

Abstract
A stochastic approach of copurification of the protease Cathepsin L that results in product fragmentation during purification processing and storage is presented. Cathepsin L was identified using mass spectroscopy, characterization of proteolytic activity, and comparison with fragmentation patterns observed using recombinant Cathepsin L. Cathepsin L existed in Chinese hamster ovary cell culture fluids obtained from cell lines expressing different products and cleaved a variety of recombinant proteins including monoclonal antibodies, antibody fragments, bispecific antibodies, and fusion proteins. Therefore, characterization its chromatographic behavior is essential to ensure robust manufacturing and sufficient shelf life. The chromatographic behaviors of Cathepsin L using a variety of techniques including affinity, cation exchange, anion exchange, and mixed mode chromatography were systematically evaluated. Our data demonstrates that copurification of Cathepsin L on nonaffinity modalities is principally because of similar retention on the stationary phase and not through interactions with product. Lastly, Cathespin L exhibits a broad elution profile in cation exchange chromatography (CEX) likely because of its different forms. Affinity purification is free of fragmentation issue, making affinity capture the best mitigation of Cathepsin L. When affinity purification is not feasible, a high pH wash on CEX can effectively remove Cathepsin L but resulted in significant product loss, while anion exchange chromatography operated in flow‐through mode does not efficiently remove Cathepsin L. Mixed mode chromatography, using Capto™ adhere in this example, provides robust clearance over wide process parameter range (pH 7.7 ± 0.3 and 100 ± 50 mM NaCl), making it an ideal technique to clear Cathepsin L. © 2018 American Institute of Chemical Engineers Biotechnol. Prog., 35: e2732, 2019
Keywords: fragmentation, host cell proteins, cysteine protease, Cathepsin L, copurification
Introduction
Chinese hamster ovary (CHO) cells are the most commonly used mammalian host cells for large‐scale clinical and commercial production of therapeutic proteins.1, 2 More than 6,000 host cell proteins (HCPs) have been found in the CHO cell proteome,3 and many HCPs are released to cell culture fluid (CCF) as the results of secretion and cell lysis caused by either normal cell death or shear stress during harvest.4 For manufacture of therapeutic proteins, adequate control of residual HCP, which carries a potential risk of immunogenicity to patients, is considered critically important.5, 6 Maintaining high cell viability in upstream processing and robust HCP clearance in downstream operations are crucial to ensure patient safety.4 HCPs are monitored as process‐related impurities and the limit established for biotherapeutics is guided by regulations and historical precedent.5 Most biotechnology products reviewed and approved by the U.S. food and drug administration (FDA) contain HCP levels of <100 ppm (parts per million).2
In recent years, many studies have focused on HCP clearance in downstream processing,7, 8, 9, 10 including the use of novel HCP detection methods (such as two‐dimensional gel and mass spectrometry) in addition to more traditional enzyme‐linked immunosorbent assay (ELISA) detection methods.5, 11 A wash strategy during a chromatography step is often utilized to facilitate effective HCP removal.1, 5, 7, 12 Washes containing sodium chloride,10 urea,7 guanidine hydrochloride,8 arginine,13 caprylate,12 and isopropanol7 have been shown to effectively remove HCP during Protein A chromatography. For a variety of reasons use of affinity chromatography is often not practical for purification of recombinant protein therapeutics. These include low‐binding capacity, high cost, lack of an effective cleaning procedure, or short lifetime. Nonaffinity capture methods (e.g., ion exchange and mixed mode) have been evaluated in several studies and separation of HCPs is often found to be challenging because of the nonspecific nature of these separation techniques.14, 15, 16, 17 Therefore, an in‐depth understanding of the HCP chromatographic behavior would provide valuable insight for the rational design of purification processes.18
Besides the immunogenicity concern, HCPs can also compromise product quality and shelf‐life by resulting in degradation of protein or excipient, which leads to fragmentation, aggregation, and particle formation.12, 19, 20, 21 The presence of HCPs exhibiting proteolytic activity (i.e., proteases) in the drug product significantly affects the long‐term stability of protein therapeutics. There are several different families of proteases, such as aspartic proteases, cysteine proteases, serine proteases, metalloproteases, threonine proteases, glutamic proteases, asparagine proteases, and so forth.22 CHO cells express many proteases as part of normal functioning which may copurify with the protein of interest and result in fragmentation. Multiple cases have been reported, including the Cathepsin D catalyzed fragmentation of an Fc‐fusion protein20 and two monoclonal antibodies (mAbs),21 dipeptidyl peptidase 3 cleavage of recombinant human acid alpha glucosidase,23 and the metalloprotease fragmentation of recombinant factor VIII.24 Residual Cathepsin D remained in the final drug substance in the three cases even when Protein A affinity purification was implemented in the downstream processes. Bee et al. proposed interactions between Cathepsin D and the protein of interest to explain the observed copurification.21
In this study, we report product fragmentation caused by copurification of the cysteine protease, Cathepsin L. Cathepsin L is intrinsically different from Cathepsin D, such as different protease family, protein size, hydrophobicity, and charge. These differences have major implications for bioprocess development and manufacturing. Cathepsin L was observed in cell culture supernatants obtained from different CHO cell lines, manifesting proteolytic activity against a variety of non‐mAb recombinant therapeutic proteins. The protease was identified and characterized using orthogonal techniques. The cleavage sites on a model protein by the CHO Cathepsin L were also compared with those of the recombinant murine Cathepsin L. We then systematically investigate chromatographic techniques for robust removal of all forms of Cathepsin L.
Materials and Methods
Chemicals, monoclonal antibodies, column resins and membranes
The chemicals used in this study were obtained from J.T. Baker (Phillipsburg, NJ). Recombinant antibody fragments (Fabs), mAbs, fusion proteins, and bispecific antibodies were produced in‐house using standard CHO cell culture techniques. Recombinant murine Cathepsin L (His‐tagged) was purchased from Sino Biological, Inc. (Gaithersburg, MD). The protease inhibitors used in the study were purchased from ThermoFisher Scientific (Grand Island, NY). Chromatographic resins MabSelect SuRe™ Protein A, Capto™ SP ImpRes, and Capto™ adhere were from GE Healthcare (Piscataway, NJ); POROS™ HQ, XQ, XS, and CaptureSelect™ CH1 (IgG‐CH1, specifically designed the purification of recombinant Fab fragments and IgGs) resins were from ThermoFisher Scientific; Toyopearl® GigaCap S650 M and NH2‐750F resins were from Tosoh Bioscience (King of Prussia, PA); Eshmuno® S and Fractogel® EMD SO3[M] resins were from EMD Millipore (Burlington, MA), Nuvia™ HR‐S and C‐prime resins were from Bio‐Rad (Hercules, CA), MEP® HyperCel were from Pall (New York, NY), Natriflo® HD‐Q membrane cassette was from Natrix (Burlington, ON, Canada).
Chromatography instrument and operations
Chromatographic experiments were carried out using an ÄKTA Avant controlled with Unicorn Software version 6.4 (GE Healthcare). Instantons conductivity, pH, and ultraviolet (UV)280 at the column outlet were monitored by in‐line probes on the Avant. Sample protein concentration was measured off‐line using a NanoDrop 2000 (Thermo Fisher Scientific, Wilmington, DE) with the corresponding extinction coefficient. Resins were packed into 0.66 cm inner diameter (ID) Omnifit columns (Diba Industries, Danbury, CT) or 1.15 cm ID Vantage L laboratory columns (Millipore, Billerica, MA) to a bed height of 19 ± 3 cm and operated at 300 cm/h.
Purification by Protein A, CH1 affinity, and MEP® HyperCel was performed as follows: three column volumes (CV) of 50 mM Tris–HCl, pH 7.4 for equilibration prior to sample loading; 30 mg protein per mL resin load challenge; 3 CV of 50 mM Tris–HCl, pH 7.4 for postloading equilibration; 5 CV of 25 mM NaAc, pH 3.6 for elution. The elution product was collected when UV280 is greater than 200 mAU.
