

Abstract
The concept that subthalamic nucleus deep brain stimulation (STN DBS) may be disease modifying in Parkinson's disease (PD) is controversial. Several clinical trials that enrolled subjects with late‐stage PD have come to disparate conclusions on this matter. In contrast, some clinical studies in early‐ to midstage subjects have suggested a disease‐modifying effect. Dopaminergic innervation of the putamen is essentially absent in PD subjects within 4 years after diagnosis, indicating that any neuroprotective therapy, including STN DBS, will require intervention within the immediate postdiagnosis interval. Preclinical prevention and early intervention paradigms support a neuroprotective effect of STN DBS on the nigrostriatal system via increased brain‐derived neurotrophic factor (BDNF). STN DBS‐induced increases in BDNF provide a multitude of mechanisms capable of ameliorating dysfunction and degeneration in the parkinsonian brain. A biomarker for measuring brain‐derived neurotrophic factor‐trkB signaling, though, is not available for clinical research. If a prospective clinical trial were to examine whether STN DBS is disease modifying, we contend the strongest rationale is not dependent on a preclinical neuroprotective effect per se, but on the myriad potential mechanisms whereby STN DBS‐elicited brain‐derived neurotrophic factor‐trkB signaling could provide disease modification. © 2018 The Authors. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society.
Keywords: brain‐derived neurotrophic factor, deep brain stimulation, disease modification, Parkinson's disease, subthalamic nucleus
The use of subthalamic nucleus deep brain stimulation (STN DBS) to treat the cardinal motor signs of Parkinson's disease (PD) has increased dramatically since its first use was reported in 1994.1 DBS is a vetted, safe, and efficacious neurosurgical therapy for PD.2 Once considered a treatment of last resort where patients underwent neurosurgery approximately 10 to 16 years postdiagnosis,3 STN DBS now is U.S. Food and Drug Administration approved for use as early as 4 years after diagnosis and may be superior to medical therapy at that time.4 With a trend toward implanting earlier in the course of the disease, questions remain as to whom will best benefit from additional and earlier years of stimulation treatment. The neurologist‐patient discussion must weigh symptomatic benefit versus neurosurgical risks over a now longer interval.
There is a strong prevalent opinion in the neurological community that STN DBS is not disease modifying in PD.5 In light of the lack of direct evidence, this is an appropriate stance in counseling patients considering STN DBS. However, from a scientific perspective, this conclusion may be premature in light of an evolved PD literature. The preclinical evidence still supports pursuing a clinical trial with the addition of newer studies. In addition, we will argue that previous trials that assessed the disease‐modifying potential of STN DBS were flawed in their designs. Further, we will emphasize that stimulation‐induced increases in brain‐derived neurotrophic factor (BDNF)6, 7, 8, 9, 10 provide multiple mechanistic avenues for STN DBS to promote the survival of the nigrostriatal system, promote functionality of the basal ganglia‐cortical circuitry, and decrease α‐synuclein (α‐syn) aggregation in the parkinsonian brain.
Current Wisdom on Trial Design for Disease Modification in PD — Timing Is Everything
Dysfunction and degeneration of the nigrostriatal system begin long before diagnosis and the ability to intervene. At the time of motor symptom onset and on prompt diagnosis, it has been estimated that half of striatal dopamine content has been depleted along with 30% of nigral dopamine neurons.11 However, it was not until the work by Kordower and colleagues published in 201312 that the early magnitude of putaminal denervation in the disease process was fully appreciated (schematically represented in Fig. 1). Specifically, this study demonstrated that 50% putaminal denervation had already occurred at the time of diagnosis and that this denervation progressed to approximately 90% loss within 4 years after diagnosis. This loss of putaminal innervation preceded cell body loss; about 50% of melanin‐containing nigral dopamine neurons remained at 4 years postdiagnosis, and fewer than a third retained their tyrosine hydroxylase (TH) phenotype. This important revelation of the scope of early dopaminergic terminal loss suggests that neuroprotective therapies that target the nigrostriatal system cannot be adequately evaluated with a clinical trial population with disease duration longer than this 4‐year time frame.
Figure 1.
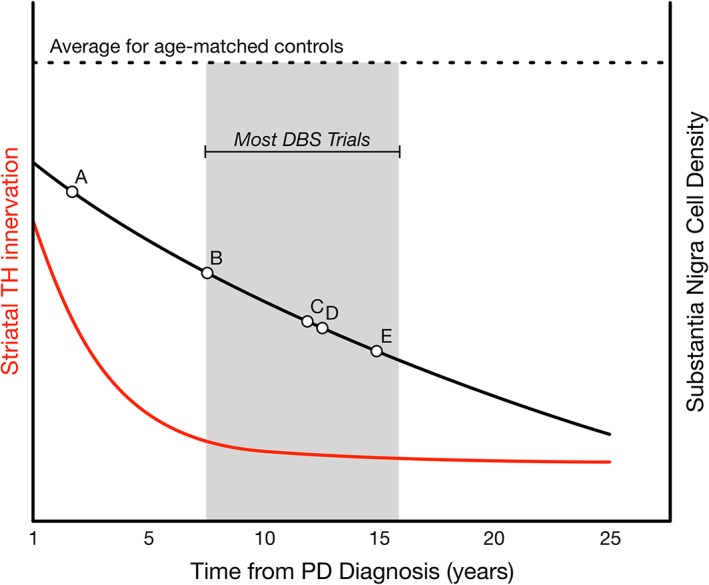
Timing of nigrostriatal degeneration (adapted from Kordower et al [2013]12. Average time course for degeneration since diagnosis of PD (x axis) is plotted for both putaminal TH immunoreactivity (left y axis, red line) and number of melanized neurons in the substantia nigra (right y axis, black line) compared with the average from age‐matched controls (dashed line). The gray box brackets the window during which the majority of trials examining STN DBS have occurred (compare with Table 1). For illustration, points A, B, C, D, and E correspond to the studies conducted by Charles et al,36 Schuepbach et al,4 Tagliati et al,21 Hilker et al,22 and Pal et al,34, 35 respectively.
Current wisdom for design of clinical trials to assess for disease modification now includes this perspective on timing.13 More recent clinical trials enrolled early‐stage PD subjects (eg, the NET‐PD trials14, 15, 16, 17, and the confound of diagnostic uncertainty in this population was addressed by enrolling a large population. As a separate issue for detecting an effect, early‐stage PD subjects also exhibit a long‐term effect of l‐dopa that may take months to wash out, so the trial design can take this symptomatic effect into account, for example, through a delayed‐start design.13 Other trial designs aimed to detect development of disability over a long period16 or detect a change in slopes between treatment groups. Of importance, neuroprotection per se is not assessed directly by trial end points, as there is no method to do so; rather, the clinical consequences of an intervention—neuroprotective or not—are used. Ultimately, trial design for neuroprotective therapies for PD is still an area of active research, but standard practice now is to enroll early‐stage PD subjects prior to the degeneration of the nigrostriatal system.
Completed Trials Assessing STN DBS Disease‐Modifying Potential
Several clinical studies have investigated whether STN DBS has the ability to slow or halt the progression of PD. However, the common thread in all these studies was that motor symptom progression was examined in subjects who were in late‐stage PD, that is, about 10 years after diagnosis. Many of these investigations have been retrospective studies, evaluating symptomatic progression in subjects receiving STN DBS and making comparisons with either expectations of symptom progression or best‐matched, medication‐only cohorts in either the ON or OFF states (or both). One limitation of examination of OFF‐state symptoms is the unknown impact of the washout period, which for medication ranged from 12 hours to 7 days (Table 1)—that is, what is truly OFF for either medication or DBS? Given the long disease durations and variability in washout, it is not surprising that results have been mixed. Four retrospective analyses showed STN DBS could maintain subjects’ off‐medication motor signs several years after electrode implantation.18, 19, 20, 21 In contrast, a prospective study showed equivalent disease progression, as measured by striatal fluorodopa uptake in subjects receiving or not receiving STN DBS.22
Table 1.
