

Abstract
The pharmacokinetic (PK) and clinical implications of combining metformin with rifampicin are relevant to increasing numbers of patients with diabetic tuberculosis (TB) across the world and are yet unclear. We assessed the impact of rifampicin on metformin PKs and its glucose‐lowering effect in patients with diabetic TB by measuring plasma metformin and blood glucose during and after TB treatment. Rifampicin increased metformin exposure: plasma area under the plasma concentration‐time curve from time point 0 to the end of the dosing interval (AUC 0–τ) and peak plasma concentration (Cmax) geometric mean ratio (GMR; during vs. after TB treatment) were 1.28 (90% confidence interval (CI) 1.13–1.44) and 1.19 (90% CI 1.02–1.38; n = 22). The metformin glucose‐lowering efficacy did not change (Δglucose − Cmax; P = 0.890; n = 18). Thus, we conclude that additional glucose monitoring in this population is not warranted. Finally, 57% of patients on metformin and rifampicin, and 38% of patients on metformin alone experienced gastrointestinal adverse effects. Considering this observation, we advise patients to take metformin and rifampicin with food and preferably separated in time. Clinicians could consider metoclopramide if gastrointestinal adverse effects occur.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Metformin PKs is largely driven by drug transporter proteins. Rifampicin is a known inducer of the expression of these transporters. In healthy volunteers, rifampicin previously led to higher exposure levels and enhanced glucose‐lowering action.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study describes the impact of rifampicin on metformin PKs and its glucose‐lowering effect in patients with diabetic TB.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study was the first to evaluate the effect of rifampicin on metformin exposure and activity in patients with diabetic TB. The study provides the key leads for treatment optimization of this growing group of patients.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
We found that the PK interaction between rifampicin and metformin does not require additional glucose monitoring, in contrast to previous results in healthy volunteers. Because of a high incidence (> 50%) of gastrointestinal adverse events, we advise patients with diabetic TB to take metformin and rifampicin separated in time, and together with food and possibly metoclopramide, to alleviate gastrointestinal adverse events as much as possible.
Tuberculosis (TB) remains a leading cause of morbidity and mortality in developing countries. In 2017, an estimated 10.0 million people developed active TB and 1.6 million patients died.1 At the same time, ~415 million people had diabetes mellitus (DM). The DM prevalence is growing rapidly, especially in low‐income and middle‐income countries, where TB is endemic. Furthermore, DM increases the risk of developing active TB.2, 3 Overall, there is an increasing number of TB cases attributable to DM, namely 10% in 2010, and 15% in 2013.4
Patients with concurrent TB and DM face a higher risk of TB treatment failure, relapse after cure, and death.5 DM management in patients with TB is also problematic. The TB drug rifampicin may affect blood glucose concentrations and induce hyperglycemia by augmenting intestinal absorption of glucose or reducing insulin sensitivity.6, 7 More importantly, rifampicin increases the clearance of most oral antidiabetic drugs that are commonly used in low‐income to middle‐income countries.8, 9 For example, sulphonylureas are metabolized in the liver by cytochrome P450 enzymes, of which rifampicin is a very potent inducer.9, 10
To overcome the effects of rifampicin on treatment and maintenance of glycemic control, metformin has been proposed as a good alternative to other oral antidiabetic drugs, as it is not metabolized in the liver. Moreover, metformin is the first choice antidiabetic according to type 2 DM treatment guidelines.11 The drug is relatively cheap, widely available, and not associated with weight gain or hypoglycemia.12, 13 The main disadvantage of metformin is the risk for lactic acidosis and the high frequency of gastrointestinal adverse effects.14, 15 The oral absorption, hepatic uptake, and renal excretion of metformin are largely mediated by organic cation transporters (OCTs) and multidrug and toxin extrusion protein 1 and 2K (MATE1 and MATE2K), all members of the solute carrier family (Figure 1).14 It is eliminated unchanged in the urine with a half‐life of ~5 hours.14 Its renal clearance is greater than that of creatinine, indicating that tubular secretion contributes to its elimination.16
Figure 1.
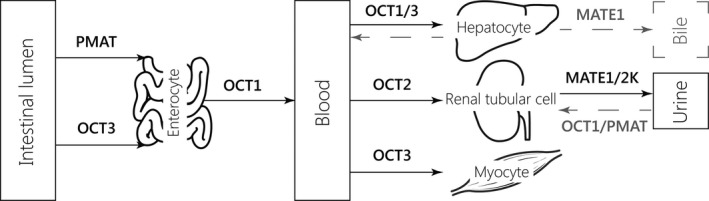
Schematic overview of the most important transporters involved in the absorption, hepatic uptake, and excretion of metformin.14, 38, 39, 40, 41, 42 The physiological significance of the transporters depicted in gray is unclear.14 MATE, multidrug and toxin extrusion; OCT, organic cation transporter; PMAT, plasma membrane monoamine transporter.
There is very limited data on co‐administration of metformin and rifampicin and the need for dose adjustments in patients with TB‐DM. Rifampicin is an agonist of the pregnane X receptor (PXR), a transcription factor that upregulates a large number of genes involved in xenobiotic detoxification, including drug‐metabolizing enzymes and drug transporters.17 In rats, the PXR agonist pregnenolone‐16‐carbonitrile upregulated the expression of OCT1 in the liver and OCT2 in the kidneys, which significantly reduced metformin plasma exposure.18 In healthy volunteers, rifampicin caused altered metformin absorption kinetics, leading to higher exposure levels and enhanced glucose‐lowering action.19 Extrapolation of these results to complex patients with TB‐DM should be made cautiously, because disease status can alter transporter expression levels,20, 21, 22 and patients with diabetes have altered glucose regulation compared with healthy volunteers.
