapitant by the start of a new chemotherapy cycle.
When assessing NK1 receptor antagonist PK/pharmacodynamic relationships, 90% receptor occupancy in the striatum was indicated as a recognized threshold that may correlate with efficacy.38 The netupitant NK1 receptor occupancy in the striatum estimated by positron emission tomography was 92.5% at 6 hours (the time to Cmax) after single 300‐mg oral netupitant.19, 28 However, when a sigmoidal PK/pharmacodynamic maximum efficacy (Emax) model was used to predict the time necessary to achieve 90% NK1 receptor occupancy in the striatum region, this indicated that, following a single dose of 300‐mg netupitant, 90% NK1 receptor occupancy was already achieved at 2.23 hours after administration, that is, before achieving Cmax.20, 39, 40, 41 Considering that the earliest time to first emetic episode calculated for NEPA in a pivotal trial in patients (N = 135) receiving cisplatin19 was 8.0 h and the mean time to first emetic episode for NEPA was 114.4 hours, these results suggest flexibility in the timing of NEPA administration prior to chemotherapy. That is, shifting the timing of NEPA administration closer to the time of chemotherapy initiation seems unlikely to negatively impact efficacy. This hypothesis, to be validated in a clinical trial, as allowing flexibility in timing of dosing may further enhance the convenience of this new antiemetic combination.
Conclusions
These summarized data show the complementarity of netupitant and palonosetron PK profiles and explain the lack of netupitant and palonosetron PK interactions between the 2 drugs at the absorption, distribution, metabolic, or excretory levels. This, together with their synergistic effects on NK1 receptor antagonism, supports the pharmacologic rationale for combining these 2 antiemetic agents as 1 oral fixed combination targeting 2 critical CINV‐associated pathways. A major advantage of this oral fixed combination is the convenience of administration, which may reduce potential dosing errors and increase treatment compliance by patients. Furthermore, the observed therapeutic effects in preventing CINV during the acute and delayed phases support the clinical use of the fixed combination NEPA for overall CINV control.
Acknowledgments
The authors thank the patients, investigators, and study teams at each of the participating centers. Editorial and medical writing assistance was provided by Oana Draghiciu, PhD, CMPP, TRM Oncology, The Hague, The Netherlands, The authors are fully responsible for all content and editorial decisions for this manuscript.
Data “Accessibility” Statement
Data supporting the findings of this study are not available.
Declaration of Conflicting Interest
A.B. is an employee of Helsinn Healthcare SA. J.G. declares no conflict of interest.
Funding
This study was funded by Helsinn Therapeutics (US), Inc, Iselin, New Jersey.
References
- 1. Bloechl‐Daum B, Deuson RR, Mavros P, Hansen M, Herrstedt J. Delayed nausea and vomiting continue to reduce patients’ quality of life after highly and moderately emetogenic chemotherapy despite antiemetic treatment. J Clin Oncol. 2006;24(27):4472–4478. [DOI] [PubMed] [Google Scholar]
- 2. Hesketh PJ. Chemotherapy‐induced nausea and vomiting. N Engl J Med. 2008;358(23):2482–2494. [DOI] [PubMed] [Google Scholar]
- 3. Lesurtel M, Soll C, Graf R, Clavien PA. Role of serotonin in the hepato‐gastrointestinal tract: an old molecule for new perspectives. Cell Mol Life Sci. 2008;65(6):940–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Girish C, Manikandan S. Aprepitant: a substance P antagonist for chemotherapy‐induced nausea and vomiting. Indian J Cancer. 2007;44(1):25–30. [DOI] [PubMed] [Google Scholar]
- 5. Hesketh PJ, Van Belle S, Aapro M, et al. Differential involvement of neurotransmitters through the time course of cisplatin‐induced emesis as revealed by therapy with specific receptor antagonists. Eur J Cancer. 2003;39(8):1074–1080. [DOI] [PubMed] [Google Scholar]
- 6. Navari RM. Palonosetron for the prevention of chemotherapy‐induced nausea and vomiting in patients with cancer. Future Oncol. 2010;6(7):1073–1084. [DOI] [PubMed] [Google Scholar]
- 7. Navari RM. Management of chemotherapy‐induced nausea and vomiting: focus on newer agents and new uses for older agents. Drugs. 2013;73(3):249–262. [DOI] [PubMed] [Google Scholar]
- 8. Roila F, Molassiotis A, Herrstedt J, et al. 2016 MASCC and ESMO guideline update for the prevention of chemotherapy‐ and radiotherapy‐induced nausea and vomiting and of nausea and vomiting in advanced cancer patients. Ann Oncol. 2016;27(suppl 5):v119–v133. [DOI] [PubMed] [Google Scholar]
- 9. Hesketh PJ, Bohlke K, Lyman GH, et al. Antiemetics: American Society of Clinical Oncology focused guideline update. J Clin Oncol. 2016a;34(4):381–386. [DOI] [PubMed] [Google Scholar]
- 10. National Comprehensive Cancer Network . NCCN clinical practice guidelines in oncology (NCCN Guidelines®). Antiemesis. Version 1.2018. http://www.nccn.org/professionals/physician_gls/pdf/antiemesis.pdf. Published March 14, 2018. Accessed April 23, 2018.