Clinical trials assessing STN DBS
| Author (year) | Age (SD) at surgery | Disease duration at time of surgery in years (SD) | ON/OFF assessments? | Washout period? | Conclusion(s) |
|---|---|---|---|---|---|
| Charles et al. (2014)36 | 60 (6.8) | 2.2 (1.4)c | MedOFF/ON, StimOFF/ON | 7 days for meds and stim | No difference in UPDRS (total) or in part III from STN DBS compared with medication controls over 2 years. |
| Weaver et al (2017)24 | 65.6 (7.6) | >5 | None | N/A | DBS (STN or GPi) associated with increased survival compared with matched controls. |
| Schuepbach et al (2013)4 and Lhommee et al (2018)125 | 52.9 (6.6) | 7.3 (3.1) | MedOFF/ON, StimOFF/ON | 12‐48 hours for meds, 2 hours for stim | PDQ‐39, UPDRS‐II, ‐III, and ‐IV improved compared with medication control group; PDQ‐39 also improved compared with baseline in DBS group. Improved behavioral outcomes in DBS group compared with medical therapy alone. |
| Yamada et al (2009)29 | 65.7 (7.8) | 9.8 (5.6) | MedOFF/ON, StimON | 12 hours for meds | Shorter disease duration at surgery associated with better postoperative S&E ADLs. |
| Dafsari et al (2017)30 | 53.2‐72.3§ | 10.5‐11.1d | MedON, StimON | N/A | PDQ‐8 improved with STN DBS over 5 months, with larger effect size associated with younger age at time of surgery. |
| Dafsari et al (2018)126 | 62.3 (7.8) | 10.9 (4.8) | MedON, StimON | N/A | Improvement in nonmotor symptom scale over 2 years (compared with baseline) with bilateral STN DBS. |
| Toft et al (2011)127 | 60.3 (7.8) | 11.0 (4.8) | MedOFF/ON, StimON | Not reported | Annual increase of 3.2 points on UPDRS‐III scale after STN DBS surgery and a survival of 97% and 90% at 3 and 5 years postoperation, respectively. |
| Ngoga et al (2014)23 | 60 (53‐63)a | 11.0 (8.8‐13.0)a | None | N/A | Longer survival and less likely to enter a residential care home with STN DBS compared with medical management. |
| Trager et al (2016)33 | 61.6 (8.0) | 11.0 (3.5) | MedOFF, StimON/OFF | 12‐72 hours for meds, 60 minutes for stim | Improved UPDRS‐III and reduced beta‐band power with StimOFF after 12 months of DBS compared with baseline. |
| Tagliati et al (2010)21 | 60 (12) | 12 (4) | MedOFF/ON, StimOFF/ON | ∼12 hours for meds, 30 minutes for stim | UPDRS‐III stable when off medication at baseline compared with off medication and off stimulation over 3‐4 years. |
| Aviles‐Olmos (2014)128 | 52.8 (10.1) | 12.3 (4) | MedOFF/ON, StimON | 12 hours for meds | Over 8 years, STN DBS improved UPDRS‐III versus baseline when off medication, and on medication, UPDRS‐III scores declined over time. |
| Merola et al (2012)25 | 54.7‐65.5d | 12.5‐19.2d | MedOFF/ON, StimOFF/ON | Overnight for meds, 1 hour for stim | Lower incidence of medication‐ or stimulation‐resistant symptoms in young‐onset PD compared with non‐young‐onset PD over 5 years postsurgery. |
| Hilker et al (2005)22 | 59.8 (7.2) | 12.6 (4.2) | MedOFF/ON, StimON | 12 hours for meds | No change in rate of decline of F‐dopa uptake in caudate nucleus or putamen in association with STN DBS compared with rates in the literature (no matched medication group in study). |
| Merola et al (2014)31 | 60.11 (5.62) | 12.94 (2.15) | MedOFF/ON, StimOFF/ON | 12 hours for meds, 1 hour for stim | Decreased off time and disability from dyskinesia with STN DBS but no difference in UPDRS‐III between DBS and medication control group over about 6 years. |
| Lezcano et al (2016)129 | 61.3 (7.4) | 13.2 (5.7) | MedOFF/ON, StimON | Not reported | Improved UPDRS‐II and ‐III and S&E ADL scores over 5 years compared with baseline off medication. Worse UPDRS‐III versus baseline when on medication. |
| Castrioto et al (2011)26 | 52.9 (7.9) | 13.4 (4.8) | MedOFF/ON, StimOFF/ON | Overnight for meds, 1 hour for stim | UPDRS‐III improved compared with baseline when assessed off medication at 10 years. |
| Fasano et al (2010)130 | 56.9 (7.2) | 13.7 (4.8) | MedOFF/ON, StimON | Overnight for meds | Over 8 years, STN DBS improved UPDRS‐III versus baseline but not relative to 5 years postsurgery, and UPDRS‐II significantly worsened from year 5 to year 8. |
| Rocha et al (2014)131 | 60 (8) | 14 (range, 5‐48) | None | N/A | Survival of 99% and 94% at 3 and 5 years, respectively, with DBS (mixed GPi and STN). |
| Rodriguez‐Oroz et al (2005)20 | 59.8 (9.8) | 14.1 (5.9) | MedOFF/ON, StimOFF/ON | ∼12 hours for meds, 1‐2 hours for stim | UPDRS‐II and UPDRS‐III improved compared with baseline when assessed off medication, on stimulation over 3‐4 years. |
| Krack et al (2003)18 | 55 (7.5) | 14.6 (5.0) | MedON/OFF, Stim ON | 8‐12 hours for meds | UPDRS‐III and S&E improved compared with baseline when assessed off medication over 5 years. |
| Pal et al (2017)34 and Pal et al (2018)35 | ≈ 72 | ≈14.6 | MedOFF/ON, StimON but not specifically reported | Overnight for meds | Increased α‐synuclein density scores, equivalent loss of pigmented nigral neurons, and equivalent putaminal dopamine and dopamine metabolite whole‐tissue content with STN DBS compared with medically treated controls. |
| Hilker et al (2003)132 | 61.8 (4.9) | 15.3 (4.4) | MedOFF, StimON/OFF | 12 hours, allowed 1 adjunctive dose | Using [11C]raclopride PET, no change in binding between on and off stimulation. |
| Bang Henriksen et al (2016)133 | 59.7 (7.7) | 15.7 (6.0) | None | N/A | Postsurgery, 70% of STN DBS subjects survived 10 years (25 total years’ disease duration). |
| Zibetti et al (2011)134 | 61.4 (6.0) | 16.4 (4.9) | MedOFF/ON, StimOFF/ON | Overnight for meds, 1 hour for stim | Over 9 years, STN DBS improved UPDRS‐III versus baseline without improvement in UPDRS‐II and some with cognitive decline. |
| Rodriguez‐Oroz et al (2004)19 | 62 | “Advanced PD” | MedOFF, StimON/OFF | Overnight for meds, 2 hours for stim | UPDRS‐II and UPDRS‐III improved compared with baseline when assessed off medication, on stimulation over 4 years. |
| Lilleeng et al (2014)32 | 64 (6) | 18 (9)b | MedON, StimON | N/A | No change in time to death or in rate of decline by UPDRS‐III when assessed on medication and on stimulation over several years compared with age‐matched group on medication alone. |
| Strafella et al (2003)135 | ≈59 (9.0) | 32.6 (5.9)b | MedON, StimON/OFF | No stim overnight | Using [11C]raclopride PET, no change in binding between on and off stimulation. |
Median (quartiles).
Reported as UPDRS‐III on medication (SD).
Measured in years from start of medication use, not time since diagnosis.
Three groups compared, with means of youngest and oldest groups displayed.
S&E ADLs, Schwab and England activities of daily living.
Not examining disease modification per se but providing corroborating evidence, STN DBS in late‐stage PD improved survival and decreased the likelihood of entering a residential care home,23, 24 and when performed in younger patients with late‐stage PD, benefits were sustained even 10 years after implantation.25, 26 Shorter disease duration improves outcomes from DBS in primary dystonia27 and young‐onset DYT1 dystonia.28 Similarly, when examining PD patients from mid‐ to late‐stage disease who elected STN DBS, postoperative independence measured by activities of daily living was greater in those with a shorter disease duration,29 and a better quality of life, measured by Parkinson's Disease Questionnaire‐8 (PDQ‐8), after STN DBS was observed with younger age at time of surgery.30 These results indicate enhanced tolerability for the surgery itself with younger age and earlier disease stage, implying when there is greater cognitive reserve.
A retrospective cohort study found over several years of follow‐up that late‐stage PD subjects treated with STN DBS had improved outcomes regarding motor fluctuations, OFF time, and dyskinesia, but they did not find any difference overall in motor outcome—measured by Unified Parkinson's Disease Rating Scale part III (UPDRS‐III) in both OFF and ON conditions—between subjects treated with DBS or medical therapy alone, concluding no disease‐modifying effect.31 Similarly, another retrospective cohort study of a similar design reported no disease‐modifying effect of STN DBS, although it included patients with severe motor complications who were likely in late‐stage PD as disease duration was specifically not reported.32 Of note, a study that used beta‐band power, which is associated with PD motor symptoms, showed reduced power after 1 year of DBS even after the stimulator was turned off, suggesting a disease‐modifying effect on the circuit.33 The most recent retrospective cohort study examining the disease‐modifying potential of STN DBS examined α‐syn density, pigmented nigral neurons, and putaminal dopamine tissue content in late‐stage PD subjects (14.5 years) and found no differences compared with medically treated controls.34, 35
The common thread in all these aforementioned studies is that disease modification, whether measured by motor symptom or pathology progression, was examined in subjects who were in late‐stage PD, that is, about 10 years after diagnosis. Because the majority of loss of putaminal innervation occurs by 4 years post diagnosis,11, 12 it is unreasonable to assess the question of disease modification in the context of a PD subject in whom dopaminergic putaminal innervation had long been absent (Fig. 1). Simply put, it is not possible to protect what is no longer there. Granted, a few pigmented neurons may remain, but they no longer contribute to circuit integrity. A more recent clinical trial has employed STN DBS at an earlier time in the disease course (ie, about 7 to 8 years postdiagnosis),4 but this trial also did not enroll subjects early enough in disease duration to overcome this hurdle in experimental design. The only existing PD cohort that may be able to address the question of disease modification is at Vanderbilt University, although current follow‐up may be too short and the cohort too small to provide any definitive evidence.36 Ergo, the available clinical data (Table 1) are insufficient to evaluate whether STN DBS is disease modifying. Of note, in a retrospective analysis of a subset of the Vanderbilt data, a neuroprotective signal was present. Using a “clinically important worsening” measure—defined as both a 3‐point increase in UPDRS part III and a 1‐point increase in part IV—early‐stage subjects treated with STN DBS had a reduced risk of worsening compared with those subjects managed with medical therapy alone.37 Although an appropriately designed (and powered) clinical trial has yet to be completed, the aforementioned pilot trial may serve as a template for a future one.
Preclinical Evidence for STN DBS‐Mediated Neuroprotection
Several laboratories have examined the effects of STN DBS and whether it is neuroprotective using a variety of animal models of PD that collectively provide support to pursue a clinical trial (Table 2). The neurotoxicant models of PD are able to produce a severely dopamine‐depleted striatum and a modestly progressive loss of nigral somata, and these models have been used extensively to investigate the molecular and morphological effects of STN DBS. In rats, STN DBS used immediately after 6‐hydroxydopamine (6‐OHDA) administration results in a doubling of the remaining tyrosine hydroxylase immunoreactive neurons in the SNpc compared with rats without activated electrodes,38 and when STN DBS is activated 1 or 2 weeks after 6‐OHDA, the SNpc neurons that remain are protected from further degeneration.39, 40, 41 Similar results have been found in nonhuman primate models of PD using 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) with either pretreatment with STN DBS or waiting 6 days after MPTP administration.42
Table 2.