In summary, the pharmacokinetic (PK) and clinical implications of combining metformin with rifampicin are relevant to increasing numbers of patients with TB with type 2 DM across the world and are yet unclear. We, therefore, assessed the effect of rifampicin on the steady‐state PK parameters and glucose‐lowering effect of metformin in patients with TB‐DM.
Results
Subjects
From the TANDEM TB‐DM cohort, 57 patients were eligible and asked for informed consent, of which eventually 24 patients (12 women and 12 men) participated in the study. For further details on eligibility, inclusion, and exclusion of patients see Supplementary Files S1–S4 . Nineteen patients were on 500 mg of metformin (test dose during PK day), of which two patients were once daily, nine patients were twice daily, four patients were thrice daily, and three patients were thrice daily in combination with 850 mg doses. One subject switched from twice daily 500 mg to thrice daily 500 mg in between the PK sessions. This was corrected for by comparing area under the plasma concentration‐time curve (AUC) across the entire dosing interval. Five patients were taking 850 mg of metformin (test dose during PK day) all thrice daily. Median fasting blood glucose during the first screen was 152 (range: 91–329) mg/dL and estimated glomerular filtration rate (eGFR) amounted to 101 (range: 65–141) mL/min. Median age at both sessions was 51 years (range: 24–63 years). Body mass index remained comparable between sampling sessions 1 and 2, with 23 (14–34) kg/m2 and 24 (16–35) kg/m2, respectively, supporting within‐subject comparability of the two sessions (P = 0.339).
Effect of rifampicin on metformin PKs
Steady‐state metformin PKs were assessed for subjects who underwent PK sampling in the first (n = 24) and second session (n = 22; Table 1). Rifampicin increased metformin exposure: for rifampicin + metformin (during TB treatment) relative to metformin alone (after TB treatment) the geometric mean ratio (GMR) estimates of area under the plasma concentration‐time curve from time point 0 to the end of the dosing interval (AUC 0–τ) and peak plasma concentration (Cmax) were 1.28 (90% confidence interval (CI) 1.13–1.44) and 1.19 (90% CI 1.03–1.38), respectively. Tubular secretion of metformin remained comparable (P = 0.777; Table 1 and Figure 2 a–c). Creatinine clearance was slightly reduced (−4 mL/min) after rifampicin discontinuation (P = 0.005; Table 1). The PK parameters of rifampicin are shown in Supplementary File S2 and demonstrate significant exposure to the perpetrator drug.
Table 1.
Comparison of steady‐state metformin pharmacokinetic parameters and renal elimination parameters with and without co‐administration of rifampicin
| Parameter | With rifampicin (n = 24) | Without rifampicin (n = 22) | P value or GMR estimate (90% CI) (n = 22) |
|---|---|---|---|
| AUC0–τ (mg/L × hour) | 15.4 (7.8–32.8) | 12.1 (8.1–21.6) | 1.28 (1.13–1.44)a |
| Cmax (mg/L) | 2.3 (1.25–5.22) | 2.0 (1.10–3.86) | 1.19 (1.03–1.38)a |
| Tmax (hour) − median | 1.5 (1–4) | 1.5 (0–6) | |
| CL/F (L/hour) | 36 (21–69) | 47 (34–73) | <0.001b |
| V d/F (L) | 332 (201–681) | 310 (87–477) | 0.586b |
| t1/2 (hour) | 6.4 (4.2–17.2) | 4.6 (1.6–6.6) | 0.002b |
| AUC0–8 hour (mg/L × hour) | 13.4 (7.8–32.8) | 10.9 (7.4–11.2) | 0.001 |
| AUC0–τ (mg/L × hour) –excluding sessions with ≥ 20% extrapolation | 15.1 (7.8–32.8) | 12.2 (8.1–21.6) | 0.083 (n = 14) |
| CLcreatinine (mL/min)c | 110 (88–155) | 106 (74–148) | 0.005b |
| CL0–8 hour,renal (mL/min)d | 309 (90–625) | 332 (126–659) | 0.836b |
| CL0–8 hour,secretion (mL/min)d | 205 (–1–494) | 219 (52–533) | 0.777b |
| CL0–τ,secretion (mL/min)d | 212 (27–494) | 216 (52–533) | 0.856b |
Metformin plasma concentrations were extrapolated from 8–12 or 24‐hours postdose to calculate AUC0–τ for patients on once or twice daily metformin, respectively. Pharmacokinetic parameters are expressed as geometric mean (range), unless stated otherwise. AUC0–τ, area under the plasma concentration‐time curve from time point 0 to the end of the dosing interval; CI, confidence interval; CL, clearance; Cmax, maximum plasma concentration; F, bioavailability; GMR, geometric mean ratio; t1/2, half‐life; Tmax, time to maximum plasma concentration; V d, volume of distribution.
aEvaluation of a pharmacokinetic interaction by means of the bioequivalence approach. bPaired‐samples t test on log‐transformed pharmacokinetic parameters. cSession comparison of creatinine clearance based on n = 21 individuals. dDue to miscollection of urine at home between 8 and 24 hours postdose for five individuals, we calculated renal clearance and tubular secretion from observed urine collections up to 8 hours after metformin intake (CL0–8 hour,renal (mL/min) and CL0–8 hour,secretion; n = 21), next to tubular secretion based on confirmed collections during the full time interval (CL0–τ,secretion) (n = 16).
Figure 2.
