

Summary
Background
Biosimilar versions of widely prescribed drugs, including the tumour‐necrosis factor antagonist infliximab, are becoming increasingly available. As biosimilars are not identical copies of reference products, evidence may be required to demonstrate that switching between a reference biologic and biosimilars is safe and efficacious. To establish interchangeability, US Food and Drug Administration guidance states that studies must demonstrate that biosimilars remain equivalent or non‐inferior to a reference product after multiple switches between products.
Aims
To investigate the evidence evaluating the safety and efficacy of switching between reference and biosimilar infliximab in patients with inflammatory disorders, including Crohn's disease, ulcerative colitis, rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis.
Methods
Published studies presenting data on switching between reference and biosimilar infliximab were identified by searching the MEDLINE database. Congress abstracts were identified by searching the EMBASE database and manually searching abstracts from relevant congresses.
Results
A total of 113 journal articles and 149 abstracts were found. Of these, 70 were considered relevant and included in this analysis. Most of the publications were uncontrolled, observational studies. Data from six randomised, controlled trials were identified. In general, the evidence revealed no clinically important efficacy or safety signals associated with switching.
Conclusions
While available data have not identified significant risks associated with a single switch between reference and biosimilar infliximab, the studies available currently report on only single switches and were mostly observational studies lacking control arms. Additional data are needed to explore potential switching risks in various populations and scenarios.
1. INTRODUCTION
Biologics are drugs, such as growth factors and monoclonal antibodies that are produced in a living system. These agents have been increasingly used to treat a wide‐range of diseases.1, 2 Unlike small molecules, which are produced by chemical synthesis and have well‐defined structures, biologics are often structurally heterogeneous, as each molecule may have subtle differences in tertiary and quaternary structure.3 Although biosimilars will usually have identical amino‐acid sequences to the reference product, there is a possibility of altered glycosylation as a result of their production in different cell lines.3 These structural changes can affect the pharmacology, pharmacodynamic (PD) effect, and immunogenicity of a biologic drug.4 As the properties of a biologic agent may be highly dependent on complex manufacturing procedures, a well‐defined and controlled manufacturing process is critical to maintain therapeutic consistency.4
Patents on widely prescribed biologics, such as entanercept, adalimumab, and infliximab have recently expired and biosimilar versions of these products have been developed (Table 1).5, 6, 7 The US Food and Drug Administration (FDA), the European Medicines Agency (EMA), and other global regulatory agencies have issued guidance on the development of biosimilars.6, 10, 11 The FDA defines a biosimilar as “the biological product is highly similar to the reference product notwithstanding minor differences in clinically inactive components,” and that “there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product”.11 The EMA states that “similarity to the reference medicinal product in terms of quality characteristics, biological activity, safety, and efficacy based on a comprehensive comparability exercise needs to be established”.10
Table 1.
| Reference product | Biosimilar | Synonyms | Date of FDA approval |
|---|---|---|---|
| Infliximab | Infliximab‐dyyb | CT‐P13 | 5 April 2016 |
| Infliximab‐abda | SB2 | 21 April 2017 | |
| Infliximab‐qbtx | PF‐06438179, GP1111 | 13 December 2017 | |
| Adalimumab | Adalimumab‐atto | ABP 501 | 23 September 2016 |
| Adalimumab‐adbm | BI 695501 | 25 August 2017 | |
| Etanercept | Etanercept‐szzs | GP2015 | 30 August 2016 |
FDA, Food and Drug Administration; TNF, tumour‐necrosis factor.
In addition to the need to demonstrate functional and structural similarities of a biosimilar with the reference product in preclinical studies, the FDA and EMA require that several key clinical studies be performed.10, 11 Pharmacology studies need to demonstrate that a biosimilar and reference product have similar pharmacological properties in human subjects through evaluation of pharmacokinetic (PK) and PD parameters that are relevant to the licenced use of the reference product.10, 11 Clinical assessments using validated assays that can detect anti‐drug antibodies (ADAs) are used to evaluate potential differences between the biosimilar and reference product in the incidence and severity of human immune responses.10, 11 Studies are also needed to demonstrate comparable safety and efficacy in clinically relevant patient populations.10, 11 When biosimilarity is demonstrated for one of the approved indications of the reference product, approval can be extrapolated to other clinical indications of the reference product with scientific justification.10, 11
While biosimilars may be approved for the same indications as the reference product, they are not necessarily interchangeable with the reference product, meaning that the biosimilar cannot be directly substituted for the reference product in the same manner as a generic small molecule. As biosimilars are not identical to the reference product, additional evidence may be required to demonstrate that switching or alternating therapy between the reference product or multiple biosimilars is safe and efficacious, partially due to concerns over the potential risk of immunogenicity that exists due to possible differences in epitopes between the biosimilar and the reference product.12 The FDA has recently issued guidance on the interchangeability of biosimilars, which indicates the need for a dedicated switching study prior to approval of a biosimilar as an interchangeable product.13 The guidance recommends that switching studies should have at least three switches between products for at least two exposure periods with each drug.13 Currently, no biosimilars have been designated as interchangeable by the FDA.8
Infliximab is a tumour necrosis factor (TNF) blocker indicated for the treatment of Crohn's disease (CD), paediatric CD, ulcerative colitis (UC), paediatric UC, rheumatoid arthritis (RA) in combination with methotrexate, ankylosing spondylitis (AS), psoriatic arthritis (PsA), and plaque psoriasis (PsO).14 Initially approved in 1998, infliximab has been used to treat over 2.6 million patients and has a well‐established long‐term safety and efficacy profile.14, 15 In recent years, multiple infliximab biosimilars have been approved by both the FDA and the EMA and are commercially available in multiple global markets.6, 8
The first infliximab biosimilar, CT‐P13, was approved in the EU in 2013 and the US in 2016.6, 8 The bioequivalence of CT‐P13 with reference infliximab has been demonstrated in two randomised, double‐blind, phase 3 studies of patients with AS (PLANETAS) and RA (PLANETRA).16, 17 Similarly, the bioequivalence of two other FDA‐approved infliximab biosimilars (SB2 and PF‐06438179) have also been demonstrated in patients with RA.18, 19 Additional infliximab biosimilars have been approved in other countries, including BOW015 (in India) and NI‐071 (in Japan), or are in clinical development. In summary, the biosimilar environment is becoming crowded and complex with the potential for switching among different products based upon economic pressures becoming highly likely in the near future. The purpose of this review is to critically analyse the existing evidence regarding the safety and efficacy of switching between reference and biosimilar infliximab one or more times.