Cation exchange chromatography (CEX) with step elution was operated under the following conditions: 3 CV of 50 mM NaAc, pH 4.5 for equilibration before sample loading; 30 mg protein per mL resin load challenge with the feed adjusted to pH 4.5 and 10 mS/cm; 3 CV of 50 mM NaAc, pH 4.5 for postloading equilibration; 3 CV of 50 mM Tris–HCl, 40 mM NaCl (or other modifier described separately), pH 7.4 for postload wash; 7 CV of 50 mM NaAc buffer with NaCl at pH 4.5 for elution. The NaCl concentration used for elution was dependent on the resin used, where Fractogel® SO3[M], Eshmuno® S, and POROS™ XS used 200 mM NaCl; Nuvia™ C‐prime used 260 mM NaCl, and Nuvia™ HR‐S used 350 mM NaCl. Flow‐through, wash peak, and elution product were collected when UV280 is greater than 100 mAU.
CEX and mixed mode (using Nuvia™ C‐prime) chromatography with linear gradient elution (LGE) was operated under the following conditions: 3 CV of buffer with no NaCl for equilibration before sample loading; 30 mg protein per mL resins load challenge; 3 CV of equilibration buffer for postloading equilibration; LGE from 0 to 500 mM NaCl over 20 CV. CEX LGE was operated at different pH values where the equilibration and elution buffers were 50 mM NaAc (pH 4.5 and 5.5), 50 mM sodium phosphate (pH 6.0), or 50 mM Tris–HCl (pH 7.4 and pH 8.0). Elution product was fractionated on 1 CV basis. Anion exchange chromatography (AEX) and mixed mode (using Capto™ adhere) chromatography was operated under the following conditions: 3 CV of 50 mM Tris–HCl, pH 7.4 for equilibration before sample loading; 30 mg protein per mL resin load challenge with the feed adjusted to pH 7.4 and 9 mS/cm; 3 CV of 50 mM Tris–HCl, pH 7.4 for postloading chase. In instances where operating conditions were varied to understand the design space, pH values were adjusted with 1 M acetic acid or 1 M Tris base pH 11 and NaCl concentrations were adjusted with 4 M NaCl. Flow‐through product was collected when UV280 is greater than 100 mAU.
Analytical size‐exclusion chromatography and protease activity measurement
Analysis of purity and fragment level was monitored with high performance size exclusion chromatography (HP‐SEC) using a TSK‐GEL G3000SWXL column (7.8 mm × 30 cm) from Tosoh Bioscience (King of Prussia, PA) with an Agilent 1200 HPLC system (Agilent Technologies, Santa Clara, CA). The column was operated at a flow rate of 1 mL/min with a mobile phase consisting of 100 mM sodium phosphate, 0.5 M NaCl, pH 6.8. Samples were 0.22 μm filtered and injected at a mass load of 250 μg. The eluted protein was monitored by absorbance at 280 nm and sample purity was estimated by integrating the chromatograms.
Protease activity was evaluated by monitoring fragmentation using HP‐SEC. Unless otherwise stated, the sample containing protease was incubated with Fab A (2.5 mg/mL) at pH 3.4 ± 0.1 and 37°C for the desired time, and then the sample was neutralized to pH 7.0 ± 0.5 and analyzed by HP‐SEC. In the experiments evaluating Cathepsin L removal on AEX and Capto™ adhere, the remaining protease level is calculated by the following formula: (fragment level caused by the purified product/fragment level caused by the load) × 100%.
Fragment analysis by intact mass and cleavage site analysis by peptide mapping
Fragments and their cleavage sites were analyzed using reversed‐phase high performance chromatography (RP‐HPLC) coupled to a mass spectrometer. For fragment identification, samples separated using a Waters ACQUITY UPLC system (Milford, MA) equipped with a Waters BEH C4 column (2.1 mm × 100 mm, 300 Å, 1.7 μm). Samples were prepared by dilution to 1 mg/mL with water and the separation was carried out in a linear gradient from 100% mobile Phase A (0.1% formic acid [FA] and 0.01% triflouracetic acid [TFA] in water) to 100% mobile Phase B (0.1% FA and 0.01% TFA in acetonitrile) operated at 0.2 mL/min. Fragments were identified using a quadrupole and orthogonal time‐of‐flight type mass spectrometer (Synapt G2) from Waters. Mass spectrometric data are collected at a m/z range of 600–4,500. The accurate masses of the fragments and intact Fab A were obtained through the deconvolution of the mass data, identification, and quantitation using the MassLynx MaxEnt1 software package from Waters.
For the cleavage site identification, the samples were separated on a Waters UPLC system equipped with a Waters BEH C18 column (2.1 mm × 150 mm, 300 Å, 1.7 μm). The sample was prepared by diluting to 1 mg/mL with water and the separation is done using a linear gradient from 100% mobile Phase A (0.02% TFA in water) to 100% mobile Phase B (0.02% TFA in acetonitrile) at 0.2 mL/min. Separated fragments were monitored using a UV detector and identified using an OrbiTrap Fusion mass spectrometer from ThermoFisher Scientific in positive ion mode. Each fragment is identified by its mass (corresponding to the amino acid composition). The mass was determined using MS spectrum and the fragmentation masses were determined using tandem mass spectrum.
HCP quantitation and identification
HCPs in all samples were quantified by in‐house ELISA using sheep anti‐CHO HCP antibodies. HCP level is calculated based on a standard curve using HCPs prepared from null CHO cells.
The HCP profile in an enriched sample containing the protease was evaluated using reversed‐phase ultra‐performance liquid chromatography (RP‐UPLC) coupled with tandem MS. liquid chromatography with tandem mass spectrometry (LC–MS/MS) analysis was carried out after digesting samples with trypsin, separating digested peptides on RP‐UPLC, and then identifying samples using mass spectrometer. For digestion, 100 μg protein was denatured and reduced in 50 mM Tris–HCl 8 M guanidine, 50 mM dithiothreitol (DTT), pH 7.4 at 37°C for 30 min. Samples were then alkylated by adding 500 mM iodoacetamide at ambient temperature with a 30‐min incubation in the dark. Proteins were precipitated by adding prechilled ethanol for 2 h at −20°C. After centrifugation and removal of ethanol, the precipitated proteins were dried and then reconstituted in 6 M urea and 100 mM Tris–HCl, pH 7.6. Sequencing grade Trypsin (Catalog number V5280); Promega, Madison, WI) was then added to the sample and proteins were digested overnight at 37°C. For peptide separation, 15 μL sample was injected onto a Water Acquity BEH C18 column (2.1 mm × 150 mm; 300 Å, 1.7 μm) Elution was performed with a linear gradient from 0 to 42% B over 70 min (mobile Phase A: 0.1% FA in water; mobile Phase B: 0.1% FA in acetonitrile) at a flow rate of 200 μL/min. The UPLC was coupled via a standard ESI source to a Synapt G2 mass spectrometer (Waters) and data were collected in MSe mode. The identity of each HCP present was determined with the Protein Lynx Global Server (PLGS 2.4 version 24) software (Waters) by searching mass spectral data against a FASTA protein sequence database from the UniProt portal (www.uniprot.org). A minimum of two tryptic peptides was used to positively identify each HCP. Proteins identified were checked individually against the UniProt database using accession IDs to determine biological functions (proteolytic activity). Score and probability values were used to avoid identification of false positives.