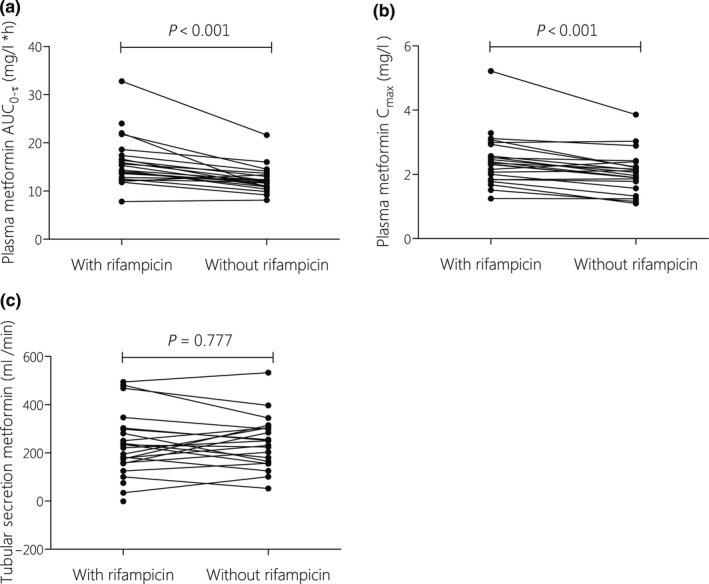
Individual changes in steady‐state metformin pharmacokinetic parameters, AUC 0–τ (a), Cmax (b) and tubular secretion (c), with and without co‐administration of rifampicin. Metformin plasma concentrations were extrapolated from 8 until 12–24 hours to calculate AUC 0–τ for patients taking metformin once or twice daily. Tubular secretion was based on observed urine collections up to 8 hours after metformin intake. Data were assessed with paired‐samples t test on the log‐transformed parameters. AUC0–τ, area under the plasma concentration‐time curve from time point 0 to the end of the dosing interval; Cmax, maximum plasma concentration. ETH, ethambutol; IC, informed consent; INH, isoniazid; MTF, metformin; PK‐GC, pharmacokinetic‐glucose curve sampling session; PZA, pyrazinamide; RIF, rifampicin.
Effect of rifampicin on the glucose‐lowering efficacy of metformin
Blood glucose concentrations for both sessions are displayed in Supplementary File S5 . Fasting blood glucose concentrations differed slightly (15% reduction) during and after rifampicin treatment, but this did not reach statistical significance (P = 0.081, n = 18; Table 2). Similarly, the ability of metformin to reduce maximum blood glucose levels (Gmax) and the AUC of glucose was not significantly different when measured during and after TB treatment (Table 2 and Figure 3).
Table 2.
The glucose‐lowering effect of metformin with and without co‐administration of rifampicin
| Parameter | With rifampicin (n = 21) | Without rifampicin (n = 18) | P value |
|---|---|---|---|
| Baseline blood glucose (mg/dL) | 127 (81–283) | 148 (75–353) | 0.081a |
| G‐AUC0–3 hour (mg/dL × hour) | 777 (523–1,488) | 791 (509–1,459) | 0.890a |
| Gmax (mg/dL) | 330 (223–597) | 347 (214–595) | 0.441a |
Data are expressed as geometric mean (minimum–maximum). G‐AUC0–3 hour, area under the blood glucose concentration‐time curve; Gmax, maximum blood glucose concentration.
Paired‐samples t test on log‐transformed blood glucose parameters for n = 18.
Figure 3.
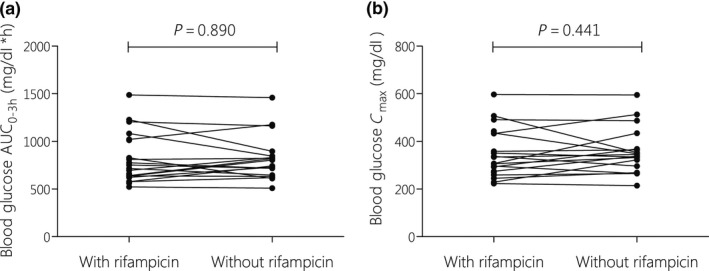
Individual changes in G‐AUC (a) and Gmax (b), after a 75‐g glucose challenge, with and without co‐administration of rifampicin. Data were assessed with paired‐samples t test on log‐transformed blood glucose parameters (n = 18). AUC, area under the blood concentration‐time curve from; Gmax, maximum blood glucose concentration.
Drug adherence and adverse events
Patients’ adherence to metformin and TB drugs was good. Mean percentages of drug adherence prior to the first session, according to physician assessment, pill count, self‐assessment, and diary were 100% (n = 24), 99% (n = 24), 100% (n = 24), and 92% (n = 19), respectively. Adherence was 100% (n = 22), 96% (n = 22), 100% (n = 22), and 100% (n = 17) for the second session, respectively. We expect any nonadherence to be of minor importance, because we actively stimulated drug intake during the final 3 days before PK sampling by personally contacting each patient.
Of the 24 patients who underwent PK sampling, 13 patients experienced gastrointestinal adverse event(s) when they took both metformin and rifampicin (57%; during TB treatment), and 8 patients experienced gastrointestinal adverse event(s) when they took only metformin (38%; after TB treatment; Table 3). These additional gastrointestinal events could be attributed to more cases of nausea and vomiting (Table 3). Data do not include the cases that were excluded from the analysis due to vomiting on the PK‐glucose curve (GC) days (Supplementary File S4 ). This occurred mainly prior to the addition of metoclopramide and separation of intake of metformin and TB drugs; four out of the first six patients vomited during PK‐GC sampling session 1 (67%), with n = 2 during PK‐session 1 and n = 4 during GC‐session 1. All adverse events were grades 1 or 2.