2. METHODS
2.1. Search strategy
Several sources were searched to identify appropriate journal articles and congress abstracts. An electronic search of the MEDLINE database was conducted to identify relevant journal articles on switching between reference infliximab and biosimilars. Search strings included various terms for biosimilars and biosimilar candidates: SB2, CT‐P13, GP‐2018, ABP‐710, BCD‐055, PF‐06438179, NI‐071, BOW015, and RTPR‐015. The search string also contained known synonyms for these molecules. Alternative terms for switching included substituting, interchanging, and variations on these terms. See Methods S1 for full search string. Publication date filters were set to included publications from 1 January 2004 to 30 January 2018.
Applicable congress abstracts were searched from major US and EU rheumatology, gastroenterology, and dermatology congresses comprising the American Academy of Dermatology, the American College of Gastroenterology, the American College of Rheumatology, Digestive Disease Week, European Academy of Dermatology and Venereology, European Crohn's and Colitis Organisation, European League Against Rheumatism, and United European Gastroenterology Week. The EMBASE database was searched for relevant results for these conferences from 2012 to 2017 using search terms similar to those described for journal articles. Abstract books not available in the EMBASE database were manually searched.
2.2. Selection criteria and evaluation of evidence
In order to focus on primary analyses of switch studies, several criteria were applied. Review articles, editorials, and other opinion pieces were excluded from this analysis. Journal articles or congress abstracts not focused on infliximab, not discussing an infliximab switch, or not presenting safety, efficacy, or immunogenicity data specifically from an infliximab switch were also excluded. Data were only included if efficacy, safety, or immunogenicity specifically surrounding an infliximab switch were available. Thus, studies that only evaluated switch from any reference product to any biosimilar were not included. Additionally, data from studies only reporting on treatment patterns of biosimilars were not included. Duplicate abstracts and abstracts describing data that was subsequently published in a journal article, were excluded. Only articles with full text available in English were evaluated.
Results were reviewed on the basis of several criteria, including type of study (eg, randomised controlled trial [RCT], observational studies, retrospective analysis) and patient populations. Several study designs were considered, including single‐transition studies, crossover studies, and full interchangeability studies (Figure 1).12 Efficacy, safety, and immunogenicity endpoints were assessed from each study. The presence of a control arm was declared if the switch was controlled or if a historical cohort was included. Thus, comparing the effects of a biosimilar in treatment naïve patients to patients that switched from reference infliximab was not considered a controlled switch.
Figure 1.
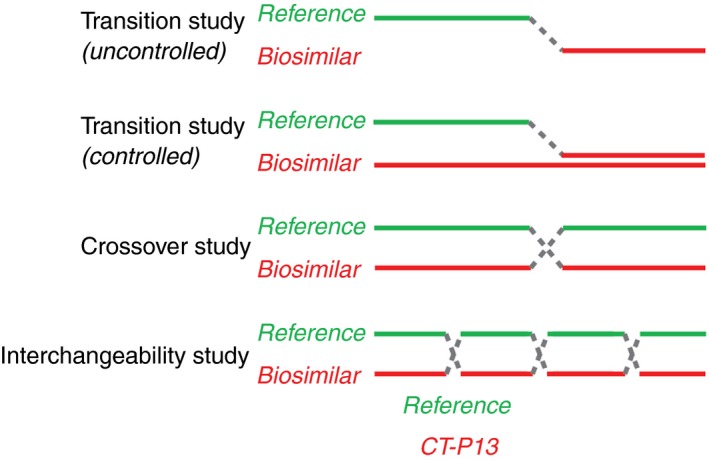
Examples of potential switching study designs12
2.3. Evaluation of RCTs
The reporting of all RCTs were evaluated against the 2010 Consolidated Standards of Reporting Trials (CONSORT) checklist;20 the CONSORT statement includes a 25‐item checklist that provides guidance for reporting RCTs.
3. RESULTS
3.1. Search results
A total of 113 journal articles were obtained from the MEDLINE search (Figure 2). Of these, 77 journal articles were excluded (full article not available in English [n = 1], review article or position statement [n = 59], not relevant [n = 17]) and 36 were included in this review. The EMBASE search revealed 68 abstracts and manual searching found an additional 81 abstracts. A total of 115 abstracts were excluded (duplicate [n = 28], superseded by journal article [n = 18], review article or position statement [n = 5], not relevant [n = 64]) and 34 were included in this review. Thus, a total of 70 entries, investigating the interchangeability biosimilars were included.
Figure 2.

Literature search results
3.2. Randomised controlled trials
A total of 13 publications presenting results from six RCTs, including open‐label extensions (OLEs) or sub‐group analyses of these trials were identified (Figure 3;21, 22, 23, 24, 25, 26, 27, 28, 29 Table 2; Table S1). All of them were single‐transition studies, and none of these RCTs described a multiple‐switch scenario or switches between biosimilars. Three different biosimilars were tested in these trials (SB2, CT‐P13, BOW015), mainly in patients with RA, but also in patients with AS, PsA, UC, CD, and PsO. Two of these studies were dedicated single‐transition studies, while the remaining four were studies where switching only occurred in the OLE. In one study (NOR‐SWITCH), switches occurred in one group in the randomised phase and in the other group in the OLE.
Figure 3.

Study designs for (A) the SB2 transition trial,21 (B) the CT‐P13 Japanese RA study,24 (C) the PLANETRA OLE,25 (D) the PLANETAS OLE,26 (E) the BOW015 phase 3 OLE,27, 28, 29 and (F) NOR‐SWITCH and the NOR‐SWITCH OLE.22, 23 In the NOR‐SWITCH study, the full analysis set had 241 and 240 patients in the reference and switch arms, respectively. Numbers shown in the figure represent the per‐protocol population from the main study. AS, ankylosing spondylitis; CD, Crohn's disease; PsA, psoriatic arthritis; PsO, psoriasis, RA, rheumatoid arthritis, UC, ulcerative colitis
Table 2.