Results and Discussion
Fragmentation of Fab A during downstream processing
Figure 1A shows a downstream process that was developed for manufacturing a recombinant Fab (Fab A, molecular weight 48 kD) expressed using CHO cell culture. Affinity Captureselect™ CH1 resin that can specifically bind to the CH1 domain in the heavy chain was evaluated but it showed low binding capacity (~7 mg/mL) for Fab A. CH1 rein was not chosen for because of the low‐binding capacity that leads to high manufacturing cost. Fab A has a Kyte–Doolitle hydrophobicity index value of −0.34. Hydrophobicity interaction chromatography option was not chosen mainly because of the need of high salt (500 mM sodium citrate) for enabling binding. High salt and resultant dilution were determined as significant challenge during our fit‐to‐plant analysis for large scale manufacturing. Fab A has a basic pI of 9.6. CEX was evaluated and met the desires, such as product purity, binding capacity, and ease of manufacturing. CEX resins (Fractogel® SO3[M]) is selected for capturing (Step 1) Fab A from the clarified CCF and removes a majority of the process‐related impurities such as HCP, DNA, and cell culture media components, while Step 2 the low pH treatment step (pH 3.5 ± 0.1 and 60 min) is a dedicated viral inactivation step. Step 3 is an AEX column (POROS™ HQ) operated in flow‐through mode that removes process‐related impurities (HCP and DNA) and also serves as an orthogonal viral clearance step. The final chromatography step is a CEX column (Nuvia™ HR‐S) operated in bind‐and‐elute mode that was designed to product‐related impurities (such as aggregates). The nanofiltration and ultrafiltration/diafiltration steps are for removing potential viruses and final formulation, respectively. The drug substance (DS) produced by this process met product quality specifications suitable for use in clinical studies.
Figure 1.
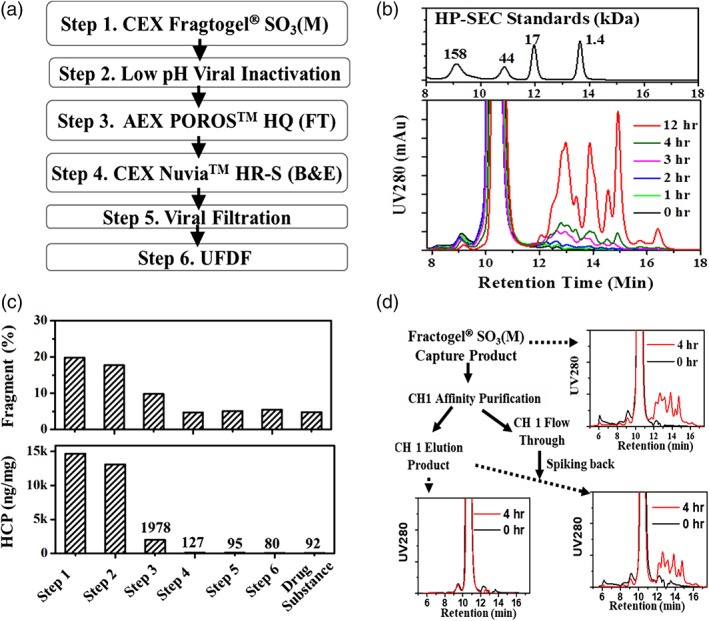
Fragmentation during purification of Fab A. (A) The initial purification process flow for Fab A. (B) Time‐dependent fragmentation of Fab A visualized with HP‐SEC profiles during incubation at pH 3.4. (C) HCP and fragment levels (after pH 3.5 incubation) in purification intermediates. (D) Fragmentation (after pH 3.5 incubation) visualized with HP‐SEC profiles for Fractogel® SO3(M) capture product, Fractogel® SO3(M) product further purified with CH1 affinity chromatography, and Fractogel® SO3(M) product further purified with CH1 affinity chromatography and spiked with CH1 unbound fraction.
For the low pH viral inactivation step, the targeted pH and incubation time were selected based on historical in‐house viral inactivation data (not shown) and literature.25 Typically, lower pH and longer incubation time than the target condition are tested at bench‐scale to support a wider operational window for manufacturing, for example, pH 3.4 for 12 h hold. Figure 1B shows the HP‐SEC profile obtained for extended incubation of Fab A at pH 3.4. As can be seen in the figure, the 0‐h sample showed a small aggregate peak (2.2%), a monomer peak (~97.8%) and negligible late eluting fragment peaks. The 1‐h sample showed a similar HP‐SEC profile to the 0‐h sample; however, the longer incubation time (2, 3, 4, and 12‐h) samples showed a decreased monomer peak and multiple late eluting species, suggesting that the sample was fragmented. The level of fragments also increased with the incubation time: 4‐h incubation generated ~5% fragments and 12‐h incubation led to a much higher level of fragments (~40%). Eight distinct peaks on the HP‐SEC profile were observed for the 12‐h sample, suggesting the presence of at least eight different fragment species with estimated molecular weight ranging from 15 to <1 kD (as indicated by the molecular weight standards). This time‐ and pH‐sensitive fragmentation is a significant risk for manufacturing and storage as fragmentation results in lower purity and concomitant product loss. Therefore, identification of the fragmentation mechanism was crucial to ensure successful large‐scale manufacturing and acceptable long‐term stability.
To determine whether the fragmentation was exacerbated by a particular purification step or solution condition, the purified intermediates and DS were adjusted to pH 3.4 and then incubated at 37°C for 3 h. As shown in the top panel in Figure 1C, all tested samples showed fragmentation. Importantly, the fragmentation rate decreases as Fab A progresses through the process, suggesting that a host related protease being cleared by the process is the likely root cause.
To test the supposition that a protease was responsible, we carried out an affinity purification and spiking study as depicted in Figure 1D. In brief, the Fractogel® SO3(M) capture product (which has high level of HCP and showed fragmentation at low pH) was purified by CaptureSelect™ CH1 affinity chromatography, which should selectively capture Fab A through specific interactions between the resin and the CH1 domain on Fab A while allowing the protease and other HCPs to flow‐through the column. Compared to the captured product, the CH1 purified Fab A sample has a much lower HCP level and fragmentation was no longer observed after incubating at low pH. When this CH1 purified sample was spiked with the CH1 flow‐through (unbound) fraction (which has a high level of HCPs and the putative protease), the spiked sample showed fragmentation after low pH incubation. These results suggest that the putative protease is present in the CH1 unbound fraction but not in the CH1 elution product. Proteases that generate fragments profiles similar to those observed in Figures 1B,D have been reported to result in fragmentation of DS during storage.12, 23 Select proteases have been reported to bind with the product of interest, resulting in copurification.10, 12 In this case, the postulated protease does not interact or interacts weakly with Fab A as it was removed by the CH1 affinity purification. The protease appears to copurify with Fab A on the cation exchanger Fractogel SO3(M) and the subsequent chromatography steps do not completely remove the protease, resulting in DS that is susceptible to fragmentation. This represents a significant threat to storage of the DS because short‐term stability under accelerated conditions is usually employed as a prediction of long‐term stability.
Identification of the protease
Obtaining the optimal pH for proteolytic activity often provides an important clue for protease identification.20 Moreover, information on the pH‐dependent behavior of the protease aids in the rational selection of purification and formulation conditions. To elucidate the impact of pH on protease activity, samples of Fab A CEX captured product were prepared at pH values ranging from 3.5 to 7.0, held for 0–4 h, and then analyzed by HP‐SEC. Figure 2A summarizes the effect of pH and hold time on the fragmentation rate. As can be seen in the figure, fragmentation occurs much faster at low pH and at elevated temperatures. For example, at pH 3.5 at room temperature, conditions which are common for a low pH viral inactivation step,25 fragmentation proceeds at a rate of 0.6% per hour. Interestingly, fragmentation did not occur at pH 7.0 at both 25°C and 37°C, suggesting that the protease is nearly inactive under neutral pH conditions. At pH 5.0 and 6.0, while the fragmentation was barely detectable at 25°C, it was pronounced at 37°C. The results suggest that the presence of the proteolytic activity under mildly acidic conditions (commonly used for formulation) still poses a significant threat to long‐term product stability.
Figure 2.