Table 3.
Number of patients experiencing adverse events according to Common Terminology Criteria for Adverse Events version 4.032
| System organ class | With rifampicin (n = 23) | Without rifampicin (n = 21) |
|---|---|---|
| All adverse events (grade 1 or 2) | 17 (74%) | 14 (64%) |
| Gastrointestinal disorders (grade 1 or 2) | 13 (57%) | 8 (38%) |
| Nausea | 9 (39%) | 5 (24%) |
| Grade 1 | 8 (35%) | 5 (24%) |
| Grade 2 | 1 (4%) | 0 (0%) |
| Vomiting | 7 (30%) | 4 (19%) |
| Grade 1 | 6 (26%) | 4 (19%) |
| Grade 2 | 1 (4%) | 0 (0%) |
| Diarrhea | 3 (13%) | 3 (14%) |
| Abdominal discomfort | 0 (0%) | 2 (10%) |
| Nervous system disorders | ||
| Headache | 9 (39%) | 6 (29%) |
| Grade 1 | 8 (35%) | 6 (29%) |
| Grade 2 | 1 (4%) | 0 (0%) |
| Musculoskeletal tissue disorders | ||
| Myalgia | 8 (35%) | 4 (19%) |
| Grade 1 | 8 (35%) | 2 (10%) |
| Grade 2 | 0 (0%) | 2 (10%) |
| Metabolism and nutrition disorders | ||
| Loss of appetite or anorexia | 7 (30%) | 3 (14%) |
| Respiratory disorders | ||
| Cough | 5 (22%) | 8 (38%) |
| General disorders | ||
| Flu | 6 (26%) | 3 (14%) |
| Fever | 1 (4%) | 1 (5%) |
| Edema | 0 (0%) | 1 (5%) |
| Skin and subcutaneous tissue disorders | ||
| Pruritus | 2 (9%) | 0 (0%) |
| Skin ulcer or ulcus | 0 (0%) | 2 (10%) |
| Rash | 2 (9%) | 1 (5%) |
Number (%) of subjects with adverse events up to and including pharmacokinetic‐glucose curve sampling session sampling session 1 (with rifampicin) and up to and including session 2 (without rifampicin). Grade 3 or 4 adverse events were not observed.
Discussion
This study was the first to evaluate the effect of rifampicin on metformin exposure and activity in patients with TB‐DM. We observed an increase in metformin exposure, with an AUC0–τ GMR (exposure during vs. after TB treatment) of 1.28 (90% CI 1.13–1.44), suggesting a PK interaction (bio‐inequivalence) when metformin is co‐administered with rifampicin.23 This interaction did not result in a statistically significant change in the glucose‐lowering effect of metformin.
Strikingly, renal clearance and tubular secretion were unaffected by rifampicin in our study. Because metformin is also not metabolized, we hypothesize that the higher metformin total and peak exposures are the result of increased metformin absorption. Metformin has relatively poor oral absorption with a bioavailability of 55%,14 leaving substantial space for absorption increases. OCT1, OCT3, and plasma membrane monoamine transporter (PMAT) are the relevant metformin transporters in the small intestine, with PMAT being the most abundant.14, 24 Rifampicin‐induced upregulation of these transporters is probably responsible for the increased absorption assumed in our study. Indeed, rifampicin increased OCT1 expression in blood cells in healthy volunteers.19 Further study is needed to evaluate the inductive effect of rifampicin on tissue (e.g., intestinal) expression of metformin transporters to confirm if this mechanism explains the rifampicin‐metformin interaction.
The effect of rifampicin on systemic exposure to metformin is consistent with the findings of Cho et al.19 in healthy volunteers, although the PK interaction in our study seems larger (14–18% vs. 17–30%). This difference may be explained by the enhanced renal clearance and tubular secretion in the healthy volunteer study, whereas we did not identify such an elimination effect. Disease status may have altered expression levels of transporters important for metformin elimination, such as OCT2, MATE1, or MATE2K. Information on the transcriptional regulation or inducibility of the MATE family in humans is lacking.
Importantly, increased metformin exposure in our study was not associated with any clinically relevant or statistically significant increase in the glucose‐lowering effect of metformin. Several explanations can be brought forward. First, rifampicin may have differential effects on expression of the various drug transporters in the intestine, liver, and kidneys. In humans, the liver has a central role in the regulation of systemic glucose, but also the kidneys contribute to glucose uptake, gluconeogenesis, and glucose utilization.25, 26 Theoretically, altered OCT1 and OCT2 expressions influence local metformin exposure and, thus, overall drug efficacy. Unaffected expression would be in contrast with findings in rats, where the PXR agonist pregnenolone‐16‐carbonitrile upregulated the expression of both OCT1 in the liver and OCT2 in the kidneys.18 Second, PXR itself is known to influence glucose homeostasis,27, 28 and rifampicin has been reported to induce early phase hyperglycemia possibly by enhancing glucose absorption.6 These parallel processes may have counteracted or dampened a pharmacodynamic interaction.
To reduce the risk of gastrointestinal adverse effects, intake of metformin and rifampicin was separated in time and patients were provided with metoclopramide after a high incidence of vomiting in the initial patients (see Supplementary Files S4 and S6 ). Without these preventive measures we would not have been able to complete the study. Nonetheless, (mild) gastrointestinal adverse events remained extremely common, especially among patients with TB‐DM on TB treatment and metformin (57%), with more nausea and vomiting compared with patients using metformin alone. Preventive actions should be considered when treating patients with TB‐DM with metformin. Normally, it is advised to take TB drugs on an empty stomach, as food is known to limit their bioavailability,29 but they can also be taken with food if gastrointestinal adverse effects occur.
