

Abstract
Aneuploidy is a very rare and tissue‐specific event in normal conditions, occurring in a low number of brain and liver cells. Its frequency increases in age‐related disorders and is one of the hallmarks of cancer. Aneuploidy has been associated with defects in the spindle assembly checkpoint (SAC). However, the relationship between chromosome number alterations, SAC genes and tumor susceptibility remains unclear. Here, we provide a comprehensive review of SAC gene alterations at genomic and transcriptional level across human cancers and discuss the oncogenic and tumor suppressor functions of aneuploidy. SAC genes are rarely mutated but frequently overexpressed, with a negative prognostic impact on different tumor types. Both increased and decreased SAC gene expression show oncogenic potential in mice. SAC gene upregulation may drive aneuploidization and tumorigenesis through mitotic delay, coupled with additional oncogenic functions outside mitosis. The genomic background and environmental conditions influence the fate of aneuploid cells. Aneuploidy reduces cellular fitness. It induces growth and contact inhibition, mitotic and proteotoxic stress, cell senescence and production of reactive oxygen species. However, aneuploidy confers an evolutionary flexibility by favoring genome and chromosome instability (CIN), cellular adaptation, stem cell‐like properties and immune escape. These properties represent the driving force of aneuploid cancers, especially under conditions of stress and pharmacological pressure, and are currently under investigation as potential therapeutic targets. Indeed, promising results have been obtained from synthetic lethal combinations exploiting CIN, mitotic defects, and aneuploidy‐tolerating mechanisms as cancer vulnerability.
Keywords: aneuploidy, carcinogenesis, cancer therapy, spindle assembly checkpoint
Abbreviations
- APC
APC, WNT signaling pathway regulator
- APC/C
anaphase‐promoting complex/cyclosome
- APP
Amyloid Beta Precursor Protein
- BUB1
BUB1 Mitotic Checkpoint Serine/Threonine Kinase
- BUB3
BUB3, Mitotic Checkpoint Protein
- BUBR1/BUB1B
BUB1 Mitotic Checkpoint Serine/Threonine Kinase B
- CCP1
cytochrome‐c peroxidase
- CDC20
Cell Division Cycle 20
- CDK1
Cyclin Dependent Kinase 1
- CENP‐E
centromere protein E
- CIN
chromosome instability
- DSB
double strand break
- FISH
Fluorescence In Situ Hybridization
- GIN
genomic instability
- hPSCs
human pluripotent stem cells
- KNL1
Kinetochore Scaffold 1
- KRAS
KRAS proto‐oncogene
- MAD1/MAD1L1
MAD1 Mitotic Arrest Deficient Like 1
- MAD2/MAD2L1
Mitotic Arrest Deficient 2 Like 1
- MCC
mitotic checkpoint complex
- MEFs
murine embryonic fibroblasts
- MHC
major histocompatibility complex
- MPS1
TTK Protein Kinase
- MVA
Mosaic Variegated Aneuploidy
- MYO1
myosin 1
- NDC80
NDC80, Kinetochore Complex Component
- PLK1
Polo Like Kinase 1
- PPP2R1A
protein phosphatase 2 scaffold subunit alpha
- PTEN
phosphatase and tensin homolog
- RB
RB Transcriptional Corepressor 1
- ROS
Reactive oxygen species
- SAC
spindle assembly checkpoint
- SKY
Spectral Karyotyping
- tg
trasgenic
- TP53
Tumor Protein P53
- UBP6
ubiquitin‐specific protease UBP6
- UTH1
SUN family protein UTH1.
Aneuploidy: A Normal and Abnormal Condition
Normal human diploid cells contain 23 pairs of chromosome (44 autosomes and two sex chromosomes). In some circumstances, the number of whole chromosomes is altered, a condition known as aneuploidy. Aneuploidy is physiological during cellular development in some tissues (e.g., in liver and brain), probably because of its contribution to cellular diversity, which provides a selective advantage in response to injuries. Binucleated and polyploid hepatocytes are detectable in mice few weeks after birth.1 They can revert to diploidy and become aneuploid,2 while diploid liver cells can increase their ploidy. This dynamic mechanism, defined as “ploidy‐conveyor,”3 generates aneuploidy and has also been reported in human hepatocytes.4 Under conditions of stress, hepatocytes acquire specific aneuploidies enabling them to resist to chronic liver injuries.5 Moreover, aneuploid neurons, retaining functional activity,6 have been observed in developing and adult murine models7, 8 and in the human brain.9, 10 The majority of studies have reported a prevalence of aneuploid cells exceeding 50% and 20% in the liver3, 4 and brain,7, 10, 11, 12 respectively. However, SKY and FISH approaches, which have been used for karyotype analysis, may overestimate aneuploidy. Indeed, a recent single cell sequencing study revealed that even in liver and brain tissues, aneuploidy accounted for less than 5% of all cells13 under physiological conditions. In different tissues, aneuploidy is associated with aging and age‐related disorders. Mice expressing reduced levels of the spindle assembly checkpoint (SAC) component BUBR1, which is mutated in the majority of patients with Mosaic Variegated Aneuploidy (MVA) syndrome, develop progressive aneuploidy and age‐related defects including cataracts, loss of subcutaneous fat, skeletal muscle wasting, lordokyphosis, impaired wound healing14, 15, 16 and cerebral degeneration,17 with deficits in neural progenitor proliferation and maturation.18 In oocytes, the frequency of chromosome segregation errors in meiosis I increase with maternal aging,19 along with a decrease in BUBR1 protein.20 The aneuploid condition may also favor neurodegeneration during aging. The APP gene, which encodes for the protein forming amyloid β plaques in Alzheimer's disease, is located on chromosome 21. Individuals with Down's syndrome frequently develop this neurodegenerative disorder by the age of 40,21 and buccal cells from patients with Alzheimer's disease frequently carry trisomy of chromosomes 21 or 17, where many susceptibility genes are located.22
These findings suggest that a low frequency of aneuploid cells can be tolerated13 or may even be advantageous under specific conditions in nonmalignant tissues,23 whereas increased rates of aneuploidy can become pathogenic, as observed in neurodegenerative diseases22 and in cancer.24 Theodor Boveri initially suggested that an abnormal chromosome number causes tumorigenesis.24 Over the past 100 years, a number of studies have investigated the cellular and molecular events that cause aneuploidy and studied its potential involvement in cancer development. Here, we describe SAC gene alterations across tumors and their link with neoplastic transformation. We also focus on the complex relationship between aneuploidy and cancer, including the oncogenic and tumor suppressor functions of the abnormal chromosome number and its therapeutic potential.
The Spindle Assembly Checkpoint in Aneuploidy Generation and Cancer
Aneuploidy in mitotically dividing cells can result from numerous defects, including mitotic slippage, cytokinesis failure, spindle multipolarity, defective kinetochore‐microtubule attachments, perturbed microtubule dynamics, cohesion defects, and impaired SAC function.25, 26 The SAC prevents entry into anaphase and premature chromosome segregation until all kinetochores are properly attached to the mitotic spindle. This function is achieved through assembly of the mitotic checkpoint complex (MCC), the SAC effector, which inhibits the activity of the anaphase‐promoting complex/cyclosome (APC/C)CDC20 .27 Briefly, when the SAC is satisfied, the MCC is disassembled and APC/CCDC20 drives ubiquitination and proteolytic degradation of cyclin B1 and securin. These events induce mitotic exit and sister chromatid separation by degradation of the cohesin complex.
A weakened SAC may allow cells to enter anaphase in the presence of unattached or misaligned chromosomes and both copies of one chromosome may be deposited into a single daughter cell (Fig. 1). Thus, failure of the SAC machinery is an obvious candidate mechanism involved in the generation of aneuploidy during mitosis. However, the genes encoding SAC proteins (including MAD1L1, BUB1, BUB1B, CDC20, BUB3, and MAD2L1) are rarely mutated in human cancers. A mutation frequency exceeding 5% has been only detected in uterine corpus endometrial carcinoma (9.6, 7.4, 6.2, 6.4, and 7.6% of patients carrying MAD1L1, BUB1, BUB1B, CDC20, or BUB3 mutations, respectively) and colon adenocarcinoma (5.5% of patients with MAD1L1 or BUB1 mutations) according to next generation sequencing data from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/, Fig. 2). On the contrary, SAC genes are deregulated at mRNA and protein level in a number of tumors (Table 1), suggesting potential alterations of epigenetic, transcriptional and post‐transcriptional regulation. For example, mutations of oncogenic or tumor suppressor pathways can lead to deregulated SAC gene expression. There is evidence that inactivating RB mutations cause deregulation of the E2F family of transcription factors resulting in MAD2 overexpression28 and chromosome instability (CIN), which have also been detected in a p53 mutant mouse model.29 With the exception of a few reported cases of reduced expression, SAC genes are generally overexpressed in primary tumors (Table 1). High expression levels associate with elevated proliferation index and metastatic potential and predict advanced stage, reduced overall survival, disease‐free survival and recurrence‐free survival across several cancer types, including solid tumors, and hematological malignancies. This observation appears in contrast to the fact that aneuploidy occurs in cases of defective SAC. However, both increased and decreased SAC gene expression induces aneuploidy and favors tumor development, as demonstrated in mice (Table 2). The protumorigenic or antitumorigenic effect is also dependent on the specific SAC gene which is overexpressed or downregulated. For example, CDC20 overexpression impairs SAC function and favors aneuploidization in oral cancer.30 Moreover, chromosome missegregation and aneuploidy have been reported in both transgenic (tg), hypomorphic and haploinsufficient mouse models, including MAD2‐tg31 and mad2 +/−,32 BUB1‐tg,33 bub1 Δ2–3/Δ2–3,34 bub1 −/H and bub1 H/H.35 These findings suggest that expression of some SAC genes above threshold levels is required to maintain genomic stability and prevent tumorigenesis, as shown in the bub1 +/− model, characterized by higher levels of BUB1 protein compared to bub1 H/H and bub1 −/H mice and lower tumor incidence.35 However, SAC gene deficiency is also detrimental, probably due to extreme CIN.26 Indeed homozygous deletion of many SAC components causes early embryonic lethality. Conversely, the protumorigenic effect of SAC gene overexpression may be linked to delayed mitotic exit, which induces aneuploidy (e.g., MAD2 overexpression stabilizes securin and cyclin B and inhibits cytokinesis28, 31), and to potential oncogenic roles of SAC proteins outside mitosis: BUBR1 plays a role in DNA damage responses,36 CDC20 is involved in the regulation of apoptosis,37 DNA repair38 and stem‐like cell properties39, 40, 41 and MAD1 has a function at the Golgi apparatus.42 This complex scenario suggests that both increased and decreased SAC gene expression may favor tumorigenesis, depending on the threshold level, the gene functions inside and outside mitosis, the effect on chromosome stability, the cell type and its genomic background.
Figure 1.
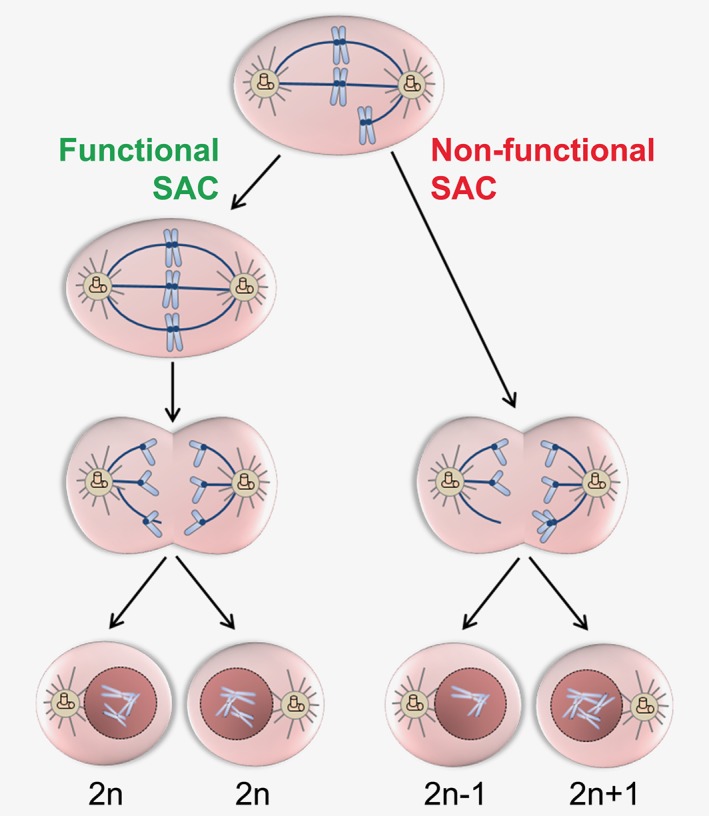
Generation of aneuploidy by non‐functional SAC. The SAC is activated by the presence of unattached or misaligned kinetochores and prevents chromosome segregation errors. A non‐functional SAC allows cells with unattached or misaligned kinetochores to proceed from metaphase to anaphase, resulting in daughter cells with an abnormal chromosome number.