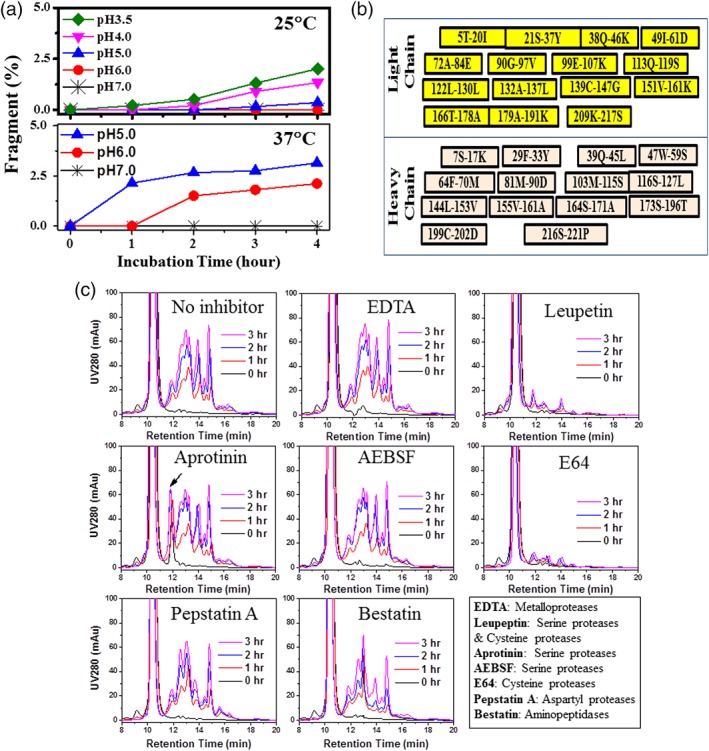
Characterization of the protease optimal pH, cleavage sites, and the effect of inhibitors. (A) The effects of pH and temperature on the protease‐induced fragmentation determined by HP‐SEC (after pH 3.5 incubation). (B) Schematic summary for the Fab A fragments identified by mass spectrometry with the N‐ and C‐terminal amino acids labeled. (C) Effect of protease inhibitors on fragmentation as visualized by HP‐SEC profiles (after pH 3.5 incubation). The legend gives the protease family for which the inhibitor is intended; inhibitor concentration was controlled to 1 mM.
Identification of the proteolytic cleavage sites can also provide insights to aid in determination of the protease class; thus, peptide mapping was carried out on the fragments that were generated by the protease. Because the fragments were small, samples were analyzed directly without further enzyme digestion. Figure 2B summarizes the identified fragments with the indicated amino acids on the N‐ and C‐terminus. The fragments were peptides containing 8–22 amino acids, and the cleavage sites at N‐ or C‐terminus include charged, aromatic, and hydrophobic amino acids. These results suggested that the fragments are caused by a low specificity protease or several different proteases.
To further narrow the protease class, we tested the effect of different protease inhibitors. Figure 2C shows the effect of different inhibitors. As can be seen in the figure, EDTA, Aprotinin, AEBSF (4‐[2‐aminoethyl] benzenesulfonyl fluoride hydrochloride), Pepstatin A, and Bestain spiked individually did not inhibit fragmentation. On the other hand, Cysteine protease inhibitors Leupeptin and E64 (N‐[trans‐Epoxysuccinyl]‐l‐leucine 4‐guanidinobutylamide) effectively inhibited fragmentation, suggesting the dominant role of a cysteine protease in the fragmentation of Fab A. Consistent with our observations, cysteine proteases are active under acidic conditions and inactive at neutral pH.22
The sample containing protease was enriched, concentrated, and then analyzed by mass spectrometry for identification of HCPs. Table 1 summarizes the HCPs that were identified in the sample. As shown in Table 1, 14 HCPs were identified and several of them have been reported to be copurify with the protein of interest in two other studies.18, 26 Among the 14 HCPs, two are proteases: Cathepsin L and D. Cathepsin L is low specificity protease and is active at pH 3.0–6.5.27 It has three forms of molecular weight of 34, 32, and 28 kD, respectively, as indicated by SDS‐PAGE.28 These facts are consistent with the above characterization results. Conversely, Cathepsin D belongs to the aspartyl protease family12 that can be specifically inhibited by Pepstatin A. Pepstatin A did not effectively inhibit the fragmentation of Fab A (Figure 2C). Therefore, the fragmentation is mainly caused by Cathepsin L. Obviously, Cathepsin D does significant contribution to the fragmentation observed above, however, whether it digests the Cathepsin L resultant fragments remains unclear and needs additional evaluation.
Table 1.
Summary of HCPs Identified in the Enriched CH1 Unbound Fraction
| Identified HCP | Description | Score† | Intensity* | Coverage (%) | Amino Acids |
|---|---|---|---|---|---|
| Cathepsin L | >gi|344,259,154|gb|EGW15258.1| | 465.6 | 6.13E+06 | 84 | 333 |
| Clusterin | >gi|344,249,681|gb|EGW05785.1| | 435.0 | 3.77E+06 | 40 | 447 |
| Metalloproteinase inhibitor 1 | >gi|344,258,664|gb|EGW14768.1| | 347.5 | 1.73E+06 | 41 | 203 |
| Inter‐alpha‐trypsin inhibitor heavy chain H5 | >gi|344,238,973|gb|EGV95076.1| | 228.1 | 2.75E+06 | 19 | 913 |
| Follistatin‐related protein 1 | >gi|344,236,558|gb|EGV92661.1| | 366.6 | 1.83E+06 | 22 | 583 |
| Sulfated glycoprotein 1 | >gi|344,242,104|gb|EGV98207.1| | 363.1 | 1.54E+06 | 36 | 249 |
| Glucose‐regulated protein | >gi|350,537,423|gb|NP_001233668.1| | 231.4 | 1.14E+06 | 15 | 654 |
| Amyloid beta A4 protein | >gi|344,251,481|gb|EGW07585.1| | 290.4 | 1.11E+06 | 15 | 433 |
| Cathepsin D | >gi|344,248,735|gb|EGW04839.1| | 203.9 | 4.42E+05 | 16 | 408 |
| Protein S100‐A11 | >gi|344,255,219|gb|EGW11323.1| | 236.5 | 8.26E+05 | 50 | 100 |
| Insulin‐like growth factor‐binding protein 4 | >gi|344,256,935|gb|EGW13039.1| | 213.2 | 1.02E+06 | 30 | 254 |
| Cornifin‐A | >gi|344,257,771|gb|EGW13875.1| | 286.4 | 4.64E+05 | 55 | 113 |
| Ganglioside GM2 activator | >gi|344,252,723|gb|EGW08827.1| | 228.3 | 5.80E+05 | 45 | 191 |
| Granulins | >gi|344,252,078|gb|EGW08182.1| | 201.7 | 2.05E+05 | 12.67 | 592 |
The total intensity is the sum of all fragment ion peak intensities from all MS/MS spectra.
The score reflects the quality of the match between the predicted and observed MS/MS peaks (peptide‐spectrum match). The score ranges from 0 to 1000. Only HCPs with score ≥200 are listed.
To confirm Cathepsin L is the root cause, the cleavage sites obtained by a recombinant Cathepsin L was compared to those of the native CHO Cathepsin L. CHO recombinant Cathepsin L was not commercially available; thus, we used a recombinant version of a murine (a species close to Chinese hamster) Cathepsin L that shares 97% sequence similarity with CHO Cathepsin L. Figure 3A shows the fragmentation obtained by mixing purified Fab A with either recombinant murine Cathepsin L or with the CHO Cathepsin L present in the CH1 unbound fraction. As shown in the figure, the recombinant murine Cathepsin L cleaved Fab A and generated a similar HP‐SEC profile to that of CHO Cathespin L. The fragments were also subjected to intact mass analysis and peptide mapping. As shown in Figure 3B, approximately 70% of the fragment peaks in the two samples are identical and peptide mapping results showed that more than 80% cleavage sites are identical. The differences between the two samples may be because of the presence of another protease (such as Cathepsin D) in the CH1 unbound fraction. Based on the totality of this evidence, we concluded that the fragmentation of Fab A under acidic pH is caused by CHO Cathepsin L. Assuming CHO Cathepsin L has similar activity to the recombinant murine Cathepsin L, CHO Cathepsin L concentration in the CH1 unbound fraction (or the CEX capture product) is approximately 250 ng/mL.
Figure 3.