Predictive validity of preclinical models for STN DBS‐mediated neuroprotection (adapted from Spieles‐Engemann et al [2010]136
| PD model | Major finding | Reference(s) | |
|---|---|---|---|
| Rat | Nonhuman primate | ||
| Intact/unlesioned | STN DBS excites STN output structures | Windels et al (2000),55 Windels et al (2003)137 | |
| STN DBS inhibits the STN | Tai et al (2003),138 Zheng et al (2011)139 | ||
| STN DBS increases subthalamic glutamate | Lee et al (2007)140 | ||
| STN DBS increases striatal DA | Paul et al (2000),141 Bruet et al (2001)142 | ||
| STN DBS increases BDNF in striatum and motor cortex | Spieles‐Engemann et al (2011)8 | ||
| STN DBS increases rpS6 and Akt phosphorylation in SNpc neurons | Fischer et al (2017)10 | ||
| 6‐OHDA, complete lesion | STN DBS inhibits the STN | Tai et al (2003),143 Shi et al (2006)144 | |
| STN DBS does not increase striatal DA | Meissner et al (2001),145 Meissner et al (2002)146 | ||
| 6‐OHDA, partial lesion | STN DBS increases striatal DA | Bruet et al (2001)142 | |
| STN DBS protects against neurotoxicant | Maesawa et al (2004),38 Temel et al (2006),39 Harnack et al (2008),40 Spieles‐Engemann et al (2010),41 Fischer et al (2017a)10 | ||
| STN DBS increases BDNF in SN and motor cortex | Spieles‐Engemann et al (2011)8 | ||
| MPTP | STN DBS inhibits the STN | Hashimoto et al (2003),147 Meissner et al (2005)148 | |
| STN DBS increases striatal DA | Zhao et al (2009)149 | ||
| STN DBS protects against neurotoxicant | Wallace et al (2007)42 | ||
| α‐Synuclein viral overexpression | STN DBS protects SNpc somata | Musacchio et al (2017)50 | |
| STN DBS does not protect SNpc somata or nigrostriatal fibers | Fischer et al (2017b)52 | ||
| STN DBS does not increase rpS6 phosphorylation in SNpc neurons | Fischer et al (2017b)52 | ||
Whereas 6‐OHDA and MPTP are neurotoxicant‐based models of PD that primarily rely on the oxidative stress component of PD pathophysiology, models using viral vector‐mediated overexpression of α‐syn should possess greater construct validity for PD.43 A large body of evidence points to α‐syn's involvement in PD, including that point mutations and multiplications of the Snca gene have been linked to onset of familial forms of PD.44, 45, 46 Subsequent discoveries of the presence of α‐syn in the hallmark protein aggregates (Lewy bodies) and dystrophic neurites of PD have linked α‐syn to sporadic forms of the disease.47 Delivery of viral vectors encoding human wild‐type or A53T α‐syn results in nigrostriatal α‐syn overexpression, degeneration of nigral somata and nigrostriatal terminals and motor dysfunction.43, 48, 49 Overexpression of α‐syn at specific titers results in progressive degeneration over several weeks to months, and the remaining neurons exhibit α‐syn‐immunoreactive inclusions and dystrophic neurites. In one laboratory's use of this model, overexpression of A53T α‐syn, a form with greater propensity for aggregation, via a high viral titer was used to examine STN DBS‐mediated neuroprotection. In this study, STN DBS protected nigral neurons50; however, nigrostriatal terminals were not examined and may not have been protected.51 Indeed, vector‐mediated overexpression of the human wild‐type form of α‐syn using a lower titer produces a more progressive model of PD in which STN DBS is not neuroprotective of nigral terminals or somata.52 Because the use of high levels of α‐syn expression to model sporadic PD may produce artifactual results not relevant to the human condition,53, 54 the construct validity of the approach of using α‐syn overexpression to model sporadic PD is less than ideal. Moreover, the predictive validity of all animal models of PD to date has been poor. One way to circumvent concerns related to preclinical models is to understand the impact of STN DBS on the brain in general, devoid of parkinsonian manipulations. We contend that the strongest rationale for a prospective clinical trial to examine the disease‐modifying potential of STN DBS is not dependent on the preclinical neuroprotective effect per se, but on the mechanism by which STN DBS achieves this.
The Role of BDNF in STN DBS‐Mediated Disease Modification
The mechanism for STN DBS‐mediated neuroprotection provides insight into how STN DBS may be disease modifying in idiopathic PD. First, it is worth stating that the early hypothesis that DBS decreased glutamate release from the STN, thereby protecting nigral neurons from excitotoxicity, is not supported because stimulation results in the propagation of action potentials leading to increased nigral glutamate levels.55, 56 More recent evidence supports a neurotrophic mechanism of neuroprotection by STN DBS, specifically via increased levels of BDNF.8, 10 Specifically, BDNF release can be driven through electrical stimulation. In neuronal cultures, high‐frequency stimulation leads to increased BDNF release.6, 57 In addition, glutamatergic signaling at N‐methyl‐d‐aspartate (NMDA) receptors can lead to increased BDNF mRNA expression.58
Brain‐Derived Neurotrophic Factor
Brained‐derived neurotrophic factor (BDNF) is a member of the neurotrophin family. All neurotrophins are secreted proteins that bind tropomyosin‐related kinase (trk) receptors, leading to dimerization and receptor activation.59, 60 The effects of BDNF were first described as a factor in glioma‐conditioned medium capable of supporting survival and fiber formation of isolated chick sensory neurons,61 findings that were replicated when examined using rat brain extracts.62 Identification of the responsible new factor was conducted by the same laboratory group63 and later became known as brain‐derived neurotrophic factor. Of particular importance to STN DBS, BDNF mRNA is expressed by numerous nuclei in the basal ganglia including the subthalamic nucleus, entopeduncular nucleus (rat homologue to the internal globus pallidus), striatal medium spiny neurons (MSNs), and dopaminergic neurons of the substantia nigra (SN).8, 64, 65, 66, 67 The receptor for BDNF, tropomyosin‐related kinase type 2 (trkB), is similarly expressed throughout the basal ganglia, including by dopaminergic neurons of the SN.68, 69 In addition, BDNF and trkB mRNA are expressed in the motor cortex.70, 71
On the firing of an action potential, the presynaptic neuron releases neurotransmitter and coreleases vesicular proBDNF into the synaptic cleft.72 The prodomain is proteolytically cleaved, thereby converting the proBDNF form into the mature form that is referred to simply as BDNF.57, 73 Then, the mature form of BDNF can act via two categories of signaling pathways on the postsynaptic neuron: the canonical and the noncanonical pathways. In the canonical pathway, BDNF binds to its high‐affinity trkB receptor. Three canonical trkB intracellular signaling cascades have been identified: (1) the mitogen‐activated protein kinase/extracellular signal related‐kinase (MAPK/ERK) cascade, (2) the phosphatidylinositol 3‐kinase/AKT (PI3K/AKT) cascade, and (3) the phospholipase C gamma (PLCγ) cascade.74, 75 Binding of BDNF to trkB triggers phosphorylation that initiates all three cascades. MAPK/ERK and PI3K/AKT signaling play key roles in both translation and trafficking of proteins, whereas PLCγ regulates intracellular Ca2+ that can drive transcription via cyclic adenosine monophosphate and protein kinase.
TrkB signaling affects neuronal survival, growth/arborization, and regulation of synaptic plasticity through mediating long‐term potentiation.76, 77 Although the time course for the signaling events is many minutes, the measurable effects take much longer, on the order of hours, as they require alterations in transcription and translation of specific genes and the production of a complement of new proteins. In the noncanonical pathway, the effects of BDNF are also mediated through TrkB, but the intracellular signaling takes a tangential path. Through PI3K‐Akt signaling and a series of phosphorylation events at the NMDA receptor 2B subunit,78, 79, 80 potentiated responses are observed. In addition, BDNF‐trkB signaling has been suggested to have effects on presynaptic dopamine release and reuptake.81 Of importance, because of the involvement of immediate phosphorylation events, the effects mediated via the noncanonical pathway occur at a much faster rate than the translational and transcriptional events in the canonical pathway. Collectively, an increase in BDNF can exert a multitude of effects on the basal ganglia.
Stimulation Results in Increased BDNF In vitro and In Vivo
As mentioned in the previous section, firing of action potentials can cause presynaptic neurons to corelease BDNF in addition to neurotransmitter.72 Specifically, action potentials elicited by high‐frequency stimulation of glutamatergic neurons is associated with release of BDNF.6 In a similar manner, DBS to the glutamatergic STN in either naive rats (unlesioned) or rats lesioned with 6‐OHDA show a stimulation‐specific increase in BDNF protein and mRNA in the nigrostriatal system and motor cortex.8 Specifically, STN DBS drives robust increases in BDNF protein in the SN, striatum, and M1 cortex and enhances Bdnf gene expression in the SN and entopeduncular nucleus (rat homologue to the internal globus pallidus; Fig. 2). Within dopaminergic neurons of the SNpc, STN DBS activates trkB signaling cascades, as measured by phosphorylation of ribosomal protein S6 and Akt.10 Further, STN DBS‐mediated neuroprotection from 6‐OHDA insult can be specifically linked to BDNF‐trkB signaling because this neuroprotection is abolished when trkB is blocked pharmacologically.10 Similarly, when α‐syn overexpression interferes with STN DBS‐mediated trkB signaling, nigrostriatal axonopathy cannot be prevented.52 In addition, other evidence supports a connection between stimulation and increased BDNF, specifically in rodent models of depression.79, 82 These preclinical studies suggest that through increased BDNF, STN DBS may—depending on the ability of trkB survival signaling to occur—provide neuroprotection of the nigrostriatal system.
Figure 2.