Figure 2.
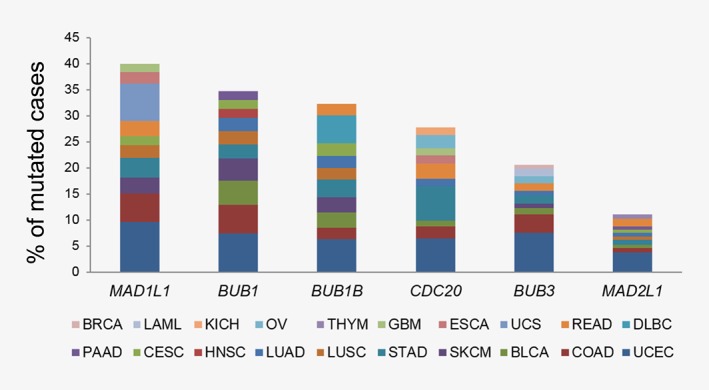
Distribution of SAC gene mutations across cancers. Frequency of patients with mutations in SAC genes from TCGA cohorts (LAML, Acute Myeloid Leukemia; BLCA, Bladder Urothelial Carcinoma; BRCA, Breast Invasive Carcinoma; CESC, Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma; COAD, Colon Adenocarcinoma; ESCA, Esophageal Carcinoma; GBM, Glioblastoma Multiforme; HNSC, Head and Neck Squamous Cell Carcinoma; KICH, Kidney Chromophobe; LUAD, Lung Adenocarcinoma; LUSC, Lung Squamous Cell Carcinoma; DLBC, Diffuse Large B‐cell Lymphoma; OV, Ovarian Serous Cystadenocarcinoma; PAAD, Pancreatic Adenocarcinoma; READ, Rectum Adenocarcinoma; SKCM, Skin Cutaneous Melanoma; STAD, Stomach Adenocarcinoma; THYM, Thymoma; UCS, Uterine Carcinosarcoma; UCEC, Uterine Corpus Endometrial Carcinoma).
Table 1.
Deregulated expression of SAC genes across tumors
| Tumor type | Expression level | Main observations | Detection method | Reference(s) |
|---|---|---|---|---|
| BUB1 | ||||
| Acute myeloid leukemia | Up | Associated with −5/del(5q) therapy‐related AML (CD34+ stem/progenitor cells) | GEM | 100 |
| Down | RT‐PCR | 101 | ||
| Breast cancer | Up | Associated with cases diagnosed at <40 years of age, estrogen‐ and progesterone‐receptor negative tumors, high grade, and poor OS and RFS | qPCR, GEM, IHC | 102, 103, 104 |
| Up | Compared to normal tissue samples | RNAseq, GEM | 105 | |
| Clear cell renal cell carcinoma | Up | Correlated with the number of genomic copy number changes and high Furhman grade | qPCR | 106 |
| Up | Compared to normal tissue samples | GEM | 105 | |
| Colon cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Endometrial cancer | +/Up | 28.6% of cases, associated with low clinical stage and histological grade; higher in nonendometrioid compared to endometrioid carcinomas | IHC, GEM | 107, 108 |
| Gastric cancer | Up | 40–84% of cases, associated with Ki‐67 expression and PCNA marker, not correlated with ploidy | qPCR, RT‐PCR | 109, 110, 111 |
| Low Freq | Associated with larger tumor size, higher incidence of lymph node metastases, distant metastases and higher UICC stage, reduced Ki‐67 protein expression and shorter survival | IHC | 112 | |
| Glioma | Up | Associated with high grade | GEM, qPCR | 113 |
| Hepatocellular carcinoma | Up | Part of a gene expression signature predicting OS and DFS | RNAseq, GEM | 114, 115 |
| Up | Compared to normal tissue samples | GEM | 105 | |
| Lung cancer | Up | Associated with adverse OS and RFS; progressive increase in expression from adenocarcinoma to squamous cell carcinoma, large cell carcinoma and the small cell subtype | qPCR, GEM | 116, 117 |
| Up | Compared to normal tissue samples | GEM | 105 | |
| Melanoma | Up | Associated with metastatic melanoma | GEM | 118 |
| Ovarian/Uterine cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Pheochromocytoma and paraganglioma | Up | Associated with metastatic tumor | RNAseq | 119 |
| Prostate cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Salivary gland tumors | Up | Associated with advanced clinical stage and Ki‐67 labeling index | qPCR and WB | 120 |
| Thyroid Carcinomas | Up | Associated with undifferentiated carcinoma | qPCR | 121 |
| BUBR1 | ||||
| Acute Myeloid Leukemia | Down | Reduced in total bone marrow cells as well as in CD34+ bone marrow cells | GEM | 122 |
| Bladder cancer | Up | Associated with CIN, aneuploidy, centrosome amplification, high histological grade, advanced pathological stage, high cell proliferation, shorter RFS and PFS | IHC | 123 |
| Breast cancer | Up | 38% of cases, associated with triple negative tumors, poor OS, DFS and disease‐specific survival, improved OS in basal‐like tumors and worse OS in luminal and untreated patients, grade 2 and 3 ductal breast cancer; correlated with high histological tumor grade, Ki‐67 proliferation index and intrachromosomal instability in ductal breast cancer | IHC, GEM, IHC, qPCR | 104, 124, 125, 126, 127, 128 |
| Up | Compared to normal tissue samples | RNAseq, GEM | 105 | |
| Clear cell renal cell carcinoma | Up | Correlated with the number of genomic copy number changes and high Furhman grade | qPCR | 106 |
| Up | Compared to normal tissue samples | GEM | 105 | |
| Colon cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Colorectal cancer | Down | Reduced in aneuploid compared to diploid cases | IHC | 129 |
| Epithelial ovarian cancer | + | Associated with advanced stage, serous histology and high grade, shorter RFS | IHC | 130 |
| Esophageal squamous cell carcinoma | Up | Ab array, GEM, IHC, WB | 131 | |
| Gallbladder cancer | Up | qPCR | 132 | |
| Gastric cancer | Up | 50–68% of cases, correlated with Ki‐67 expression, aneuploidy (debated), deep invasion, lymph node and liver metastasis, poor prognosis | IHC, qPCR | 109, 133, 134 |
| Glioma | Up | Associated with high grade and predictor of poor prognosis | GEM and qPCR | 113 |
| Hepatocellular carcinoma | Up | 45–64% of cases; associated with larger tumor size, high histological grade, advanced pathological stage, reduced OS and RFS; associated with p53 and Ki‐67 markers | qPCR, WB, IHC, RNAseq, GEM | 114, 135, 136 |
| Up | Compared to normal tissue samples | GEM | 105 | |
| Lung cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Malignant peripheral nerve sheath tumors | Up | Associated with malignant transformation of plexiform neurofibroma | GEM, IHC | 137 |
| Multiple myeloma | Up | Associated with high risk | GEM | 138 |
| Nasopharyngeal carcinoma | Up | Commonly upregulated across six different studies | GEM | 139 |
| Oral squamous cell carcinoma | Up | Controversial results across studies: upregulated in 22.4% of cases, associated with advanced stages, larger tumor size, shorter OS and HPV‐positivity (76% of cases) according to Lira et al.; associated with less advanced pathologic stage, longer OS and shorter RFS according to Rizzardi et al. | IHC, qPCR | 140, 141 |
| Ovarian cancer | Up | Associated with serous carcinomas, advanced stage, and increased cellular proliferation | IHC | 142 |
| Ovarian/Uterine cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Pancreatic cancer | Up | GEM | 143 | |
| Low | 65% of cases | IHC | 144 | |
| Pediatric adrenocortical tumors | Up | Associated with Weiss score ≥3 | qPCR | 145 |
| Pheochromocytoma and paraganglioma | Up | Associated with metastatic tumor | RNAseq | 119 |
| Primary gastrointestinal diffuse large B cell lymphoma | Up | Correlates with Ki‐67 proliferation index, not with survival | IHC | 146 |
| Prostate cancer | Up | 63% of cases, associated with reduced OS, high Gleason score, and predictor of shorter RFS | IHC, qPCR | 147, 148 |
| Up | Compared to normal tissue samples | GEM | 105 | |
| Salivary duct carcinoma | Up | 25.9% of cases, no prognostic value | IHC | 149 |
| Testicular germ cell tumor | Down | Decreased in nonseminomas compared to seminomas | IHC | 129 |
| Thyroid Carcinomas | Up | Associated with undifferentiated carcinoma, followed by advanced differentiated carcinoma | qPCR | 121 |
| Tonsillar carcinomas | + | 16% of positive cells (median number); prognostic factor in univariate survival analysis and in multivariate analyses (together with stage, age, and HPV status) | IHC | 150 |
| Upper tract urothelial carcinoma | Up | Associated with CIN, high histological grade, shorter disease‐specific survival | IHC | 151 |
| Wilms Tumors | Down | Associated with hyperdiploid or near‐or‐pseudodiploid tumor, while expression levels are increased in diploid tumors | WB | 152 |
| CDC20 | ||||
| Breast cancer | Up | Associated with aneuploidy, aggressive course, and poor OS | qPCR, IHC | 104, 153 |
| Up | Compared to normal tissue samples | RNAseq, GEM | 105 | |
| Cervical cancer | Up | 8% of low‐grade squamous intraepithelial lesions, 49.4% of high‐grade squamous intraepithelial lesions, 22.3% of squamous cell carcinomas | GEM, IHC | 154, 155 |
| Clear cell renal cell carcinoma | Up | Associated with advanced pathologic stage and shorter OS | GEM | 156 |
| Up | Compared to normal tissue samples | GEM | 105 | |
| Colon cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Colorectal cancer | Up | Associated with III and IV clinical stage, N classification, M0 classification, moderate pathologic differentiation, shorter OS; increased in liver metastasis | GEM, qPCR, IHC | 157, 158 |
| Gastric cancer | Up | Associated with increased tumor size, histological grade, lymph node involvement, TNM stage and poor OS; independent predictor of OS | qPCR, IHC | 159 |
| Glioblastoma | Up | 74.1% of cases | GEM, IHC | 160 |
| Glioma | Up | Associated with high grade | GEM, qPCR | 113 |
| Head and neck tumors | Up | WB | 30 | |
| Hepatocellular carcinoma | Up | Hub gene, associated with poor tumor differentiation, high TNM stage, P53 and Ki‐67 expression | qPCR, WB, IHC, GEM | 115, 136, 161, 162, 163 |
| Up | Compared to normal tissue samples | GEM | 105 | |
| Kidney renal clear cell carcinoma | Up | Associated with poor OS | RNAseq | 164 |
| Lung cancer | Up | 19.6% of cases, correlated with male sex, pT status, pleural invasion, nonadenocarcinoma histology, MAD2 expression, shorter OS | IHC, RNAseq, GEM | 117, 165, 166, 167 |
| Up | Compared to normal tissue samples | GEM | 105 | |
| Multiple myeloma | Up | Associated with high‐risk patients and poor prognosis | GEM | 168 |
| Myelodisplastc syndrome | Up | Increased in patients with dysmegakaryopoiesis, thrombocytopenia and high‐risk cases, associated with increased bone marrow cellularity, age, severe thrombocytopenia, three‐lineage dysplasia, complex karyotype, and worse prognosis | qPCR, IHC | 169, 170, 171 |
| Oral squamous cell carcinoma | Up | 56.9% of cases, associated with shorter OS | IHC | 172 |
| Ovarian/Uterine cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Pancreatic cancer | Up | Associated with poor differentiation, reduced RFS in pancreatic ductal adenocarcinoma | WB, GEM. IHC | 143, 173, 174 |
| Prostate cancer | Up | Associated with lower biochemical‐RFS after laparoscopic radical prostatectomy | IHC | 175 |
| Up | Compared to normal tissue samples | GEM | 105 | |
| Serous epiphelial ovarian cancer | Up | Associated with poor OS | IHC | 176 |
| Urothelial bladder cancer | Up | 59% of cases, associated with high grade, advanced age and stage, nonpapillary growth pattern and distant metastasis; predictor of poor OS and RFS | IHC | 177 |
| Uterine leiomyosarcoma | Up | GEM | 178 | |
| MAD1 | ||||
| Breast cancer | Up | 60% of cases; associated with lymph node involvement, tumor size, grade, TP53 mutations, poor OS; not associated with increased proliferation rate and estrogen receptor status | IHC, qPCR, GEM | 104, 179 |