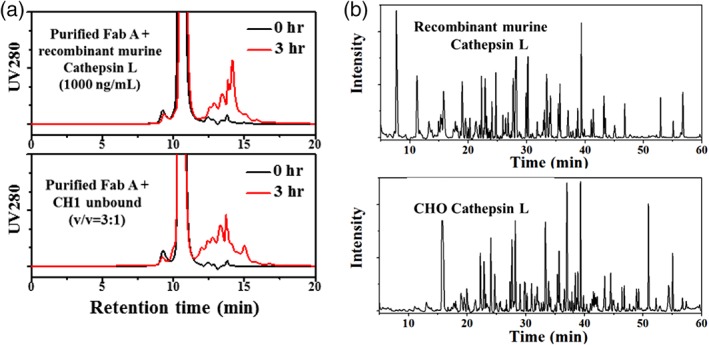
Confirmation of Cathepsin L as the root cause of the fragmentation. (A) Fragmentation visualized by HP‐SEC profiles (during pH 3.4 incubation) for Fab A cleaved by recombinant murine Cathepsin L or CHO Cathepsin L (in CH1 unbound fraction). Fab A concentration was 2.5 mg/mL and 1 mM DTT was added in the sample mixture for the recombinant murine Cathepsin L. (B) Intact mass spectroscopy ion chromatograms for Fab A fragments generated by recombinant mouse Cathepsin L or CHO Cathepsin L (in CH1 unbound fraction).
Cathepsin L specificity
Cathepsin L plays a very important role in diverse biological processes,27 thus the presence of Cathepsin L in CCFs is unlikely unique to Fab A. We evaluated several CCFs for three different CHO cell produced recombinant proteins, as well as a CCF from null CHO cells. After the recombinant proteins were depleted by the appropriate affinity chromatography modality, the CCFs were purified by Fractogel® SO3(M) and then protease activities of the eluates were evaluated by using Fab A as substrate. Figure 4A shows the fragmentation of Fab A by Cathepsin L isolated from various lots of CCFs. As shown in Figure 4A, the eluates from the four CCFs resulted in similar fragmentation of Fab A, demonstrating the presence of the same protease, Cathepsin L, in these CCFs.
Figure 4.
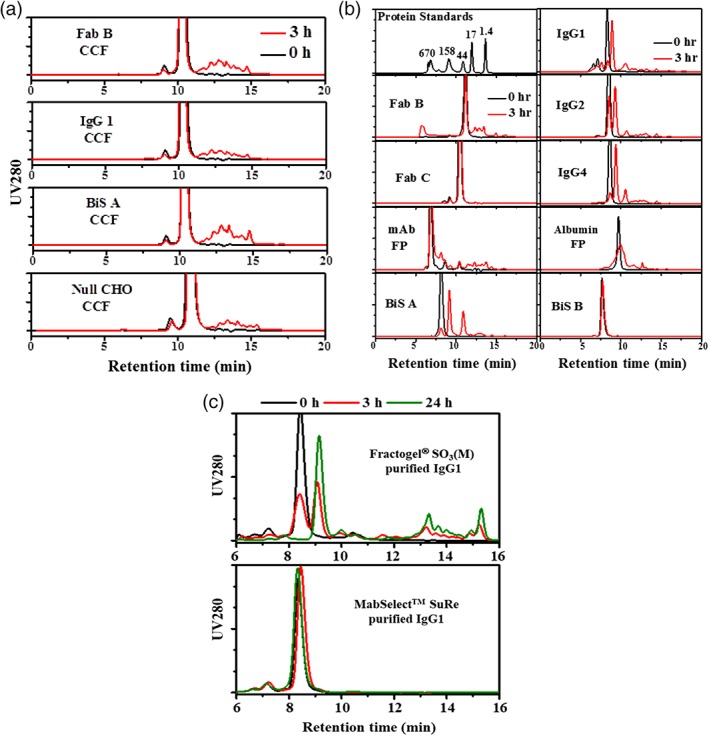
Cathepsin L exists in different CHO cell culture fluids (CCFs) and cleaves variety of recombinant proteins. (A) Fragmentation of Fab A by Cathepsin L isolated from different CHO CCFs as visualized by HP‐SEC profiles. CCFs were obtained from bioreactors operated with CHO cells expressing therapeutic proteins (Fab B, IgG1, and Bis A), purified with affinity chromatography to deplete the therapeutic protein, and then Cathepsin L was captured from the affinity unbound fraction with CEX. (B) Fragmentation (during pH 3.4 incubation) visualized by HP‐SEC profiles for Fabs (B and C), mAbs (IgG1, 2 and 4), Bi‐specific (BIS A and B), a mAb‐fusion protein (mAb FP) and an albumin‐fusion protein (albumin FP). (C) Fragmentation (during pH 3.4 incubation) visualized by HP‐SEC profiles for IgG1 captured from CCF by Fractogel® EMD SO3(M) and MabSelect™ SuRe.
Owing to its low specificity, Cathepsin L is able to cleave a diversity of proteins,27 therefore, CHO Cathepsin L should be considered as a potential risk to many of the CHO expressed molecular classes used as biotherapeutics, including various antibody‐like formats and fusion proteins. Figure 4B shows the HP‐SEC fragmentation of three mAbs with different subclass (IgG1, IgG2, and IgG4), two Fabs, two bispecific antibodies (Bis A and B), a mAb‐fusion protein, and an albumin‐fusion protein that were incubated with the CHO Cathepsin L in the CH1 unbound fraction (from Fab A purification). As shown in Figure 4B, fragments were observed in most of the tested proteins except Fab C and Bis B. The results confirm that CHO Cathepsin L can cleave a variety of recombinant proteins. Additionally, CHO Cathepsin L seems to have preferential cleavage sites on mAbs of different subclass as similar fragment profiles were seen for three tested mAbs (IgG1, 2, and 4).
It is unclear whether Cathepsin L copurifies with other recombinant proteins on Fractogel® SO3(M) or whether it binds to a protein and purifies as a complex. To address this, the CCF containing a mAb (IgG1) was purified separately by CEX (using Fractogel® SO3(M) resin) and by Protein A affinity (using MabSelect™ SuRe resin) similar to the CH1 purification of Fab A. Figure 4C shows the resulting HP‐SEC fragmentation profiles obtained during low pH incubation of the elution products, respectively. As can be seen in Figure 4C, Fractogel® SO3(M) purified IgG1 proteins showed fragmentation but the sample purified by Protein A chromatography did not. The results suggest that Cathepsin L also copurifies with IgG1 on Fractogel® SO3(M) and similar to Fab A, IgG1 does not interact with Cathepsin L.
Based on these results, it is likely that CHO Cathepsin L naturally exists in CCFs and is a common issue for the purification of CHO cell expressed recombinant proteins when nonaffinity capture is used.
Chromatographic behavior of Cathepsin L on CEX
To test whether copurification of Cathepsin L is unique to Fractogel® SO3(M), several different resins, including CEX, mixed mode, and affinity, were evaluated with the CCF for Fab A. Figure 5A shows HP‐SEC profiles obtained for Fab A during low pH incubation postcapture with various resins. As shown in Figure 5A, the elution products from the two CEX resins (Eshmuno® S and POROS™ XS) showed similar fragmentation while the Fab A purified by CH1 affinity resin did not show fragmentation. The elution products of the two mixed mode resins (Nuvia™ C‐Prime, and MEP® HyperCel) showed fragmentation but at much lower level than seen in the CEX capture product. The cation exchangers (Fractogel® SO3(M), Eshmuno® S, and POROS™ XS) and C‐prime utilized similar operating conditions, and CH1 and MEP® HyperCell were operated similarly using conditions different to those for the cation exchangers. These results indicated that the copurification of Cathepsin L is associated with nonaffinity capture, and shows some sensitivity to the mode or mechanism of the purification.
Figure 5.