A limitation of this study is its sequential design; a clinically significant interaction might have led to adjustment of the metformin dose by the attending physician. Indeed, in three of the first seven subjects, the dosing interval of metformin had been changed after the first PK‐GC session. Two of these patients could be measured in a third session, after switching back to the dosing schedule of their first PK‐GC session. Furthermore, bias as a consequence to the fixed sequence design could have been introduced by differences in comedication and food within‐patient between sampling sessions. To avoid such interference, we standardized the timing and content of meals, as well as fixed the timing of intake of comedication (if any) to assure patients were sampled under comparable conditions throughout the study. Overall, we feel that our measures have been sufficient to prevent any confounding of the metformin‐rifampicin drug‐drug interaction and were, most importantly, mimicking the real‐life clinical situation.
Another limitation is the use of creatinine clearance as estimation for the fraction metformin eliminated from the body by glomerular filtration (eGFR). It is well known that creatinine is freely filtered by the glomerulus, however, active secretion by the proximal tubule also occurs.30 This possibly explains the slight change in creatinine clearance after stopping rifampicin (−4 mL/min; P = 0.005). The impact of this finding is minor; creatinine secretion is only a small contributor to creatinine clearance, ~10–20% of the GFR,31 whereas renal clearance of metformin can be up to 4–11 times as great as creatinine clearance.
As another limitation of our study, we cannot exclude an effect of isoniazid on the PKs and efficacy of metformin, as it was always co‐administered with rifampicin in our study. However, to our knowledge, interactions of isoniazid with drug transporters have not been reported in literature. Similarly, we do not anticipate interactions with metformin drug transporters caused by other concomitant drugs used by patients in this study.
Finally, we would like to note here that the average percentage of extrapolation for twice‐daily dosing (from AUC0–8 hour until AUC0–τ was 21%; range: 15–26%), which is within acceptable limits for reliable extrapolation. Average percentage of extrapolation for once‐daily dosing was 29% (range: 14–36%), which may be considered bordering/exceeding reliable extrapolation. However, this only concerned two patients in the entire study. The comparison for nonextrapolated total exposures (AUC0–8 hour; i.e., with vs. without rifampicin), was similarly significant (P = 0.001; n = 22) as for AUC0–τ (Table 1, Figure 2). The comparison for AUC0–τ, excluding subjects with an AUC extrapolation > 20% for at least one sampling session, showed less significance (P = 0.083), probably because of a loss in power (n = 14), because geometric means and ranges were similar to AUC0–τ in the entire group (Table 1).
To summarize, we found that rifampicin exposure increases plasma metformin concentrations, without affecting its glucose lowering effect, suggesting that additional monitoring of glycemic control may not be necessary when treating patients with TB‐DM. We observed a high incidence of gastrointestinal adverse effects, and, therefore, we advise patients to take metformin and rifampicin with food, and preferably separated in time. In addition, physicians could consider combining TB‐DM treatment with metoclopramide as an anti‐emetic when patients are experiencing gastrointestinal adverse effects.
Methods
Subjects
Study subjects were Indonesian patients with pulmonary TB and type 2 DM that had been included in the TANDEM study (ClinicalTrials.gov: NCT02106039) at the University of Padjadjaran, Bandung, Indonesia. Patients were enrolled if they were between 18 and 65 years of age; were taking metformin, irrespective of other blood glucose control drugs; were treated with the standard Indonesian continuation phase TB regimen containing rifampicin and isoniazid thrice weekly; had a stable renal function, classified as an eGFR of > 60 mL/min; and signed an informed consent. Patients were excluded if they had an alanine aminotransferase > 3 × upper limit of normal; had inadequately controlled blood glucose concentrations, classified as an average fasting blood glucose and 2‐hour postprandial glucose ≥ 300 mg/dL; and were pregnant or lactating. The study was reviewed and approved by the Independent Ethics Committee, Faculty of Medicine, University of Padjadjaran, Bandung, Indonesia. All procedures were in accordance with the Helsinki Declaration of 1975 (as revised in 1983) and Good Clinical Practice.
After enrollment, patients underwent a physical screening just prior to the first sampling session, to test stable adequate liver (alanine aminotransferase < 3× upper normal limit) and kidney function (eGFR > 60 mL/min, calculated from plasma creatinine according to the Chronic Kidney Disease Epidemiology Collaboration formula32, 33); to confirm adequately controlled blood glucose concentrations and to perform a pregnancy test, to ensure all inclusion criteria and exclusion criteria were still met, and patients were stable over time.
Study design
This was an open‐label, one‐arm (within‐subject), two‐period, fixed‐order PK and pharmacodynamic interaction study. Figure 4 shows the schematic overview of the study design. Sampling occurred in two sessions to enable the assessment of metformin exposure when patients were on rifampicin and isoniazid during the last week of TB treatment, and after completion of TB treatment with a 1‐month washout period in between.9 All patients were switched from thrice weekly to daily intake of TB drugs for at least > 7 days prior to the first PK sampling session, to match standard World Health Organization guidelines (dashed lines, Figure 4). The doses of rifampicin (450 or 675 mg) and isoniazid (300 mg) were once daily. The doses of metformin (500 or 850 mg) were once, twice, or thrice daily. In between the two sessions, an intermediate screening visit was planned to assess patient status once more.