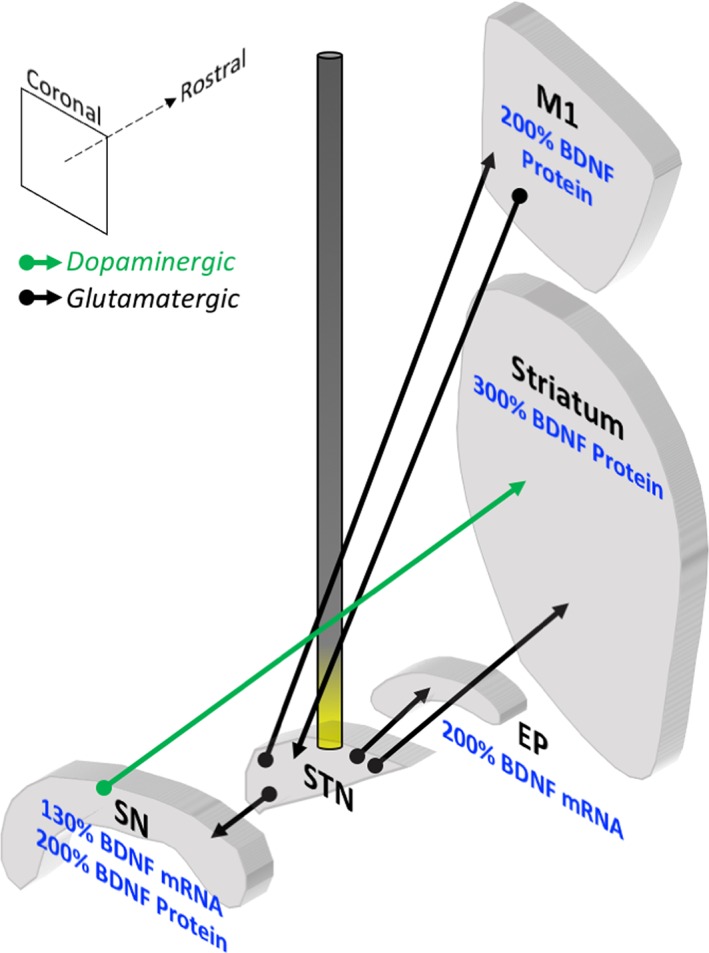
STN DBS increases BDNF in the basal ganglia in PD animal models. Coronal sections of select basal ganglia structures in the rat are depicted in 3 dimensions relative to one another, and an electrode stimulating the STN is illustrated. Effects of high‐frequency stimulation of the STN on BDNF levels in the rat are noted. STN DBS increases BDNF mRNA in the SN and entopeduncular nucleus (EP, rodent homologue to primate GPi). STN DBS also increases BDNF protein in the primary motor cortex (M1) and the striatum of unlesioned animals and the SN of lesioned animals. The green arrow represents dopaminergic fibers; the black arrows represent glutamatergic fibers. Data summarized from Spieles‐Engemann et al (2011).8
Potential Functional Effects of Stimulation‐Induced BDNF
Our previous STN DBS research illustrated a clear causal link between stimulation of the STN and neuroprotection of nigral dopamine neurons from 6‐OHDA insult. This neuroprotective effect mirrors what has been shown previously. BDNF application to mesencephalic dopamine neurons in vitro protects against 1‐methyl‐4‐phenylpyridinium (MPP+)‐ or 6‐OHDA‐induced cell death.83 BDNF also has been shown to be neuroprotective in vivo against MPP+, resulting in decreased loss of nigral dopamine neurons,84, 85 results that were essentially replicated in nonhuman primates.86 Further, BDNF can augment neurite outgrowth from transplanted embryonic DA neurons in a 6‐OHDA rodent model.87 These prosurvival, pro‐outgrowth effects of BDNF represent the classic mechanism whereby DBS can be disease modifying. Indeed, the finding that BDNF levels can be reduced in the brains of PD subjects88, 89 lends further credence to the concept that elevated BDNF could be neuroprotective.
The goal of disease modification in PD typically refers to the prevention of worsening of motor symptoms via preservation of nigrostriatal circuitry. However, there are additional compensatory mechanisms beyond maintenance of neural architecture90—that is, mechanisms capable of maintaining or augmenting dopaminergic transmission at a cellular level—that could be harnessed by BDNF‐trkB signaling. BDNF‐trkB signaling is associated with increased dopamine release, tyrosine hydroxylase synthesis, enhanced dopamine turnover, and increased dopamine neuron activity.91, 92, 93, 94, 95, 96, 97 Indeed, preclinical and clinical studies have suggested that STN DBS can alter dopamine transmission and dopamine receptors,98 effects to which enhanced BDNF‐trkB signaling may contribute. In addition to augmented survival and function of nigral dopamine neurons, STN DBS‐induced increases in BDNF have the potential to exert other disease‐modifying effects in both the nigrostriatal system and the motor cortex.8 BDNF plays a critical role in the maintenance and remodeling of neuronal circuits. BDNF is a critical modulator of gamma aminobutyric acid‐ergic and glutamatergic synapses. BDNF facilitates long‐term potentiation and mediates use‐dependent plasticity.72, 99, 100 Within the striatum, BDNF has the potential to exert a multitude of effects. BDNF plays an essential role in the maintenance of postsynaptic spine density of striatal MSNs that are the targets of dopaminergic innervation.101 Loss of MSN spines has been demonstrated in both preclinical models of dopamine depletion and postmortem PD patients.102, 103 Striatal MSN spines are the site of interaction for nigral dopamine, glutamatergic cortical, and thalamic neurons, and this interaction is necessary for normal basal ganglia functioning. In the face of dysfunctional and degenerating circuits, BDNF may mitigate the aberrant plasticity present in PD104 and maintain native circuit integrity. Such a role for BDNF has been described, albeit indirectly: in PD subjects, a BDNF variant conferring decreased BDNF release105 is associated with earlier development of l‐dopa‐induced dyskinesia in a gene dose‐dependent manner.106
Lastly, augmented trkB signaling resulting from STN DBS‐induced BDNF may have the potential to attenuate the aggregation of α‐syn. One preclinical report described decreased accumulation of α‐syn aggregates in the enteric nervous system following a pharmacologically induced increase in BDNF.107 The same study observed that blockade of trkB signaling increased α‐syn aggregation. This finding adds a new dimension to the disease‐modifying potential of elevated BDNF induced by STN DBS. Using STN DBS to bolster BDNF levels in a manner that is spatially and temporally controlled by the native circuit(s) provides a multitude of mechanisms capable of positively modifying dysfunction and degeneration in the parkinsonian brain (Fig. 3).
Figure 3.
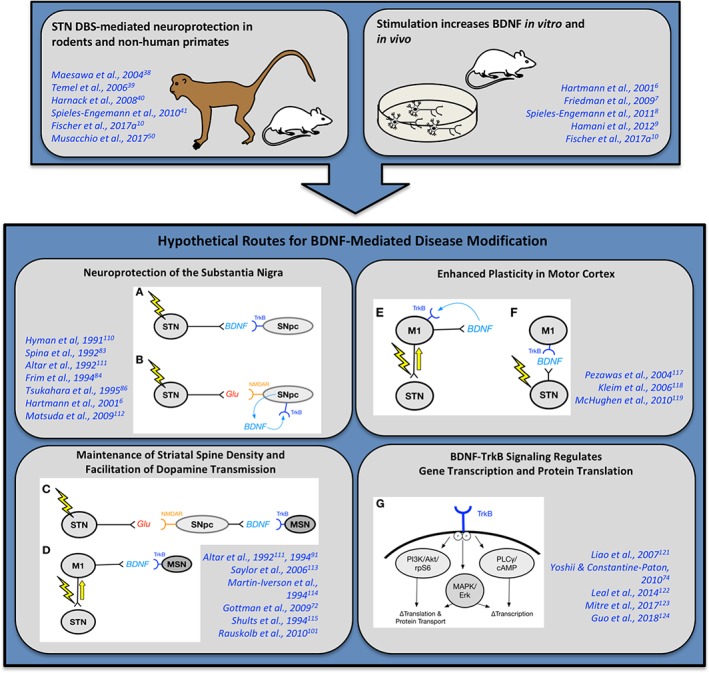
Hypothetical routes for BDNF‐mediated disease modification. Preclinical studies have demonstrated STN DBS‐mediated neuroprotection in rodents and nonhuman primates.10, 38, 39, 40, 41, 50 In addition, high‐frequency stimulation increases BDNF in vitro and in vivo.6, 7, 8, 9, 10 In light of these preclinical studies, there are several hypothetical routes for BDNF‐mediated disease modification. The SNpc may be protected directly (A,B).6, 83, 84, 86, 110, 111, 112 (A) DBS increases STN activity, increases activity‐dependent release of BDNF at the SNpc and binding to TrkB for a trophic effect. (B) DBS increases STN activity, increases glutamate (Glu) release at the SNpc and binding to NMDA receptors (NMDAR). SNpc activation results in production of BDNF transcript, translation, and local release of BDNF that binds to TrkB for an autocrine/paracrine trophic effect. BDNF may maintain striatal spine density and facilitate dopamine transmission (C,D).72, 91, 101, 111, 113, 114, 115 (C) DBS increases STN activity, increases glutamate (Glu) release at the SNpc and binding to NMDA receptors (NMDAR). SNpc activation results in production of BDNF transcript, translation, and activity‐dependent release of BDNF that binds to TrkB on striatal medium spiny neurons (MSNs) for a trophic effect, including maintenance of spine density. (D) DBS results in antidromic activation of corticosubthalamic projections from the motor cortex (M1)116 and subsequent activity‐dependent release of BDNF via corticostriatal fibers to bind to TrkB on MSNs for an ultimately trophic effect. BDNF may enhance M1 plasticity (E,F).117, 118, 119 (E) DBS results in antidromic activation of corticosubthalamic projections from the M1 and activity‐dependent release of BDNF by cortical neurons in an autocrine/paracrine manner, thereby enhancing plasticity. (F) DBS activates STN activity and through subthalamocortical projections found in the rat120 releases BDNF in the M1 and enhancing plasticity. Of importance, BDNF‐trkB signaling exerts powerful effects on intracellular signaling pathways (G).74, 121, 122, 123, 124 (G) Intraneuronal changes with some shown in the STN DBS paradigm specifically,10 where TrkB phosphorylation results in phosphorylation of Akt and ribosomal protein S6 (rpS6), as well as MAPK/Erk and PLCγ/cAMP signaling pathways that have been shown to result in changes in transcription, translation, and protein transport.