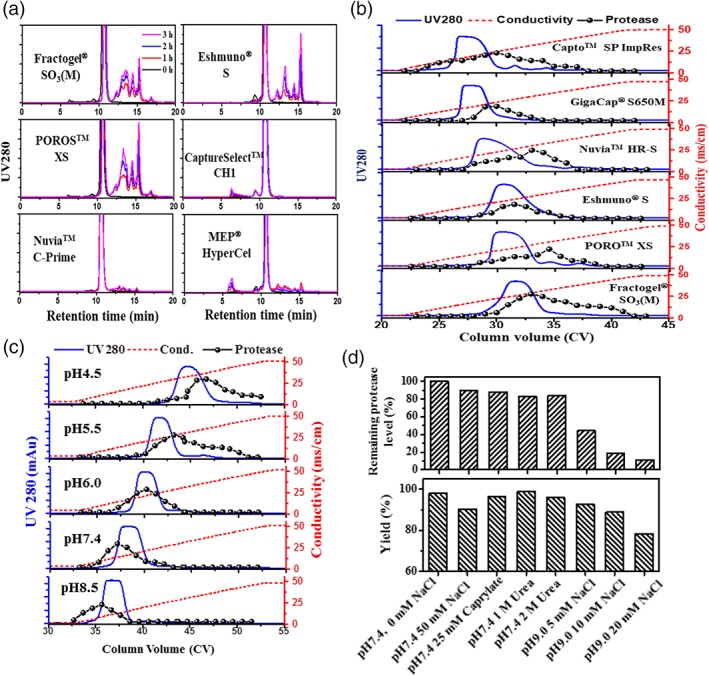
Chromatographic behavior of Cathepsin L in bind‐and‐elute chromatography. (A) Fragmentation (during pH 3.4 incubation) of Fab A visualized by HP‐SEC profiles after purification from CCF by cation exchange, affinity and mixed mode chromatography. (B) Elution profiles of Fab A and Cathepsin L on different cation exchanger resins eluted in a 0–500 mM NaCl, 20 CV linear gradient at pH 5.0. (C) Elution profiles of Fab A and Cathepsin L on Fractogel® SO3(M) in a 0–500 mM NaCl, 20 CV linear gradient at different pH values. (D) The effects of washes on elution product protease level and step yield observed during Fab A purification with Fractogel® SO3(M). The elution product was acidified to pH 3.4 and incubated at 37°C for 3 h and fragment level was measured by HP‐SEC.
CEX is a common choice for capture of recombinant proteins when affinity options are not available or are cost prohibitive.18 Therefore, evaluation of chromatographic behaviors of Cathepsin L on different cation exchange resins should provide useful information for rational design of a nonaffinity purification process. Figure 5B shows the elution profile (LGE from 0 to 500 mM NaCl at pH 5.0) of Fab A on six CEX resins as well as the proteolytic activity measured in fractions collected during the elution. The protease activity is shown as the fragment level (evaluated by HP‐SEC) that each collected fraction caused at low pH for 3 h using Fab A as the substrate. As can be seen in Figure 5B, Cathepsin L was eluted over a wide range of NaCl concentrations and, and the elution of Cathepsin L overlapped with that of Fab A on all tested resins. Nuvia™ HR‐S and POROS™ XS showed slightly better separation than other resins, but even for those two cases, significant overlap of elution peaks was observed, which would make it difficult to fully remove the Cathepsin L. The wide elution profile of Cathepsin L is not well understood, but it may be associated with its several formats as indicated by literature.28
The elution behavior of Cathepsin L was also evaluated under different pH conditions to better understand the potential to remove Cathepsin L during CEX capture. Especially, CHO Cathepsin L has a predicted pI of 6.8, which is lower than that (9.6) of Fab A. Figure 5C shows the elution profiles of Fab A on Fractogel® SO3(M). As shown in Figure 5C, the elution peak of Cathepsin L became sharper at higher pH; however, the elution of Cathepsin L still overlaps with that of Fab A. As expected, both Fab A and Cathepsin L were more strongly retained at lower pH, but an interesting trend was observed where the protease eluted ahead of the Fab A peak at higher pH, while Fab A eluted ahead of the protease at lower pH. These results suggested that implementation of a wash step at high pH (such as pH 8.5) or elution at low pH (such as pH 4.5) may offer advantages for separating Cathepsin L from Fab A.
For Fab A, at pH 4.5, a minimum of 300 mM NaCl is needed for elution from Fractogel® SO3(M), which is unfavorable from process perspective because dilution would be required to lower the salt concentration prior to subsequent purification steps (e.g., flow‐through AEX). Instead, washes were tested at a higher pH to remove Cathepsin L while keeping Fab A bound to the resin. Figure 5D shows the remaining protease level (by comparing to that in the load) and step yields obtained during the wash study for Fab A on Fractogel® SO3(M). As shown in Figure 5D, the wash condition at pH 7.4 with or without 50 mM NaCl did not reduce Cathepsin L levels significantly; however, the washes at pH 9.0 containing 5–20 mM NaCl were effective for removing Cathepsin L. Only 50%, 20%, and 10% Cathepsin L remained after 5, 10, and 20 mM NaCl washes, respectively. 20 mM NaCl at pH 9.0 was the most effective wash but the Cathepsin L removal came at the expense of step yield, suggesting that a delicate balance must be struck between purity and yield. Moreover, the optimal operating window may be narrow, which may require setting narrow parameter ranges in a manufacturing setting. Perhaps not surprisingly, the nonionic washes containing caprylate or urea were not effective for removing Cathepsin L because these washes are often employed when HCPs interact with the recombinant protein of interest, which is not the case for Fab A and Cathepsin L as previously discussed.
Capto™ adhere in flow‐through mode provides good clearance on Cathepsin L
AEX operated in flow‐through mode is widely used for host cell protein removal,2 and improved HCP removal usually is achieved when the load has a lower salt concentration. In the case of Fab A, the CEX captured product contains high salt (200 mM NaCl) and would require significant dilution to achieve the desired HCP removal on a subsequent AEX column. Because large dilution steps can be challenging for large‐scale manufacturing facilities because of product‐hold tank volume limitations, salt tolerant AEX (POROS™ XQ, Toyopearl® NH2‐750F, NatriFlo® HD‐Q) and mixed mode AEX (Capto™ adhere) resins were evaluated alongside traditional AEX resin (POROS™ HQ) for the removal of Cathepsin L. Figure 6A shows the remaining protease level (by comparing fragmentation generated by purified product and load) and step yield obtained during AEX and mixed mode purification of Fab A. As shown in Figure 6A, all resins evaluated showed significant Cathepsin L removal and good step yield (>90%). Superior Cathepsin L clearance was observed in the salt tolerant resins compared to a traditional AEX resin; however, the mixed mode (anion exchange and hydrophobic interaction) resin, Capto™ adhere, showed the best Cathepsin L removal and no protease activity was detected in the purified product. The results consistent with the fact that CHO Cathepsin L has relatively higher hydrophobicity index (−0.46) than Fab A (−0.34).
Figure 6.
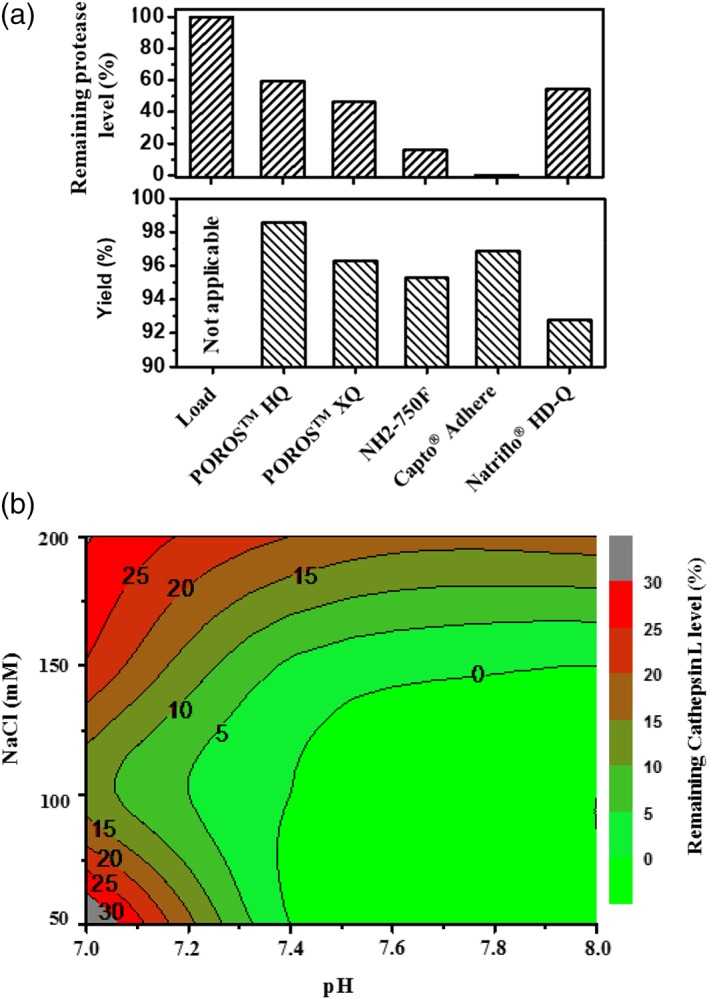
Chromatographic behavior of Cathepsin L in flow‐through chromatography. (A) Removal of Cathepsin L on different polish resins operated in flow‐through mode with load adjusted to pH 7.4 and 8 mS/cm. (B) Summary for Cathepsin L removal on Capto adhere under different pH and NaCl concentrations.
