

Summary
The CRISPR/Cas9 system has been extensively applied for crop improvement. However, our understanding of Cas9 specificity is very limited in Cas9‐edited plants. To identify on‐ and off‐target mutation in an edited crop, we described whole genome sequencing (WGS) of 14 Cas9‐edited cotton plants targeted to three genes, and three negative (Ne) control and three wild‐type (WT) plants. In total, 4188–6404 unique single‐nucleotide polymorphisms (SNPs) and 312–745 insertions/deletions (indels) were detected in 14 Cas9‐edited plants compared to WT, negative and cotton reference genome sequences. Since the majority of these variations lack a protospacer‐adjacent motif (PAM), we demonstrated that the most variations following Cas9‐edited are due either to somaclonal variation or/and pre‐existing/inherent variation from maternal plants, but not off‐target effects. Of a total of 4413 potential off‐target sites (allowing ≤5 mismatches within the 20‐bp sgRNA and 3‐bp PAM sequences), the WGS data revealed that only four are bona fide off‐target indel mutations, validated by Sanger sequencing. Moreover, inherent genetic variation of WT can generate novel off‐target sites and destroy PAMs, which suggested great care should be taken to design sgRNA for the minimizing of off‐target effect. These findings suggested that CRISPR/Cas9 system is highly specific for cotton plants.
Keywords: cotton, CRISPR/Cas9, off‐target, whole genome sequencing, somaclonal variation, pre‐existing/inherent variation
Introduction
The CRISPR (clustered regularly interspaced short palindromic repeat)‐associated protein 9 (Cas9) is a bacteria immune system that has facilitated for genome editing in plant biotechnology and functional genomics research (Jiang et al., 2013; Lawrenson et al., 2015; Li et al., 2015; Liang et al., 2014; Mao et al., 2013; Miao et al., 2013; Wang et al., 2018). The Stretococcus pyogenes Cas9 (SpCas9) use ~20‐nt of a single guide RNA (sgRNA) to recognize with canonical NGG and non‐canonical NAG or NGA protospacer‐adjacent motif (PAM) sequence (Jinek et al., 2012). Once the CRISPR/Cas9 system has been introduced into the cell, it will induce the DNA double strand breaks (DSBs) and result in on‐ and off‐target mutations through non‐homologous end joining (NHEJ). The Cas9 protein can bind and cleave genomic sites that are homologous to sgRNA, which may result in unwanted off‐target mutations (Jinek et al., 2012). For diploid plant species, such as Arabidopsis and rice, the mutation profile is relatively simple, with four potential editing types: homozygous mutations, biallelic mutations, heterozygous mutations and no mutation (Feng et al., 2014). However, polyploid species such as cotton potentially have more complex mutation profiles (Wang et al., 2018). Off‐target mutations in CRISPR/Cas9‐edited cells can further add complexity to the mutation analysis, and this is emerging as the major concern for this promising technology.
In the early stages of Cas9 research, unusually high frequencies of off‐target mutations were detected in human cancer cell lines (Fu et al., 2013). The highly sensitive genome‐wide methods have been developed and applied in the detection of off‐target mutations in animal and human cell lines (Cameron et al., 2017; Hu et al., 2016; Kim et al., 2015; Kuscu et al., 2014; Ran et al., 2015; Tsai et al., 2015, 2017). With determination of the crystal structure of Cas9 and optimization of guide RNA design, off‐target mutations have been significantly decreased (Jinek et al., 2014). Recent reports show a moderate off‐target mutation frequency and this frequency is closely linked to the sgRNA's specificity. The off‐target number ranges from 0 to up to 150 mutations per genome in human cell lines (Frock et al., 2015). All these methods have been developed for the off‐target detection in animal or human cells, and then seldom have been successfully applied to study plant genome editing.
Actually, the off‐target effect is rare in plants. Several previous reports claimed that there is no mutation of potential off‐target sites in Cas9‐edited Arabidopsis (Feng et al., 2014), rice (Zhang et al., 2014) and tomato (Nekrasov et al., 2017). However, these studies exhibited several defects: (i) The number of the sequencing samples is low and absent the effective control; (ii) Moreover, these reports did not investigate the genetic variation of the maternal plants used for genetic transformation; and (iii) the detected potential off‐target sites were limited. Therefore, it is quite necessary to detect off‐target mutation at a genome‐wide level. Recently, a highly sensitive screen for genome‐wide CRISPR/Cas9 nuclease off‐target effect was reported in maize genome editing using CIRCLE‐seq method (Lee et al., 2018). A very meaningful off‐target analysis was conducted in Cas9‐ and Cpf1‐edited rice through whole genome sequencing (Tang et al., 2018). The authors analyzed the control with multiple background and nuclease edited rice suggested that off‐target mutations caused by Cas9 and Cpf1 nucleases are negligible when compared to somaclonal variations and spontaneous mutations.
Compared to these diploid species, cotton (Gossypium hirsutum) contains a more complex genetic structure and larger genome. Cotton is an allotetraploid (2n = 4x = 52, AADD) with a genome size of 2.5 Gb (Zhang et al., 2015). Moreover, G. hirsutum has homologous residings on each At‐ and Dt‐subgenome, suggesting that four alleles for each homologous pair of genes in the cotton genome. Allotetraploidy of cotton potentially will significantly complicate the mutation profile as well as the off‐target effects in CRISPR/Cas9‐edited plants. During the long‐term tissue culture process, some genetic variability is known as somaclonal variation, and can lead to genetic change at high frequency (Jin et al., 2008). These variations are induced by its long‐time maintenance under artificial conditions, leading to disorganized cell division in vitro and by the exposure of propagated tissue to high concentrations of plant growth regulators (Jiang et al., 2011). We therefore aimed to distinguish between potential off‐target mutations of CRISPR/Cas9 and somaclonal variation or/and pre‐existing/inherent variation from maternal plants in regenerated transgenic crop plants.
Here, we report results from a large‐scale whole genome sequencing (WGS) CRISPR/Cas9 library in three target genes. The WGS results indicated that the pre‐existing/inherent genetic or/and somaclonal variations during plant tissue culture process is more than the CRISPR off‐target effects. We demonstrated that the CRISPR/Cas9 system did not cause larger number of off‐target mutations in cotton plants. Furthermore, pre‐existing/inherent genetic variation of transgenic maternal plants can generate novel off‐targets. These findings also provided insights into great care should be taken when selecting a suitable reference genome to design sgRNA.
Results
Whole genome sequencing of CRISPR/Cas9‐edited cotton plants, WT and negative plants
Recently, we established the CRISPR/Cas9 (Streptococcus pyogenes Cas9) system in cotton with high efficiency. Interestingly, no off‐target mutations were detected by targeted deep sequencing in the top 26 potential off‐target sites of four sgRNAs, suggesting that the CRISPR/Cas9 system is an efficient and reliable tool for allotetraploid cotton genome editing (Wang et al., 2018). To further investigate the specificity of CRISPR/Cas9 system on the genome‐wide scale in cotton genome editing, three target genes – encoding the AP2/B3‐like transcription factor (AP2), MYB44 transcription factor (MYB44) and nucleotide‐binding (NB)‐ARC domain‐containing disease resistance protein (ARC) were edited using CRISPR/Cas9 in this study (Li et al., 2016). We performed whole genome sequencing in T0 generation plants and T1 lines with an approximate 35× sequencing depth (Table S1). To distinguish between Cas9‐edited mutations, somaclonal variations due to the tissue culture process and pre‐existing/inherent variations, three wild‐type (WT, G. hirsutum cultivar JIN668), three independent T0 negative (Ne) transgenic cotton plants undergoing tissue culture and plant generation but without the T‐DNA insertion (no CRISPR/Cas9 component), 12 independent T0 Cas9‐edited plants, and two T1 lines from AP2 T0 parent were sequenced (Figure 1). For each gene, two sgRNAs were designed and sgRNAs target sites are shown in Table S2. Therefore, six sgRNAs were investigated for on‐target and off‐target mutations analysis.