Keys for a Well‐Designed STN DBS Trial for Disease Modification
In light of the preclinical evidence supporting STN DBS‐mediated neuroprotection and the myriad mechanisms whereby STN DBS‐elicited BDNF‐trkB signaling could provide disease modification, a prospective clinical trial to assess whether STN DBS may modify the course of PD progression is warranted. Of importance, the trial should only include subjects who are truly early‐stage PD, specifically fewer than 3 or 4 years since diagnosis, and as discussed above, employ a trial design that takes into account this unique time frame. With the Vanderbilt clinical trial experience in mind,36 it is certainly feasible to recruit and retain early‐stage PD subjects for such an endeavor. Of note, enrolling early‐stage PD subjects reduces a noted selection bias108 that has affected the validity of DBS studies that included subjects with advanced disease, where exclusion criteria for surgery include factors that negatively affect survival and are more prevalent in the late‐stage PD population. The confound of symptomatic benefit from instrumenting the STN versus stimulation should be addressed, possibly by inclusion of a “sham” group in which electrodes are placed but not activated. Given that the course of PD progression has been determined as a decrease by 2 points on the UPDRS (total score) per year,16 the study should be powered appropriately to detect a change in slope or a similar measure of progression; this will likely require a study of several years’ duration. Of note, direct measurement of a neuroprotective effect is not required: STN DBS may be disease modifying through a neuroprotection‐independent mechanism, and the result of any neuroprotection would improve the disease course if it were clinically meaningful. Lastly, as such a clinical trial will likely represent a large financial investment, it would be prudent to gather specimens and imaging data for testing other hypotheses using the database that is created. Since PD likely represents the convergence of several unique etiologies,109 the ability to retrospectively analyze the data set, even if the study does not support a disease‐modifying role for PD, cannot go underappreciated.
Conclusions
There is sufficient preclinical evidence to support a clinical trial examining the disease‐modifying potential of STN DBS, likely via BDNF‐mediated effects and perhaps extending beyond strictly neuroprotective mechanisms. To date, an appropriately designed and statistically powered trial that enrolls early‐stage PD subjects has yet to be conducted. The pilot Vanderbilt trial may serve as a template for a larger multicenter trial to assess if STN DBS is disease modifying when applied to early‐stage PD subjects whose neural circuitry and physiology still may be most capable of responding to the effects of BDNF.
Author Roles
D.L.F. and C.E.S. conceived the study, wrote the first draft, and reviewed, critiqued, and approved the final manuscript.
Relevant conflicts of interest/financial disclosures: D.L.F. and C.E.S. have no disclosures.
Funding agencies: Support was provided by the St. Mary's Foundation (Grand Rapids, MI).
References
- 1. Benabid AL, Pollak P, Gross C, et al. Acute and long‐term effects of subthalamic nucleus stimulation in Parkinson's disease. Stereotact Funct Neurosurg 1994;62(1‐4):76‐84. [DOI] [PubMed] [Google Scholar]
- 2. Okun MS. Deep‐brain stimulation for Parkinson's disease. N Engl JMed 2012;367(16):1529‐1538. [DOI] [PubMed] [Google Scholar]
- 3. Mansouri A, Taslimi S, Badhiwala JH, et al. Deep brain stimulation for Parkinson's disease: meta‐analysis of results of randomized trials at varying lengths of follow‐up. J Neurosurg 2018:128(4):1199‐1213. [DOI] [PubMed] [Google Scholar]
- 4. Schuepbach WM, Rau J, Knudsen K, et al. Neurostimulation for Parkinson's disease with early motor complications. N Engl J Med 2013;368(7):610‐622. [DOI] [PubMed] [Google Scholar]
- 5. Hariz M. There is no credible rational for deep brain stimulation in very early Parkinson's disease! Parkinsonism Relat Disord 2015;21(3):345‐346. [DOI] [PubMed] [Google Scholar]
- 6. Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high‐frequency stimulation of glutamatergic synapses. The EMBO J 2001;20(21):5887‐5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Friedman A, Frankel M, Flaumenhaft Y, et al. Programmed acute electrical stimulation of ventral tegmental area alleviates depressive‐like behavior. Neuropsychopharmacology 2009;34(4):1057‐1066. [DOI] [PubMed] [Google Scholar]
- 8. Spieles‐Engemann AL, Steece‐Collier K, Behbehani MM, et al. Subthalamic nucleus stimulation increases brain derived neurotrophic factor in the nigrostriatal system and primary motor cortex. J Parkinsons Dis 2011;1(1):123‐136. [PMC free article] [PubMed] [Google Scholar]
- 9. Hamani C, Machado DC, Hipolide DC, et al. Deep brain stimulation reverses anhedonic‐like behavior in a chronic model of depression: role of serotonin and brain derived neurotrophic factor. Biol Psychiatry 2012;71(1):30‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fischer DL, Kemp CJ, Cole‐Strauss A, et al. Subthalamic nucleus deep brain stimulation employs trkB signaling for neuroprotection and functional restoration. J Neurosci 2017;37(28):6786‐6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 2010;67(6):715‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kordower JH, Olanow CW, Dodiya HB, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 2013;136(Pt 8):2419‐2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Olanow CW, Kieburtz K, Katz R. Clinical approaches to the development of a neuroprotective therapy for PD. Exp Neurol 2017;298(Pt B):246‐251. [DOI] [PubMed] [Google Scholar]
- 14. NINDS NET‐PD Investigators . A randomized clinical trial of coenzyme Q10 and GPI‐1485 in early Parkinson disease. Neurology 2007;68(1):20‐28. [DOI] [PubMed] [Google Scholar]
- 15. NINDS NET‐PD Investigators . A randomized, double‐blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology 2006;66(5):664‐671. [DOI] [PubMed] [Google Scholar]
- 16. Writing Group for the NINDS Exploratory Trials in Parkinson Disease (NET‐PD) Investigators , Kieburtz K, Tilley BC, Elm JJ, et al. Effect of creatine monohydrate on clinical progression in patients with Parkinson disease: a randomized clinical trial. JAMA 2015;313(6):584‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. NINDS Exploratory Trials in Parkinson Disease (NET‐PD) FS‐ZONE Investigators . Pioglitazone in early Parkinson's disease: a phase 2, multicentre, double‐blind, randomised trial. Lancet Neurol 2015;14(8):795‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krack P, Batir A, Van Blercom N, et al. Five‐year follow‐up of bilateral stimulation of the subthalamic nucleus in advanced Parkinson's disease. New Engl J Med 2003;349(20):1925‐1934. [DOI] [PubMed] [Google Scholar]
- 19. Rodriguez‐Oroz MC, Zamarbide I, Guridi J, et al. Efficacy of deep brain stimulation of the subthalamic nucleus in Parkinson's disease 4 years after surgery: double blind and open label evaluation. J Neurol Neurosurg Psychiatry 2004;75(10):1382‐1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rodriguez‐Oroz MC, Obeso JA, Lang AE, et al. Bilateral deep brain stimulation in Parkinson's disease: a multicentre study with 4 years follow‐up. Brain 2005;128(Pt 10):2240‐2249. [DOI] [PubMed] [Google Scholar]
- 21. Tagliati M, Martin C, Alterman R. Lack of motor symptoms progression in Parkinson's disease patients with long‐term bilateral subthalamic deep brain stimulation. Int J Neurosci 2010;120(11):717‐723. [DOI] [PubMed] [Google Scholar]
- 22. Hilker R, Portman AT, Voges J, et al. Disease progression continues in patients with advanced Parkinson's disease and effective subthalamic nucleus stimulation. J Neurol Neurosurg Psychiatry 2005;76(9):1217‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ngoga D, Mitchell R, Kausar J, Hodson J, Harries A, Pall H. Deep brain stimulation improves survival in severe Parkinson's disease. J Neurol Neurosurg Psychiatry 2014;85(1):17‐22. [DOI] [PubMed] [Google Scholar]
- 24. Weaver FM, Stroupe KT, Smith B, et al. Survival in patients with Parkinson's disease after deep brain stimulation or medical management. Mov Disord 2017;32(12):1756‐1763. [DOI] [PubMed] [Google Scholar]
- 25. Merola A, Zibetti M, Artusi CA, et al. Subthalamic nucleus deep brain stimulation outcome in young onset Parkinson's disease: a role for age at disease onset? J Neurol Neurosurg Psychiatry 2012;83(3):251‐257. [DOI] [PubMed] [Google Scholar]
- 26. Castrioto A, Lozano AM, Poon YY, Lang AE, Fallis M, Moro E. Ten‐year outcome of subthalamic stimulation in Parkinson disease: a blinded evaluation. Arch Neurol 2011;68(12):1550‐1556. [DOI] [PubMed] [Google Scholar]
- 27. Isaias IU, Alterman RL, Tagliati M. Outcome predictors of pallidal stimulation in patients with primary dystonia: the role of disease duration. Brain 2008;131(Pt 7):1895‐1902. [DOI] [PubMed] [Google Scholar]
- 28. Markun LC, Starr PA, Air EL, Marks WJ Jr, Volz MM, Ostrem JL. Shorter disease duration correlates with improved long‐term deep brain stimulation outcomes in young‐onset DYT1 dystonia. Neurosurgery 2012;71(2):325‐330. [DOI] [PubMed] [Google Scholar]