To further understand the capability of Capto™ adhere to remove Cathepsin L, a study was carried out to evaluate the effects of pH and NaCl concentration on Cathepsin L clearance and is summarized in Figure 6B. pH values less than 7.0 were not tested as CHO Cathespin L has pI at 6.8. As can be seen in Figure 6B, Cathepsin L clearance improves at higher pH and at lower NaCl concentrations and a nondetectable level of the protease can be achieved (i.e., no fragmentation is observed) under wide pH operating ranges (pH 7.4–8.0) and NaCl concentrations (50–100 mM). Moreover, step yield within these operating conditions was consistently above 90% (data not shown), confirming that Capto™ adhere is a robust option for Cathepsin L removal.
Removal of the fragments caused by Cathepsin L
In addition to evaluation of Cathepsin L process clearance, we also examined the two chromatographic purification steps in the downstream process for fragment removal to address product purity concerns. The flow‐through mode POROS™ HQ or Capto™ adhere was unable to remove these fragments (data not shown), therefore, we focused on developing Nuvia™ HR‐S and obtained robust conditions for fragment removal. Figure 7A shows the chromatographic profile of Nuvia™ HR‐S, operated in bind‐and‐elute mode with an optimized salt wash (40 mM NaCl). As shown in Figure 7A, protein was observed in the unbound fraction, wash peak, and elution peak, and each was collected and analyzed. As shown in Figure 7B, a majority of the small fragments are in the unbound fraction and the slightly larger fragments are in the wash peak sample, while the elution peak sample contains no fragment other than the intact Fab A. Therefore, the Nuvia™ HR‐S step with the optimized salt wash condition can effectively remove the fragments generated by the protease, mitigating the concern on product purity.
Figure 7.
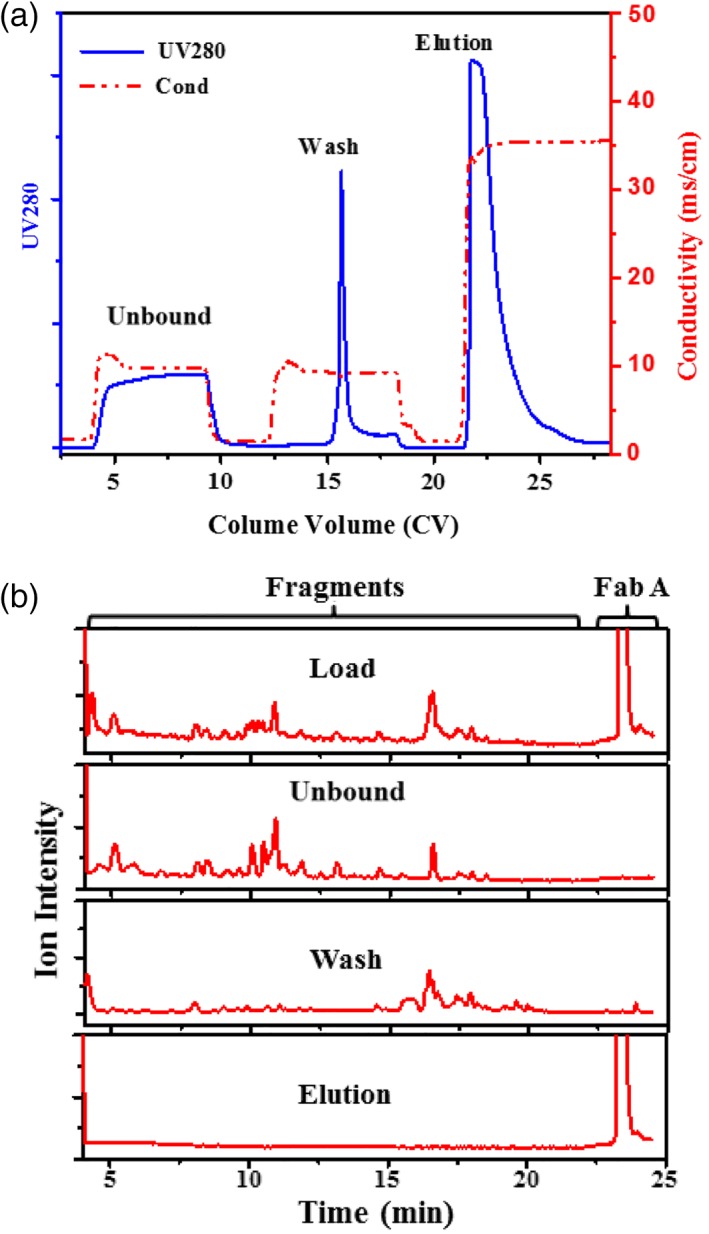
Removal of the protease induced fragments. (A) Separation of the protease induced fragments by Nuvia™ HR‐S. Wash buffer, 50 mM Tris–HCl, 40 mM NaCl pH 7.4; elution buffer, 50 mM NaAc, 310 mM NaCl. (B) MS ion chromatograms for the Nuvia™ HR‐S purification fractions.
Summary and Conclusion
In this work, we report fragmentation of a recombinant Fab, observed during a low pH viral inactivation step, that is caused by the presence of the protease Cathepsin L, that copurifies with the Fab. Cathepsin L was found in the CCFs of multiple CHO expressed recombinant proteins and was shown to be nonspecific and cleave many other recombinant proteins including a different Fab, three mAbs, a mAb‐fusion protein, a bispecific antibody, and an albumin‐fusion protein. Moreover, the Cathepsin L was demonstrated to exhibit proteolytic activity over a mildly acidic pH range of 3.5–6.0 at 25°C, making it a stability concern for purification and storage of therapeutic proteins.
Affinity chromatography was shown to selectively remove the Cathepsin L, which is different to the reported copurification of Cathepsin D.12 When nonaffinity capture was employed, the protease copurifies with the recombinant protein of interest and must be removed in subsequent chromatographic steps. The use of a high pH wash on CEX was shown to be an effective protease removal option but led to product loss making it less attractive for large‐scale manufacturing. A more attractive solution for Cathepsin L removal was found to be mixed mode (anion exchange and hydrophobic interactions) chromatography using Capto™ adhere, which was shown to effectively reduce Cathepsin L to nondetectable levels in flow‐through mode within a wide operating window (pH 7.4–8.0, 50–100 mM NaCl). Similar copurification of CHO Cathepsin L on a CEX (Fractogel® S) with a therapeutic mAb was also reported by Wan et al.29 In that case, Cathepsin L was effectively removed by hydrophobic interaction chromatography (polish step).29 Along with our findings, the hydrophobic property of CHO Cathepsin L may be an advantage that can be used in its separation with other proteins of interest. Additionally, Cathepsin L level in cell lines is potentially different, therefore, cell line screening to be part of the mitigation strategy for the Cathespsin L issue.
Lastly, CEX using Nuvia™ HR‐S with a moderate salt wash was found to be an effective tool to remove Fab fragments that were generated during earlier process steps. The findings of this work should be helpful for rational design a purification process that controlled Cathepsin L levels and delivered high quality product suitable for clinical use.