| Up | Compared to normal tissue samples | RNAseq, GEM | 105 | |
| Chromophobe renal cell carcinoma | Down | qPCR | 180 | |
| Clear cell renal cell carcinoma | Down | qPCR | 106 | |
| Up | Compared to normal tissue samples | GEM | 105 | |
| Colon cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Gastric carcinoma | Down | 47.1% of adenomas and 60.5% carcinomas, associated with advanced carcinomas and intestinal type | 2‐DE, pPCR, IHC, WB | 181, 182 |
| Glioma | Up | Associated with high grade | GEM and qPCR | 113 |
| Hepatocellular carcinoma | Down | Associated with tumor recurrence after surgical resection | WB | 183 |
| Up | Compared to normal tissue samples | GEM | 105 | |
| Lung cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Ovarian/Uterine cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Small cell lung cancer | + | 39.8% of primary tumors and 46.9% of lymph node metastasis; associated with high tumor‐node‐metastasis stage and International Association for the Study of Lung Cancer stage, increased tumor size and recurrence, shorter OS and RFS | IHC | 184 |
| MAD2 | ||||
| Breast cancer | Up | 28.4% of cases; associated with age <50 years, HER‐2 and P53 positivity, luminal B and HER‐2 subtypes, estrogen and progesterone‐receptor negative tumors, high grade and poor OS and RFS; overexpressed in invasive ductal breast carcinoma | IHC, GEM, qPCR | 103, 104, 128, 185 |
| Down | Associated with HER‐2 overexpression in ductal breast carcinoma | IHC | 125 | |
| Up | Compared to normal tissue samples | RNAseq, GEM | 105 | |
| Cervical cancer | Up | 2% of low‐grade squamous intraepithelial lesions, 67.1% of high‐grade squamous intraepithelial lesions, 52.4% of squamous cell carcinomas; correlated with patient age <60 years, non‐keratinizing histologic type and a lesser degree of stromal invasion in squamous cell carcinoma cases | IHC | 154 |
| Clear cell renal cell carcinoma | Up | qPCR | 106 | |
| Up | Compared to normal tissue samples | GEM | 105 | |
| Colon cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Colorectal cancer | Up | 75% of cases, associated with increased stage, poor differentiation, presence of lymph node metastasis, and reduced survival after excision | IHC, GEM, qPCR | 157, 186 |
| Endometrial cancer | + | 85.7% of cases, associated with high clinical stage and histological grade | IHC | 107 |
| Esophageal squamous cell carcinoma | Up | Associated with low histological grade | AbM, GEM, IHC, WB | 131 |
| Gastric cancer | Up | Associated with poor differentiation, and presence of lymph node metastasis | IHC | 187 |
| Glioma | Up | Associated with high grade | GEM and qPCR | 113 |
| Hepatocellular carcinoma | Up | Associated with histologic grade progression and low OS | qPCR, IHC, WB, GEM | 115, 136, 188 |
| Up | Compared to normal tissue samples | GEM | 105 | |
| High‐grade serous epithelial ovarian cancer | Down | Associated with reduced PFS | IHC | 189 |
| Lung cancer | Up | 26.3% of cases, associated with male sex, tumor progression, visceral or parietal pleural invasion, nonadenocarcinoma, histological classification, smoking history and shorter OS and RFS; independent prognostic factor in multivariate analysis; associated with CDC20 expression in nonsmall cell lung cancer | IHC, GEM | 117, 165, 190, 191 |
| Up | Compared to normal tissue samples | GEM | 105 | |
| Malignant pleural mesothelioma | Up | Potentially correlated with reduced OS | GEM, qPCR, WB, IHC | 192 |
| Multiple myeloma | Down | Hub gene | GEM | 193 |
| Myelodysplastic syndrome | Down | Decreased in patients with 2 or 3 cytopenias and hypoplastic cases, associated with high frequency of chromosomal alterations and high mortality rate | qPCR | 169 |
| Up | Associated with increased bone marrow cellularity and age, severe thrombocytopenia, poor prognosis | IHC, qPCR | 170, 171 | |
| Nasopharyngeal carcinoma | Up | Commonly upregulated across six different studies | GEM | 139 |
| Oral squamous cell carcinoma | Up | 36.7% of cases; associated with advanced stages, larger tumor size, poor differentiation histological grade, lymph nodes involvement, high Ki‐67 labeling index, shorter OS | IHC, qPCR | 141, 194 |
| Osteosarcoma | Up | Associated with low differentiation and high clinical stage, earlier metastasis and poor OS | IHC | 195 |
| Ovarian cancer | Down | Associated with increased cellular proliferation, shorter OS, and RFS | IHC | 142, 196 |
| Up | 52.3% of cases of high‐grade serous carcinoma, where low expression predicts inferior PFS; overexpressed in malignant mucinous ovarian cancer compared to non‐malignant and benign lesions | IHC | 197, 198 | |
| Ovarian/Uterine cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Papillary renal cell carcinoma | Up | qPCR | 180 | |
| Primary gastrointestinal diffuse large B cell lymphoma | Up | Associated with Ki‐67 proliferation index and lower DFS | IHC | 199 |
| Prostate cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Salivary duct carcinoma | Up | 55.6% of cases, no prognostic value | IHC | 149 |
| Soft‐tissue sarcoma | Up | 52% of translocation‐associated (TA, atypical or high‐grade morphology, such as round cell liposarcoma and fibrosarcomatous dermatofibrosarcoma protuberans) and 66% of non‐TA sarcoma; associated with multipolar mitoses and anaphase bridges | IHC | 200 |
| Testicular germ cell tumor | Down | Decreased nuclear expression and increased cytoplasmic levels in seminomas, decreased in nonseminomas compared to seminomas | IHC | 129, 201 |
| Thyroid carcinomas | Up | Associated with undifferentiated carcinoma, followed by advanced differentiated carcinoma | qPCR | 121 |
| Tonsillar carcinomas | + | 27% of positive cells (median number) | IHC | 150 |
| Urothelial bladder cancer | Up | 51% of cases, associated with high grade, advanced stage and nonpapillary growth pattern, predictor of poor OS | IHC | 177 |
| BUB3 | ||||
| Breast cancer | Up | Significantly overexpressed when amplified in triple negative breast cancer | qPCR, GEM | 104, 202 |
| Up | Compared to normal tissue samples | RNAseq, GEM | 105 | |
| Clear cell renal cell carcinoma | Up | Compared to normal tissue samples | GEM | 105 |
| Colon cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Gastric cancer | Up | 79% of cases, correlates with Ki‐67 expression, does not correlate with ploidy | qPCR | 109 |
| Hepatocellular carcinoma | Up | Compared to normal tissue samples | GEM | 105 |
| Lung cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Ovarian/Uterine cancer | Up | Compared to normal tissue samples | GEM | 105 |
| Prostate cancer | Up | Compared to normal tissue samples | GEM | 105 |
Up, overexpressed; down, downregulated; +, positive; low freq, low frequency; low, low expression; del, deletion; AML, acute myeloid leukemia; OS, overall survival; DFS, disease‐free survival; RFS, recurrence‐free survival; PFS, progression‐free survival; PCNA, proliferating cell nuclear antigen; UICC, International Union Against Cancer; HPV, human papillomavirus; pT, pathological tumor progression; TNM, tumor/node/metastasis; IHC, immunohistochemistry; GEM, gene expression microarray; qPCR, quantitative polymerase chain reaction; RT‐PCR, reverse transcription‐polymerase chain reaction; WB, western blotting; RNAseq, RNA sequencing; 2‐DE, two‐dimensional gel electrophoresis.
Table 2.
Mouse models with SAC gene overexpression or downregulation showing evidence of increased/reduced predisposition to tumor development
| Mouse model | Phenotype | Reference |
|---|---|---|
| MAD2‐tg |
|
31 |
| mad2 +/− |
|
32 |
| mad1 +/− |
|
203 |
| BUB1‐tg |
|
33 |
| bubR1 +/− |
|
204 |
| bubR1 K243R/+ |
|
205 |
| bub1 Δ2–3/Δ2–3 |
|
34 |
| BUBR1‐tg |
|
14 |
| bub1 −/H bub1 H/H bub1 +/− |
|
35 |
| bub3 +/− |
|
206 |
| rae1 +/− bub3+/− |
|
207 |
| cdc20 +/AAA |
|
208 |
| cdc20 +/−, cdc20 H/H, cdc20 −/H |
|
209 |
Tg, transgenic; H, hypomorphic allele.
Tumor‐Protecting and Tumor‐Promoting Effects of Aneuploidy
The complex and much debated relationship between aneuploidy and cancer fosters a very active research field. There is evidence to suggest that aneuploidy can exert an antitumorigenic or a protumorigenic effect (Fig. 3). Studies on yeast strains, murine and human cells have shown that aneuploidy impairs the proliferative capacity of nonmalignant cells and that the phenotype is independent of the identity of the individual chromosomes, while being potentially proportional to its size.43, 44, 45, 46, 47, 48 Changes in chromosome copy number result in transcriptomic alterations and gene‐dosage effects at the proteomic level, as extensively reviewed by Ried et al. 49 These in turn lead to an imbalance in the cellular protein composition, which may saturate the protein folding and degradation machineries, thus eliciting a proteotoxic stress response and altering the redox anabolic homeostasis, leading to increased reactive oxygen species (ROS).50, 51 These findings indicate that aneuploidy is generally disadvantageous for cells and there is ample evidence of the negative effect of aneuploidy on the fitness of nonmalignant cells (Fig. 3 a). First, aneuploidy is an extremely rare event in normal conditions, even in brain and liver.13 Second, trisomic murine embryonic fibroblasts (MEFs) show contact inhibition properties, proliferation arrest in low‐serum medium, lack of clonogenic capacity and senescence features after 7–10 passages in culture.52 Third, trisomic cells can revert to the euploid state by losing extra chromosomes both in vitro and in vivo in order to improve their growth capacity.52 Accordingly, human fibroblasts from patients with constitutional triploidy show moderate levels of somatic mosaicism due to the progressive accumulation of cells that undergo whole chromosome loss.53 Moreover, constitutional aneuploidy per se is not sufficient to generate tumor‐like CIN.53 Aneuploid cells modulate their metabolic and transcriptional programs to improve their fitness. Indeed, aneuploidy is associated with higher glucose and/or glutamine consumption46, 48 and with changes in the expression of proteins involved in cell cycle, ribosome biogenesis, endoplasmic reticulum, Golgi apparatus, lysosomes, membrane metabolism, the major histocompatibility complex (MHC) protein complex and antigen processing, DNA replication, transcription, energy production, and response to stress.44, 45, 54 These pathways are deregulated in several aneuploid cell lines, although the specific combinations of genes displaying altered expression differ.51
Figure 3.