Summary of switch data from RCTs
| Study/treatment group | Efficacy | AE incidence after switch (n/N) | ADA incidence after switch (n/N) |
|---|---|---|---|
| Rheumatoid arthritis, spondyloarthritis, psoriatic arthritis, ulcerative colitis, Crohn's disease, and psoriasis | |||
| NOR‐SWITCH22 | Disease worseninga, n/N (%) | ||
| Reference → CT‐P13 | 61/206 (29.6) | 164/240 (68.3) | 19/240 (7.9) |
| Reference → reference | 53/202 (26.2) | 168/241 (69.7) | 17/241 (7.1) |
| NOR‐SWITCH OLE23 | Disease worseninga, n/N (%) | ||
| CT‐P13 → CT‐P13 | 32/190 (16.8) | Similar across groupsb | 3/197 (1.5) |
| Reference → CT‐P13 | 20/173 (11.6) | 5/183 (2.7) | |
| Rheumatoid arthritis | |||
| SB2 transition trial21 | ACR20/50/70 rate, % | ||
| Reference → SB2 | 63.5/37.6/22.4 | 34/94 (36.2) | 6/41 (14.6)c |
| Reference → reference | 68.8/47.3/31.2 | 36/101 (35.6) | 7/47 (14.9)c |
| SB2 → SB2 | 68.3/40.6/25.6 | 81/201 (40.3) | 11/78 (14.1)c |
| Japanese RA study OLE24 | DAS28, mean (SD) | ||
| CT‐P13 → CT‐P13 | 3.166 (1.533) | 34/38 (89.5) | 5/32 (15.6) |
| Reference → CT‐P13 | 3.955 (1.751) | 29/33 (87.9) | 4/23 (17.4) |
| PLANETRA OLE25 | ACR20/50/70 rate, % | TEAEs | |
| CT‐P13 → CT‐P13 | 71.7/48.0/24.3 | 85/159 (53.5) | 64/159 (40.3) |
| Reference → CT‐P13 | 71.8/51.4/26.1 | 77/143 (53.8) | 64/143 (44.8) |
| BOW015 study OLE27, 28, 29 | |||
| BOW015 → BOW015 | ACR20 rate similar across groupsc | Not reported | Not reported |
| Reference → BOW015 | |||
| Ankylosing spondylitis | |||
| PLANETAS OLE26 | ASAS 20/40 rate, % | TEAEs | |
| CT‐P13 → CT‐P13 | 80.7/63.9 | 44/90 (48.9) | 21/90 (23.3) |
| Reference → CT‐P13 | 76.9/61.5 | 60/84 (71.4) | 23/84 (27.4) |
ADA, anti‐drug antibody; ACR, American College of Rheumatology; AE, adverse event; ASAS, Assessment of Spondyloarthritis International Society; DAS‐28 ESR, Disease Activity Score 28‐joint erythrocyte sedimentation rate; OLE, open label extension; RCT, randomized, controlled trial; SD, standard deviation; TEAE, treatment‐emergent AE.
Based on disease‐specific composite measures or a consensus between investigator and patient.
Specific values not available.
Excluding patients with ADA at baseline.
The SB2 transition trial was a 78‐week randomised, double‐blind, phase 3 single transition study comparing the safety and efficacy of reference infliximab with the biosimilar SB2 in patients with moderate to severe RA despite methotrexate (Figure 3A).21 In the initial phase of the study, patients were randomised 1:1 to receive either reference infliximab or SB2. At week 54, patients receiving reference infliximab were re‐randomised 1:1 to receive reference infliximab (INF/INF) or SB2 (INF/SB2) up to week 78. At the transition, a total of 94 patients were in the INF/SB2 group, 101 in the INF/INF group, and 201 in the SB2/SB2 group. Overall American College of Rheumatology (ACR) 20, ACR50, and ACR70 response rates during the transition period were similar across all three groups. The overall incidence of adverse events (AEs) during the transition period was similar in the INF/SB2 (36.2%), INF/INF (35.6%), and SB2/SB2 (40.3%) groups. Furthermore, among the patients that were negative for ADA up until the transition at week 54, 14.6% of patients in the INF/SB2 developed ADA in the transition period, compared with 14.9% in the INF/INF group and 14.1% in the SB2/SB2 group. These data do not indicate a change in safety, efficacy, or immunogenicity in patients that underwent a single switch from reference infliximab to SB2 for 24 weeks compared to those that remained on SB2 or reference infliximab.
The CT‐P13 Japanese RA switch study was a single‐arm OLE of a phase 1/2 study comparing the safety and efficacy of CT‐P13 with reference infliximab in Japanese patients with RA (Figure 3B).24 All patients being treated with CT‐P13 (maintenance group; n = 38) or reference infliximab (switch group; n = 33) in the 54‐week main study were switched to CT‐P13 in the OLE. At week 134, the mean (SD) 28‐joint count (DAS28) scores were 3.166 (1.533) and 3.955 (1.751) in the maintenance and switch groups, respectively. The incidence of AEs was comparable in the maintenance (34/38, 89.5%) and switch (29/33, 87.9%) groups during the OLE. However, the number of AEs leading to discontinuation were higher in the switch arm (8/33, 24.2%) than the maintenance arm (4/38, 10.5%). At week 134, the number of patients that were ADA positive was comparable in the maintenance (5/32, 15.6%) and switch (4/23, 17.4%) groups.
PLANETRA was a 54‐week, randomised, parallel‐group, multicentre, phase 3 study comparing the safety and efficacy of CT‐P13 with reference infliximab in patients with RA (Figure 3C). In the OLE, all patients were switched to CT‐P13 while remaining blinded to the initial study treatment.25 At week 102, ACR20, ACR50, and ACR70 response rates were 71.7%, 48.0%, and 24.3% in the maintenance group compared to 71.8%, 51.4%, and 21.6% in the switch group. The incidence of treatment emergent adverse events (TEAEs) was comparable in the maintenance (85/159, 53.5%) and switch group (77/143, 53.8%). At week 102, the number of patients that were ADA positive was similar in the maintenance (64/159, 40.3%) and switch (64/143, 44.8%) groups (P = 0.48).