Acknowledgments
We would like to thank Christopher Boswell, Robert Stadelman, Jeremy Springall, and Arsala Wallace for their contributions and discussions. We also thank Jie Zhu, Ellen O'Connor, Dana Motabar, Frank Bartnik, and Ramin Samadani for providing materials used in the study.
Literature Cited
- 1. Kim JY, Kim YG, Lee GM. CHO cells in biotechnology for production of recombinant proteins: current state and further potential. Appl Microbiol Biotechnol. 2012;93:917–930. [DOI] [PubMed] [Google Scholar]
- 2. Chon JH, Zarbis‐Papastoitsis G. Advances in the production and downstream processing of antibodies. N Biotechnol. 2011;28:458–463. [DOI] [PubMed] [Google Scholar]
- 3. Baycin‐Hizal D, Tabb DL, Chaerkady R, Chen L, Lewis NE, Nagarajan H, Sarkaria V, Kumar A, Wolozny D, Colao J, Jacobson E, Tian Y, O'Meally RN, Krag SS, Cole RN, Palsson BO, Zhang H, Betenbaugh M. Proteomic analysis of Chinese hamster ovary cells. J Proteome Res. 2012;11:5265–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hogwood CE, Bracewell DG, Smales CM. Measurement and control of host cell proteins (HCPs) in CHO cell bioprocesses. Curr Opin Biotechnol. 2014;30:153–160. [DOI] [PubMed] [Google Scholar]
- 5. Bracewell DG, Francis R, Smales CM. The future of host cell protein (HCP) identification during process development and manufacturing linked to a risk‐based management for their control. Biotechnol Bioeng. 2015;112:1727–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jawa V, Joubert MK, Zhang Q, Deshpande M, Hapuarachchi S, Hall MP, Flynn GC. Evaluating immunogenicity risk due to host cell protein impurities in antibody‐based biotherapeutics. AAPS J. 2016;18:1439–1452. [DOI] [PubMed] [Google Scholar]
- 7. Shukla AA, Hinckley P. Host cell protein clearance during protein A chromatography: development of an improved column wash step. Biotechnol Prog. 2008;24:1115–1121. [DOI] [PubMed] [Google Scholar]
- 8. Sisodiya VN, Lequieu J, Rodriguez M, McDonald P, Lazzareschi KP. Studying host cell protein interactions with monoclonal antibodies using high throughput protein A chromatography. Biotechnol J. 2012;7:1233–1441. [DOI] [PubMed] [Google Scholar]
- 9. Chollangi S, Parker R, Singh N, Li Y, Borys M, Li Z. Development of robust antibody purification by optimizing protein‐A chromatography in combination with precipitation methodologies. Biotechnol Bioeng. 2015;112:2292–2304. [DOI] [PubMed] [Google Scholar]
- 10. Hunter AK, Wang X, Suda EJ, Herberg JT, Shell RE, Thomas KE, Dufield RL, Gustafson ME, Mozier NM, Ho SV. Separation of product associating E. coli host cell proteins OppA and DppA from recombinant apolipoprotein A‐I(Milano) in an industrial HIC unit operation. Biotechnol Prog. 2009;25:446–453. [DOI] [PubMed] [Google Scholar]
- 11. Farrell A, Mittermayr S, Morrissey B, Mc Loughlin N, Navas Iglesias N, Marison IW, Bones J. Quantitative host cell protein analysis using two dimensional data independent LC–MS(E). Anal Chem. 2015;87:9186–9193. [DOI] [PubMed] [Google Scholar]
- 12. Bee JS, Tie L, Johnson D, Dimitrova MN, Jusino KC, Afdahl CD. Trace levels of the CHO host cell protease Cathepsin D caused particle formation in a monoclonal antibody product. Biotechnol Prog. 2015;31:1360–1369. [DOI] [PubMed] [Google Scholar]
- 13. Sun, S. Arginine Wash in Protein Purification Using Affinity Chromatography 2008.
- 14. Ng PK, Snyder MA. pH‐based cation exchange chromatography in the capture and elution of monoclonal antibodies. J Sep Sci. 2012;35:29–35. [DOI] [PubMed] [Google Scholar]
- 15. Follman DK, Fahrner RL. Factorial screening of antibody purification processes using three chromatography steps without protein A. J Chromatogr A. 2004;1024:79–85. [DOI] [PubMed] [Google Scholar]
- 16. Maria S, Joucla G, Garbay B, Dieryck W, Lomenech AM, Santarelli X, Cabanne C. Purification process of recombinant monoclonal antibodies with mixed mode chromatography. J Chromatogr A. 2015;1393:57–64. [DOI] [PubMed] [Google Scholar]
- 17. Pezzini J, Joucla G, Gantier R, Toueille M, Lomenech AM, Le Senechal C, Garbay B, Santarelli X, Cabanne C. Antibody capture by mixed‐mode chromatography: a comprehensive study from determination of optimal purification conditions to identification of contaminating host cell proteins. J Chromatogr A. 2011;1218:8197–8208. [DOI] [PubMed] [Google Scholar]
- 18. Levy NE, Valente KN, Lee KH, Lenhoff AM. Host cell protein impurities in chromatographic polishing steps for monoclonal antibody purification. Biotechnol Bioeng. 2016;113:1260–1272. [DOI] [PubMed] [Google Scholar]
- 19. Dixit N, Salamat‐Miller N, Salinas PA, Taylor KD, Basu SK. Residual host cell protein promotes polysorbate 20 degradation in a sulfatase drug product leading to free fatty acid particles. J Pharm Sci. 2016;105:1657–1666. [DOI] [PubMed] [Google Scholar]
- 20. Robert F, Bierau H, Rossi M, Agugiaro D, Soranzo T, Broly H, Mitchell‐Logean C. Degradation of an fc‐fusion recombinant protein by host cell proteases: identification of a CHO Cathepsin D protease. Biotechnol Bioeng. 2009;104:1132–1141. [DOI] [PubMed] [Google Scholar]
- 21. Gao SX, Zhang Y, Stansberry‐Perkins K, Buko A, Bai S, Nguyen V, Brader ML. Fragmentation of a highly purified monoclonal antibody attributed to residual CHO cell protease activity. Biotechnol Bioeng. 2011;108:977–982. [DOI] [PubMed] [Google Scholar]
- 22. Oda K. New families of carboxyl peptidases: serine‐carboxyl peptidases and glutamic peptidases. J Biochem. 2012;151:13–25. [DOI] [PubMed] [Google Scholar]
- 23. Migani D, Smales CM, Bracewell DG. Effects of lysosomal biotherapeutic recombinant protein expression on cell stress and protease and general host cell protein release in Chinese hamster ovary cells. Biotechnol Prog. 2017;33:666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sandberg H, Lutkemeyer D, Kuprin S, Wrangel M, Almstedt A, Persson P, Ek V, Mikaelsson M. Mapping and partial characterization of proteases expressed by a CHO production cell line. Biotechnol Bioeng. 2006;95:961–971. [DOI] [PubMed] [Google Scholar]
- 25. Durno L, Tounekti O. Viral inactivation: low pH and detergent. PDA J Pharm Sci Technol. 2015;69:163–172. [DOI] [PubMed] [Google Scholar]
- 26. Joucla G, Le Senechal C, Begorre M, Garbay B, Santarelli X, Cabanne C. Cation exchange versus multimodal cation exchange resins for antibody capture from CHO supernatants: identification of contaminating host cell proteins by mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;942–943:126–133. [DOI] [PubMed] [Google Scholar]
- 27. Mason RW, Massey SD. Surface activation of pro‐Cathepsin L. Biochem Biophys Res Commun. 1992;189:1659–1666. [DOI] [PubMed] [Google Scholar]
- 28. Hashimoto Y, Kondo C, Katunuma N. An active 32‐kDa Cathepsin L is secreted directly from HT 1080 fibrosarcoma cells and not via lysosomal exocytosis. PLoS ONE. 2015;10:e0145067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wan, M. ; Avgerinos, G. ; Zarbis‐Papastoitsis G. Purified antibody composition (2014) U.S. Patent No. US8916153 B2 AbbVie Biotechnology Ltd., Hamilton U.S. Patent Office.