Figure 1.
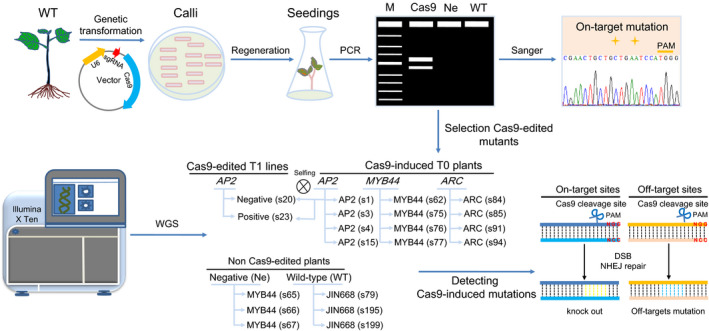
The whole genome sequencing analysis for the on‐ and off‐target mutations in CRISPR/Cas9‐edited cotton plants. Schematic diagram of the whole procedure of CRISPR/Cas9 system for gene editing in transgenic cotton plants. Whole genome sequencing was applied to 12 plants from Cas9‐edited T0 generation, targeting three endogenous genes ( AP2, MYB44 and ARC , four T0 plants from each target gene), two plants from T1 generation, three negative plants (Ne) in T0 generation and three wild‐type (WT) control cotton plants to detect the on‐ and off‐target mutations. As for the two T1 plants, they are derived from the same transgenic AP2 T0 plant. Cas9‐negative plants contained the edited target site without the T‐DNA (no CRISPR/Cas9 fragment). Cas9‐positive plants contained the edited target site as well as the T‐DNA (with CRISPR/Cas9 fragment). The number in the brackets e.g. AP2 (s1) represents the plant or line number used for the WGS.
Detection of on‐target mutations at the genome‐wide scale
First, we tested on‐target site mutations in six sgRNAs of CRISPR/Cas9‐edited lines by Sanger sequencing (Figure 2a–c). The result showed that the efficiency of gene editing at each on‐target site was 70.1%–100%. Multiple types of mutations were detected at the specific on‐target sites (Figure S1a and Table S3). This editing efficiency is comparable to the efficiency (66%–100%) when editing exogenous transgene Discosoma red fluorescent protein (dsRed2) and an endogenous cotton gene GhCLA1 in our recent report (Wang et al., 2018). We found no bias on the editing efficiency between At‐ and Dt‐subgenome loci based on WGS data, but different mutation patterns (Figure S1b). Further, analysis of the WGS data clearly reveals specific on‐target mutations in the CRISPR/Cas9‐edited transgenic cotton plants but not in WT, nor in negative control plants (Figures 2d and S2–S4). We detected 11 mutation types in the two sgRNAs of AP2, nine mutation types in the two sgRNAs sites of MYB44 and four mutation types in the two ARC sgRNAs between WGS and Sanger sequencing. The WGS and Sanger sequencing data revealed that most of Cas9‐generated mutations are deletions (Figure 2e–g). We found a positive linear correlation (Pearson's correlation coefficient, R 2 = 0.69, P < 0.001) between WGS and Sanger sequencing for the two sgRNAs mutation frequencies of the AP2 gene (Figure S5). These results confirmed that six on‐target sites exhibited multiple mutation types and different mutation frequencies from three target genes.
Figure 2.

Whole genome sequencing and Sanger sequencing confirm on‐target mutations. (a–c) Sanger sequencing validate six sgRNA mutations of three endogenous genes. The orange and red lines represent the sgRNA and PAM sequences, respectively. The labelling on the side of the sequence alignment represent the each clones, and on top label represent on‐target genome coordinate, respectively. Black arrows indicate sgRNAs direction. (d) Whole genome sequencing analysis at sgRNA2 target region of MYB44 in three wild‐type, three negative plants and four Cas9‐edited MYB44 plants by Integrative Genomics Viewer (IGV). The number in the square brackets e.g. [0–45] represent the WGS supporting sequence reads with on‐target sites and the pileup strip represent Cas9‐edited MYB44 plants a heterozygous deletion of 43‐bp in exon 1 of MYB44. Red arrows indicate DNA cleavage sites in different Cas9‐edited cotton plants. Orange and red box represent sgRNA sequence and PAM sequence, respectively. The sequences alignment of other five sgRNAs region in the three endogenous target genes are showed in Figures S2–S4. (e–g) The comparison of WGS and Sanger sequencing mutation frequencies for on‐target site. The ‘d’, ‘i’, ‘s’ represent the deletion, insertion and SNP genotypes, respectively. The ‘no’ indicate no editing and other mutation types at the on‐target site.
Inherent genetic variation or/and somaclonal variation in Cas9‐edited plants, WT and negative plants
To evaluate the potential off‐target mutations of CRISPR/Cas9 in cotton plants, two standard computational methods were applied to detect all the variations, including SNPs and indels in the edited plants, negative and WT control plants compared with the cotton reference genome (Zhang et al., 2015; Figure 3a). Compared to the reference genome (G. hirsutum cultivar: TM‐1), the number of SNPs and indels in three WT plants (JIN668) are approximately 1 210 509–1 277 072 and 135 845–152 535, respectively. There are in a similar range to the genetic variation seen among different cotton cultivars (Wang et al., 2017; Table S4). To investigate pre‐existing mutations in the three WT cotton plants, we identified 28 054 SNPs and four indels with high confidence. The data suggested that the pre‐existing mutation ratio is ~2.8 × 10−6, which is much higher than in rice (Tang et al., 2018). However, there is no significant difference of variation number when comparing to Cas9‐edited lines and negative lines, whereas there is considerable genetic variation among different individual WT plants (Table 1). Analysis of the variation in Cas9‐edited and negative plants compared with WT plants as well as cotton TM‐1 reference genome found that the average SNP number was 65 152 and indel number was 14 415 (Table 1). An average 34.68% SNPs and 50.57% indels in negative control plants were found to overlap with variations in Cas9 (+) plants, suggesting that these variations might be caused by somaclonal variation during tissue culture, or/and inherent variations and the mutations induced by Cas9 endonuclease (Table 1). For example, several variations in the AP2, MYB44, ARC transgenic plants were observed due to somaclonal variation or/and inherent variation. These variations located in the regions without the homology with the sgRNAs (Figure S6). The 15 210–32 412 SNPs and 6316–8040 indels present in Cas9‐edited cotton plants, but not in WT, nor in negative control plants (Table 1). After filtering the shared variation, 4188–6404 SNPs and 312–745 indels were detected in AP2‐, MYB44‐, ARC‐edited plants and all these variations were only present in the Cas9‐edited cotton plants (Table 1). We found a small number of sample‐specific variations among Cas9 transgenic cotton plants (Figure S7). When these variations were annotated against the cotton reference genome, most variations occurs in intergenic region (Student's t‐test, P < 2.86e‐11; Table S5). For the SNP mutation types, a slight preponderance of transitions over transversions was detected (Chi‐square test, P < 0.001), in which C to T (19.35%–22.46%) and G to A (18.55%–23.08%) were the most two frequent mutations in independent Cas9‐edited plants (Figure 3b). The most abundant indels were 1~2‐bp in length (Figure 3c). Surprisingly, the flanking 20‐bp regions of these variation sites did not show any homology with the six sgRNAs.
Figure 3.