- 29. Yamada K, Hamasaki T, Kuratsu J. Subthalamic nucleus stimulation applied in the earlier vs. advanced stage of Parkinson's disease ‐ retrospective evaluation of postoperative independence in pursuing daily activities. Parkinsonism Relat Disord 2009;15(10):746‐751. [DOI] [PubMed] [Google Scholar]
- 30. Dafsari HS, Reker P, Stalinski L, et al. Quality of life outcomes after subthalamic stimulation in Parkinson's disease depends on age. Mov Disord 2018;33(1):99‐107. [DOI] [PubMed] [Google Scholar]
- 31. Merola A, Rizzi L, Zibetti M, et al. Medical therapy and subthalamic deep brain stimulation in advanced Parkinson's disease: a different long‐term outcome? J Neurol Neurosurg Psychiatry 2014;85(5):552‐559. [DOI] [PubMed] [Google Scholar]
- 32. Lilleeng B, Bronnick K, Toft M, Dietrichs E, Larsen JP. Progression and survival in Parkinson's disease with subthalamic nucleus stimulation. Acta Neurol Scand 2014;130(5):292‐298. [DOI] [PubMed] [Google Scholar]
- 33. Trager MH, Koop MM, Velisar A, et al. Subthalamic beta oscillations are attenuated after withdrawal of chronic high frequency neurostimulation in Parkinson's disease. Neurobiol Dis 2016;96:22‐30. [DOI] [PubMed] [Google Scholar]
- 34. Pal GD, Ouyang B, Serrano G, et al. Comparison of neuropathology in Parkinson's disease subjects with and without deep brain stimulation. Mov Disord 2017;32(2):274‐277. [DOI] [PubMed] [Google Scholar]
- 35. Pal G, Ouyang B, Verhagen L, et al. Probing the striatal dopamine system for a putative neuroprotective effect of deep brain stimulation in Parkinson's disease. Mov Disord 2018;33(4):652‐654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Charles D, Konrad PE, Neimat JS, et al. Subthalamic nucleus deep brain stimulation in early stage Parkinson's disease. Parkinsonism Relat Disord 2014;20(7):731‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hacker ML, Tonascia J, Turchan M, et al. Deep brain stimulation may reduce the relative risk of clinically important worsening in early stage Parkinson's disease. Parkinsonism Relat Disord 2015;21(10):1177‐1183. [DOI] [PubMed] [Google Scholar]
- 38. Maesawa S, Kaneoke Y, Kajita Y, et al. Long‐term stimulation of the subthalamic nucleus in hemiparkinsonian rats: neuroprotection of dopaminergic neurons. J Neurosurg 2004;100(4):679‐687. [DOI] [PubMed] [Google Scholar]
- 39. Temel Y, Visser‐Vandewalle V, Kaplan S, et al. Protection of nigral cell death by bilateral subthalamic nucleus stimulation. Brain Res 2006;1120(1):100‐105. [DOI] [PubMed] [Google Scholar]
- 40. Harnack D, Meissner W, Jira JA, Winter C, Morgenstern R, Kupsch A. Placebo‐controlled chronic high‐frequency stimulation of the subthalamic nucleus preserves dopaminergic nigral neurons in a rat model of progressive Parkinsonism. Exp Neurol 2008;210(1):257‐260. [DOI] [PubMed] [Google Scholar]
- 41. Spieles‐Engemann AL, Behbehani MM, Collier TJ, et al. Stimulation of the rat subthalamic nucleus is neuroprotective following significant nigral dopamine neuron loss. Neurobiol Dis 2010;39(1):105‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wallace BA, Ashkan K, Heise CE, et al. Survival of midbrain dopaminergic cells after lesion or deep brain stimulation of the subthalamic nucleus in MPTP‐treated monkeys. Brain 2007;130(Pt 8):2129‐2145. [DOI] [PubMed] [Google Scholar]
- 43. Fischer DL, Gombash SE, Kemp CJ, et al. Viral vector‐based modeling of neurodegenerative disorders: Parkinson's disease. Methods Mol Biol 2016;1382:367‐382. [DOI] [PubMed] [Google Scholar]
- 44. Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha‐synuclein gene identified in families with Parkinson's disease. Science 1997;276(5321):2045‐2047. [DOI] [PubMed] [Google Scholar]
- 45. Singleton AB, Farrer M, Johnson J, et al. alpha‐Synuclein locus triplication causes Parkinson's disease. Science 2003;302(5646):841. [DOI] [PubMed] [Google Scholar]
- 46. Ibáñez P, Bonnet AM, Debarges B, et al. Causal relation between alpha‐synuclein gene duplication and familial Parkinson's disease. Lancet 2004;364(9440):1169‐1171. [DOI] [PubMed] [Google Scholar]
- 47. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha‐synuclein in Lewy bodies. Nature 1997;388(6645):839‐840. [DOI] [PubMed] [Google Scholar]
- 48. Gombash SE, Manfredsson FP, Kemp CJ, et al. Morphological and behavioral impact of AAV2/5‐mediated overexpression of human wildtype alpha‐synuclein in the rat nigrostriatal system. PLoS One 2013;8(11):e81426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ulusoy A, Decressac M, Kirik D, Bjorklund A. Viral vector‐mediated overexpression of alpha‐synuclein as a progressive model of Parkinson's disease. Progr Brain Res 2010;184:89‐111. [DOI] [PubMed] [Google Scholar]
- 50. Musacchio T, Rebenstorff M, Fluri F, et al. STN‐DBS is neuroprotective in the A53T alpha‐synuclein Parkinson's disease rat model. Ann Neurol 2017;81(6):825‐836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fischer DL, Manfredsson FP, Sortwell CE. Can STN DBS protect both nigral somata and innervation of the striatum? Ann Neurol 2017;82(6):865. [DOI] [PubMed] [Google Scholar]
- 52. Fischer DL, Manfredsson FP, Kemp CJ, et al. Subthalamic nucleus deep brain stimulation does not modify the functional deficits or axonopathy induced by nigrostriatal alpha‐synuclein overexpression. Sci Rep 2017;7(1):16356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Su X, Fischer DL, Li X, Bankiewicz K, Sortwell CE, Federoff HJ. Alpha‐synuclein mRNA is not increased in sporadic PD and alpha‐synuclein accumulation does not block GDNF signaling in Parkinson's disease and disease models. Mol Ther 2017;25(10):2231‐2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhou J, Broe M, Huang Y, et al. Changes in the solubility and phosphorylation of alpha‐synuclein over the course of Parkinson's disease. Acta Neuropathol 2011;121(6):695‐704. [DOI] [PubMed] [Google Scholar]
- 55. Windels F, Bruet N, Poupard A, et al. Effects of high frequency stimulation of subthalamic nucleus on extracellular glutamate and GABA in substantia nigra and globus pallidus in the normal rat. Eur J Neurosci 2000;12(11):4141‐4146. [DOI] [PubMed] [Google Scholar]
- 56. Boulet S, Lacombe E, Carcenac C, et al. Subthalamic stimulation‐induced forelimb dyskinesias are linked to an increase in glutamate levels in the substantia nigra pars reticulata. J Neurosci 2006;26(42):10768‐10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nagappan G, Zaitsev E, Senatorov VV Jr, Yang J, Hempstead BL, Lu B. Control of extracellular cleavage of ProBDNF by high frequency neuronal activity. Proc Natl Acad Sci U S A 2009;106(4):1267‐1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bustos G, Abarca J, Campusano J, Bustos V, Noriega V, Aliaga E. Functional interactions between somatodendritic dopamine release, glutamate receptors and brain‐derived neurotrophic factor expression in mesencephalic structures of the brain. Brain Res Brain Res Rev 2004;47(1‐3):126‐144. [DOI] [PubMed] [Google Scholar]
- 59. Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci 1995;18:223‐253. [DOI] [PubMed] [Google Scholar]
- 60. Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 2001;24:677‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Barde YA, Lindsay RM, Monard D, Thoenen H. New factor released by cultured glioma cells supporting survival and growth of sensory neurones. Nature 1978;274(5673):818. [DOI] [PubMed] [Google Scholar]
- 62. Barde YA, Edgar D, Thoenen H. Sensory neurons in culture: changing requirements for survival factors during embryonic development. Proc Natl Acad Sci U S A 1980;77(2):1199‐1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Barde YA, Edgar D, Thoenen H. Purification of a new neurotrophic factor from mammalian brain. EMBO J 1982;1(5):549‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang X, Andren PE, Svenningsson P. Repeated l‐DOPA treatment increases c‐fos and BDNF mRNAs in the subthalamic nucleus in the 6‐OHDA rat model of Parkinson's disease. Brain Res 2006;1095(1):207‐210. [DOI] [PubMed] [Google Scholar]
- 65. Seroogy KB, Gall CM. Expression of neurotrophins by midbrain dopaminergic neurons. Exp Neurol 1993;124(1):119‐128. [DOI] [PubMed] [Google Scholar]
- 66. Sauer H, Wong V, Bjorklund A. Brain‐derived neurotrophic factor and neurotrophin‐4/5 modify neurotransmitter‐related gene expression in the 6‐hydroxydopamine‐lesioned rat striatum. Neuroscience 1995;65(4):927‐933. [DOI] [PubMed] [Google Scholar]
- 67. Friedman WJ, Olson L, Persson H. Cells that express brain‐derived neurotrophic factor mRNA in the developing postnatal rat brain. Eur J Neurosci 1991;3(7):688‐697. [DOI] [PubMed] [Google Scholar]
- 68. Numan S, Seroogy KB. Expression of trkB and trkC mRNAs by adult midbrain dopamine neurons: a double‐label in situ hybridization study. J Comp Neurol 1999;403(3):295‐308. [DOI] [PubMed] [Google Scholar]
- 69. Freeman AY, Soghomonian JJ, Pierce RC. Tyrosine kinase B and C receptors in the neostriatum and nucleus accumbens are co‐localized in enkephalin‐positive and enkephalin‐negative neuronal profiles and their expression is influenced by cocaine. Neuroscience 2003;117(1):147‐156. [DOI] [PubMed] [Google Scholar]