Figure 4.
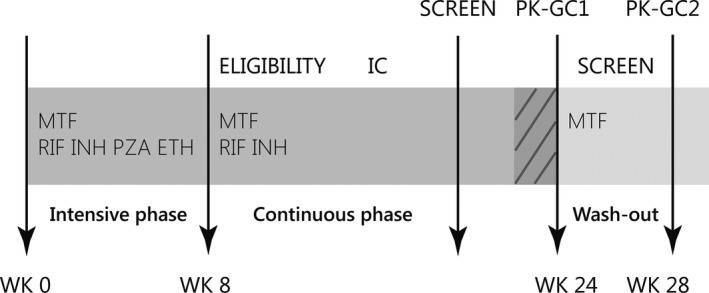
Schematic overview of the study design in weeks from the start of tuberculosis (TB) treatment (wk 0). The TB treatment period (week 0–24) is colored dark gray. Patients were enrolled if they were continuation phase of TB treatment. Daily intake of TB drugs (for > 7days) is indicated by dashed lines. In between sessions, there was at least a 1‐month washout period, after which any induction caused by rifampicin was expected to have dissipated.9 AUC, area under the plasma concentration‐time curve; Cmax, maximum plasma concentration.
The primary objective of this study was to evaluate the effect of rifampicin on the steady‐state PKs of metformin. This was assessed with two 8‐hour PK curves measured in plasma, together with urine collections during the dosing interval for estimation of renal clearance and tubular secretion. A PK sampling time period of 8 hours was selected, because the most frequent daily dosing was thrice daily metformin, which approximates a maximum of an 8‐hour per dosing interval. Using the sample size nomograms by Diletti et al.,34 we estimated that 24 patients would be needed to conclude lack of an interaction (bioequivalence) with a power of 80% and a conservative estimate of the intra‐individual coefficient of variation in metformin exposure of 25%. As a secondary objective, we evaluated the effect of rifampicin on the glucose‐lowering effect of metformin by sampling two GCs following ingestion of 75 g of glucose on an empty stomach. Reported signs and symptoms were graded according to Common Terminology Criteria for Adverse Events 4.0.35 Information on adverse events was collected throughout the study, starting from immediately after the screening visit, during all PK‐GC days, and as part of the intermediate screening. The study was approved by the ethical review board of the Hasan Sadikin Hospital and Medical Faculty of Universitas Padjadjaran, Bandung, Indonesia.
Blood and urine collection during sampling sessions
Each of the two sampling sessions consisted of 2 days, one PK day and one GC day. During the PK days, metformin and rifampicin/isoniazid were initially taken together, and with food (n = 6), but this resulted in a high incidence of vomiting (n = 4 in total; 2/6 patients during PK1 and another 2/6 during GC1). Subsequently, patients were provided with oral metoclopramide (10 mg) 1 hour before taking metformin, and the TB drugs were ingested 3 hours after metformin. Patients were ordered to first take their breakfast and immediately afterward their metformin. Metoclopramide was the preferred anti‐emetic, as there may be a drug‐drug interaction between rifampicin and domperidone36 and there is less experience with the use of other anti‐emetics in this setting. Eight serial blood samples were drawn at 0 (predose), 1, 2, 3, 4, 5, 6, and 8 hours after witnessed ingestion of metformin with a standardized breakfast. During the PK day, cumulative urine from all subjects was collected for 8 hours in 2‐hour intervals. Patients taking metformin once or twice daily received a urine container to collect their urine at home between the 8‐hour until the 12–24‐hour interval. Additional blood was withdrawn to assess plasma creatinine concentrations for the estimation of creatinine clearance.
On the second morning of each session, metoclopramide and metformin were taken once more, this time on an empty stomach. A total of 75 g of glucose was ingested on an empty stomach, at 2.5 hours after metformin intake when metformin concentrations are expected to be at their maximum (Cmax). Glucose concentrations were measured just before and at ½, 1, 1½, 2, 2½, and 3 hours after ingestion of glucose. See Supplementary File S6 for a schematic overview of the PK‐GC sampling days.
Study drugs
All patients received 500 or 850 mg metformin tablets (Glucophage) from PT Merck (Jakarta, Indonesia). TB drugs (rifampicin (450 mg) and isoniazid (300 mg)) were from PT Kimia Farma (Bandung, Indonesia), formulated in separate tablets. The bioequivalence of the rifampicin tablets and an international reference standard was established previously.37 Metoclopramide (Metolon) 10 mg was provided by PT Bernofarm (Sidoarjo, Indonesia). Drug adherence was monitored from 1 week prior to the first PK session until the end of the study, using physician assessment, pill count, self‐assessment, and a patient‐kept diary.
Bio‐analysis and PK evaluation
Plasma (metformin and rifampicin) and urine (metformin) concentration analyses were performed with validated ultraperformance liquid chromatography assays (Supplementary Files S1 and S2 ). Noncompartmental PK analyses were applied to calculate metformin and rifampicin PK parameters using Phoenix WinNonlin version 6.3 (Pharsight, Mountain View, CA; Supplementary Files S2 and S3). The net tubular secretion of metformin was calculated by subtracting creatinine clearance from metformin renal clearance (total amount excreted divided by AUC0–τ; Supplementary File S3 for full details).
To assess changes in the glucose lowering effect of metformin, basal/fasting blood glucose and Gmax were determined directly from the GC. The glucose AUC until 3 hours (G‐AUC0–3 hour) was calculated using the Linear Trapezoidal Linear Interpolation rule using Phoenix WinNonlin.