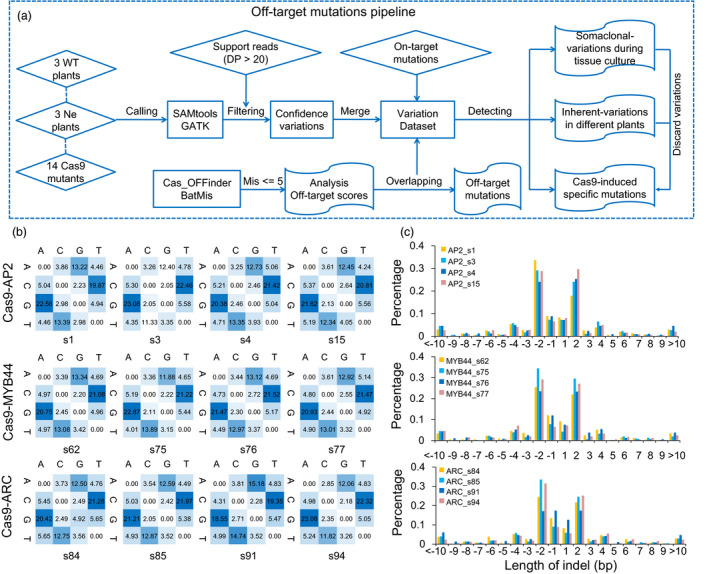
Genome‐wide analysis of variations in Cas9‐edited cotton plants during tissue culture process. (a) The bioinformatics pipeline for the off‐target mutations analysis. (b) Heatmap represents the percentage of specific mutation type in Cas9‐edited transgenic cotton plants. (c) Length of indels in different Cas9‐edited plants.
Table 1.
The summary of total variations in wild‐type, negative and CRISPR/Cas9‐edited cotton plants
| Description | Lines vs Ref | Plants vs Ref/WT | Plants vs Ref/WT/Ne | Private variations | ||||
|---|---|---|---|---|---|---|---|---|
| Plants | SNP | Indel | SNP | Indel | SNP | Indel | SNP | Indel |
| WT (s79) | 1 211 622 | 149 327 | – | – | – | – | – | – |
| WT (s195) | 1 214 683 | 152 535 | – | – | – | – | – | – |
| WT (s199) | 1 210 509 | 148 567 | – | – | – | – | – | – |
| Negative (s65) | 1 203 206 | 149 636 | 69 242 | 13 080 | – | – | – | – |
| Negative (s66) | 1 217 124 | 148 842 | 62 882 | 13 431 | – | – | – | – |
| Negative (s67) | 1 209 155 | 135 845 | 82 264 | 19 840 | – | – | – | – |
| Cas9‐AP2 (s1) | 1 266 777 | 150 156 | 61 648 | 13 870 | 15 210 | 6935 | 4188 | 500 |
| Cas9‐AP2 (s3) | 1 284 258 | 150 421 | 68 483 | 14 940 | 18 540 | 7736 | 4893 | 527 |
| Cas9‐AP2 (s4) | 1 270 814 | 150 405 | 67 517 | 13 691 | 19 415 | 6707 | 5976 | 549 |
| Cas9‐AP2 (s15) | 1 260 704 | 149 367 | 61 976 | 14 292 | 16 704 | 7282 | 4345 | 495 |
| Cas9‐AP2 (s20) | 1 262 125 | 149 979 | 62 580 | 14 224 | 32 591 | 7950 | 6096 | 777 |
| Cas9‐AP2 (s23) | 1 258 810 | 149 834 | 66 866 | 14 204 | 32 412 | 8040 | 5984 | 859 |
| Cas9‐MYB44 (s62) | 1 259 373 | 149 594 | 64 312 | 13 508 | 21 368 | 6596 | 6404 | 532 |
| Cas9‐MYB44 (s75) | 1 268 973 | 150 803 | 59 756 | 14 353 | 17 179 | 7123 | 2807 | 312 |
| Cas9‐MYB44 (s76) | 1 261 093 | 150 637 | 61 338 | 13 064 | 18 726 | 6316 | 4595 | 484 |
| Cas9‐MYB44 (s77) | 1 261 356 | 150 166 | 61 323 | 13 599 | 18 845 | 6684 | 5604 | 660 |
| Cas9‐ARC (s84) | 1 269 877 | 151 413 | 66 408 | 14 202 | 25 447 | 6884 | 5839 | 593 |
| Cas9‐ARC (s85) | 1 277 072 | 149 570 | 65 577 | 15 603 | 25 114 | 7852 | 5397 | 745 |
| Cas9‐ARC (s91) | 1 245 699 | 145 824 | 64 627 | 14 312 | 26 249 | 7035 | 4405 | 378 |
| Cas9‐ARC (s94) | 1 264 623 | 149 644 | 60 793 | 14 837 | 22 497 | 7406 | 4660 | 578 |
The ‘lines vs Ref’ represent the variation of each plant compared to TM‐1 reference genome using SAMtools and GATK tools (Table S4). The ‘lines vs Ref/WT’ represent the variations of each plant aligned to wild‐type (WT) and TM‐1 reference genome. Similarly, the ‘lines vs Ref/WT/Ne’ represent the variation of each Cas9‐edited transgenic plants aligned to WT, negative plants. Sample‐specific variations in three WT plants have the same genotype as three negative plants, but differ from each Cas9‐edited plants. Sample‐specific variations (including tissue culture variations, or/and inherent variations or/and Cas9‐edited mutations) were annotated by ANNOVAR (Table S5).
Low frequency off‐target mutations detected in Cas9‐edited plants
To detect all the potential off‐target mutations, six sgRNA and their PAM sequences were aligned with the TM‐1 reference genome using CRISPR‐P and Cas‐OFFinder software (Bae et al., 2014; Liu et al., 2017). With ≤5 mismatches in the sgRNA and PAM sequences, there were 3296 (PAM: NGG), 410 (PAM: NAG) and 707 (PAM: NGA) potential off‐target sites identified (Table S6, Figure S8 and Appendix S1). A very low off‐target mutation frequency (four indels, PAM: NGG) was detected in different CRISPR/Cas9 transgenic lines by WGS (Table 2). Interestingly, there was no mutation detected by WGS at the most likely 10 off‐target sites (Table S7), according to the scoring system of off‐target sites by a high‐throughput analysis in mammalian cells (Hsu et al., 2013).
Table 2.
Identification of off‐target mutations in Cas9‐edited plants by whole genome sequencing
| Cas9‐edited plants/sgRNA | Cas9 mutations/No. of NGG sites (Ratio%) | Cas9 mutations/No. of NAG sites (Ratio%) | Cas9 mutations/No. of NGA sites (Ratio%) |
|---|---|---|---|
| AP2 (sgRNA1) | 0/441 (0.00) | 0/57 (0.00) | 0/155 (0.00) |
| AP2 (sgRNA2) | 0/765 (0.00) | 0/55 (0.00) | 0/83 (0.00) |
| MYB44 (sgRNA1) | 0/683 (0.00) | 0/55 (0.00) | 0/151 (0.00) |
| MYB44 (sgRNA2) | 2/182 (1.10) | 0/8 (0.00) | 0/15 (0.00) |
| ARC (sgRNA1) | 2/341 (0.59) | 0/66 (0.00) | 0/54 (0.00) |
| ARC (sgRNA2) | 0/884 (0.00) | 0/169 (0.00) | 0/249 (0.00) |
Sanger sequencing was used to confirm these off‐target mutations with PAM (NGG) site. The Cas9‐edited AP2 lines had no off‐target mutation detectable by WGS. In the MYB44 Cas9‐edited plants, two off‐target sites were detected in the promoter region of dicarboxylate diiron protein (Crd1; Gh_D01G1828, OFTM1) and the first exon of MYB77 (Gh_D06G0115, OFTM2; Figure S9). The WGS data revealed the length frequencies of deletion at Crd1 and MYB77 off‐target sites (Figure 4a,b, middle panel). The indels were 1~4‐bp in length at Crd1 off‐target sites and 1‐bp at the MYB77 off‐target sites. We also detected ARC off‐target mutations and revealed 1‐bp deletions in two off‐target sites (OFTM3/4; Figures 4c,d and S10). The Cas9‐generated 1‐bp deletion at two off‐target sites in the non‐coding region was also validated by Sanger sequencing in both Cas9‐edited and WT plants (Figure 4c,d). We also found the deletion frequencies varied in independent Cas9‐edited plants. These results indicated that the low frequency off‐target mutations were low in Cas9‐edited cotton plants, even there were some unpredicted mutations in the regions that were not homologous to sgRNA target sites.