PLANETAS was a 54‐week, randomised, parallel‐group, multicentre study comparing the safety and efficacy of CT‐P13 with reference infliximab in patients with AS (Figure 3D). In the OLE, all patients were switched to CT‐P13 while remaining blinded to the initial study treatment.26 At week 102, Assessment of Spondyloarthritis International Society (ASAS) 20 and ASAS40 response rates were 80.7% and 63.9% in the maintenance group compared to 76.9% and 61.5% in the switch group. The incidence of TEAEs were lower in the maintenance group (44/90, 48.9%) than the switch group (60/84, 71.4%). However, no statistical comparisons of the incidence of TEAEs between groups were performed (ie, not a pre‐specified endpoint). The rates of TEAEs leading to discontinuation were similar in both groups. At week 102, the number of patients that were ADA positive was similar in the maintenance (21/90, 23.3%) and switch groups (23/84, 27.4%; P = 0.60).
In another phase 3 study, patients with severe to moderate RA receiving a stable dose of methotrexate were randomised 2:1 to with BOW015 or reference infliximab for 16 weeks (Figure 3E). In the OLE, all patients were switched to BOW015 until week 54.27, 28, 29 At week 54, primary (ACR20) and secondary (eg, C‐reactive protein, tender joint count, swollen joint count) endpoints were comparable across both treatment groups.27 Measures of disease activity and disability were also similar across both treatment groups.28 ACR20 response rates were similar in the maintenance (N = 104) and switch groups (N = 53) in both severe and moderate RA patient subgroups.29 Safety and immunogenicity data were not reported.
NOR‐SWITCH was a randomised, double‐blind, non‐inferiority, phase 4 transition trial with 52 weeks of follow‐up (Figure 3F). Adult patients with RA, spondyloarthritis, PsA, UC, CD, and PsO on stable treatment with reference infliximab for at least 6 months were randomised 1:1 to continue on reference infliximab or switch to the biosimilar CT‐P13 for 52 weeks with no change in dosing regimen.22 Data were collected from 19 gastroenterology departments, 16 rheumatology departments, and five dermatology departments in 25 Norwegian hospitals.22 Of the 481 patients in the full analysis set, 155 (32%) had CD, 93 (19%) had UC, 91 (19%) had AS, 77 (16%) had RA, 30 (6%) had PsA, and 35 (7%) had PsO.22 The primary endpoint of the study was proportion of patients with pre‐defined disease worsening during the 52‐week follow‐up period based on disease‐specific composite measures or a consensus between investigator and patient leading to major change in treatment.22 Disease worsening occurred in 26.2% (53/202) and 29.6% (61/206) of the patients in the reference infliximab and CT‐P13 groups, respectively. The adjusted‐risk difference was −4.4 with a 95% confidence interval (CI) of −12.7 to 3.9, which was within the pre‐specified 95% CI non‐inferiority margin of 15%.22 Non‐inferiority was not demonstrated in any of the diagnostic subgroups, except for spondyloarthritis, although the study was not powered for specific disease evaluation.22 Additionally, the number of patients with TEAEs were similar across the two treatment groups (reference infliximab, 70%; CT‐P13, 68%) and the incidence of ADA (excluding patients positive at baseline) were comparable (reference infliximab, 7%; CT‐P13, 8%).22, 30
Further exploratory analyses of safety, efficacy, and immunogenicity, in spondyloarthritis, RA, CD, and UC patient subgroups in NOR‐SWITCH were also conducted.31, 32, 33 In the largest of these cohorts (CD; N = 129), disease worsening occurred in more frequently in the switch arm (36.5%) than the maintenance arm (21.2%; 95% CI of adjusted difference, −29.3% to 0.7%).33 Among the other patient subgroups, disease worsening, immunogenicity, and safety profiles were similar across the reference infliximab and CT‐P13 groups.31, 32 However, these sub‐studies were not powered to evaluate non‐inferiority.31, 32, 33
In the OLE of NOR‐SWITCH, all patients were switched to CT‐P13 for an additional 26 weeks.23 At week 52, disease worsening occurred in 11.6% of patients that switched from reference infliximab to CT‐P13 and 16.8% of patients that maintained CT‐P13 treatment (risk difference [95% CI], −5.9% [−12.9, 1.1]).23 During the extension phase, the rate of ADA and AEs were similar in both treatment groups.23
The quality of RCT reporting was assessed against the 2010 CONSORT standards (Table S2). All the publications were generally consistent with CONSORT guidelines, reporting on the majority of the CONSORT recommendation items. The phase 3 study of BOW015 has not been reported in full publication and the abstracts were not included in the CONSORT evaluation.
3.3. Observational studies
A total of 53 publications reported on switching to infliximab biosimilars in observational studies (Table S3). All studies described a single transition from reference to biosimilar infliximab. The majority (n = 47), reported on switching from reference infliximab to CT‐P13, while the remaining publications (n = 6) did not specify the biosimilar. No studies were found on switching to other specific biosimilars (eg, SB2, BOW015, PF‐06438791, NI‐071). A concurrent control arm (eg, a group that maintained treatment with reference infliximab) was reported in only three of the publications, which are described in greater detail here. There were limited commonalities among these observational studies, with variable assessments, follow‐up times, and patient populations.
In one controlled prospective cohort analysis, immunogenicity was compared in patients that switched from reference infliximab to CT‐P13 (n = 18) to those that maintained treatment with reference infliximab (n = 30) or CT‐P13 (n = 52).34 During follow‐up, 43% (13/30) of patients in the reference infliximab maintenance group, 17% (9/52) in the CT‐P13 maintenance group, and 67% (12/18) in the switch group developed ADA (P = 0.5). Notably, the follow‐up time in this abstract was not well‐defined, the study was inadequately powered to detect differences in ADA rates, and assignment to treatment was not randomised.
In a large, controlled review of a Turkish healthcare administrative database, safety was compared in patients that switched from reference infliximab to CT‐P13 (n = 136) to those that continued to receive reference infliximab (n = 1388).35 The adjusted incidence rate ratio (95% CI) for AEs between the groups was 0.67 (0.19, 2.30), indicating no statistically significant difference. However, the rate of discontinuations was higher in the switch group (13.2 per 1000 patient‐years [PY]) than the reference infliximab maintenance group (1.52 per 1000 PY). After adjusting for baseline characteristics, patients in the switch group were significantly more likely to discontinue treatment than those in the infliximab maintenance group (hazard ratio [HR], 5.53 [95% CI, 4.01, 7.63]). Of those patients that discontinued CT‐P13 treatment, 79% switched back to reference infliximab. Reasons for discontinuation were not captured in this study.