Statistical analysis
Patient characteristics, metformin PKs, glucose data, and adverse events were presented descriptively for all patients included in the study. A bioequivalence approach was used to evaluate the metformin‐rifampicin interaction. To conclude the absence of an interaction, or bioequivalence,34 the 90% CI of the GMRs of AUC‐session 1:AUC‐session 2 and Cmax‐session 1: Cmax‐session 2 should be between 0.80 and 1.25. Further conclusions with regard to the presence of an interaction were drawn as described by Williams et al.23 To assess differences between sampling sessions in patient characteristics, metformin elimination, and glucose exposures, either Related‐Samples Wilcoxon Signed Rank tests or paired‐samples t tests on log‐transformed plasma/blood parameters were performed. All statistical analyses were performed with IBM SPSS Statistics 22 for Windows. The P values < 0.05 were judged significant.
Funding
This work was supported by the European Union's Seventh Framework Programme FP7/2007‐2013 (Grant Agreement No. 305279), which funded the TANDEM project on tuberculosis and diabetes (www.tandemfp7.eu).
Conflicts of Interest
The authors declared no competing interests for this work.
Author Contributions
L.H.M.t.B., V.Y., R.L., N.S., E.v.E.‐B.K., J.B.K., D.M.B., P.S., R.v.C., B.A., R.E.A., and R.R. wrote the article. L.H.M.t.B., V.Y., R.L., N.S., E.v.E.‐B.K., J.B.K., D.M.B., P.S., R.v.C., B.A., R.E.A., and R.R. designed the research. L.H.M.t.B., V.Y., R.L., N.S., E.v.E.‐B.K., P.S., R.E.A., and R.R. performed the research. L.H.M.t.B., V.Y., R.L., N.S., E.v.E.‐B.K., J.B.K., D.M.B., P.S., R.v.C., B.A., R.E.A., and R.R. analyzed the data.
Supporting information
Supplementary File S1. Metformin concentration analyses.
Supplementary File S2. Rifampicin concentration analysis and pharmacokinetic evaluation.
Supplementary File S3. Pharmacokinetic evaluation of metformin.
Supplementary File S4. Eligibility, inclusion, and exclusion of patients.
Supplementary File S5. Blood glucose curves.
Supplementary File S6. Schematic overview of the PK‐GC sampling days.
References
- 1. WHO . Global tuberculosis report 2018. <http://www.who.int/tb/publications/global_report/en/> (2018).
- 2. Jeon, C.Y. & Murray, M.B. Diabetes mellitus increases the risk of active tuberculosis: a systematic review of 13 observational studies. PLoS Med. 5, e152 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Riza, A.L. et al Clinical management of concurrent diabetes and tuberculosis and the implications for patient services. Lancet Diabetes Endocrinol. 2, 740–753 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lonnroth, K. , Roglic, G. & Harries, A.D. Improving tuberculosis prevention and care through addressing the global diabetes epidemic: from evidence to policy and practice. Lancet Diabetes Endocrinol. 2, 730–739 (2014). [DOI] [PubMed] [Google Scholar]
- 5. Baker, M.A. et al The impact of diabetes on tuberculosis treatment outcomes: a systematic review. BMC Med. 9, 81 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takasu, N. et al Rifampicin‐induced early phase hyperglycemia in humans. Am. Rev. Respir. Dis. 125, 23–27 (1982). [DOI] [PubMed] [Google Scholar]
- 7. Waterhouse, M. , Wilson, C. , White, V.L. & Chowdhury, T.A. Resolution of insulin‐requiring diabetes after cessation of chemotherapy for tuberculosis. J. R. Soc. Med. 98, 270–271 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ruslami, R. , Aarnoutse, R.E. , Alisjahbana, B. , van der Ven, A.J. & van Crevel, R. Implications of the global increase of diabetes for tuberculosis control and patient care. Trop. Med. Int. Health 15, 1289–1299 (2010). [DOI] [PubMed] [Google Scholar]
- 9. Niemi, M. , Backman, J.T. , Fromm, M.F. , Neuvonen, P.J. & Kivisto, K.T. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin. Pharmacokinet. 42, 819–850 (2003). [DOI] [PubMed] [Google Scholar]
- 10. Burman, W.J. , Gallicano, K. & Peloquin, C. Comparative pharmacokinetics and pharmacodynamics of the rifamycin antibacterials. Clin. Pharmacokinet. 40, 327–341 (2001). [DOI] [PubMed] [Google Scholar]
- 11. IDF . Global Guideline for Type 2 Diabetes. <https://www.idf.org/e-library/guidelines.html> (2012).
- 12. Intensive blood‐glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) group. Lancet. 352, 837–853 (1998). [PubMed] [Google Scholar]
- 13. Hermann, L.S. , Schersten, B. , Bitzen, P.O. , Kjellstrom, T. , Lindgarde, F. & Melander, A. Therapeutic comparison of metformin and sulfonylurea, alone and in various combinations. A double‐blind controlled study. Diabetes Care 17, 1100–1109 (1994). [DOI] [PubMed] [Google Scholar]
- 14. Graham, G.G. et al Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 50, 81–98 (2011). [DOI] [PubMed] [Google Scholar]
- 15. Summary of Product Characteristics Glucophage 500 mg and 850 mg. Updated January 23, 2015 by Merck. <https://www.medicines.org.uk/emc/medicine/1043>.