Figure 4.
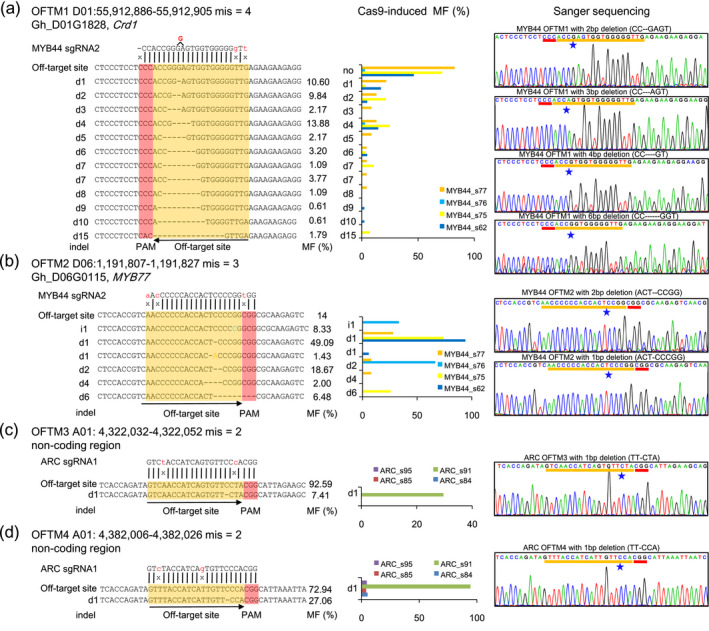
The identification of potential off‐target mutations in Cas9‐edited cotton plants by whole genome sequencing and Sanger sequencing. (a) Off‐target mutations in promoter region of the MYB44 gene. (b) Off‐target mutations in exon region of the MYB44 gene. (c,d) Off‐target mutations in non‐coding regions of ARC gene. The left panel showed the different off‐target mutation types. Mismatch (on‐target site vs off‐target site) nucleotides are showed in ‘x’, the PAM and off‐target region are showed in red and orange rectangular. The on top of left panel represent the each off‐target genome coordinate. Black arrows represent sgRNA transcription direction. The OFTM represent the off‐target site mutation in Cas9‐edited cotton plants. The Cas9‐edited MF represents the mutation frequency (MF) in different Cas9‐edited cotton plants compared to WT plants. The MF in the left panel showed the average mutation frequency per plant. The middle panel exhibited indel frequency in different Cas9‐edited plants. The right panel illustrated the Sanger sequencing data at off‐target sites. The stars in the right panel represent the cleavage sites.
Genetic variation among cultivars can generate novel off‐target sites
As mentioned previously, the WT used for genetic transformation is G. hirsutum cultivar JIN668, which contains more than 1 million high‐confidence SNPs and indels compared to the cotton reference genome (Table 1). To investigate the new off‐target sites or PAMs created by these inherent genetic variations in the WT, we reanalyzed the aforementioned predicted 4413 potential off‐target sites from six sgRNAs. When comparing the genetic variation in maternal WT with the reference genome, more than 61 SNPs and indels were identified in the 4413 potential off‐target sites, which can alter on‐ and off‐target variations by increasing and decreasing the number of mismatches with sgRNAs as well as PAM sites. Therefore, we defined these off‐target sites with variations in the WT genomes as ‘Novel’ potential off‐targets. For example, one PAM (NGG) generated a novel site (NCG) at the target site of AP2 gene (Table S8). Moreover, there were 39, 6 and 10 novel off‐target sites identified with NGG, NAG and NGA ‘PAM’ site, respectively, at six sgRNA target sites in the AP2, MYB44 and ARC genes (Tables 3 and S8). These results demonstrated that there is considerable genetic variability in the maternal cultivar used for genetic transformation, which will affect the accuracy of sgRNAs if these sgRNAs are designed based on a different reference genome.
Table 3.
Newly generated off‐target sites or PAMs by genetic variations in the WT plants
| Target | AP2 | MYB44 | ARC | |||
|---|---|---|---|---|---|---|
| sgRNA | sgRNA1 | sgRNA2 | sgRNA1 | sgRNA2 | sgRNA1 | sgRNA2 |
| Off‐target site (NGG) | 4/441 | 7/765 | 12/683 | 4/182 | 4/341 | 8/884 |
| PAM site (NGG) | 0/441 | 3/765 | 1/683 | 0/182 | 0/341 | 1/884 |
| Off‐target site (NAG) | 0/57 | 2/55 | 0/55 | 1/8 | 0/66 | 3/169 |
| PAM site (NAG) | 0/57 | 0/55 | 0/55 | 0/8 | 0/66 | 0/169 |
| Off‐target site (NGA) | 2/155 | 2/83 | 3/151 | 0/15 | 0/54 | 3/249 |
| PAM site (NGA) | 0/155 | 0/83 | 0/151 | 0/15 | 1/54 | 0/249 |
The number in front of the ‘/’ represent the novel off‐target site in WT and number behind the ‘/’ represent the total potential off‐target sites in reference genome, respectively.
Investigation of spontaneous mutations and the inheritance of Cas9‐edited mutations
To investigate and estimate the level of spontaneous mutations across generation, we analyzed the WGS data from T0 to T1 generation. For this purpose, three WT plants, three Negative plants, and 12 T0 Cas9‐edited plants were analyzed based on the WGS data to exclude Cas9‐induced mutations and pre‐existing mutations. After filtering the pre‐existing and Cas9‐induced mutations, 466 SNPs and 77 indels were identified as spontaneous mutations from T0 (s1) to T1 (s20, s23) in this experiment. The inherited mutation rate was ~5.43 × 10−8 per site per tetraploid genome from T0 to T1, which is consistent with previous report in rice with the spontaneous mutation rates (~5.4 × 10−8; Tang et al., 2018) and maize (2.17~3.87 × 10−8; Yang et al., 2017).
To test the inheritance of on‐target and off‐target mutations in the Cas9‐edited plants, transgenic AP2 plants were evaluated at on‐ and off‐target sites from the T0 (s1) to T1 (s20, s23) generations. The result showed that the on‐target mutation in Cas9‐edited AP2 T0 plant could be transmitted to T1 progeny at two sgRNAs loci (Table 4 and Figure S11). More importantly, there was no new editing detected at the target sites in T1 progenies. As expected, we did not detect any off‐target mutations (SNPs/indels) in either T0 and T1 generation from the WGS data, suggesting that the inheritance of Cas9‐edited mutation is very faithful between different cotton generations.
Table 4.