- 70. Wong J, Webster MJ, Cassano H, Weickert CS. Changes in alternative brain‐derived neurotrophic factor transcript expression in the developing human prefrontal cortex. Eur J Neurosci 2009;29(7):1311‐1322. [DOI] [PubMed] [Google Scholar]
- 71. Romanczyk TB, Weickert CS, Webster MJ, Herman MM, Akil M, Kleinman JE. Alterations in trkB mRNA in the human prefrontal cortex throughout the lifespan. Eur J Neurosci 2002;15(2):269‐280. [DOI] [PubMed] [Google Scholar]
- 72. Gottmann K, Mittmann T, Lessmann V. BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp Brain Res 2009;199(3‐4):203‐234. [DOI] [PubMed] [Google Scholar]
- 73. Mowla SJ, Pareek S, Farhadi HF, et al. Differential sorting of nerve growth factor and brain‐derived neurotrophic factor in hippocampal neurons. J Neurosci 1999;19(6):2069‐2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yoshii A, Constantine‐Paton M. Postsynaptic BDNF‐TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol 2010;70(5):304‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yoshii A, Constantine‐Paton M. Postsynaptic localization of PSD‐95 is regulated by all three pathways downstream of TrkB signaling. Front Synaptic Neurosci 2014;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cobb MH, Cobb MH. MAP kinase pathways. Prog Biophys Mol Bio 1999;71(3‐4):479‐500. [DOI] [PubMed] [Google Scholar]
- 77. Airaksinen MS, Saarma M, Airaksinen MS, Saarma M. The GDNF family: Signalling, biological functions and therapeutic value. Nat Rev Neurosci 2002;3(5):383‐394. [DOI] [PubMed] [Google Scholar]
- 78. Chen W, Walwyn W, Ennes HS, Kim H, McRoberts JA, Marvizon JC. BDNF released during neuropathic pain potentiates NMDA receptors in primary afferent terminals. Eur J Neurosci 2014;39(9):1439‐1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nakai T, Nagai T, Tanaka M, et al. Girdin phosphorylation is crucial for synaptic plasticity and memory: a potential role in the interaction of BDNF/TrkB/Akt signaling with NMDA receptor. J Neurosci 2014;34(45):14995‐15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li J, McRoberts JA, Ennes HS, et al. Experimental colitis modulates the functional properties of NMDA receptors in dorsal root ganglia neurons. Am J Physiol Gastrointest Liver Physiol 2006;291(2):G219‐G228. [DOI] [PubMed] [Google Scholar]
- 81. Bosse KE, Maina FK, Birbeck JA, et al. Aberrant striatal dopamine transmitter dynamics in brain‐derived neurotrophic factor‐deficient mice. J Neurochem 2012;120(3):385‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gersner R, Toth E, Isserles M, Zangen A. Site‐specific antidepressant effects of repeated subconvulsive electrical stimulation: potential role of brain‐derived neurotrophic factor. Biol Psychiatry 2010;67(2):125‐132. [DOI] [PubMed] [Google Scholar]
- 83. Spina MB, Hyman C, Squinto S, Lindsay RM. Brain‐derived neurotrophic factor protects dopaminergic cells from 6‐hydroxydopamine toxicity. Ann N Y Acad Sci 1992;648:348‐350. [DOI] [PubMed] [Google Scholar]
- 84. Frim DM, Uhler TA, Galpern WR, et al. Implanted fibroblasts genetically‐engineered to produce brain‐derived neurotrophic factor prevent 1‐methyl‐4‐phenylpyridinium toxicity to dopaminergic‐neurons in the rat. Proc Natl Acad Sci U S A 1994;91(11):5104‐5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yoshimoto Y, Lin Q, Collier TJ, et al. Astrocytes retrovirally transduced with BDNF elicit behavioral improvement in a rat model of Parkinson's disease. Brain Res 1995;691(1‐2):25‐36. [DOI] [PubMed] [Google Scholar]
- 86. Tsukahara T, Takeda M, Shimohama S, et al. Effects of brain‐derived neurotrophic factor on 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐induced parkinsonism in monkeys. Neurosurgery 1995;37(4):733‐739. [DOI] [PubMed] [Google Scholar]
- 87. Yurek DM, Lu W, Hipkens S, et al. BDNF enhances the functional reinnervation of the striatum by grafted fetal dopamine neurons. Exp Neurol 1996;137(1):105‐118. [DOI] [PubMed] [Google Scholar]
- 88. Parain K, Murer MG, Yan Q, et al. Reduced expression of brain‐derived neurotrophic factor protein in Parkinson's disease substantia nigra. Neuroreport 1999;10(3):557‐561. [DOI] [PubMed] [Google Scholar]
- 89. Howells DW, Porritt MJ, Wong JY, et al. Reduced BDNF mRNA expression in the Parkinson's disease substantia nigra. Exp Neurol 2000;166(1):127‐135. [DOI] [PubMed] [Google Scholar]
- 90. Blesa J, Trigo‐Damas I, Dileone M, Del Rey NL, Hernandez LF, Obeso JA. Compensatory mechanisms in Parkinson's disease: Circuits adaptations and role in disease modification. Exp Neurol 2017;298(Pt B):148‐161. [DOI] [PubMed] [Google Scholar]
- 91. Altar CA, Boylan CB, Fritsche M, et al. Efficacy of brain‐derived neurotrophic factor and neurotrophin‐3 on neurochemical and behavioral deficits associated with partial nigrostriatal dopamine lesions. J Neurochem 1994;63(3):1021‐1032. [DOI] [PubMed] [Google Scholar]
- 92. Knusel B, Winslow JW, Rosenthal A, et al. Promotion of central cholinergic and dopaminergic neuron differentiation by brain‐derived neurotrophic factor but not neurotrophin 3. Proc Natl Acad Sci U S A 1991;88(3):961‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Beck KD, Knusel B, Hefti F. The nature of the trophic action of brain‐derived neurotrophic factor, des(1‐3)‐insulin‐like growth factor‐1, and basic fibroblast growth factor on mesencephalic dopaminergic neurons developing in culture. Neuroscience 1993;52(4):855‐866. [DOI] [PubMed] [Google Scholar]
- 94. Hyman C, Juhasz M, Jackson C, Wright P, Ip NY, Lindsay RM. Overlapping and distinct actions of the neurotrophins BDNF, NT‐3, and NT‐4/5 on cultured dopaminergic and GABAergic neurons of the ventral mesencephalon. J Neurosci 1994;14(1):335‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zhou J, Bradford HF, Stern GM. The stimulatory effect of brain‐derived neurotrophic factor on dopaminergic phenotype expression of embryonic rat cortical neurons in vitro. Brain Res Dev Brain Res 1994;81(2):318‐324. [DOI] [PubMed] [Google Scholar]
- 96. Blochl A, Sirrenberg C. Neurotrophins stimulate the release of dopamine from rat mesencephalic neurons via Trk and p75Lntr receptors. J Biol Chem 1996;271(35):21100‐21107. [DOI] [PubMed] [Google Scholar]
- 97. Shen RY, Altar CA, Chiodo LA. Brain‐derived neurotrophic factor increases the electrical activity of pars compacta dopamine neurons in vivo. Proc Natl Acad Sci U S A 1994;91(19):8920‐8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Stefani A, Trendafilov V, Liguori C, Fedele E, Galati S. Subthalamic nucleus deep brain stimulation on motor‐symptoms of Parkinson's disease: Focus on neurochemistry. Progr Neurobiol 2017;151:157‐174. [DOI] [PubMed] [Google Scholar]
- 99. Cirillo J, Hughes J, Ridding M, Thomas PQ, Semmler JG. Differential modulation of motor cortex excitability in BDNF Met allele carriers following experimentally induced and use‐dependent plasticity. Eur J Neurosci 2012;36(5):2640‐2649. [DOI] [PubMed] [Google Scholar]
- 100. Schinder AF, Poo M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci 2000;23(12):639‐645. [DOI] [PubMed] [Google Scholar]
- 101. Rauskolb S, Zagrebelsky M, Dreznjak A, et al. Global deprivation of brain‐derived neurotrophic factor in the CNS reveals an area‐specific requirement for dendritic growth. J Neurosci 2010;30(5):1739‐1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ingham CA, Hood SH, van Maldegem B, Weenink A, Arbuthnott GW. Morphological changes in the rat neostriatum after unilateral 6‐hydroxydopamine injections into the nigrostriatal pathway. Exp Brain Res 1993;93(1):17‐27. [DOI] [PubMed] [Google Scholar]
- 103. McNeill TH, Brown SA, Rafols JA, Shoulson I. Atrophy of medium spiny I striatal dendrites in advanced Parkinson's disease. Brain Res 1988;455(1):148‐152. [DOI] [PubMed] [Google Scholar]
- 104. Bastide MF, Meissner WG, Picconi B, et al. Pathophysiology of L‐dopa‐induced motor and non‐motor complications in Parkinson's disease. Progr Neurobiol 2015;132:96‐168. [DOI] [PubMed] [Google Scholar]
- 105. Chen ZY, Patel PD, Sant G, et al. Variant brain‐derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity‐dependent secretion of wild‐type BDNF in neurosecretory cells and cortical neurons. J Neurosci 2004;24(18):4401‐4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Foltynie T, Cheeran B, Williams‐Gray CH, et al. BDNF val66met influences time to onset of levodopa induced dyskinesia in Parkinson's disease. J Neurol Neurosurg Psychiatry 2009;80(2):141‐144. [DOI] [PubMed] [Google Scholar]
- 107. Vidal‐Martinez G, Vargas‐Medrano J, Gil‐Tommee C, et al. FTY720/Fingolimod Reduces Synucleinopathy and Improves Gut Motility in A53T Mice: CONTRIBUTIONS OF PRO‐BRAIN‐DERIVED NEUROTROPHIC FACTOR (PRO‐BDNF) AND MATURE BDNF. The J Biol Chem 2016;291(39):20811‐20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Contarino MF, Marinus J, van Hilten JJ. Does deep brain stimulation of the subthalamic nucleus prolong survival in Parkinson's Disease? Mov Disord 2018;33(6):947‐949. [DOI] [PubMed] [Google Scholar]
- 109. Espay AJ, Schwarzschild MA, Tanner CM, et al. Biomarker‐driven phenotyping in Parkinson's disease: A translational missing link in disease‐modifying clinical trials. Mov Disord 2017;32(3):319‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hyman C, Hofer M, Barde YA, et al. BDNF is a neurotrophic factor for dopaminergic‐neurons of the substantia‐nigra. Nature 1991;350(6315):230‐232. [DOI] [PubMed] [Google Scholar]