- 11. Hesketh PJ, Kris MG, Basch E, et al. Antiemetics: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2017;35(28):3240–3261. [DOI] [PubMed] [Google Scholar]
- 12. Grunberg S, Chua D, Maru A, et al. Single‐dose fosaprepitant for the prevention of chemotherapy‐induced nausea and vomiting associated with cisplatin therapy: randomized, double‐blind study protocol—EASE. J Clin Oncol. 2011;29(11):1495–1501. [DOI] [PubMed] [Google Scholar]
- 13. Ruddy K, Mayer E, Partridge A. Patient adherence and persistence with oral anticancer treatment. CA Cancer J Clin. 2009;59(1):56–66. [DOI] [PubMed] [Google Scholar]
- 14. Aapro M, Rugo H, Rossi G, et al. A randomized phase III study evaluating the efficacy and safety of NEPA, a fixed‐dose combination of netupitant and palonosetron, for prevention of chemotherapy‐induced nausea and vomiting following moderately emetogenic chemotherapy. Ann Oncol. 2014;25(7):1328–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lorusso V. Management of chemotherapy‐induced nausea and vomiting by risk profile: role of netupitant/palonosetron. Ther Clin Risk Manag. 2016;12:917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lorusso V, Karthaus M, Aapro M. Review of oral fixed‐dose combination netupitant and palonosetron (NEPA) for the treatment of chemotherapy‐induced nausea and vomiting. Future Oncol. 2015;11(4):565–577. [DOI] [PubMed] [Google Scholar]
- 17. Stathis M, Pietra C, Rojas C, Slusher BS. Inhibition of substance P–mediated responses in NG108‐15 cells by netupitant and palonosetron exhibit synergistic effects. Eur J Pharmacol. 2012;689(1‐3):25–30. [DOI] [PubMed] [Google Scholar]
- 18. Thomas AG, Stathis M, Rojas C, Slusher BS. Netupitant and palonosetron trigger NK1 receptor internalization in NG108‐15 cells. Exp Brain Res. 2014;232(8):2637–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hesketh PJ, Rossi G, Rizzi G, et al. Efficacy and safety of NEPA, an oral combination of netupitant and palonosetron, for prevention of chemotherapy‐induced nausea and vomiting following highly emetogenic chemotherapy: a randomized dose‐ranging pivotal study. Ann Oncol. 2014;25(7):1340–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aapro M, Spinelli T, Palmas M, Bernareggi A. Association between NK1 receptor occupancy (RO) of netupitant (NETU) and efficacy of NEPA, the fixed antiemetic combination of NETU and palonosetron (PALO). Support Care Cancer. 2015;23(suppl 1):abstract 11‐09‐P. [Google Scholar]
- 21. Gralla RJ, Bosnjak SM, Hontsa A, et al. A phase III study evaluating the safety and efficacy of NEPA, a fixed‐dose combination of netupitant and palonosetron, for prevention of chemotherapy‐induced nausea and vomiting over repeated cycles of chemotherapy. Ann Oncol. 2014;25(7):1333–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Akynzeo® (netupitant and palonosetron) [FDA prescribing information]. Lugano, Switzerland: Helsinn Therapeutics, Inc.; 2018. [Google Scholar]
- 23. Akynzeo® 300 mg/0.5 mg hard capsules. Summary of product characteristics, European Medicine Agency; 2015.
- 24. US Department of Health and Human Services . Guidance for industry: bioanalytical method validation. https://www.fda.gov/downloads/drugs/guidances/ucm368107.pdf. Published September 2013. Accessed April 23, 2018.
- 25. European Medicines Agency . Committee for Medicinal Products for Human Use (CHMP). Guideline on bioanalytical method validation. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf. Published July 21, 2011. Accessed April 23, 2018.
- 26. Aloxi® (palonosetron HCl) [prescribing information]. Dublin, Ireland: Helsinn Birex Pharmaceuticals; 2015.
- 27. Drugbank . Palonosetron. https://www.drugbank.ca/drugs/DB00377. Published April 22, 2018. Accessed April 23, 2018.