The inheritance of Cas9‐edited mutations from T0 to T1 in AP2 Cas9‐edited plants
| AP2 sgRNA1 | Type | s1 (T0, Cas9+) | s20 (T1, Cas9−) | s23 (T1, Cas9+) | |||
|---|---|---|---|---|---|---|---|
| D13 | D12 | D13 | D12 | D13 | D12 | ||
| ATGGTTGCATCCTGCCTAAAAGG | no | 0 | 0 | 0 | 0 | 0 | 0 |
| ATGGTTGCATCCTGCCTTAAAAGG | i1 | 0 | 0 | 0 | 0 | 0 | 0 |
| ATGGTTGCATCCTG‐‐TAAAAGG | d2 | 18 | 7 | 0 | 3 | 26 | 11 |
| ATGGTTGCATCCTGC‐‐AAAAGG | d2 | 23 | 41 | 54 | 27 | 11 | 31 |
| AP2 sgRNA2 | D13 | A12 | D13 | A12 | D13 | A12 | |
| CCTAGCAAAGTCCGATGGTATAA | no | 24 | 22 | 32 | 29 | 0 | 27 |
| CCTAGCAAAAGTCCGATGGTATAA | i1 | 0 | 0 | 2 | 3 | 0 | 0 |
| CCTAGATTAGTCCGATGGTATAA | s3 | 0 | 0 | 0 | 0 | 0 | 0 |
| CCTAGCA‐AGTCCGATGGTATAA | d1 | 1 | 13 | 1 | 0 | 21 | 0 |
| CCTAGCA‐‐GTCCGATGGTATAA | d2 | 0 | 0 | 0 | 0 | 0 | 0 |
| CCTAGC‐‐‐GTCCGATGGTATAA | d3 | 1 | 0 | 0 | 1 | 0 | 0 |
Discussion
To increase Cas9 accuracy by protein engineering, marked improvements have been achieved as a result of decoding the crystal structure of Cas9, identifying Cas9 orthologs, identifying CRISPR variants and single/paired nickases (Tycko et al., 2016). Although these strategies allow engineering of the Cas9 protein, higher specificity also can be obtained by improving the sgRNAs computational tools to guide RNA prediction.
However, recent research from Schaefer et al. describes 117 small insertions and deletions (indels) and 1397 single‐nucleotide variations (SNVs) using whole genome sequencing in two Cas9‐treated mice, which were absent in the uncorrected control (Schaefer et al., 2017). Although several groups questioned this research both in the mouse sample number (only two CRISPR‐treated mice were used) and the some group concerns about the method for the off‐target analysis, the frequency of off‐target mutations following CRISPR/Cas9 gene editing are of current interest (Kim et al., 2018; Lareau et al., 2018; Wilson et al., 2018).
In this study, we adopted the most widely used SpCas9 for genome editing. Since it shows higher on‐target editing efficiency and undetectable off‐target effects in cotton genome editing. We demonstrated that the most variations in Cas9‐edited cotton plants were due either to somaclonal variation or/and pre‐existing/natural variation from maternal plants, but not off‐target effects, which is consistent with previous report in rice (Tang et al., 2018). To investigate pre‐existing mutations in the three WT plants, we get high confidence 28 054 SNPs and four indels, the pre‐existing mutations is ~2.8 × 10−6, but much higher that the pre‐existing mutation rate in rice (Tang et al., 2018). This is maybe due to the excellent rice reference genome, but in cotton the considerable pre‐existing variations from one of subgenome or heterozygous and genome assembling error. Through sequencing reasonable Cas9‐edited cotton plants, we detected four bona fide off‐target mutations. Based on the off‐target homology and score, we revealed that highly homologous sequences have the potential Cas9 cleavage sites in seed sequence of the protospacer (Figure S8b). These findings suggest that CRISPR/Cas9 system is quite reliable and specific in plants.
The sgRNA design may largely determine the likelihood of off‐target effect. We revealed that genetic variations from maternal plants can alter PAMs and increase the risk of off‐target effects. Recently, two groups demonstrated that human genetic variation can alter the landscape of on‐target and potential off‐target sites genome‐wide, by creating and breaking their canonical PAM sequence in the CRISPR/Cas9 system (Lessard et al., 2017; Scott and Zhang, 2017). The current standard procedure is to design sgRNAs against the standard reference genome of relevance (i.e. Col‐0 in Arabidopsis, Nipponbare in rice and TM‐1 in cotton). However, in many plant species, a high quality reference genome is not available and usually the cultivars/genotypes with reference genome may not be widely used for genetic transformation (e.g. JIN668 rather than TM‐1 of cotton, and Zhonghua 11 rather than Nipponbare of rice, are widely used for genetic transformation). Any variations in the particular genomes of plant cultivars, the animal cell line or tissue, or strain of bacteria can adversely affect sgRNA specificity. In this study, more than one million SNPs and indels were found in WT plants compared with the reference genome, which generated novel off‐target sites and dramatically affected sgRNA specificity. Therefore, an exquisite level of understanding of genotypic variation through WGS analysis becomes an important initial step in sgRNA design, to increase specificity for CRISPR/Cas9 applications.
Once a reference genome for cultivars/cell/lines/strains of interest is available, the next important step is to choose appropriate tool for sgRNA design and off‐target prediction. To date, more than 10 computational tools [including CRISPR Design Tool (Hsu et al., 2013), and Cas‐OFFinder (Bae et al., 2014)] have been developed, mainly for animal genome editing (Tycko et al., 2016). We used the CRISPR‐P web tool to design sgRNA (Liu et al., 2017), which is widely used for the guide RNA design and the off‐target prediction for plant species (Ma et al., 2015; Soyk et al., 2017; Zhang et al., 2017). Moreover, it supports the design of guide sequences for various Cas9 orthologs as well as various CRISPR‐Cas systems like Cpf1. The undetectable off‐target effect in our previous report (Wang et al., 2018) and lower off‐target mutations imply that the CRISPR‐P is reliable for sgRNA design in plant species. Taken together, when designing sgRNAs, great care should be taken to consider the genetic variation from cultivar with different genetic background to minimize off‐target effect.
Currently, several methods are available to detect the off‐target mutation caused by sequence‐specific nucleases (SSNs) including targeted sequencing, Digenome‐seq (Kim et al., 2015), GUIDE‐seq (Tsai et al., 2015), SITE‐seq (Cameron et al., 2017), BLESS‐seq (Ran et al., 2015), ChIP‐seq (Kuscu et al., 2014), LAM‐HTGTS and whole genome sequencing (Hu et al., 2016). All these methods have strengths and weaknesses. These methods can detect genome‐wide direct labeling of DSBs, but only detect the DSBs present at the time of labeling (Zischewski et al., 2017). Among these methods, the WGS is very convenient and effective for identifying not only small indels and SNPs but also structural variations, such as major deletions, rearrangements, inversions and duplications (Veres et al., 2014). Therefore, a relatively high sequencing depth (>50) is required, which means high cost. The major limitation of the method is that it is difficult to detect the low frequency mutations in low sequencing depth.
In summary, the WGS data revealed thousands of novel SNPs/indels in Cas9‐edited cotton plants compared to WT, negative and cotton reference genome sequence. However, when compared to the sgRNAs, we found out that the majority of these variations loci lacked PAM sequence. We only detected four off‐target mutations in Cas9‐edited plants from 4413 predicted off‐targets based on the WGS data and these mutations were validated by Sanger sequencing. Notably, 61 potential off‐target sites and PAM were generated by genetic variation in WT plants. Also, there is no evidence of off‐target mutations from T0 to T1 generation. We therefore suggested that most variations in Cas9‐edited plants is induced by somaclonal variation during the tissue culture process, pre‐existing variation across germline, or/and inheritance from the maternal plants.
Experimental procedures
Computational sgRNA design and selection
The sgRNAs were designed according to CRISPR‐P web tool (Liu et al., 2017). The AP2, MYB44 and ARC target genes were selected for further analysis (Li et al., 2016). First, we found all possible sgRNA sequences in specific target gene with GC content (40%~60%) and on‐target score (score value > 0.6). For the off‐target effect, the number of off‐target site mismatches was <5 with on‐target site. Then, the off‐target scores were calculated based on the scoring system of off‐target sites from previous research (Hsu et al., 2013). The detail steps of optimal CRISPR sgRNAs were filtered, including the sgRNA genome position, on‐target score, off‐target score and the number of potential off‐targets. For each target gene, two sgRNAs were designed in the coding region, namely sgRNA1 and sgRNA2. For the polyploid plants, we need to consider the homologous gene pair of At‐ and Dt‐subgenome to increase the specificity of the sgRNA designing, i.e. sgRNA1 and sgRNA2 of AP2, and ARC have two target loci in At‐ and Dt‐subgenome, respectively. The sgRNA1 and sgRNA2 of MYB44 have one target site in At‐subgenome.