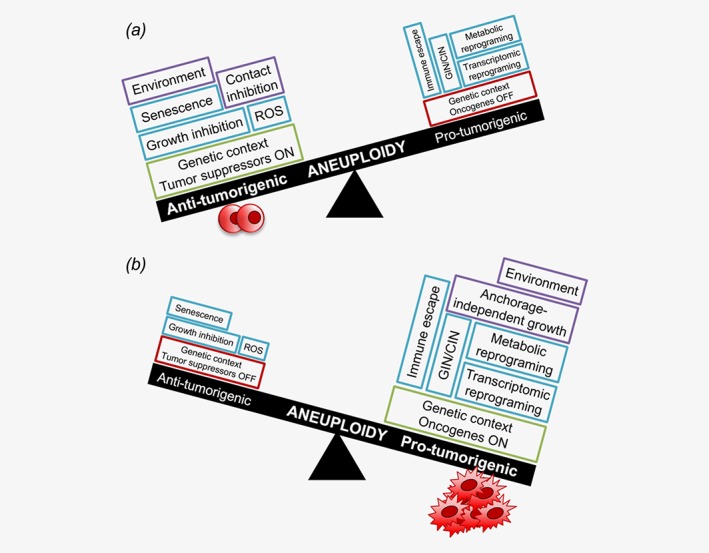
The complex relationship between aneuploidy and cancer. (a) Aneuploidy‐related growth and contact inhibition, ROS production, cell senescence can cooperate with environmental conditions and tumor suppressor activity to inhibit malignant transformation (round shaped cells represent nontransformed cells). (b) When prosurvival and protumorigenic events induced by aneuploidy (anchorage‐independent growth, transcriptional and metabolic reprogramming, GIN, CIN, immune escape) synergize with activation of oncogenes and favorable environmental conditions, cells carrying an aberrant chromosome number undergo malignant transformation (irregular shaped cells represent malignant cells; ROS, reactive oxygen species; CIN, chromosomal instability; GIN, genomic instability).
These observations, mostly obtained from trisomic models, indicate that single‐chromosome aneuploidy is not sufficient per se to induce malignant transformation52 but rather has antitumorigenic properties. Accordingly, individuals with Down's syndrome display a reduced incidence of solid tumors, including breast, lung, and prostate cancers, suggesting that trisomy 21 is a protective event against malignant transformation in those tissues.55 Moreover, some mouse models with deregulated SAC genes that develop aneuploidy have a decreased rate of tumorigenesis (Table 2), even under various oncogenic backgrounds. Recently, Benezra's group showed that individual chromosome loss in tetraploid MEFs may drive tumorigenesis by favoring anchorage‐independent growth, DNA damage, and CIN.56 These results are highly relevant within the context of tumors with increased ploidy and may suggest a difference between the tumorigenic potential of chromosome gain and loss. However, single‐chromosome loss did not induce transformation of diploid MEFs, indicating a ploidy‐specific effect. Moreover, single‐chromosome loss hampered the proliferative capacity of diploid hematopoietic cells, in line with previous studies reporting a negative effect of single‐chromosome gain under normal ploidy.
Despite the detrimental effect of chromosome number alterations on cellular fitness, aneuploidy is one of the hallmarks of cancer, a disease of cells undergoing uncontrolled proliferation. According to the Mitelman Database, about 90% of solid tumors and 50% of hematological neoplasms are aneuploid.57 How can this be reconciled with the findings described so far in this section?
Although in vitro culturing of fibroblasts from patients with constitutional aneuploidy have shown that whole chromosome gains do not confer levels of CIN comparable to those observed in cancer cells,53 several studies indicate a correlation or a causal relationship between aneuploidy and genome/CIN. A correlation between the two phenomena has been observed in monosomic and trisomic models. Amniocytes from trisomic fetuses display a higher incidence of random aneuploidy,58 and lymphocytes from patients with Turner's syndrome or constitutional autosomal trisomy (of chromosome 21, 18, or 13) are prone to develop nonchromosome specific aneuploidy under phytohemagglutinin stimulation.59, 60 Moreover, genomic instability (GIN) is proportional to the degree of aneuploidy in transformed Chinese hamster embryo cells.61 GIN and CIN are key features of the adaptive response driven by aneuploidy and favoring malignant transformation (Fig. 3 b). Single‐chromosome aneuploidy is sufficient to induce CIN, an increase in double strand break (DSB) during DNA replication and defective DSB repair, resulting in a “mutator phenotype” in yeasts.62 Indeed, yeast strains with high proliferative capacity display aneuploidy‐tolerating mutations,63 including both strain‐specific genetic lesions and common mutations shared between different aneuploid strains. Common lesions mainly target the ubiquitin‐proteasome machinery, with recurrent loss‐of‐function mutations in the gene encoding the deubiquitinating enzyme UBP6. Therefore, aneuploidy is maintained and propagated through the positive selection of cells that evolve and become fitter. Increased DNA damage, genomic rearrangements and replication stress have also been reported in human cells as a consequence of aneuploidy.64 In particular, chromosome segregation errors (specifically chromatin bridges, which typically arise as a result of DNA damage) can lead to the accumulation of postmitotic DNA damage due to the “trapping” of chromatin bridges in the cleavage furrow.65 Moreover, trisomic colon cancer cells display a higher rate of chromosome missegregation than that of euploid ones,66 and the capacity of accurate chromosome segregation decreases in a discontinuous way compared to chromosome number changes.67 Indeed, yeast cells with a ploidy between 1.5 and 2 are more susceptible to chromosome missegregation than those with a near haploid karyotype.67 This evidence suggests that additional numerical and structural chromosomal aberrations exacerbate genomic complexity in aneuploid cells. It was recently suggested that replicative stress caused by aneuploidy64 and oncogenic alterations targeting TP53, RB, 29 or KRAS 68 induce CIN,69 even in the absence of mutations in genes involved in chromosome segregation or mitotic checkpoint. For example, in immortalized colonic epithelial cells that acquire an extra copy of chromosome 7 under the selective pressure of serum‐free culture conditions, the expression of oncogenic KRAS or the depletion of TP53 predispose to the acquisition of trisomy 20.70 Thus, the progressive accumulation of mutations, translocations and/or copy number variants improves cell tolerability toward the negative consequences of the altered chromosome number and promotes cell growth, as demonstrated in yeasts, trisomic MEFs52 and human cell lines.64 Moreover, aneuploidy confers an evolutionary flexibility that may contribute, along with oncogenic events, to the cellular heterogeneity observed in cancer and to the aggressive phenotype of advanced malignancies with complex karyotypes.
Aneuploidy itself is an evolutionary strategy to compensate for the deletion of evolvable genes in yeasts. Although, these genes regulate essential cellular processes (e.g., Golgi vescicle trafficking, nuclear transport and nuclear pore complex, protein targeting to endoplasmic reticulum), the cells can survive their loss by developing alternative strategies, including altered gene dosage induced by aneuploidy.71 For example, aneuploidization can correct failure of cytokinesis in MYO1‐deficient yeasts.72 Altered levels of transcription factors encoded by aneuploid chromosomes induce changes in the expression of downstream genes involved in cytokinesis. Different patterns of aneuploidy, arising in the absence of MYO1, converge to common targets capable of restoring cytokinesis.
Specific biological and metabolic properties contribute to the adaptive response induced by aneuploidy (Fig. 3 b). Aneuploid cells show heightened anchorage‐independent growth and migration capacity (e.g., in a colorectal model23). In human pluripotent stem cells (hPSCs), aneuploidy inhibits differentiation propensity and apoptosis, increases proliferation and favors the formation of teratomas characterized by a gene expression profile resembling that of germ cell tumors.43 Aneuploid cells can also redistribute their resources to overcome functional defects.73 For example, yeast cells use aneuploidy to survive telomerase insufficiency by increasing the expression of the telomerase components at the expense of ribosome synthesis.73 The deregulated expression of genes that are involved in oxidative phosphorylation and that protect from oxidative stress also contributes to the tumor‐promoting effect of aneuploidy. Yeasts can adapt to deficiency of all thiol peroxidases, the enzymes that alleviate oxidative stress‐mediated DNA damage, by acquiring an extra copy of chromosome XI.74 This allows the removal of hydrogen peroxide through increased expression of the mitochondrial proteins CCP1 and UTH1 and enforced respiration. Therefore, the evolutionary flexibility induced by aneuploidy is not only an attempt to survive a disadvantageous chromosome number, but also a favorable condition in some settings. A recent study suggested a role of aneuploidy in immune escape75 (Fig. 3 b). Most aneuploid tumors show decreased neoantigen load, possibly mediated by limited neoantigen generation and presentation through the MHC complex. This, in turn, results in decreased immune cell infiltration and makes aneuploid neoplasms less responsive to immunomodulating agents.
Overall, evidence obtained in nonmalignant cells and cancer models indicates that aneuploidy, which is detrimental per se, can be beneficial and even favor the development and selection of aggressive malignant clones by enabling cells to modulate independent pathways simultaneously and to explore a wide phenotypic landscape.
Aneuploidy and Cancer: Cell Type, Genomic Background and Environmental Conditions Matter
Although Down's syndrome patients have a 10‐fold lower solid tumor‐related mortality than the general population, they are more likely to develop leukemia.55 A gain of chromosome 21 is a common event in sporadic leukemia, is the most frequent karyotypic alteration in acute lymphoblastic leukemia, but is rare in glioblastoma, breast, and colorectal cancer.76 These observations are suggestive of a tissue‐ and chromosome‐specific oncogenic effect of aneuploidy. Accordingly, malignant transformation does not occur randomly in the majority of transgenic and knock‐out mouse models of aneuploidy (Table 2). MAD2 overexpression specifically increases the susceptibility to hepatoma and hepatocellular carcinoma, lung adenoma, fibrosarcoma, and lymphoma.31 Reduced BUB1 expression favors the development of thymic lymphoma and colon cancer in tp53 +/─ and apc Min/+ mice, respectively, but suppresses prostatic intraepithelial neoplasia in pten +/─ mice and does not alter the frequency of pituitary tumors and sarcoma formation in the rb +/─ model.77 Moreover, cultured hPSCs tend to acquire trisomy of chromosome 12, which is the most common chromosomal aberration in germ cell tumors,43 and trisomy of chromosome 13, which confers a distinctive cytokinesis failure phenotype to colon cancer cells.66
Although the spectrum and degree of aneuploidy differ among tumors, many human cancers share recurrent aneuploidies.78 It is currently believed that tumor‐specific aneuploidies coexist with recurrent aneuploidies across tumors. Using a computational approach, Davoli et al. showed that chromosome number alterations do not occur randomly: a selective pressure forces the acquisition of oncogenes and the loss of tumor suppressors,79 as observed in yeast cells that overcome MYO1 deficiency by preferential gain of specific chromosomes.72
Aneuploidy also improves cellular adaptation to specific biological and environmental features. In nonpathological conditions aneuploidy is well tolerated under “population flush” effects,80 when rapid cell expansion is needed (e.g., during embryogenesis) and in nonregenerating tissues, as brain and liver, in which the nonproliferative cellular status is protective against the potentially dangerous consequences of aneuploidy. On the contrary, aneuploidy is physiologically selected against in tissues that undergo self‐renewal, including the hematopoietic compartment, skin, and intestines.81 However, aneuploidy improves the survival rate under conditions of stress, including extreme temperature or pH, lack of nutrients, and incubation with chemotherapeutic or antifungal agents in budding yeasts.44 Similarly, trisomic colorectal cancer cell lines have a proliferative advantage over euploid cells under hypoxic conditions, chemotherapeutic pressure and in conditions of serum starvation.23 Serum‐free conditions also favor the acquisition of trisomy 7 in immortalized human colonic epithelial cells.70 The pathogen Candida Albicans develops aneuploidy, mainly consisting in chromosome 5 trisomy (or gain of an isochromosome composed of the two left arms of chromosome 5), as an adaptive strategy to resist fluconazole, commonly used to treat fungal infections.82 The aneuploid strain has an increased growth rate compared to the wild‐type strain under fluconazole pressure, but this advantage is lost in the absence of the drug. Moreover, the accumulation of multiple trisomies of chromosome 3–7 during fluconazole treatment increases resistance, while also having a low fitness cost under nonselective conditions in this model.83 Similarly, Chen et al. showed that dynamic karyotype changes allow yeast cells to survive drug exposure.84
This complex scenario recapitulates tumor biology given that both cancer and leukemia stem cells localize mainly in hypoxic niches. If we consider aneuploid cells as a premalignant state, their genomic plasticity confers the ability to evolve to a malignant phenotype in order to tolerate adverse environmental conditions. The DNA replication stress, which fuels defective chromosome condensation and segregation in aneuploid hPSCs,69 may also propagate GIN in cancer stem cells. Karyotypic heterogeneity may in turn result in phenotypic variations, thus allowing specific aneuploid cells to be fitter under conditions of stress, including oncogene withdrawal and pharmacological treatment. CIN induced by MAD2 overexpression sustains tumor progression and recurrence upon oncogene inactivation in Kras G12D models of breast85 and lung68 cancer, respectively, through activation of alternative oncogenic signaling pathways. In human and murine medulloblastoma, GIN and aneuploidy are common features of the malignant clone driving relapse, which originates from a minor clone present at diagnosis and selected by therapy.86 Similarly, CIN and aneuploidy characterize chemotherapy‐resistant subclones, giving rise to metastases in breast cancer.87 This suggests that aneuploidy, when causing moderate levels of genetic instability, can improve adaptation to the microenvironmental conditions in a specific tumor site, without compromising cell viability.