- 16. Campbell, R.K. , Jr. White, J.R. & Saulie, B.A. Metformin: a new oral biguanide. Clin. Ther. 18, 360–371 (1996); discussion 359. [DOI] [PubMed] [Google Scholar]
- 17. Chen, J. & Raymond, K. Roles of rifampicin in drug‐drug interactions: underlying molecular mechanisms involving the nuclear pregnane X receptor. Ann. Clin. Microbiol. Antimicrob. 5, 3 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maeda, T. et al Effect of pregnane X receptor ligand on pharmacokinetics of substrates of organic cation transporter Oct1 in rats. Drug Metab. Dispos. 35, 1580–1586 (2007). [DOI] [PubMed] [Google Scholar]
- 19. Cho, S.K. et al Rifampin enhances the glucose‐lowering effect of metformin and increases OCT1 mRNA levels in healthy participants. Clin. Pharmacol. Ther. 89, 416–421 (2011). [DOI] [PubMed] [Google Scholar]
- 20. Xu, C. et al The altered renal and hepatic expression of solute carrier transporters (SLCs) in type 1 diabetic mice. PLoS One 10, e0120760 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Atilano‐Roque, A. , Roda, G. , Fogueri, U. , Kiser, J.J. & Joy, M.S. Effect of disease pathologies on transporter expression and function. J. Clin. Pharmacol. 56(suppl. 7), S205–S221 (2016). [DOI] [PubMed] [Google Scholar]
- 22. Staudinger, J.L. Disease, drug metabolism, and transporter interactions. Pharm. Res. 30, 2171–2173 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Williams, R.L. , Chen, M.L. & Hauck, W.W. Equivalence approaches. Clin. Pharmacol. Ther. 72, 229–237 (2002). [DOI] [PubMed] [Google Scholar]
- 24. Zhou, M. , Xia, L. & Wang, J. Metformin transport by a newly cloned proton‐stimulated organic cation transporter (plasma membrane monoamine transporter) expressed in human intestine. Drug Metab. Dispos. 35, 1956–1962 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gerich, J.E. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabet. Med. 27, 136–142 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gerich, J.E. Physiology of glucose homeostasis. Diabetes Obes. Metab. 2, 345–350 (2000). [DOI] [PubMed] [Google Scholar]
- 27. Spruiell, K. , Richardson, R.M. , Cullen, J.M. , Awumey, E.M. , Gonzalez, F.J. & Gyamfi, M.A. Role of pregnane X receptor in obesity and glucose homeostasis in male mice. J. Biol. Chem. 289, 3244–3261 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ihunnah, C.A. , Jiang, M. & Xie, W. Nuclear receptor PXR, transcriptional circuits and metabolic relevance. Biochim. Biophys. Acta 1812, 956–963 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin, H.C. , Yu, M.C. , Liu, H.J. & Bai, K.J. Impact of food intake on the pharmacokinetics of first‐line antituberculosis drugs in Taiwanese tuberculosis patients. J. Formos. Med. Assoc. 113, 291–297 (2014). [DOI] [PubMed] [Google Scholar]
- 30. Ciarimboli, G. et al Proximal tubular secretion of creatinine by organic cation transporter OCT2 in cancer patients. Clin. Cancer Res. 18, 1101–1108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Breyer, M.D. & Qi, Z. Better nephrology for mice – and man. Kidney Int. 77, 487–489 (2010). [DOI] [PubMed] [Google Scholar]
- 32. Inker, L.A. et al Estimating glomerular filtration rate from serum creatinine and cystatin C. N. Engl. J. Med. 367, 20–29 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Levey, A.S. et al A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 150, 604–612 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Diletti, E. , Hauschke, D. & Steinijans, V.W. Sample size determination for bioequivalence assessment by means of confidence intervals. Int. J. Clin. Pharmacol. Ther. Toxicol. 29, 1–8 (1991). [PubMed] [Google Scholar]
- 35. Common Terminology Criteria for Adverse Events 4.0 (CTCAE) . <http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm>.
- 36. Devandla, A. , Yamsani, S.K. & Yamsani, M.R. Effect of rifampicin pretreatment on the oral bioavailability of domperidone in healthy human volunteers. Drug Metab. Pers. Ther. 30, 257–261 (2015). [DOI] [PubMed] [Google Scholar]
- 37. van Crevel, R. et al Bioavailability of rifampicin in Indonesian subjects: a comparison of different local drug manufacturers. Int. J. Tuberc. Lung Dis. 8, 500–503 (2004). [PubMed] [Google Scholar]
- 38. Kusuhara, H. et al Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin. Pharmacol. Ther. 89, 837–844 (2011). [DOI] [PubMed] [Google Scholar]
- 39. Muller, J. , Lips, K.S. , Metzner, L. , Neubert, R.H. , Koepsell, H. & Brandsch, M. Drug specificity and intestinal membrane localization of human organic cation transporters (OCT). Biochem. Pharmacol. 70, 1851–1860 (2005). [DOI] [PubMed] [Google Scholar]
- 40. Nies, A.T. et al Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology 50, 1227–1240 (2009). [DOI] [PubMed] [Google Scholar]
- 41. Tzvetkov, M.V. et al The effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metformin. Clin. Pharmacol. Ther. 86, 299–306 (2009). [DOI] [PubMed] [Google Scholar]
- 42. Motohashi, H. & Inui, K. Organic cation transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney. AAPS J. 15, 581–588 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File S1. Metformin concentration analyses.
Supplementary File S2. Rifampicin concentration analysis and pharmacokinetic evaluation.
Supplementary File S3. Pharmacokinetic evaluation of metformin.
Supplementary File S4. Eligibility, inclusion, and exclusion of patients.
Supplementary File S5. Blood glucose curves.
Supplementary File S6. Schematic overview of the PK‐GC sampling days.