Vector construction
The CRISPR/Cas9 vector used in this report is modified from pRGEB32‐GhU6.9 according to our recent report (Wang et al., 2018). These two sgRNAs were integrated in a single vector, which included the fragments containing tRNA‐sgRNA1 and gRNA‐tRNA‐sgRNA2 fusion using pGTR as template, namely PTG (Xie and Yang, 2013), and then these two fragments were fused together with an overlapping extension PCR. The PTG fragment was ligated to pRGEB32‐GhU6.9‐NPT II expression vector, which was transformed into Agrobacterium tumefaciens strain EHA105 for stable cotton transformation.
Agrobacterium‐mediated transformation in cotton
Elite cotton cultivar (Gossypium hirsutum: Jin668) was used as the transformation receptor as described in our previous protocol (Li et al., 2018). Putative transgenic T0 plants were obtained and screened by Polymerase Chain Reaction (PCR) analysis. All the positive and negative plants then were transferred to a greenhouse to generate T1 seeds.
On‐target analysis of CRISPR/Cas9‐edited plants
DNA from T0 and T1 mutated plants were subjected to PCR for the positive identification of mutants induced through using Cas9 forward and reverse primers (Table S9). Selected positive Cas9‐edited plants were amplified via PCR and the PCR products were ligated to pGEMT‐Easy vector for TA cloning with T4 DNA ligase (Promega, Madison). These clones were used for Sanger sequencing to detect on‐target mutations caused by Cas9 nuclease. On‐target site mutations are showed through WGS data in Figures S2–S4 using Integrative Genomics Viewer (IGV) tools (Robinson et al., 2011). The sequences were aligned using ClustalW between Cas9‐edited and WT plants.
Genomic DNA isolation and library construction for Whole Genome Sequencing
Genomic DNA was extracted from young leaves of the three WT plants, three negative plants, 14 Cas9‐edited lines using Plant Genomic DNA kit (TIANGEN, Cat. #DP305‐03). For each sample, at least 5 μg DNA was used to construct sequencing library according to Illumina TruSeq DNA Sample Prep Kit (San Diego, CA). Final libraries were sequenced on an Illumina HiSeq X Ten (Paired‐end 150 bp) with an average 35× sequencing depth at the Novogene company (Beijing, China).
Bioinformatics analysis for variant calling
The reference allotetraploid cotton genome (Gossypium hirsutum, TM‐1) and its annotation were downloaded from CottonGen (https://www.cottongen.org/). Scaffolds with sequence lengths less than 2000‐bp were discarded from further analysis. First, the raw reads were filtered with Trimmomatic (Version 0.32, MINLEN:75; Bolger et al., 2014). Then, clean reads were mapped TM‐1 genome (Zhang et al., 2015) using BWA (Li and Durbin, 2009) with default parameters. The SNPs were called with SAMtools (Li et al., 2009) software and Genome Analysis Toolkit (GATK; Mckenna et al., 2010; ‐stand_call_conf 30.0). For indels, the BAM files were realigned using the GATK with (‐T RealignerTargetCreator, IndelRealigner) to get accurate indel. To obtain high‐quality SNPs and indels, common variations detected by GATK and SAMtools using GATK software merged the (‐T Selectvariations) at least depth 20× for each site was retained for further analysis. Finally, the VCF files were merged from different groups with VCFtools (Danecek et al., 2011). The SNPs and indels were filtered with parameter (QD < 20.0 || ReadPosRankSum < −8.0 || FS > 10.0 || QUAL < $MEANQUAL). The final private variations were annotated by ANNOVAR software (Wang et al., 2010).
Genome‐wide prediction off‐target cleavage sites and detection their mutations
The six sgRNAs were aligned against TM‐1 reference genome using BatMis and Cas‐OFFinder algorithm with up five mismatched as described (Bae et al., 2014; Tennakoon et al., 2012). The BatMis program was employed to perform sgRNA alignment with whole reference genome sequences. The off‐target sites were divided into NGG, NAG, NGA and others according to PAM site type using custom Perl script. The detail protocol for analysis in Cas9‐induced off‐target variations, somaclonal variations and inherent variations were detected. The pipeline is showed in Figure 3a. First, the variations present in three WT plants, where three negative plants have same genotype but differs from Cas9‐edited plants. All Cas9 lines were separated for further analysis. This step can discard somaclonal variation due to the tissue culture process or/and plant inherent variations from three negative plants. Secondly, analysis of independent Cas9‐edited plants variations compared to WT plants to filter inherent variations and individual plant tissue culture variations. This allows the identification of unique variations in each Cas9‐edited plant, which were called private variations. Finally, the candidate off‐target sites flanking 20‐bp were used to search the corresponding SNP/indel variations of Cas9‐edited plants to generate a likely off‐target mutation. The potential off‐target mutations were visualized in WT, negative, and Cas9‐edited plants by IGV tool to confirm the Cas9 nuclease induced mutations.
Analysis genetic variation of potential off‐targets and PAMs between WT and cotton reference genome
In total, the 4413 off‐target sites with canonical PAMs (PAM = NGG or NAG or NGA) were used to analyze the inherent genetic variation impacts Cas9 endonucleases specificity. The WT (s79, s195, s199) variations (SNPs and indels, showed in Table 1) calling was performed with SAMtools and GATK, which then overlapped with off‐target and PAM sequences with the BEDTools package (Quinlan and Hall, 2010).
Conflict of interest
The authors have declared that no competing interests exist.
Supporting information
Figure S1 The compared genome editing efficiency in Cas9‐edited plants.
Figure S2 Confirmation of the AP2 on‐target mutation in two sgRNAs from WT, negative and CRISPR/Cas9 plants based on the WGS data.
Figure S3 Confirmation of the MYB44 on‐target mutation in two sgRNA loci from WT, negative and CRISPR/Cas9 plants based on the WGS data.
Figure S4 Confirmation of the ARC on‐target mutation in two sgRNA loci from WT, negative and CRISPR/Cas9 plants based on the WGS data.
Figure S5 Scatterplot of on‐target site correlation of sanger sequencing and Whole genome sequencing.
Figure S6 The somaclonal variation or/and inherent genetic variation were detected in AP2, MYB44, ARC Cas9‐edited plants and WT by IGV.
Figure S7 The common and private SNPs/indels in four Cas9‐edited lines of AP2, MYB44, ARC.
Figure S8 Genome‐wide prediction off‐target sites.
Figure S9 The potential off‐target mutations in MYB44 Cas9‐edited cotton plants.
Figure S10 The potential off‐target mutations in ARC Cas9‐edited cotton plants.
Figure S11 Confirmation of the AP2 mutations of inheritance from T0 to T1 plants in two sgRNA loci by WGS data.
Table S1 The depth of whole genome sequencing data.
Table S2 Summary of target genes in CRISPR/Cas9 editing.
Table S3 The different on‐target site mutation frequency of AP2, MYB44, ARC in each target site by Sanger sequencing.
Table S4 The summary of variants calling by the SAMtools and GATK.
Table S5 Genomic distribution of private variations in Cas9‐edited plants.
Table S6 Summary of the off‐target sites across six sgRNA of three target genes.
Table S7 Identification of most off‐target site mutations in CRISPR/Cas9 edited plants.
Table S8 Summary of new off‐targets and PAMs in WT plants.
Table S9 Primers used for on‐targets and off‐targets.
Appendix S1 The number of potential off‐target sites were identified by CRISPR‐P and Cas‐OFFinder software with allowing up five mismatch for six sgRNAs.
Acknowledgements
This research was supported by grants from National R&D Project of Transgenic Crops of Ministry of Science and Technology of China (2016ZX08010001‐006), National Key Research and Development Plan (2016YFD0100203‐9) and Fundamental Research Funds for the Central Universities (2013PY064, 2662015PY028, 2662015PY091 and 0900206328) to Dr. Shuangxia Jin.