Therapeutic Potential of Aneuploidy in Cancer Patients
Recent studies have shown that exacerbating chromosome missegregation rates in aneuploid cells by combining heterozygous deletions of the centromere‐linked motor protein cenp‐E and of the SAC component mad2, results in high CIN levels and leads to tumor suppression in mice due to the induction of cell death.88 High CIN levels sustain tumor‐initiating cells while depleting mature tumor cells.89 For example, CENP‐E reduction in apc Min/+ mice does not inhibit the formation of intestinal tumors but hampers their progression.89 This suggests a dual relationship between aneuploid malignancies and antitumor therapeutic strategies: aneuploidy can be a cancer strength or an Achilles’ heel.
According to two different clinical trials on metastatic melanoma, aneuploidy promotes cancer immune escape and correlates with bad prognosis in response to immune checkpoint blockade agents.75 However, chromosomally unstable tumors, such as those with aneuploidy, may be induced to mitotic catastrophe by drugs interfering with chromosome segregation, in particular by enhancing the chromosome missegregation rate.90, 91 These include compounds that disrupt microtubule dynamics either by inducing overpolymerization (stabilizing drugs, e.g., taxanes) or by reducing polymerization (destabilizing drugs, e.g., vinblastine), or drugs that disrupt the kinetochore‐microtubule attachment, correction of misattachments (e.g., Aurora B inhibitors) or SAC activity. For example, the microtubule‐stabilizing drug paclitaxel kills breast cancer cells by inducing chromosome missegregation on multipolar spindles.92 These preclinical tests need to be substantiated by clinical studies. Recently, a clinical trial to compare paclitaxel response with CIN level in breast cancer patients began its recruitment phase (NCT03096418, https://clinicaltrials.gov). The downside of mitotic drugs is their severe bone marrow toxicity. However, this can be prevented by ad hoc combination therapies, including a chemotherapy backbone, aimed at tumor debulking, disease eradication, and a reduction in side effects. The controversial role of the mitotic checkpoint in the response to antimitotic drugs93 should also be taken into account when designing clinical trials. A successful approach can be built on the concept of synthetic lethality, which refers to the simultaneous perturbation of two genes resulting in the death of the cell or the organism. Certain drugs can cause lethality in malignant cells carrying structural or functional alterations in specific genes or pathways. For example, cancer cell lines with defective chromatid cohesion are resistant to paclitaxel but highly responsive to SAC inhibition,94 and knock‐down of Ppp2r1a is lethal in MAD2 overexpressing tumors.95 These “lethal” combinations should be exploited to target aneuploidy‐supporting cellular functions. Indeed, in addition to their neutropenic effects, mitotic drugs are not expected to be effective against tumors showing a negative correlation between CIN and survival. The strength of these aneuploid tumors resides in their increased tolerability toward stress conditions, their genomic complexity and stem cell‐like quiescence, which probably favor resistance to chemotherapy and maintenance of proliferative capacity,96 driving progression to a very aggressive phenotype.
The aneuploid cell‐dependence on chaperone pathways and heightened protein turnover suggest additional therapeutic potential, exploiting proteotoxic stress as aneuploidy‐related vulnerability. Aneuploid MEFs, hPSCs, human embryonic stem cells, colorectal cancer cell lines and induced (i)PSCs from trisomy 21 fibroblasts are sensitive to compounds, inhibiting protein folding (17‐(Allylamino)‐17‐demethoxygeldanamycin), or inducing energy stress (aminoimidazole carboxamide ribonucleotide) although a comparison between aneuploid and euploid cells, led to different results among the models.43, 97, 98, 99 Moreover, hPSCs with trisomy 12 showed enhanced sensitivity to drugs targeting DNA replication, including etoposide, cytarabine and gemcitabine hydrochloride, compared to euploid cells.43 Taken together, these findings indicate that the aneuploid condition offers a therapeutic window for specific antitumor treatment strategies.
Conclusions
An improved understanding of the molecular mechanisms of aneuploidy and of its consequences on cell physiology has provided important insights into the complex relationship between chromosome number alterations and cancer. Aneuploidy can increase malignant cell strength while also creating vulnerability to specific conditions or therapeutic interventions. The tissue type, genetic background and microenvironment play a pivotal role in the match. However, the genetic determinants of the protumorigenic or antitumorigenic effects of aneuploidy and their interplay with the biology of the cell of origin remain unclear. Altered expression of SAC components is a common feature across cancer types and minority of cases carry mutated SAC genes, which may interfere with chromosome segregation fidelity. However, chromosome segregation represents a rapid event in the eukaryotic cell cycle. Cells exiting the quiescent G0 phase accumulate mass, activate signaling pathways, replicate the genome and prepare for mitotic division through G1, S and G2 phases. Dysfunction of cellular components involved in these stages through either mutations, copy number alterations, epigenetic modifications, or deregulated expression may compromise mitotic fidelity and favor aneuploidy. Thus, the identification of genomic patterns that associate and/or synergize with aneuploid phenotypic profiles in promoting tumor development might be a prerequisite to any therapeutic decision, along with the definition of chromosome missegregation frequencies inducing adaptive levels of CIN. These approaches will unravel the relationship between genetic variability, drug resistance and acquisition of stem cell characteristics, while also defining lineage‐specific vulnerabilities for aneuploid tumors. Such knowledge, complemented by the availability of rationally designed targeted agents that have produced promising results, will serve as a map for personalized synthetic lethal therapeutic strategies against aneuploid tumors.
Conflict of interest: Dr. Martinelli reports personal fees from Amgen, personal fees from Ariad/Incyte, personal fees from Pfizer, personal fees and nonfinancial support from Roche, personal fees from Celgene, personal fees from Janssen, personal fees from Jazz Pharmaceuticals, personal fees from ABBVIE, personal fees from Novartis, nonfinancial support from Daiichi Sankyo, nonfinancial support from Shire, outside the submitted work. The other authors have nothing to disclose.
References
- 1. Gupta S. Hepatic polyploidy and liver growth control. Semin Cancer Biol 2000;10:161–71. [DOI] [PubMed] [Google Scholar]
- 2. Faggioli F, Vezzoni P, Montagna C. Single‐cell analysis of ploidy and centrosomes underscores the peculiarity of normal hepatocytes. PloS One 2011;6:e26080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Duncan AW, Taylor MH, Hickey RD, et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 2010;467:707–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Duncan AW, Hanlon Newell AE, Smith L, et al. Frequent aneuploidy among normal human hepatocytes. Gastroenterology 2012;142:25–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Duncan AW, Hanlon Newell AE, Bi W, et al. Aneuploidy as a mechanism for stress‐induced liver adaptation. J Clin Invest 2012;122:3307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kingsbury MA, Friedman B, McConnell MJ, et al. Aneuploid neurons are functionally active and integrated into brain circuitry. Proc Natl Acad Sci USA 2005;102:6143–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rehen SK, McConnell MJ, Kaushal D, et al. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc Natl Acad Sci USA 2001;98:13361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang AH, Kaushal D, Rehen SK, et al. Chromosome segregation defects contribute to aneuploidy in normal neural progenitor cells. J Neurosci 2003;23:10454–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yurov YB, Iourov IY, Monakhov VV, et al. The variation of aneuploidy frequency in the developing and adult human brain revealed by an interphase FISH study. J Histochem Cytochem 2005;53:385–90. [DOI] [PubMed] [Google Scholar]
- 10. Yurov YB, Iourov IY, Vorsanova SG, et al. Aneuploidy and confined chromosomal mosaicism in the developing human brain. PloS One 2007;2:e558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pack SD, Weil RJ, Vortmeyer AO, et al. Individual adult human neurons display aneuploidy: detection by fluorescence in situ hybridization and single neuron PCR. Cell Cycle 2005;4:1758–60. [DOI] [PubMed] [Google Scholar]
- 12. Rehen SK, Yung YC, McCreight MP, et al. Constitutional aneuploidy in the normal human brain. J Neurosci 2005;25:2176–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Knouse KA, Wu J, Whittaker CA, et al. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci USA 2014;111:13409–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baker DJ, Dawlaty MM, Wijshake T, et al. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat Cell Biol 2013;15:96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wijshake T, Malureanu LA, Baker DJ, et al. Reduced life‐ and healthspan in mice carrying a mono‐allelic BubR1 MVA mutation. PLoS Genet 2012;8:e1003138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baker DJ, Jeganathan KB, Cameron JD, et al. BubR1 insufficiency causes early onset of aging‐associated phenotypes and infertility in mice. Nat Genet 2004;36:744–9. [DOI] [PubMed] [Google Scholar]
- 17. Hartman TK, Wengenack TM, Poduslo JF, et al. Mutant mice with small amounts of BubR1 display accelerated age‐related gliosis. Neurobiol Aging 2007;28:921–7. [DOI] [PubMed] [Google Scholar]
- 18. Yang Z, Jun H, Choi CI, et al. Age‐related decline in BubR1 impairs adult hippocampal neurogenesis. Aging Cell 2017;16:598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nagaoka SI, Hassold TJ, Hunt PA. Human aneuploidy: mechanisms and new insights into an age‐old problem. Nat Rev Genet 2012;13:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Riris S, Webster P, Homer H. Digital multiplexed mRNA analysis of functionally important genes in single human oocytes and correlation of changes in transcript levels with oocyte protein expression. Fertil Steril 2014;101:857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wisniewski KE, Wisniewski HM, Wen GY. Occurrence of neuropathological changes and dementia of Alzheimer's disease in Down's syndrome. Ann Neurol 1985;17:278–82. [DOI] [PubMed] [Google Scholar]
- 22. Thomas P, Fenech M. Chromosome 17 and 21 aneuploidy in buccal cells is increased with ageing and in Alzheimer's disease. Mutagenesis 2008;23:57–65. [DOI] [PubMed] [Google Scholar]
- 23. Rutledge SD, Douglas TA, Nicholson JM, et al. Selective advantage of trisomic human cells cultured in non‐standard conditions. Sci Rep 2016;6:22828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boveri T, Boveri M. The origin of malignant tumorsed. Baltimore: The Williams & Wilkins Company, 1929. ix, 119 [Google Scholar]
- 25. Nicholson JM, Cimini D. How mitotic errors contribute to karyotypic diversity in cancer. Adv Cancer Res 2011;112:43–75. [DOI] [PubMed] [Google Scholar]
- 26. Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer 2005;5:773–85. [DOI] [PubMed] [Google Scholar]
- 27. Musacchio A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol 2015;25:R1002–18. [DOI] [PubMed] [Google Scholar]
- 28. Hernando E, Nahle Z, Juan G, et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 2004;430:797–802. [DOI] [PubMed] [Google Scholar]
- 29. Schvartzman JM, Duijf PH, Sotillo R, et al. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 2011;19:701–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mondal G, Sengupta S, Panda CK, et al. Overexpression of Cdc20 leads to impairment of the spindle assembly checkpoint and aneuploidization in oral cancer. Carcinogenesis 2007;28:81–92. [DOI] [PubMed] [Google Scholar]