- 111. Altar CA, Boylan CB, Jackson C, et al. Brain‐derived neurotrophic factor augments rotational behavior and nigrostriatal dopamine turnover in vivo. Proc Natl Acad Sci U S A 1992;89(23):11347‐11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Matsuda N, Lu H, Fukata Y, et al. Differential activity‐dependent secretion of brain‐derived neurotrophic factor from axon and dendrite. J Neurosci 2009;29(45):14185‐14198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Saylor AJ, Meredith GE, Vercillo MS, Zahm DS, McGinty JF. BDNF heterozygous mice demonstrate age‐related changes in striatal and nigral gene expression. Exp Neurol 2006;199(2):362‐372. [DOI] [PubMed] [Google Scholar]
- 114. Martin‐Iverson MT, Todd KG, Altar CA. Brain‐derived neurotrophic factor and neurotrophin‐3 activate striatal dopamine and serotonin metabolism and related behaviors: interactions with amphetamine. J Neurosci 1994;14(3 Pt 1):1262‐1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Shults CW, Matthews RT, Altar CA, Hill LR, Langlais PJ. A single intramesencephalic injection of brain‐derived neurotrophic factor induces persistent rotational asymmetry in rats. Exp Neurol 1994;125(2):183‐194. [DOI] [PubMed] [Google Scholar]
- 116. Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science 2009;324(5925):354‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Pezawas L, Verchinski BA, Mattay VS, et al. The brain‐derived neurotrophic factor val66met polymorphism and variation in human cortical morphology. J Neurosci 2004;24(45):10099‐10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kleim JA, Chan S, Pringle E, et al. BDNF val66met polymorphism is associated with modified experience‐dependent plasticity in human motor cortex. Nat Neurosci 2006;9(6):735‐737. [DOI] [PubMed] [Google Scholar]
- 119. McHughen SA, Rodriguez PF, Kleim JA, et al. BDNF val66met polymorphism influences motor system function in the human brain. Cereb Cortex 2010;20(5):1254‐1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Degos B, Deniau JM, Le Cam J, Mailly P, Maurice N. Evidence for a direct subthalamo‐cortical loop circuit in the rat. Eur J Neurosci 2008;27(10):2599‐2610. [DOI] [PubMed] [Google Scholar]
- 121. Liao L, Pilotte J, Xu T, et al. BDNF induces widespread changes in synaptic protein content and up‐regulates components of the translation machinery: an analysis using high‐throughput proteomics. J Proteome Res 2007;6(3):1059‐1071. [DOI] [PubMed] [Google Scholar]
- 122. Leal G, Comprido D, Duarte CB. BDNF‐induced local protein synthesis and synaptic plasticity. Neuropharmacology 2014;76(Pt C):639‐656. [DOI] [PubMed] [Google Scholar]
- 123. Mitre M, Mariga A, Chao MV. Neurotrophin signalling: novel insights into mechanisms and pathophysiology. Clin Sci (Lond) 2017;131(1):13‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Guo W, Nagappan G, Lu B. Differential effects of transient and sustained activation of BDNF‐TrkB signaling. Dev Neurobiol 2018;78(7):647‐659. [DOI] [PubMed] [Google Scholar]
- 125. Lhommee E, Wojtecki L, Czernecki V, et al. Behavioural outcomes of subthalamic stimulation and medical therapy versus medical therapy alone for Parkinson's disease with early motor complications (EARLYSTIM trial): secondary analysis of an open‐label randomised trial. Lancet Neurol 2018;17(3):223‐231. [DOI] [PubMed] [Google Scholar]
- 126. Dafsari HS, Silverdale M, Strack M, et al. Nonmotor symptoms evolution during 24 months of bilateral subthalamic stimulation in Parkinson's disease. Mov Disord 2018;33(3):421‐430. [DOI] [PubMed] [Google Scholar]
- 127. Toft M, Lilleeng B, Ramm‐Pettersen J, et al. Long‐term efficacy and mortality in Parkinson's disease patients treated with subthalamic stimulation. Mov Disord 2011;26(10):1931‐1934. [DOI] [PubMed] [Google Scholar]
- 128. Aviles‐Olmos I, Kefalopoulou Z, Tripoliti E, et al. Long‐term outcome of subthalamic nucleus deep brain stimulation for Parkinson's disease using an MRI‐guided and MRI‐verified approach. J Neurol Neurosurg Psychiatry 2014;85(12):1419‐1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Lezcano E, Gomez‐Esteban JC, Tijero B, et al. Long‐term impact on quality of life of subthalamic nucleus stimulation in Parkinson's disease. J Neurol 2016;263(5):895‐905. [DOI] [PubMed] [Google Scholar]
- 130. Fasano A, Romito LM, Daniele A, et al. Motor and cognitive outcome in patients with Parkinson's disease 8 years after subthalamic implants. Brain 2010;133:2664‐2676. [DOI] [PubMed] [Google Scholar]
- 131. Rocha S, Monteiro A, Linhares P, et al. Long‐term mortality analysis in Parkinson's disease treated with deep brain stimulation. Parkinsons Dis 2014;2014:717041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Hilker R, Voges J, Ghaemi M, et al. Deep brain stimulation of the subthalamic nucleus does not increase the striatal dopamine concentration in parkinsonian humans. Mov Disord 2003;18(1):41‐48. [DOI] [PubMed] [Google Scholar]
- 133. Bang Henriksen M, Johnsen EL, Sunde N, Vase A, Gjelstrup MC, Ostergaard K. Surviving 10 years with deep brain stimulation for Parkinson's disease‐‐a follow‐up of 79 patients. Eur J Neurol 2016;23(1):53‐61. [DOI] [PubMed] [Google Scholar]
- 134. Zibetti M, Merola A, Rizzi L, et al. Beyond nine years of continuous subthalamic nucleus deep brain stimulation in Parkinson's disease. Mov Disord 2011;26(13):2327‐2334. [DOI] [PubMed] [Google Scholar]
- 135. Strafella AP, Sadikot AF, Dagher A. Subthalamic deep brain stimulation does not induce striatal dopamine release in Parkinson's disease. Neuroreport 2003;14(9):1287‐1289. [DOI] [PubMed] [Google Scholar]
- 136. Spieles‐Engemann AL, Collier TJ, Sortwell CE. A functionally relevant and long‐term model of deep brain stimulation of the rat subthalamic nucleus: advantages and considerations. Eur J Neurosci 2010;32(7):1092‐1099. [DOI] [PubMed] [Google Scholar]
- 137. Windels F, Bruet N, Poupard A, Feuerstein C, Bertrand A, Savasta M. Influence of the frequency parameter on extracellular glutamate and gamma‐aminobutyric acid in substantia nigra and globus pallidus during electrical stimulation of subthalamic nucleus in rats. J Neurosci Res 2003;72(2):259‐267. [DOI] [PubMed] [Google Scholar]
- 138. Tai CH, Boraud T, Bezard E, Bioulac B, Gross C, Benazzouz A. Electrophysiological and metabolic evidence that high‐frequency stimulation of the subthalamic nucleus bridles neuronal activity in the subthalamic nucleus and the substantia nigra reticulata. FASEB J 2003;17(13):1820‐1830. [DOI] [PubMed] [Google Scholar]
- 139. Zheng F, Lammert K, Nixdorf‐Bergweiler BE, Steigerwald F, Volkmann J, Alzheimer C. Axonal failure during high frequency stimulation of rat subthalamic nucleus. J Physiol 2011;589(Pt 11):2781‐2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lee KH, Kristic K, van Hoff R, et al. High‐frequency stimulation of the subthalamic nucleus increases glutamate in the subthalamic nucleus of rats as demonstrated by in vivo enzyme‐linked glutamate sensor. Brain Res 2007;1162:121‐129. [DOI] [PubMed] [Google Scholar]
- 141. Paul G, Reum T, Meissner W, et al. High frequency stimulation of the subthalamic nucleus influences striatal dopaminergic metabolism in the naive rat. Neuroreport 2000;11(3):441‐444. [DOI] [PubMed] [Google Scholar]
- 142. Bruet N, Windels F, Bertrand A, Feuerstein C, Poupard A, Savasta M. High frequency stimulation of the subthalamic nucleus increases the extracellular contents of striatal dopamine in normal and partially dopaminergic denervated rats. J Neuropathol Exp Neurol 2001;60(1):15‐24. [DOI] [PubMed] [Google Scholar]
- 143. Dickerson BC, Eichenbaum H. The Episodic Memory System: neurocircuitry and disorders. Neuropsychopharmacology 2010;35(1):86‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Shi LH, Luo F, Woodward DJ, Chang JY. Basal ganglia neural responses during behaviorally effective deep brain stimulation of the subthalamic nucleus in rats performing a treadmill locomotion test. Synapse 2006;59(7):445‐457. [DOI] [PubMed] [Google Scholar]
- 145. Meissner W, Reum T, Paul G, et al. Striatal dopaminergic metabolism is increased by deep brain stimulation of the subthalamic nucleus in 6‐hydroxydopamine lesioned rats. Neurosci Lett 2001;303(3):165‐168. [DOI] [PubMed] [Google Scholar]
- 146. Meissner W, Harnack D, Paul G, et al. Deep brain stimulation of subthalamic neurons increases striatal dopamine metabolism and induces contralateral circling in freely moving 6‐hydroxydopamine‐lesioned rats. Neurosci Lett 2002;328(2):105‐108. [DOI] [PubMed] [Google Scholar]
- 147. Hashimoto T, Elder CM, Okun MS, Patrick SK, Vitek JL. Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. J Neurosci 2003;23(5):1916‐1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Meissner W, Leblois A, Hansel D, et al. Subthalamic high frequency stimulation resets subthalamic firing and reduces abnormal oscillations. Brain 2005;128:2372‐2382. [DOI] [PubMed] [Google Scholar]
- 149. Zhao XD, Cao YQ, Liu HH, Li FQ, You BM, Zhou XP. Long term high frequency stimulation of STN increases dopamine in the corpus striatum of hemiparkinsonian rhesus monkey. Brain Res 2009;1286:230‐238. [DOI] [PubMed] [Google Scholar]