Data availability
The potential off‐target sites used for mutations analysis are available as Appendix S1. All sequencing data used for the off‐target mutations are available from the author. High‐throughput sequencing data have been deposited in the NCBI Sequence Read Archive (SRA) BioProject ID: PRJNA380842.
References
- Bae, S. , Park, J. and Kim, J.S. (2014) Cas‐OFFinder: a fast and versatile algorithm that searches for potential off‐target sites of Cas9 RNA‐guided endonucleases. Bioinformatics, 30, 1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron, P. , Fuller, C.K. , Donohoue, P.D. , Jones, B.N. , Thompson, M.S. , Carter, M.M. , Gradia, S. et al. (2017) Mapping the genomic landscape of CRISPR‐Cas9 cleavage. Nat. Methods, 14, 600–606. [DOI] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C.A. , Banks, E. , DePristo, M.A. , Handsaker, R.E. et al. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Z. , Mao, Y. , Xu, N. , Zhang, B. , Wei, P. , Yang, D.L. , Wang, Z. et al. (2014) Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas‐induced gene modifications in Arabidopsis . Proc. Natl Acad. Sci. USA, 111, 4632–4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frock, R.L. , Hu, J. , Meyers, R.M. , Ho, Y.J. , Kii, E. and Alt, F.W. (2015) Genome‐wide detection of DNA double‐stranded breaks induced by engineered nucleases. Nat. Biotechnol. 33, 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. , Foden, J.A. , Khayter, C. , Maeder, M.L. , Reyon, D. , Joung, J.K. and Sander, J.D. (2013) High‐frequency off‐target mutagenesis induced by CRISPR‐Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, P.D. , Scott, D.A. , Weinstein, J.A. , Ran, F.A. , Konermann, S. , Agarwala, V. , Li, Y. et al. (2013) DNA targeting specificity of RNA‐guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. , Meyers, R.M. , Dong, J. , Panchakshari, R.A. , Alt, F.W. and Frock, R.L. (2016) Detecting DNA double‐stranded breaks in mammalian genomes by linear amplification‐mediated high‐throughput genome‐wide translocation sequencing. Nat. Protoc. 11, 853–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, C. , Mithani, A. , Gan, X. , Belfield, E.J. , Klingler, J.P. , Zhu, J.K. , Ragoussis, J. et al. (2011) Regenerant Arabidopsis lineages display a distinct genome‐wide spectrum of mutations conferring variant phenotypes. Curr. Biol. 21, 1385–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, W. , Zhou, H. , Bi, H. , Fromm, M. , Yang, B. and Weeks, D.P. (2013) Demonstration of CRISPR/Cas9/sgRNA‐mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 41, e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, S. , Mushke, R. , Zhu, H. , Tu, L. , Lin, Z. , Zhang, Y. and Zhang, X. (2008) Detection of somaclonal variation of cotton (Gossypium hirsutum) using cytogenetics, flow cytometry and molecular markers. Plant Cell Rep. 27, 1303–1316. [DOI] [PubMed] [Google Scholar]
- Jinek, M. , Chylinski, K. , Fonfara, I. , Hauer, M. , Doudna, J.A. and Charpentier, E. (2012) A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science, 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek, M. , Jiang, F. , Taylor, D.W. , Sternberg, S.H. , Kaya, E. , Ma, E. , Anders, C. et al. (2014) Structures of Cas9 endonucleases reveal RNA‐mediated conformational activation. Science, 343, 1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Bae, S. , Park, J. , Kim, E. , Kim, S. , Yu, H.R. , Hwang, J. et al. (2015) Digenome‐seq: genome‐wide profiling of CRISPR‐Cas9 off‐target effects in human cells. Nat. Methods, 12, 237–243. [DOI] [PubMed] [Google Scholar]
- Kim, S. , Park, J. , Kim, D. , Kim, K. , Bae, S. , Schlesner, M. and Kim, J.S. (2018) Response to “Unexpected mutations after CRISPR–Cas9 editing in vivo”. Nat. Methods, 15, 239–240. [DOI] [PubMed] [Google Scholar]
- Kuscu, C. , Arslan, S. , Singh, R. , Thorpe, J. and Adli, M. (2014) Genome‐wide analysis reveals characteristics of off‐target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 32, 677–683. [DOI] [PubMed] [Google Scholar]
- Lareau, C. , Clement, K. , Hsu, J. , Pattanayak, V. , Joung, J. , Aryee, M. and Pinello, L. (2018) Response to “Unexpected mutations after CRISPR–Cas9 editing in vivo”. Nat. Methods, 15, 238–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrenson, T. , Shorinola, O. , Stacey, N. , Li, C. , Ostergaard, L. , Patron, N. , Uauy, C. et al. (2015) Induction of targeted, heritable mutations in barley and Brassica oleracea using RNA‐guided Cas9 nuclease. Genome Biol. 16, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K. , Zhang, Y. , Kleinstiver, B.P. , Guo, J.A. , Aryee, M.J. , Miller, J. , Malzahn, A. et al. (2018) Activities and specificities of CRISPR/Cas9 and Cas12a nucleases for targeted mutagenesis in maize. Plant Biotechnol. 10.1111/pbi.12982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard, S. , Francioli, L. , Alfoldi, J. , Tardif, J.C. , Ellinor, P.T. , MacArthur, D.G. , Lettre, G. et al. (2017) Human genetic variation alters CRISPR‐Cas9 on‐ and off‐targeting specificity at therapeutically implicated loci. Proc. Natl Acad. Sci. USA, 114, E11257–E11266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. and Ruan, J. (2009) The sequence alignment‐map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. , Liu, Z.B. , Xing, A. , Moon, B.P. , Koellhoffer, J.P. , Huang, L. , Ward, R.T. et al. (2015) Cas9‐guide RNA directed genome editing in soybean. Plant Physiol. 169, 960–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Zhu, L. , Hull, J.J. , Liang, S. , Daniell, H. , Jin, S. and Zhang, X. (2016) Transcriptome analysis reveals a comprehensive insect resistance response mechanism in cotton to infestation by the phloem feeding insect Bemisia tabaci (whitefly). Plant Biotechnol. J. 14, 1956–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Wang, M. , Li, Y. , Zhang, Q. , Lindsey, K. , Daniell, H. , Jin, S. et al. (2018) Multi‐omics analyses reveal epigenomics basis for cotton somatic embryogenesis through successive regeneration acclimation (SRA) process. Plant Biotechnol. J. 10.1111/pbi.12988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, Z. , Zhang, K. , Chen, K. and Gao, C. (2014) Targeted mutagenesis in Zea mays using TALENs and the CRISPR/Cas system. J. Genet. Genomics, 41, 63–68. [DOI] [PubMed] [Google Scholar]
- Liu, H. , Ding, Y. , Zhou, Y. , Jin, W. , Xie, K. and Chen, L.L. (2017) CRISPR‐P 2.0: an improved CRISPR‐Cas9 tool for genome editing in plants. Mol. Plant, 10, 530–532. [DOI] [PubMed] [Google Scholar]
- Ma, X. , Zhang, Q. , Zhu, Q. , Liu, W. , Chen, Y. , Qiu, R. , Wang, B. et al. (2015) A robust CRISPR/Cas9 system for convenient, high‐efficiency multiplex genome editing in monocot and dicot plants. Mol. Plant, 8, 1274–1284. [DOI] [PubMed] [Google Scholar]