In a small, controlled, single‐centre retrospective, observational study of patients with RA, PsA, and AS, safety and efficacy were compared in those that switched from reference infliximab to CT‐P13 (n = 7) to those that maintained treatment with reference infliximab (n = 6).36 Switching did not appear to be related to changes in safety or efficacy in these patients.
Most of the uncontrolled studies did not report any changes in safety, efficacy, or immunogenicity following a switch to biosimilar infliximab. While several uncontrolled studies did report high rates of failure or discontinuation after transition (Table S3), this was not consistently observed across studies. Two independent studies indicated a high failure rate (23% [12/53]; 27% [36/131]) of CT‐P13 in patients with spondyloarthritis after switching from reference infliximab.37, 38
3.4. Case series/reports
Two case studies were identified regarding AEs of lichenoid drug eruption and serum sickness‐like disease reported in patients with CD and RA, respectively, after switching to CT‐P13 (Table S4).39, 40 A case series of three patients with Behçet's disease reported on disease relapse after switching from reference infliximab to CT‐P13.41 An additional case series was found which indicated that safety and efficacy was maintained in nine patients with IBD that switched from reference infliximab to CT‐P13.42
4. DISCUSSION
While many studies (N = 70) have investigated the safety, efficacy, or immunogenicity of switching from reference to biosimilar infliximab, the overall quality of the evidence is weak, as the majority of publications were based on uncontrolled observational studies that only investigated a single switch. Notably, only six RCTs were identified that involved switching from reference to biosimilar infliximab. FDA guidance states that biosimilar interchangeability can be demonstrated using either an equivalence study design or a non‐inferiority study design.13 However, the sample size requirements for these studies are large if clinically meaningful differences are to be excluded with a high degree of confidence. In our opinion, this criterion has not been met in the switching studies that were identified in this review. Equivalence studies are especially problematic because although they can evaluate the possibility that a biosimilar actually has superior efficacy to the reference product (a so‐called “bio‐better”),43 the very large sample sizes mandated by this design make them, for the most part, impractical. None of the RCTs identified reported on equivalence of switching and only one study (NOR‐SWITCH) reported on non‐inferiority of switching.22 In many of the RCTs, a switch from reference infliximab to biosimilar occurred in an OLE which were not powered to detect non‐inferiority or equivalence.
While the results of most of the uncontrolled, observational studies suggested that switching between reference and biosimilar infliximab products is safe and efficacious, the lack of a control arm, where patients maintain treatment with reference infliximab, makes it difficult to appropriately interpret the results. The largest controlled study found was an observational study of 1524 patients, which showed the rate of discontinuation was significantly higher in patients that switched from reference infliximab to CT‐P13 compared to those maintained on reference infliximab.35 However, these results should also be interpreted with caution as it was not a randomised study and the reasons for switching patients were not defined. It is possible that factors related to the decision to make a switch, for example (eg, disease activity) may have cofounded the results.
Different patient groups may also have different responses to switching between reference and biosimilar infliximab. For example, two independent studies indicated a high failure rate of CT‐P13 in patients with spondyloarthritis following a switch from reference infliximab.37, 38 While this trend was not observed in most other patient populations, it has been suggested that the nocebo effect (eg, perceived or unexplained detrimental therapeutic effect) may influence the success of a non‐medical infliximab switch.44
FDA guidance states that interchangeability studies should have at least three switches between products for at least two exposure periods with each drug.13 Multiple switching may be more reflective of real‐world situations where patients may repeatedly switch between the biosimilar and the reference product.12 Switching among biosimilars is likely to occur in the real‐world, and it may be important to consider the appropriateness of such scenarios. Frequent switching between biosimilars and reference biologics products may trigger an immunological response.45 All of the studies discussed here reported on only a single switch from reference infliximab to a biosimilar and may not be reflective of switching in a real‐world situation.
Anti‐drug antibodies can develop in patients who are being treated with biologics, including infliximab.46 Therapeutic antibodies can bind to ADAs, forming immune complexes which may ultimately result in lower trough drug concentrations and decreased efficacy.47, 48 Some studies indicate that ADAs may develop in approximately half of patients during long‐term infliximab treatment.49, 50, 51 Theoretically, subtle differences in the manufacturing process or glycosylation pattern of a biosimilar and the reference product may result in different immunological response.52 However, no clinically significant differences in the immunogenicity of biosimilars and reference products have been reported in the “one way” switching studies that have been performed to date.53 Whether performance of multiple switches between products is a risk factor for immunogenicity is unknown and randomised, controlled non‐inferiority studies featuring multiple switches between reference product and biosimilar are lacking. However, an ongoing study investigating the switching of reference infliximab with NI‐071 has been reported to be designed to demonstrate interchangeability.54 While studies evaluating multiple switches have not been completed for infliximab biosimilars, they have been conducted for biosimilars of other TNF blockers (eg, etanercept and adalimumab).55, 56, 57
A recently published consensus document by the Task Force on the Use of Biosimilars to Treat Rheumatological Diseases states that the currently available scientific evidence indicates that a single switch from a reference to a biosimilar is safe and effective.53 However, the lack of evidence to support switching between different biosimilars or multiple switches was also noted. It was suggested that post‐marketing pharmacovigilance should be carried out using registries and long‐term, observational studies to evaluate the safety and efficacy of multiple switches in the real world.
Overall, the evidence presented is this review is generally supportive of the safety and efficacy of one‐time switching between reference and biosimilar infliximab. However, after conducting this systematic review of the literature, we concluded that there was insufficient data to perform a meaningful meta‐analysis at this time. Additionally, no studies have investigated multiple switching scenarios or switching between biosimilars, which may be representative of real‐world situations. Higher quality data based upon the performance of multiple‐switch studies are needed to validate the concept of interchangeability.
AUTHORSHIP
Guarantor of the article: BF, GL, GRL.
Author contributions: BF, GL, GRL: conception and design. BF, GL, CM, GRL: critical revision of the manuscript for important intellectual content and final approval.