- 31. Sotillo R, Hernando E, Diaz‐Rodriguez E, et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 2007;11:9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Michel LS, Liberal V, Chatterjee A, et al. MAD2 haplo‐insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 2001;409:355–9. [DOI] [PubMed] [Google Scholar]
- 33. Ricke RM, Jeganathan KB, van Deursen JM. Bub1 overexpression induces aneuploidy and tumor formation through aurora B kinase hyperactivation. J Cell Biol 2011;193:1049–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schliekelman M, Cowley DO, O'Quinn R, et al. Impaired Bub1 function in vivo compromises tension‐dependent checkpoint function leading to aneuploidy and tumorigenesis. Cancer Res 2009;69:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jeganathan K, Malureanu L, Baker DJ, et al. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J Cell Biol 2007;179:255–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fang Y, Liu T, Wang X, et al. BubR1 is involved in regulation of DNA damage responses. Oncogene 2006;25:3598–605. [DOI] [PubMed] [Google Scholar]
- 37. Wan L, Tan M, Yang J, et al. APC(Cdc20) suppresses apoptosis through targeting Bim for ubiquitination and destruction. Dev Cell 2014;29:377–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cho HJ, Lee EH, Han SH, et al. Degradation of human RAP80 is cell cycle regulated by Cdc20 and Cdh1 ubiquitin ligases. Mol Cancer Res 2012;10:615–25. [DOI] [PubMed] [Google Scholar]
- 39. Hadjihannas MV, Bernkopf DB, Bruckner M, et al. Cell cycle control of Wnt/beta‐catenin signalling by conductin/axin2 through CDC20. EMBO Rep 2012;13:347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mao DD, Gujar AD, Mahlokozera T, et al. A CDC20‐APC/SOX2 signaling Axis regulates human Glioblastoma stem‐like cells. Cell Rep 2015;11:1809–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xie Q, Wu Q, Mack SC, et al. CDC20 maintains tumor initiating cells. Oncotarget 2015;6:13241–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wan J, Zhu F, Zasadil LM, et al. Golgi‐localized pool of the mitotic checkpoint component Mad1 controls integrin secretion and cell migration. Curr Biol 2014;24:2687–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ben‐David U, Arad G, Weissbein U, et al. Aneuploidy induces profound changes in gene expression, proliferation and tumorigenicity of human pluripotent stem cells. Nat Commun 2014;5:4825. [DOI] [PubMed] [Google Scholar]
- 44. Pavelka N, Rancati G, Zhu J, et al. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 2010;468:321–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sheltzer JM, Torres EM, Dunham MJ, et al. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci USA 2012;109:12644–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Torres EM, Sokolsky T, Tucker CM, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 2007;317:916–24. [DOI] [PubMed] [Google Scholar]
- 47. Upender MB, Habermann JK, McShane LM, et al. Chromosome transfer induced aneuploidy results in complex dysregulation of the cellular transcriptome in immortalized and cancer cells. Cancer Res 2004;64:6941–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Williams BR, Prabhu VR, Hunter KE, et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 2008;322:703–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ried T, Hu Y, Difilippantonio MJ, et al. The consequences of chromosomal aneuploidy on the transcriptome of cancer cells. Biochim Biophys Acta 2012;1819:784–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 2013;12:931–47. [DOI] [PubMed] [Google Scholar]
- 51. Dephoure N, Hwang S, O'Sullivan C, et al. Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. Elife 2014;3:e03023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sheltzer JM, Ko JH, Replogle JM, et al. Single‐chromosome gains commonly function as tumor suppressors. Cancer Cell 2017;31:240–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Valind A, Jin Y, Baldetorp B, et al. Whole chromosome gain does not in itself confer cancer‐like chromosomal instability. Proc Natl Acad Sci USA 2013;110:21119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Durrbaum M, Kuznetsova AY, Passerini V, et al. Unique features of the transcriptional response to model aneuploidy in human cells. BMC Genomics 2014;15:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang Q, Rasmussen SA, Friedman JM. Mortality associated with Down's syndrome in the USA from 1983 to 1997: a population‐based study. Lancet 2002;359:1019–25. [DOI] [PubMed] [Google Scholar]
- 56. Thomas R, Marks DH, Chin Y, et al. Whole chromosome loss and associated breakage‐fusion‐bridge cycles transform mouse tetraploid cells. EMBO J 2018;37:201–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mitelman F, Johansson B, Mertens F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer, http://cgap.nci.nih.gov/Chromosomes/Mitelman 2017.
- 58. Biron‐Shental T, Liberman M, Sharvit M, et al. Amniocytes from aneuploidy embryos have enhanced random aneuploidy and signs of senescence ‐ can these findings be related to medical problems? Gene 2015;562:232–5. [DOI] [PubMed] [Google Scholar]
- 59. Reish O, Regev M, Kanesky A, et al. Sporadic aneuploidy in PHA‐stimulated lymphocytes of trisomies 21, 18, and 13. Cytogenet Genome Res 2011;133:184–9. [DOI] [PubMed] [Google Scholar]
- 60. Reish O, Brosh N, Gobazov R, et al. Sporadic aneuploidy in PHA‐stimulated lymphocytes of Turner's syndrome patients. Chromosome Res 2006;14:527–34. [DOI] [PubMed] [Google Scholar]
- 61. Duesberg P, Rausch C, Rasnick D, et al. Genetic instability of cancer cells is proportional to their degree of aneuploidy. Proc Natl Acad Sci USA 1998;95:13692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sheltzer JM, Blank HM, Pfau SJ, et al. Aneuploidy drives genomic instability in yeast. Science 2011;333:1026–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Torres EM, Dephoure N, Panneerselvam A, et al. Identification of aneuploidy‐tolerating mutations. Cell 2010;143:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Passerini V, Ozeri‐Galai E, de Pagter MS, et al. The presence of extra chromosomes leads to genomic instability. Nat Commun 2016;7:10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Janssen A, van der Burg M, Szuhai K, et al. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011;333:1895–8. [DOI] [PubMed] [Google Scholar]
- 66. Nicholson JM, Macedo JC, Mattingly AJ, et al. Chromosome mis‐segregation and cytokinesis failure in trisomic human cells. Elife 2015;4:e05068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhu J, Pavelka N, Bradford WD, et al. Karyotypic determinants of chromosome instability in aneuploid budding yeast. PLoS Genet 2012;8:e1002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sotillo R, Schvartzman JM, Socci ND, et al. Mad2‐induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature 2010;464:436–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lamm N, Ben‐David U, Golan‐Lev T, et al. Genomic instability in human pluripotent stem cells arises from replicative stress and chromosome condensation defects. Cell Stem Cell 2016;18:253–61. [DOI] [PubMed] [Google Scholar]
- 70. Ly P, Eskiocak U, Kim SB, et al. Characterization of aneuploid populations with trisomy 7 and 20 derived from diploid human colonic epithelial cells. Neoplasia 2011;13:348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liu G, Yong MY, Yurieva M, et al. Gene essentiality is a quantitative property linked to cellular Evolvability. Cell 2015;163:1388–99. [DOI] [PubMed] [Google Scholar]
- 72. Rancati G, Pavelka N, Fleharty B, et al. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 2008;135:879–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Millet C, Ausiannikava D, Le Bihan T, et al. Cell populations can use aneuploidy to survive telomerase insufficiency. Nat Commun 2015;6:8664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kaya A, Gerashchenko MV, Seim I, et al. Adaptive aneuploidy protects against thiol peroxidase deficiency by increasing respiration via key mitochondrial proteins. Proc Natl Acad Sci USA 2015;112:10685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Davoli T, Uno H, Wooten EC, et al. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017;355:eaaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nicholson JM, Cimini D. Cancer karyotypes: survival of the fittest. Front Oncol 2013;3:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Baker DJ, Jin F, Jeganathan KB, et al. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell 2009;16:475–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zack TI, Schumacher SE, Carter SL, et al. Pan‐cancer patterns of somatic copy number alteration. Nat Genet 2013;45:1134–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Davoli T, Xu AW, Mengwasser KE, et al. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013;155:948–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Carson H. The population flush and its genetic consequences In: Lewontin R, ed Population biology and evolution. Syracuse, NY: Syracuse University Press, 1968. 123–37. [Google Scholar]
- 81. Pfau SJ, Silberman RE, Knouse KA, et al. Aneuploidy impairs hematopoietic stem cell fitness and is selected against in regenerating tissues in vivo. Genes Dev 2016;30:1395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Selmecki A, Forche A, Berman J. Aneuploidy and isochromosome formation in drug‐resistant Candida albicans . Science 2006;313:367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Selmecki AM, Dulmage K, Cowen LE, et al. Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet 2009;5:e1000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chen G, Mulla WA, Kucharavy A, et al. Targeting the adaptability of heterogeneous aneuploids. Cell 2015;160:771–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rowald K, Mantovan M, Passos J, et al. Negative selection and chromosome instability induced by Mad2 overexpression delay breast Cancer but facilitate oncogene‐independent outgrowth. Cell Rep 2016;15:2679–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Morrissy AS, Garzia L, Shih DJ, et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 2016;529:351–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yates LR, Gerstung M, Knappskog S, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med 2015;21:751–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Silk AD, Zasadil LM, Holland AJ, et al. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci USA 2013;110:E4134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zasadil LM, Britigan EM, Ryan SD, et al. High rates of chromosome missegregation suppress tumor progression but do not inhibit tumor initiation. Mol Biol Cell 2016;27:1981–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kops GJ, Foltz DR, Cleveland DW. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl Acad Sci USA 2004;101:8699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Janssen A, Kops GJ, Medema RH. Elevating the frequency of chromosome mis‐segregation as a strategy to kill tumor cells. Proc Natl Acad Sci USA 2009;106:19108–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zasadil LM, Andersen KA, Yeum D, et al. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med 2014;6:229ra43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yamada HY, Gorbsky GJ. Spindle checkpoint function and cellular sensitivity to antimitotic drugs. Mol Cancer Ther 2006;5:2963–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. de Lange J, Faramarz A, Oostra AB, et al. Defective sister chromatid cohesion is synthetically lethal with impaired APC/C function. Nat Commun 2015;6:8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bian Y, Kitagawa R, Bansal PK, et al. Synthetic genetic array screen identifies PP2A as a therapeutic target in Mad2‐overexpressing tumors. Proc Natl Acad Sci USA 2014;111:1628–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kusumbe AP, Bapat SA. Cancer stem cells and aneuploid populations within developing tumors are the major determinants of tumor dormancy. Cancer Res 2009;69:9245–53. [DOI] [PubMed] [Google Scholar]