- Mao, Y. , Zhang, H. , Xu, N. , Zhang, B. , Gou, F. and Zhu, J.K. (2013) Application of the CRISPR‐Cas system for efficient genome engineering in plants. Mol. Plant, 6, 2008–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mckenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, J. , Guo, D. , Zhang, J. , Huang, Q. , Qin, G. , Zhang, X. , Wan, J. et al. (2013) Targeted mutagenesis in rice using CRISPR‐Cas system. Cell Res. 23, 1233–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasov, V. , Wang, C. , Win, J. , Lanz, C. , Weigel, D. and Kamoun, S. (2017) Rapid generation of a transgene‐free powdery mildew resistant tomato by genome deletion. Sci. Rep. 7, 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, A.R. and Hall, I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, F.A. , Cong, L. , Yan, W.X. , Scott, D.A. , Gootenberg, J.S. , Kriz, A.J. , Zetsche, B. et al. (2015) In vivo genome editing using Staphylococcus aureus Cas9. Nature, 520, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J.T. , Thorvaldsdóttir, H. , Winckler, W. , Guttman, M. , Lander, E.S. , Getz, G. and Mesirov, J.P. (2011) Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, K.A. , Wu, W.H. , Colgan, D.F. , Tsang, S.H. , Bassuk, A.G. and Mahajan, V.B. (2017) Unexpected mutations after CRISPR‐Cas9 editing in vivo . Nat. Methods, 14, 547–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, D.A. and Zhang, F. (2017) Implications of human genetic variation in CRISPR‐based therapeutic genome editing. Nat. Med. 23, 1095–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyk, S. , Lemmon, Z.H. , Oved, M. , Fisher, J. , Liberatore, K.L. , Park, S.J. , Goren, A. et al. (2017) Bypassing negative epistasis on yield in tomato imposed by a domestication gene. Cell, 169, 1142–1155. [DOI] [PubMed] [Google Scholar]
- Tang, X. , Liu, G. , Zhou, J. , Ren, Q. , You, Q. , Tian, L. , Xin, X. et al. (2018) A large‐scale whole‐genome sequencing analysis reveals highly specific genome editing by both Cas9 and Cpf1 (Cas12a) nucleases in rice. Genome Biol. 19, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennakoon, C. , Purbojati, R.W. and Sung, W.K. (2012) BatMis: a fast algorithm for k‐mismatch mapping. Bioinformatics, 28, 2122–2128. [DOI] [PubMed] [Google Scholar]
- Tsai, S.Q. , Zheng, Z. , Nguyen, N.T. , Liebers, M. , Topkar, V.V. , Thapar, V. , Wyvekens, N. et al. (2015) GUIDE‐seq enables genome‐wide profiling of off‐target cleavage by CRISPR‐Cas nucleases. Nat. Biotechnol. 33, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, S.Q. , Nguyen, N.T. , Malagon‐Lopez, J. , Topkar, V.V. , Aryee, M.J. and Joung, J.K. (2017) CIRCLE‐seq: a highly sensitive in vitro screen for genomewide CRISPR‐Cas9 nuclease off‐targets. Nat. Methods, 14, 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko, J. , Myer, V.E. and Hsu, P.D. (2016) Methods for optimizing CRISPR‐Cas9 genome editing specificity. Mol. Cell, 63, 355–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veres, A. , Gosis, B.S. , Ding, Q. , Collins, R. , Ragavendran, A. , Brand, H. , Erdin, S. et al. (2014) Low incidence of off‐target mutations in individual CRISPR‐Cas9 and TALEN targeted human stem cell clones detected by whole‐genome sequencing. Cell Stem Cell, 15, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Li, M. and Hakonarson, H. (2010) ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Tu, L. , Lin, M. , Lin, Z. , Wang, P. , Yang, Q. , Ye, Z. et al. (2017) Asymmetric subgenome selection and cis‐regulatory divergence during cotton domestication. Nat. Genet. 9, 579–587. [DOI] [PubMed] [Google Scholar]
- Wang, P. , Zhang, J. , Sun, L. , Ma, Y. , Xu, J. , Liang, S. , Deng, J. et al. (2018) High efficient multisites genome editing in allotetraploid cotton (Gossypium hirsutum) using CRISPR/Cas9 system. Plant Biotechnol. J. 16, 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, C. , Fennell, T. , Bothmer, A. , Maeder, M.L. , Reyon, D. , Cotta‐Ramusino, C. , Fernandez, C.A. et al. (2018) Response to “Unexpected mutations after CRISPR–Cas9 editing in vivo”. Nat. Methods, 15, 236–237. [DOI] [PubMed] [Google Scholar]
- Xie, K. and Yang, Y. (2013) RNA‐guided genome editing in plants using a CRISPR‐Cas system. Mol. Plant, 6, 1975–1983. [DOI] [PubMed] [Google Scholar]
- Yang, N. , Xu, X.W. , Wang, R.R. , Peng, W.L. , Cai, L. , Song, J.M. , Li, W. et al. (2017) Contributions of Zea mays subspecies mexicana haplotypes to modern maize. Nat. Commun. 8, 1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Zhang, J. , Wei, P. , Zhang, B. , Gou, F. , Feng, Z. , Mao, Y. et al. (2014) The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol. J. 12, 797–807. [DOI] [PubMed] [Google Scholar]
- Zhang, T. , Hu, Y. , Jiang, W. , Fang, L. , Guan, X. , Chen, J. , Zhang, J. et al. (2015) Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM‐1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537. [DOI] [PubMed] [Google Scholar]
- Zhang, D. , Zhang, H. , Li, T. , Chen, K. , Qiu, J.L. and Gao, C. (2017) Perfectly matched 20‐nucleotide guide RNA sequences enable robust genome editing using high‐fidelity SpCas9 nucleases. Genome Biol. 18, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zischewski, J. , Fischer, R. and Bortesi, L. (2017) Detection of on‐target and off‐target mutations generated by CRISPR/Cas9 and other sequence‐specific nucleases. Biotechnol. Adv. 35, 95–104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The compared genome editing efficiency in Cas9‐edited plants.
Figure S2 Confirmation of the AP2 on‐target mutation in two sgRNAs from WT, negative and CRISPR/Cas9 plants based on the WGS data.
Figure S3 Confirmation of the MYB44 on‐target mutation in two sgRNA loci from WT, negative and CRISPR/Cas9 plants based on the WGS data.
Figure S4 Confirmation of the ARC on‐target mutation in two sgRNA loci from WT, negative and CRISPR/Cas9 plants based on the WGS data.
Figure S5 Scatterplot of on‐target site correlation of sanger sequencing and Whole genome sequencing.
Figure S6 The somaclonal variation or/and inherent genetic variation were detected in AP2, MYB44, ARC Cas9‐edited plants and WT by IGV.
Figure S7 The common and private SNPs/indels in four Cas9‐edited lines of AP2, MYB44, ARC.
Figure S8 Genome‐wide prediction off‐target sites.
Figure S9 The potential off‐target mutations in MYB44 Cas9‐edited cotton plants.
Figure S10 The potential off‐target mutations in ARC Cas9‐edited cotton plants.
Figure S11 Confirmation of the AP2 mutations of inheritance from T0 to T1 plants in two sgRNA loci by WGS data.
Table S1 The depth of whole genome sequencing data.
Table S2 Summary of target genes in CRISPR/Cas9 editing.
Table S3 The different on‐target site mutation frequency of AP2, MYB44, ARC in each target site by Sanger sequencing.
Table S4 The summary of variants calling by the SAMtools and GATK.
Table S5 Genomic distribution of private variations in Cas9‐edited plants.
Table S6 Summary of the off‐target sites across six sgRNA of three target genes.
Table S7 Identification of most off‐target site mutations in CRISPR/Cas9 edited plants.
Table S8 Summary of new off‐targets and PAMs in WT plants.
Table S9 Primers used for on‐targets and off‐targets.
Appendix S1 The number of potential off‐target sites were identified by CRISPR‐P and Cas‐OFFinder software with allowing up five mismatch for six sgRNAs.
Data Availability Statement
The potential off‐target sites used for mutations analysis are available as Appendix S1. All sequencing data used for the off‐target mutations are available from the author. High‐throughput sequencing data have been deposited in the NCBI Sequence Read Archive (SRA) BioProject ID: PRJNA380842.