Supporting information
ACKNOWLEDGEMENTS
Declaration of personal interests: Dr. Feagan has received consulting fees from Abbott/AbbVie, Ablynx, Akebia Therapeutics, Allergan, Amgen, Applied Molecular Transport Inc., Aptevo Therapeutics, Astra Zeneca, Atlantic Pharma, Avir Pharma, Baxter Healthcare Corp., Biogen Idec, Boehringer‐Ingelheim, Bristol‐Myers Squibb, Calypso Biotech, Celgene, Elan/Biogen, EnGene, Ferring Pharma, Roche/Genentech, Galapagos, GiCare Pharma, Gilead, Given Imaging Inc., GSK, Inception IBD Inc, Ironwood Pharma, Janssen Biotech (Centocor), JnJ/Janssen, Kyowa Kakko Kirin Co Ltd., Lexicon, Lilly, Lycera BioTech, Merck, Mesoblast Pharma, Millennium, Nektar, Nestles, Nextbiotix, Novonordisk, Pfizer, Prometheus Therapeutics and Diagnostics, Progenity, Protagonist, Receptos, Roche/Genentech, Salix Pharma, Serano, Shire, Sigmoid Pharma, Synergy Pharma Inc., Takeda, Teva Pharma, TiGenix, Tillotts, UCB Pharma, Vertex Pharma, Vivelix Pharma, VHsquared Ltd., Warner‐Chilcott, Wyeth, Zealand, Zyngenia; has received grant/research support from AbbVie Inc., Amgen Inc., AstraZeneca/MedImmune Ltd., Atlantic Pharmaceuticals Ltd., Boehringer‐Ingelheim, Celgene Corporation, Celltech, Genentech Inc/Hoffmann‐La Roche Ltd., Gilead Sciences Inc., GlaxoSmithKline (GSK), Janssen Research & Development LLC., Pfizer Inc., Receptos Inc. / Celgene international, Sanofi, Santarus Inc., Takeda Development Center Americas Inc., Tillotts Pharma AG, and UCB; has served as a Abbott/AbbVie, Allergan, Amgen, Astra Zeneca, Atlantic Pharma, Avaxia Biologics Inc., Boehringer‐Ingelheim, Bristol‐Myers Squibb, Celgene, Centocor Inc., Elan/Biogen, Ferring, Galapagos, Genentech/Roche, JnJ/Janssen, Merck, Nestles, Novartis, Novonordisk, Pfizer, Prometheus Laboratories, Protagonist, Salix Pharma, Takeda, Teva, TiGenix, Tillotts Pharma AG, and UCB Pharma; has been on the Speakers Bureau for Abbott/AbbVie, JnJ/Janssen, Lilly, Takeda, Tillotts, and UCB Pharma; and is a member of the Board of Directors for Robarts Clinical Trials Inc, Western University, London. Dr. Lam has served on the Speaker Bureau and Advisory Board for Abbvie, Bristol Myers Squibb, Jannsen, and UCB. Dr. Lichtenstein has served as a consultant for Abbott Corporation/Abbvie, Actavis, Alaven, Celgene, Ferring, Gilead, Hospira, Janssen Biotech, LLC, Luitpold/American Regent, Merck, Pfizer Pharmaceuticals, Prometheus Laboratories, Inc., Romark, Salix Pharmaceuticals/Valeant, Santarus/Receptos, Shire Pharmaceuticals, Takeda, and UCB; has received research grants from Celgene, Janssen Biotech, LLC, Salix Pharmaceuticals/Valeant, Santarus/Receptos, Shire Pharmaceuticals, and UCB; and has received honoraria for Continuing Medical Education Programs from Ironwood Pharma, Luitpold/American Regent, Merck, and Romark. Dr. Ma, none.
Declaration of funding interests: Editorial assistance was provided by Scott Houck and Rebecca Slager (Ashfield Healthcare Communications, a UDG Healthcare plc company), Lyndhurst, NJ, USA, and funded by Janssen Pharmaceuticals.
Feagan BG, Lam G, Ma C, Lichtenstein GR. Systematic review: efficacy and safety of switching patients between reference and biosimilar infliximab. Aliment Pharmacol Ther. 2019;49:31–40. 10.1111/apt.14997
Brian G. Feagan, Gordon Lam and Gary R. Lichtenstein contributed equally to this work.
The Handling Editor for this article was Professor Jonathan Rhodes, and this uncommissioned review was accepted for publication after full peer‐review.
REFERENCES
- 1. Conner J, Wuchterl D, Lopez M, et al. The Biomanufacturing of Biotechnology Products. Biotechnology Entrepreneurship. Cambridge, MA: Elsevier;2014:351‐385. [Google Scholar]
- 2. Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs. 2015;7:9‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morrow T, Felcone LH. Defining the difference: what makes biologics unique. Biotechnol Healthc. 2004;1:24‐29. [PMC free article] [PubMed] [Google Scholar]
- 4. Kuhlmann M, Covic A. The protein science of biosimilars. Nephrol Dial Transplant. 2006;21(Suppl. 5):v4‐v8. [DOI] [PubMed] [Google Scholar]
- 5. Calo‐Fernandez B, Martinez‐Hurtado JL. Biosimilars: company strategies to capture value from the biologics market. Pharmaceuticals. 2012;5:1393‐1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krishnan A. Global regulatory landscape of biosimilars: emerging and established market perspectives. Biosimilars. 2015;5:19‐32. [Google Scholar]
- 7. McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs. 2011;3:209‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. US Food and Drug Administration . CDER List of Licensed Biological Products. Rockville, MD: US FDA; 2018. Accessed at https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/UCM560162.pdf [Google Scholar]
- 9. US Food and Drug Administration . Biosimilar Product Information. Rockville, MD: US Food and Drug Administration; 2018. Accessed at https://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm580432.htm [Google Scholar]
- 10. European Medicines Agency . Guideline on Similar Biological Medicinal Products Containing Biotechnology‐Derived Proteins as Active Substance: Non‐Clinical and Clinical Issues. London, UK: Canary Wharf; 2014. [Google Scholar]
- 11. US Food and Drug Administration . Guidance for Industry: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. Rockville, MD: US Food and Drug Administration; 2012. Accessed at https://www.biologicsblog.com/content/uploads/2016/09/FDA-Draft-Guidance-Scientific-Considerations.pdf [Google Scholar]
- 12. Faccin F, Tebbey P, Alexander E, Wang X, Cui L, Albuquerque T. The design of clinical trials to support the switching and alternation of biosimilars. Expert Opin Biol Ther. 2016;16:1445‐1453. [DOI] [PubMed] [Google Scholar]
- 13. US Food and Drug Administration . Considerations in Demonstrating Interchangeability with a Reference Product. Rockville, MD: US Food and Drug Administration; 2017. Accessed at https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM537135.pdf [Google Scholar]
- 14. Remicade PI. (Janssen Biotech, Inc) 2013. http://www.remicade.com/shared/product/remicade/prescribing-information.pdf
- 15. Johnson & Johnson . Two studies reveal differences in real‐world treatment patterns in rheumatoid arthritis patients taking Remicade® (infliximab) compared to Ct‐P13 (infliximab biosimilar), 2016. www.jnj.com/media-center/press-releases/two-studies-reveal-differences-in-real-world-treatment-patterns-in-rheumatoid-arthritis-patients-taking-remicade-infliximab-compared-to-ct-p13-infliximab-biosimilar [press release].