- 28. Spinelli T, Calcagnile S, Giuliano C, et al. Netupitant PET imaging and ADME studies in humans. J Clin Pharmacol. 2014;54(1):97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stoltz R, Parisi S, Shah A, Macciocchi A. Pharmacokinetics, metabolism and excretion of intravenous [l4C]‐palonosetron in healthy human volunteers. Biopharm Drug Dispos. 2004;25(8):329–337. [DOI] [PubMed] [Google Scholar]
- 30. Youdim KA, Zayed A, Dickins M, et al. Application of CYP3A4 in vitro data to predict clinical drug‐drug interactions; predictions of compounds as objects of interaction. Br J Clin Pharmacol. 2008;65(5):680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Natale JJ, Spinelli T, Calcagnile S, et al. Drug‐drug interaction profile of components of a fixed combination of netupitant and palonosetron: review of clinical data. J Oncol Pharm Pract. 2016;22(3):485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Giuliano C, Lovati E, Funk C, et al. In vitro drug‐drug interaction studies with the antiemetic drug netupitant and it major metabolites M1 and M2, involving several human cytochrome P450 isozymes. Ann Oncol. 2012;23(suppl 9):abstract 1618. [Google Scholar]
- 33. Shugarts S, Benet LZ. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm Res. 2009;26(9):2039–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Navari RM. Profile of netupitant/palonosetron (NEPA) fixed dose combination and its potential in the treatment of chemotherapy‐induced nausea and vomiting (CINV). Drug Des Devel Ther. 2014;9:155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Varubi (rolapitant) [prescribing information]. Waltham, MA: TESARO, Inc.; 2015. [Google Scholar]
- 36. Eisenberg P, MacKintosh FR, Ritch P, Cornett PA, Macciocchi A. Efficacy, safety and pharmacokinetics of palonosetron in patients receiving highly emetogenic cisplatin‐based chemotherapy: a dose‐ranging clinical study. Ann Oncol. 2004;15(2):330–337. [DOI] [PubMed] [Google Scholar]
- 37. McBride A, Bernareggi A, Spinelli T, Palmas M. Receptor occupancy (RO) of the neurokinin‐1 (NK1) receptor antagonist, netupitant, in different brain regions and relationship to antiemetic efficacy. Presented at the American Society of Health System Pharmacists (ASHP) Summer Meetings and Exhibition; June 6–10, 2015; Denver, Colorado. Poster 34‐T.
- 38. Bergström M, Hargreaves RJ, Burns DH, et al. Human positron emission tomography studies of brain neurokinin 1 receptor occupancy by aprepitant. Biol Psychiatry. 2004;55(10):1007–1012. [DOI] [PubMed] [Google Scholar]
- 39. Schwartzberg L, Bernareggi A, Parisi S, Palmas M, Rizzi G. Evaluating the administration timing of NEPA, a fixed combination of netupitant and palonosetron for prevention of chemotherapy‐induced nausea and vomiting (CINV). Support Care Cancer. 2015;23(suppl 1):abstract 11–28‐P.25315366 [Google Scholar]
- 40. Bernareggi A, Spinelli T. Neurokinin‐1 (NK1) receptor occupancy (RO) of netupitant in different brain regions: positron emission tomography (PET) study in healthy male subjects. Support Care Cancer. 2015;23(suppl 1):abstract 11‐30‐P. [Google Scholar]
- 41. Bernareggi A, Schwartzberg L, Palmas M, Parisi S, Rizzi G. Evaluating the administration timing of NEPA, a fixed combination of netupitant and palonosetron for prevention of chemotherapy‐induced nausea and vomiting (CINV). J Oncol Pharm Pract. 2016;22(2 suppl):poster 11. [Google Scholar]
- 42. Calcagnile S, Lanzarotti C, Rossi G, Henriksson A, Kammerer KP, Timmer W. Effect of netupitant, a highly selective NK1 receptor antagonist, on the pharmacokinetics of palonosetron and impact of the fixed dose combination of netupitant and palonosetron when coadministered with ketoconazole, rifampicin, and oral contraceptives. Support Care Cancer. 2013;21(10):2879–2887. [DOI] [PubMed] [Google Scholar]
- 43. Calcagnile S, Lanzarotti C, Gutacker M, Jakob‐Rodamer V, Peter Kammerer K, Timmer W. Evaluation of the effect of food and age on the pharmacokinetics of oral netupitant and palonosetron in healthy subjects: A randomized, open‐label, crossover phase 1 study. Clin Pharmacol Drug Dev. 2015;4(5):377–386. [DOI] [PubMed] [Google Scholar]
- 44. Australian public assessment report for netupitant/palonosetron (as hydrochloride). Therapeutic Goods Administration, Health Safety Regulation; 2016.