- 97. Tang YC, Williams BR, Siegel JJ, et al. Identification of aneuploidy‐selective antiproliferation compounds. Cell 2011;144:499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Li LB, Chang KH, Wang PR, et al. Trisomy correction in down syndrome induced pluripotent stem cells. Cell Stem Cell 2012;11:615–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ly P, Kim SB, Kaisani AA, et al. Aneuploid human colonic epithelial cells are sensitive to AICAR‐induced growth inhibition through EGFR degradation. Oncogene 2013;32:3139–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Qian Z, Fernald AA, Godley LA, et al. Expression profiling of CD34+ hematopoietic stem/ progenitor cells reveals distinct subtypes of therapy‐related acute myeloid leukemia. Proc Natl Acad Sci USA 2002;99:14925–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lin SF, Lin PM, Yang MC, et al. Expression of hBUB1 in acute myeloid leukemia. Leuk Lymphoma 2002;43:385–91. [DOI] [PubMed] [Google Scholar]
- 102. Johnson RH, Hu P, Fan C, et al. Gene expression in "young adult type" breast cancer: a retrospective analysis. Oncotarget 2015;6:13688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wang Z, Katsaros D, Shen Y, et al. Biological and clinical significance of MAD2L1 and BUB1, genes frequently appearing in expression signatures for breast Cancer prognosis. PloS One 2015;10:e0136246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yuan B, Xu Y, Woo JH, et al. Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin Cancer Res 2006;12:405–10. [DOI] [PubMed] [Google Scholar]
- 105. Thiru P, Kern DM, McKinley KL, et al. Kinetochore genes are coordinately up‐regulated in human tumors as part of a FoxM1‐related cell division program. Mol Biol Cell 2014;25:1983–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pinto M, Vieira J, Ribeiro FR, et al. Overexpression of the mitotic checkpoint genes BUB1 and BUBR1 is associated with genomic complexity in clear cell kidney carcinomas. Cellular Oncol 2008;30:389–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Li L, Xu DB, Zhao XL, et al. Combination analysis of Bub1 and Mad2 expression in endometrial cancer: act as a prognostic factor in endometrial cancer. Arch Gynecol Obstet 2013;288:155–65. [DOI] [PubMed] [Google Scholar]
- 108. Moreno‐Bueno G, Sanchez‐Estevez C, Cassia R, et al. Differential gene expression profile in endometrioid and nonendometrioid endometrial carcinoma: STK15 is frequently overexpressed and amplified in nonendometrioid carcinomas. Cancer Res 2003;63:5697–702. [PubMed] [Google Scholar]
- 109. Grabsch H, Takeno S, Parsons WJ, et al. Overexpression of the mitotic checkpoint genes BUB1, BUBR1, and BUB3 in gastric cancer‐‐association with tumour cell proliferation. J Pathol 2003;200:16–22. [DOI] [PubMed] [Google Scholar]
- 110. Shigeishi H, Oue N, Kuniyasu H, et al. Expression of Bub1 gene correlates with tumor proliferating activity in human gastric carcinomas. Pathobiology 2001;69:24–9. [DOI] [PubMed] [Google Scholar]
- 111. Grabsch HI, Askham JM, Morrison EE, et al. Expression of BUB1 protein in gastric cancer correlates with the histological subtype, but not with DNA ploidy or microsatellite instability. J Pathol 2004;202:208–14. [DOI] [PubMed] [Google Scholar]
- 112. Stahl D, Braun M, Gentles AJ, et al. Low BUB1 expression is an adverse prognostic marker in gastric adenocarcinoma. Oncotarget 2017;8:76329–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bie L, Zhao G, Cheng P, et al. The accuracy of survival time prediction for patients with glioma is improved by measuring mitotic spindle checkpoint gene expression. PloS One 2011;6:e25631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Agarwal R, Narayan J, Bhattacharyya A, et al. Gene expression profiling, pathway analysis and subtype classification reveal molecular heterogeneity in hepatocellular carcinoma and suggest subtype specific therapeutic targets. Cancer Genet 2017;216‐217:37–51. [DOI] [PubMed] [Google Scholar]
- 115. Yan H, Li Z, Shen Q, et al. Aberrant expression of cell cycle and material metabolism related genes contributes to hepatocellular carcinoma occurrence. Pathol Res Pract 2017;213:316–21. [DOI] [PubMed] [Google Scholar]
- 116. Zhang C, Min L, Zhang L, et al. Combined analysis identifies six genes correlated with augmented malignancy from non‐small cell to small cell lung cancer. Tumour Biol 2016;37:2193–207. [DOI] [PubMed] [Google Scholar]
- 117. Jin X, Liu X, Li X, et al. Integrated analysis of DNA methylation and mRNA expression profiles data to identify key genes in lung adenocarcinoma. Biomed Res Int 2016;2016:4369431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Riker AI, Enkemann SA, Fodstad O, et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med Genomics 2008;1:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Suh YJ, Choe JY, Park HJ. Malignancy in Pheochromocytoma or Paraganglioma: integrative analysis of 176 cases in TCGA. Endocr Pathol 2017;28:159–64. [DOI] [PubMed] [Google Scholar]
- 120. Shigeishi H, Yoneda S, Taki M, et al. Correlation of human Bub1 expression with tumor‐proliferating activity in salivary gland tumors. Oncol Rep 2006;15:933–8. [PubMed] [Google Scholar]
- 121. Wada N, Yoshida A, Miyagi Y, et al. Overexpression of the mitotic spindle assembly checkpoint genes hBUB1, hBUBR1 and hMAD2 in thyroid carcinomas with aggressive nature. Anticancer Res 2008;28:139–44. [PubMed] [Google Scholar]
- 122. Schnerch D, Schmidts A, Follo M, et al. BubR1 is frequently repressed in acute myeloid leukemia and its re‐expression sensitizes cells to antimitotic therapy. Haematologica 2013;98:1886–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Yamamoto Y, Matsuyama H, Chochi Y, et al. Overexpression of BUBR1 is associated with chromosomal instability in bladder cancer. Cancer Genet Cytogenet 2007;174:42–7. [DOI] [PubMed] [Google Scholar]
- 124. Cirak Y, Furuncuoglu Y, Yapicier O, et al. Predictive and prognostic values of BubR1 and synuclein‐gamma expression in breast cancer. Int J Clin Exp Pathol 2015;8:5345–53. [PMC free article] [PubMed] [Google Scholar]
- 125. Du J, Du Q, Zhang Y, et al. Expression of cell‐cycle regulatory proteins BUBR1, MAD2, aurora a, cyclin a and cyclin E in invasive ductal breast carcinomas. Histol Histopathol 2011;26:761–8. [DOI] [PubMed] [Google Scholar]
- 126. Maciejczyk A, Szelachowska J, Czapiga B, et al. Elevated BUBR1 expression is associated with poor survival in early breast cancer patients: 15‐year follow‐up analysis. J Histochem Cytochem 2013;61:330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Ocana A, Perez‐Pena J, Diez‐Gonzalez L, et al. Transcriptomic analyses identify association between mitotic kinases, PDZ‐binding kinase and BUB1, and clinical outcome in breast cancer. Breast Cancer Res Treat 2016;156:1–8. [DOI] [PubMed] [Google Scholar]
- 128. Scintu M, Vitale R, Prencipe M, et al. Genomic instability and increased expression of BUB1B and MAD2L1 genes in ductal breast carcinoma. Cancer Lett 2007;254:298–307. [DOI] [PubMed] [Google Scholar]
- 129. Burum‐Auensen E, Deangelis PM, Schjolberg AR, et al. Reduced level of the spindle checkpoint protein BUB1B is associated with aneuploidy in colorectal cancers. Cell Prolif 2008;41:645–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Lee YK, Choi E, Kim MA, et al. BubR1 as a prognostic marker for recurrence‐free survival rates in epithelial ovarian cancers. Br J Cancer 2009;101:504–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Uemura N, Nakanishi Y, Kato H, et al. Antibody‐based proteomics for esophageal cancer: identification of proteins in the nuclear factor‐kappaB pathway and mitotic checkpoint. Cancer Sci 2009;100:1612–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kim JH, Kim HN, Lee KT, et al. Gene expression profiles in gallbladder cancer: the close genetic similarity seen for early and advanced gallbladder cancers may explain the poor prognosis. Tumour Biol 2008;29:41–9. [DOI] [PubMed] [Google Scholar]
- 133. Ando K, Kakeji Y, Kitao H, et al. High expression of BUBR1 is one of the factors for inducing DNA aneuploidy and progression in gastric cancer. Cancer Sci 2010;101:639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Enjoji M, Iida S, Sugita H, et al. BubR1 and AURKB overexpression are associated with a favorable prognosis in gastric cancer. Mol Med Rep 2009;2:589–96. [DOI] [PubMed] [Google Scholar]
- 135. Liu AW, Cai J, Zhao XL, et al. The clinicopathological significance of BUBR1 overexpression in hepatocellular carcinoma. J Clin Pathol 2009;62:1003–8. [DOI] [PubMed] [Google Scholar]
- 136. Jin B, Wang W, Du G, et al. Identifying hub genes and dysregulated pathways in hepatocellular carcinoma. Eur Rev Med Pharmacol Sci 2015;19:592–601. [PubMed] [Google Scholar]
- 137. Stricker TP, Henriksen KJ, Tonsgard JH, et al. Expression profiling of 519 kinase genes in matched malignant peripheral nerve sheath tumor/plexiform neurofibroma samples is discriminatory and identifies mitotic regulators BUB1B, PBK and NEK2 as overexpressed with transformation. Modern Pathol 2013;26:930–43. [DOI] [PubMed] [Google Scholar]
- 138. Yang Y, Gu C, Luo C, et al. BUB1B promotes multiple myeloma cell proliferation through CDC20/CCNB axis. Med Oncol 2015;32:81. [DOI] [PubMed] [Google Scholar]
- 139. Huang C, Tang H, Zhang W, et al. Integrated analysis of multiple gene expression profiling datasets revealed novel gene signatures and molecular markers in nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev 2012;21:166–75. [DOI] [PubMed] [Google Scholar]
- 140. Lira RC, Miranda FA, Guimaraes MC, et al. BUBR1 expression in benign oral lesions and squamous cell carcinomas: correlation with human papillomavirus. Oncol Rep 2010;23:1027–36. [DOI] [PubMed] [Google Scholar]
- 141. Rizzardi C, Torelli L, Schneider M, et al. MAD2 expression in oral squamous cell carcinoma and its relationship to tumor grade and proliferation. Anticancer Res 2014;34:7021–7027. [PubMed] [Google Scholar]
- 142. McGrogan B, Phelan S, Fitzpatrick P, et al. Spindle assembly checkpoint protein expression correlates with cellular proliferation and shorter time to recurrence in ovarian cancer. Hum Pathol 2014;45:1509–19. [DOI] [PubMed] [Google Scholar]
- 143. Long J, Zhang Z, Liu Z, et al. Identification of genes and pathways associated with pancreatic ductal adenocarcinoma by bioinformatics analyses. Oncol Lett 2016;11:1391–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Ohashi A, Ohori M, Iwai K, et al. Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53‐mediated post‐mitotic apoptosis in SAC‐impaired cells. Nat Commun 2015;6:7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Borges KS, Moreno DA, Martinelli CE Jr, et al. Spindle assembly checkpoint gene expression in childhood adrenocortical tumors (ACT): overexpression of aurora kinases a and B is associated with a poor prognosis. Pediatr Blood Cancer 2013;60:1809–16. [DOI] [PubMed] [Google Scholar]
- 146. Chen F, Yang G, Xia B. Increased expression of the spindle checkpoint protein BubR1 is associated with high cell proliferation in primary gastrointestinal diffuse large B cell lymphoma. Cell Biochem Biophys 2013;66:747–52. [DOI] [PubMed] [Google Scholar]
- 147. Cirak Y, Sarsik B, Cakar B, et al. Predictive and prognostic values of tau and BubR1 protein in prostate cancer and their relationship to the Gleason score. Med Oncol 2013;30:526. [DOI] [PubMed] [Google Scholar]
- 148. Fu X, Chen G, Cai ZD, et al. Overexpression of BUB1B contributes to progression of prostate cancer and predicts poor outcome in patients with prostate cancer. OncoTargets Therapy 2016;9:2211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Ko YH, Roh JH, Son YI, et al. Expression of mitotic checkpoint proteins BUB1B and MAD2L1 in salivary duct carcinomas. J Oral Pathol Med 2010;39:349–55. [DOI] [PubMed] [Google Scholar]
- 150. Hannisdal K, Burum‐Auensen E, Schjolberg A, et al. Correlation between reduced expression of the spindle checkpoint protein BubR1 and bad prognosis in tonsillar carcinomas. Head Neck 2010;32:1354–62. [DOI] [PubMed] [Google Scholar]
- 151. Yamamoto Y, Oga A, Akao J, et al. BUBR1 overexpression predicts disease‐specific survival after nephroureterectomy in patients with upper tract urothelial carcinoma. Jpn J Clin Oncol 2016;46:754–61. [DOI] [PubMed] [Google Scholar]
- 152. Haruta M, Matsumoto Y, Izumi H, et al. Combined BubR1 protein down‐regulation and RASSF1A hypermethylation in Wilms tumors with diverse cytogenetic changes. Mol Carcinog 2008;47:660–666. [DOI] [PubMed] [Google Scholar]
- 153. Karra H, Repo H, Ahonen I, et al. Cdc20 and securin overexpression predict short‐term breast cancer survival. Br J Cancer 2014;110:2905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Kim Y, Choi JW, Lee JH, et al. MAD2 and CDC20 are upregulated in high‐grade squamous intraepithelial lesions and squamous cell carcinomas of the uterine cervix. Int J Gynecol Pathol 2014;33:517–23. [DOI] [PubMed] [Google Scholar]
- 155. Espinosa AM, Alfaro A, Roman‐Basaure E, et al. Mitosis is a source of potential markers for screening and survival and therapeutic targets in cervical cancer. PloS One 2013;8:e55975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Yuan L, Chen L, Qian K, et al. Co‐expression network analysis identified six hub genes in association with progression and prognosis in human clear cell renal cell carcinoma (ccRCC). Genomics Data 2017;14:132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Shi G, Wang Y, Zhang C, et al. Identification of genes involved in the four stages of colorectal cancer: gene expression profiling. Mol Cell Probes 2018;37:39–47. [DOI] [PubMed] [Google Scholar]
- 158. Wu WJ, Hu KS, Wang DS, et al. CDC20 overexpression predicts a poor prognosis for patients with colorectal cancer. J Transl Med 2013;11:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ding ZY, Wu HR, Zhang JM, et al. Expression characteristics of CDC20 in gastric cancer and its correlation with poor prognosis. Int J Clin Exp Pathol 2014;7:722–727. [PMC free article] [PubMed] [Google Scholar]
- 160. Marucci G, Morandi L, Magrini E, et al. Gene expression profiling in glioblastoma and immunohistochemical evaluation of IGFBP‐2 and CDC20. Virchows Archiv 2008;453:599–609. [DOI] [PubMed] [Google Scholar]
- 161. Li L, Lei Q, Zhang S, et al. Screening and identification of key biomarkers in hepatocellular carcinoma: evidence from bioinformatic analysis. Oncol Rep 2017;38:2607–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Li H, Jiang X, Zhu S, et al. Identification of personalized dysregulated pathways in hepatocellular carcinoma. Pathol Res Pract 2017;213:327–32. [DOI] [PubMed] [Google Scholar]
- 163. Li J, Gao JZ, Du JL, et al. Increased CDC20 expression is associated with development and progression of hepatocellular carcinoma. Int J Oncol 2014;45:1547–55. [DOI] [PubMed] [Google Scholar]
- 164. Gu Y, Lu L, Wu L, et al. Identification of prognostic genes in kidney renal clear cell carcinoma by RNAseq data analysis. Mol Med Rep 2017;15:1661–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Kato T, Daigo Y, Aragaki M, et al. Overexpression of CDC20 predicts poor prognosis in primary non‐small cell lung cancer patients. J Surg Oncol 2012;106:423–30. [DOI] [PubMed] [Google Scholar]
- 166. Yi J, Wei X, Li X, et al. A genome‐wide comprehensive analysis of alterations in driver genes in non‐small‐cell lung cancer. Anticancer Drugs 2018;29:10–8. [DOI] [PubMed] [Google Scholar]
- 167. Shi R, Sun Q, Sun J, Wang X, Xia W, Dong G, Wang A, Jiang F, Xu L. Cell division cycle 20 overexpression predicts poor prognosis for patients with lung adenocarcinoma. Tumour Biol 2017; 39: 1010428317692233, 101042831769223. [DOI] [PubMed] [Google Scholar]
- 168. Lub S, Maes A, Maes K, et al. Inhibiting the anaphase promoting complex/cyclosome induces a metaphase arrest and cell death in multiple myeloma cells. Oncotarget 2016;7:4062–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Borges DP, Dos Santos AWA, Paier CRK, et al. Prognostic importance of aurora kinases and mitotic spindle genes transcript levels in Myelodysplastic syndrome. Leuk Res 2018;64:61–70. [DOI] [PubMed] [Google Scholar]
- 170. Genga KR, Filho FD, Ferreira FV, et al. Proteins of the mitotic checkpoint and spindle are related to chromosomal instability and unfavourable prognosis in patients with myelodysplastic syndrome. J Clin Pathol 2015;68:381–7. [DOI] [PubMed] [Google Scholar]
- 171. Heredia FF, de Sousa JC, Ribeiro Junior HL, et al. Proteins related to the spindle and checkpoint mitotic emphasize the different pathogenesis of hypoplastic MDS. Leuk Res 2014;38:218–24. [DOI] [PubMed] [Google Scholar]
- 172. Moura IM, Delgado ML, Silva PM, et al. High CDC20 expression is associated with poor prognosis in oral squamous cell carcinoma. J Oral Pathol Med 2014;43:225–31. [DOI] [PubMed] [Google Scholar]
- 173. Li D, Zhu J, Firozi PF, et al. Overexpression of oncogenic STK15/BTAK/aurora a kinase in human pancreatic cancer. Clin Cancer Res 2003;9:991–7. [PubMed] [Google Scholar]
- 174. Chang DZ, Ma Y, Ji B, et al. Increased CDC20 expression is associated with pancreatic ductal adenocarcinoma differentiation and progression. J Hematol Oncol 2012;5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Mao Y, Li K, Lu L, et al. Overexpression of Cdc20 in clinically localized prostate cancer: relation to high Gleason score and biochemical recurrence after laparoscopic radical prostatectomy. Cancer Biomarkers 2016;16:351–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Ouellet V, Guyot MC, Le Page C, et al. Tissue array analysis of expression microarray candidates identifies markers associated with tumor grade and outcome in serous epithelial ovarian cancer. Int J Cancer 2006;119:599–607. [DOI] [PubMed] [Google Scholar]
- 177. Choi JW, Kim Y, Lee JH, et al. High expression of spindle assembly checkpoint proteins CDC20 and MAD2 is associated with poor prognosis in urothelial bladder cancer. Virchows Archiv 2013;463:681–7. [DOI] [PubMed] [Google Scholar]
- 178. Barlin JN, Zhou QC, Leitao MM, et al. Molecular subtypes of uterine leiomyosarcoma and correlation with clinical outcome. Neoplasia 2015;17:183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Ryan SD, Britigan EM, Zasadil LM, et al. Up‐regulation of the mitotic checkpoint component Mad1 causes chromosomal instability and resistance to microtubule poisons. Proc Natl Acad Sci USA 2012;109:E2205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Pinto M, Soares MJ, Cerveira N, et al. Expression changes of the MAD mitotic checkpoint gene family in renal cell carcinomas characterized by numerical chromosome changes. Virchows Archiv 2007;450:379–85. [DOI] [PubMed] [Google Scholar]
- 181. Nishigaki R, Osaki M, Hiratsuka M, et al. Proteomic identification of differentially‐expressed genes in human gastric carcinomas. Proteomics 2005;5:3205–13. [DOI] [PubMed] [Google Scholar]
- 182. Osaki M, Inoue T, Yamaguchi S, et al. MAD1 (mitotic arrest deficiency 1) is a candidate for a tumor suppressor gene in human stomach. Virchows Archiv 2007;451:771–9. [DOI] [PubMed] [Google Scholar]
- 183. Nam CW, Park NH, Park BR, et al. Mitotic checkpoint gene MAD1 in hepatocellular carcinoma is associated with tumor recurrence after surgical resection. J Surg Oncol 2008;97:567–71. [DOI] [PubMed] [Google Scholar]
- 184. Li D, Meng Q, Zhang H, et al. Mitotic arrest deficient‐like 1 is correlated with poor prognosis in small‐cell lung cancer after surgical resection. Tumour Biol 2016;37:4393–8. [DOI] [PubMed] [Google Scholar]
- 185. Sun Q, Zhang X, Liu T, et al. Increased expression of mitotic arrest deficient‐like 1 (MAD1L1) is associated with poor prognosis and insensitive to Taxol treatment in breast cancer. Breast Cancer Res Treat 2013;140:323–30. [DOI] [PubMed] [Google Scholar]
- 186. Li GQ, Zhang HF. Mad2 and p27 expression profiles in colorectal cancer and its clinical significance. World J Gastroenterol 2004;10:3218–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Wang L, Yin F, Du Y, et al. MAD2 as a key component of mitotic checkpoint: a probable prognostic factor for gastric cancer. Am J Clin Pathol 2009;131:793–801. [DOI] [PubMed] [Google Scholar]
- 188. Zhang SH, Xu AM, Chen XF, et al. Clinicopathologic significance of mitotic arrest defective protein 2 overexpression in hepatocellular carcinoma. Hum Pathol 2008;39:1827–34. [DOI] [PubMed] [Google Scholar]
- 189. Furlong F, Fitzpatrick P, O'Toole S, et al. Low MAD2 expression levels associate with reduced progression‐free survival in patients with high‐grade serous epithelial ovarian cancer. J Pathol 2012;226:746–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Kato T, Daigo Y, Aragaki M, et al. Overexpression of MAD2 predicts clinical outcome in primary lung cancer patients. Lung Cancer 2011;74:124–31. [DOI] [PubMed] [Google Scholar]
- 191. Shi YX, Zhu T, Zou T, et al. Prognostic and predictive values of CDK1 and MAD2L1 in lung adenocarcinoma. Oncotarget 2016;7:85235–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Suraokar MB, Nunez MI, Diao L, et al. Expression profiling stratifies mesothelioma tumors and signifies deregulation of spindle checkpoint pathway and microtubule network with therapeutic implications. Ann Oncology 2014;25:1184–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Wang XG, Peng Y, Song XL, et al. Identification potential biomarkers and therapeutic agents in multiple myeloma based on bioinformatics analysis. Eur Rev Med Pharmacol Sci 2016;20:810–7. [PubMed] [Google Scholar]
- 194. Teixeira JH, Silva P, Faria J, et al. Clinicopathologic significance of BubR1 and Mad2 overexpression in oral cancer. Oral Dis 2015;21:713–20. [DOI] [PubMed] [Google Scholar]
- 195. Yu L, Guo WC, Zhao SH, et al. Mitotic arrest defective protein 2 expression abnormality and its clinicopathologic significance in human osteosarcoma. APMIS 2010;118:222–9. [DOI] [PubMed] [Google Scholar]
- 196. Nakano Y, Sumi T, Teramae M, et al. Expression of the mitotic‐arrest deficiency 2 is associated with chemotherapy resistance in ovarian serous adenocarcinoma. Oncol Rep 2012;28:1200–4. [DOI] [PubMed] [Google Scholar]
- 197. Park PE, Jeong JY, Kim SZ, et al. MAD2 expression in ovarian carcinoma: different expression patterns and levels among various types of ovarian carcinoma and its prognostic significance in high‐grade serous carcinoma. Korean J Pathol 2013;47:418–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Nakano Y, Sumi T, Morishita M, et al. Mitotic arrest deficiency 2 induces carcinogenesis in mucinous ovarian tumors. Oncol Lett 2012;3:281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Chen F, Liu S, Zhou Y, et al. Mad2 overexpression is associated with high cell proliferation and reduced disease‐free survival in primary gastrointestinal diffuse large B‐cell lymphoma. Hematology 2016;21:399–403. [DOI] [PubMed] [Google Scholar]
- 200. Hisaoka M, Matsuyama A, Hashimoto H. Aberrant MAD2 expression in soft‐tissue sarcoma. Pathol Int 2008;58:329–33. [DOI] [PubMed] [Google Scholar]
- 201. Fung MK, Cheung HW, Wong HL, et al. MAD2 expression and its significance in mitotic checkpoint control in testicular germ cell tumour. Biochim Biophys Acta 2007;1773:821–32. [DOI] [PubMed] [Google Scholar]
- 202. Turner N, Lambros MB, Horlings HM, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene 2010;29:2013–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Iwanaga Y, Chi YH, Miyazato A, et al. Heterozygous deletion of mitotic arrest‐deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res 2007;67:160–6. [DOI] [PubMed] [Google Scholar]
- 204. Dai W, Wang Q, Liu T, et al. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res 2004;64:440–5. [DOI] [PubMed] [Google Scholar]
- 205. Park I, Lee HO, Choi E, et al. Loss of BubR1 acetylation causes defects in spindle assembly checkpoint signaling and promotes tumor formation. J Cell Biol 2013;202:295–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Kalitsis P, Fowler KJ, Griffiths B, et al. Increased chromosome instability but not cancer predisposition in haploinsufficient Bub3 mice. Genes Chromosomes Cancer 2005;44:29–36. [DOI] [PubMed] [Google Scholar]
- 207. Babu JR, Jeganathan KB, Baker DJ, et al. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J Cell Biol 2003;160:341–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Li M, Fang X, Wei Z, et al. Loss of spindle assembly checkpoint‐mediated inhibition of Cdc20 promotes tumorigenesis in mice. J Cell Biol 2009;185:983–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Malureanu L, Jeganathan KB, Jin F, et al. Cdc20 hypomorphic mice fail to counteract de novo synthesis of cyclin B1 in mitosis. J Cell Biol 2010;191:313–29. [DOI] [PMC free article] [PubMed] [Google Scholar]