- 16. Park W, Hrycaj P, Jeka S, et al. A randomised, double‐blind, multicentre, parallel‐group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT‐P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72:1605‐1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double‐blind, parallel‐group study to demonstrate equivalence in efficacy and safety of CT‐P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72:1613‐1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choe JY, Prodanovic N, Niebrzydowski J, et al. A randomised, double‐blind, phase III study comparing SB2, an infliximab biosimilar, to the infliximab reference product Remicade in patients with moderate to severe rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2017;76:58‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cohen SB, Alten R, Kameda H, et al. A randomized controlled trial comparing PF‐06438179/GP1111 (an infliximab biosimilar) and infliximab reference product for treatment of moderate to severe active rheumatoid arthritis despite methotrexate therapy. Arthritis Res Ther. 2018;20:155 10.1186/s13075-018-1646-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schulz KF, Altman DG, Moher D. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ. 2010;340:c332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smolen JS, Choe JY, Prodanovic N, et al. Safety, immunogenicity and efficacy after switching from reference infliximab to biosimilar SB2 compared with continuing reference infliximab and SB2 in patients with rheumatoid arthritis: results of a randomised, double‐blind, phase III transition study. Ann Rheum Dis. 2018;77:234‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jorgensen KK, Olsen IC, Goll GL, et al. Switching from originator infliximab to biosimilar CT‐P13 compared with maintained treatment with originator infliximab (NOR‐SWITCH): a 52‐week, randomised, double‐blind, non‐inferiority trial. Lancet. 2017;389:2304‐2316. [DOI] [PubMed] [Google Scholar]
- 23. Goll GL, Jorgensen KK, Sexton J, et al. Long‐term safety and efficacy of biosimilar infliximab (CT‐P13) after switching from originator infliximab: results from the 26‐week open label extension of a randomized Norwegian trial. Arthritis Rheumatol. 2017;69:1383‐1384. [Google Scholar]
- 24. Tanaka Y, Yamanaka H, Takeuchi T, et al. Safety and efficacy of CT‐P13 in Japanese patients with rheumatoid arthritis in an extension phase or after switching from infliximab. Mod Rheumatol. 2017;27:237‐245. [DOI] [PubMed] [Google Scholar]
- 25. Yoo DH, Prodanovic N, Jaworski J, et al. Efficacy and safety of CT‐P13 (biosimilar infliximab) in patients with rheumatoid arthritis: comparison between switching from reference infliximab to CT‐P13 and continuing CT‐P13 in the PLANETRA extension study. Ann Rheum Dis. 2017;76:355‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Park W, Yoo DH, Miranda P, et al. Efficacy and safety of switching from reference infliximab to CT‐P13 compared with maintenance of CT‐P13 in ankylosing spondylitis: 102‐week data from the PLANETAS extension study. Ann Rheum Dis. 2017;76:346‐354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kay J, Chopra A, Chandrashekara S, et al. AB0420 secondary efficacy outcomes from a phase 3 study support clinical equivalence between BOW015 and infliximab in patients with active rheumatoid arthritis on stable methotrexate doses. Ann Rheum Dis. 2015;74(Suppl. 2):1034. [Google Scholar]
- 28. Kay J, Chopra A, Lassen C, Shneyer L, Wyand M. FRI0117 BOW015, a biosimilar infliximab: disease activity and disability outcomes from a phase 3 active comparator study in patients with active rheumatoid arthritis on stable methotrexate doses. Ann Rheum Dis. 2015;74(Suppl. 2):462‐463. [Google Scholar]
- 29. Taylor P, Wyand M, Knight A, Costantino C, Lassen C. FRI0163 Efficacy of the biosimilar BOW015, compared to originator infliximab, initiated at moderate and severe disease activity thresholds in rheumatoid arthritis. Ann Rheum Dis. 2016;75(Suppl. 2):488‐489. [Google Scholar]
- 30. Goll G, Olsen I, Lundin K, et al. THU0700 Immunogenicity in patients switching from stable originator infliximab treatment to CT‐P13: analyses across six diseases from the 52‐week randomized nor‐switch study. Ann Rheum Dis. 2017;76(Suppl. 2):472. [Google Scholar]
- 31. Goll G, Olsen I, Bolstad N, et al. FRI0182 Disease worsening and safety in patients switching from originator infliximab to biosimilar infliximab (CT‐P13) in the nor‐switch study: explorative analysis of RA patients. Ann Rheum Dis. 2017;76(Suppl. 2):549‐550. [Google Scholar]
- 32. Goll G, Olsen I, Bolstad N, et al. THU0354 Disease worsening and safety in patients switching from originator infliximab to biosimilar infliximab (CT‐P13) in the randomized nor‐switch‐study: explorative analysis in SPA patients. Ann Rheum Dis. 2017;76(Suppl. 2):338‐339. [Google Scholar]
- 33. Jorgensen K, Olsen I, Goll G, et al. Biosimilar infliximab (CT‐P13) is not inferior to originator infliximab: explorative IBD subgroup‐analyses in Crohn's disease and ulcerative colitis from the NOR‐SWITCH trial. J Crohns Colitis. 2017;11:S62‐S63. [Google Scholar]
- 34. Fiorino G, Radice S, Gilardi D, et al. P475 Switching from infliximab originator to CT‐P13 is not related to increased immunogenicity in IBD patients: a prospective case‐control study. J Crohns Colitis. 2017;11(Suppl. 1):S320. [Google Scholar]
- 35. Phillips K, Juday T, Zhang Q, Keshishian A. SAT0172 Economic outcomes, treatment patterns, and adverse events and reactions for patients prescribed infliximab or ct‐p13 in the Turkish population. Ann Rheum Dis. 2017;76(Suppl. 2):835. [Google Scholar]
- 36. Vergara‐Dangond C, Saez Bello M, Climente Marti M, Llopis Salvia P, Alegre‐Sancho JJ. Effectiveness and safety of switching from innovator infliximab to biosimilar CT‐P13 in inflammatory rheumatic diseases: a real‐world case Study. Drugs R D. 2017;17:481‐485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Akrout W, Bosycot A, Levet‐Labry R, Gazaix‐Fontaine E, Paul M, Claudepierre P. AB0708 Transition from ongoing infliximab reference product to its biosimilar: can we talk about a failure? Ann Rheum Dis. 2017;76(Suppl. 2):1301‐1302. [Google Scholar]
- 38. Avouac J, Molto A, Abitbol V, et al. Systematic switch from innovator infliximab to biosimilar infliximab in inflammatory rheumatic diseases in daily clinical practice: the experience of Cochin hospital, Paris, France. Ann Rheum Dis. 2017;76:831‐832. [DOI] [PubMed] [Google Scholar]
- 39. Gonzalez N, Patel P, Han G. A dissimilar biosimilar? Lichenoid drug eruption induced by an infliximab biosimilar. Br J Dermatol. 2018;178:965‐968. 10.1111/bjd.15686 [DOI] [PubMed] [Google Scholar]
- 40. Scherlinger M, Schaeverbeke T, Truchetet ME. Serum sickness‐like disease after switching to biosimilar infliximab. Rheumatology. 2017;56:2032‐2034. [DOI] [PubMed] [Google Scholar]
- 41. Cantini F, Niccoli L, Nannini C, Cassara E, Kaloudi O. Rapid loss of efficacy of biosimilar infliximab in three patients with Behcet's disease after switching from infliximab originator. Eur J Rheumatol. 2017;4:288‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kang YS, Moon HH, Lee SE, Lim YJ, Kang HW. Clinical experience of the use of CT‐P13, a biosimilar to infliximab in patients with inflammatory bowel disease: a case series. Dig Dis Sci. 2015;60:951‐956. [DOI] [PubMed] [Google Scholar]
- 43. Lai Z, La Noce A. Key design considerations on comparative clinical efficacy studies for biosimilars: adalimumab as an example. RMD Open. 2016;2:e000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Boone NW, Liu L, Romberg‐Camps MJ, et al. The nocebo effect challenges the non‐medical infliximab switch in practice. Eur J Clin Pharmacol. 2018;74:655‐661. 10.1007/s00228-018-2418-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dorner T, Strand V, Cornes P, et al. The changing landscape of biosimilars in rheumatology. Ann Rheum Dis. 2016;75:974‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vincent FB, Morand EF, Murphy K, Mackay F, Mariette X, Marcelli C. Antidrug antibodies (ADAb) to tumour necrosis factor (TNF)‐specific neutralising agents in chronic inflammatory diseases: a real issue, a clinical perspective. Ann Rheum Dis. 2013;72:165‐178. [DOI] [PubMed] [Google Scholar]
- 47. vanKuijk AW , deGroot M , Stapel SO, Dijkmans BA, Wolbink GJ, Tak PP. Relationship between the clinical response to adalimumab treatment and serum levels of adalimumab and anti‐adalimumab antibodies in patients with psoriatic arthritis. Ann Rheum Dis. 2010;69:624‐625. [DOI] [PubMed] [Google Scholar]
- 48. Karmiris K, Paintaud G, Noman M, et al. Influence of trough serum levels and immunogenicity on long‐term outcome of adalimumab therapy in Crohn's disease. Gastroenterology. 2009;137:1628‐1640. [DOI] [PubMed] [Google Scholar]
- 49. Menter A, Feldman SR, Weinstein GD, et al. A randomized comparison of continuous vs. intermittent infliximab maintenance regimens over 1 year in the treatment of moderate‐to‐severe plaque psoriasis. J Am Acad Dermatol. 2007;56:31.e31‐15. [DOI] [PubMed] [Google Scholar]
- 50. Vermeire S, Noman M, van Assche G, Baert F, D'Haens G, Rutgeerts P. Effectiveness of concomitant immunosuppressive therapy in suppressing the formation of antibodies to infliximab in Crohn's disease. Gut. 2007;56:1226‐1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Baert F, Noman M, Vermeire S, et al. Influence of immunogenicity on the long‐term efficacy of infliximab in Crohn's disease. N Engl J Med. 2003;348:601‐608. [DOI] [PubMed] [Google Scholar]
- 52. Mellstedt H. Clinical considerations for biosimilar antibodies. EJC Suppl. 2013;11:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kay J, Schoels MM, Dorner T, et al. Consensus‐based recommendations for the use of biosimilars to treat rheumatological diseases. Ann Rheum Dis. 2017;77:165‐174. [DOI] [PubMed] [Google Scholar]
- 54. The Center for Biosimilars . Eye on pharma: Nichi‐Iko seeks interchangeable designation for NI‐071, 2017. www.centerforbiosimilars.com/news/eye-on-pharma-nichiiko-seeks-interchangeable-designation-for-ni071
- 55. Gerdes S, Thaci D, Griffiths CEM, et al. Multiple switches between GP2015, an etanercept biosimilar, with originator product do not impact efficacy, safety and immunogenicity in patients with chronic plaque‐type psoriasis: 30‐week results from the phase 3, confirmatory EGALITY study. J Eur Acad Dermatol Venereol. 2018;32:420‐427. 10.1111/jdv.14605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Griffiths CEM, Thaci D, Gerdes S, et al. The EGALITY study: a confirmatory, randomized, double‐blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, vs. the originator product in patients with moderate‐to‐severe chronic plaque‐type psoriasis. Br J Dermatol. 2017;176:928‐938. [DOI] [PubMed] [Google Scholar]
- 57. EU Clinical Trials Register . A phase 3 randomized, double‐blind, multicenter study to evaluate efficacy, safety, and immunogenicity of an adalimumab biosimilar (M923) and Humira® in subjects with moderate to severe chronic plaque‐type psoriasis. http://www.clinicaltrialsregister.eu/ctr-search/trial/2015-001751-76/SK.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials