

Abstract
Langerhans cell histiocytosis (LCH) is the most common histiocytosis with constitutive activation of the RAS–RAF–MEK–ERK (MAPKinase) cell signaling pathway. We analyzed 89 cases of BRAF and MAP2K1 mutations by Sanger sequencing, of which 18 cases showed that these two gene mutations are negative. Whole genome sequencing of suitable specimens in these negative cases revealed a translocation from the 3 intron of PLEKHA6 to the 13 intron of NTRK3 in one case. We identified that this translocation could cause a novel fusion mutation, PLEKHA6‐NTRK3. Overexpression of the PLEKHA6‐NTRK3 mutant in NIH 3T3 cells enhanced MAPKinase pathway activation, promote cell growth. Our result suggested that a new mutation need be included in LCH molecular screening panel to better define its prevalence in LCH.
Keywords: PLEKHA6‐NTRK3, langerhans cell histiocytosis, fusion mutation, Erk pathway
Short abstract
What's new?
Langerhans cell histiocytosis (LCH) is a rare immune and neoplastic disorder. While it is known as the most common histiocytosis with constitutive activation of the RAS‐RAF‐MEK‐ERK (MAPKinase) cell signaling pathway, its pathogenesis remains obscure. Here, whole‐genome sequencing of BRAF V600E‐negative and MAP2K1‐negative LCH cases revealed a translocation from the intron 3 of PLEKHA6 to the intron 13 of NTRK3 in one patient, identifying a novel fusion mutation. Overexpression of PLEKHA6‐NTRK3 in vitro enhanced MAPKinase pathway activation, promoting cell growth. The results support the inclusion of the fusion mutation in LCH molecular screening panel to better define its prevalence in patients.
Introduction
Langerhans cell histiocytosis (LCH) is the most common histiocytosis and is characterized by inflammatory lesions with abundant CD1a+CD207+ histiocytes, which lead to the destruction of affected tissues.1, 2, 3
In 2010, high prevalence of oncogenic V‐Raf murine sarcoma viral homolog B1 (BRAFV600E) mutations (about 55% of LCH), which is responsible for activation of the MAPKinase RAS–RAF–MEK–ERK cell signaling pathway in pathologic histiocytes, was firstly discovered in LCH cells.4 Responses to BRAF inhibitors in patients with BRAF V600E‐mutated LCH confirmed that BRAF V600E is a driver mutation in LCH.5, 6, 7 But it was found that LCH patients do not have BRAF V600E mutation, the ERK pathway was also reported to be activated in pathologic histiocytes.4, 8, 9, 10 Then the mutations in MAPkinase pathway relate genes were further identified, including MAP2K1 mutations (10–20% of LCH),11, 12, 13 β3‐αC loop deletion in the kinase domain of BRAF (6% of LCH),14 and case reports about ARAF and MAP3K1 mutation.8, 15 In addition, recurrent kinase fusions involving BRAF, ALK, and NTRK1 were found to be activated the MAPkinase pathway in LCH.14, 16
To analyze the mechanism of pathologic ERK activation in LCH, we performed Sanger sequencing across 89 pediatric LCH patients diagnosed in our hospital to specifically detect the BRAF V600E and MAP2K1 mutations. The double‐negative cases were performed by the whole‐genome sequencing to further reveal the underlying pathogenesis of LCH, and a novel translocation involving PLEKHA6 and NTRK3 was identified in one case.
Material and Methods
Patients and samples
The 89 pediatric patients were histologically confirmed LCH in Shanghai Children's Medical Center (SCMC) between September 2008 and March 2015, and the age of onset ranged from 2 month to 13 years old. The ratio of males and females were 2:1 which was consistent with previous reports.17 The bone was involved in about 91% of pediatric patients with LCH, followed by soft tissue (8%), liver (8%), skin (6%) and lymph node (6%), and other organs. Clinical staging data were available for all patients, most patients were SS‐LCH (85%) (Table 1).
Table 1.
Clinical features of patients with LCH
| Total | BRAF mutated | MAP2K1 mutated | PLEKHA6‐NTRK3 | |
|---|---|---|---|---|
| n | 89 | 37/89(41.6%) | 34/89(38.2%) | 1/89(1%) |
| Age(y)* | 2(0.2–13) | 2(0.3–11) | 2(0.2–11) | 2 |
| Gender, n(%) | ||||
| Male | 59(66.3%) | 24 | 24 | 1 |
| Female | 30(33.7%) | 13 | 10 | 0 |
| Disease site, n (%) | ||||
| Bone | 81(91%) | 33(89%) | 32(94%) | 1(100%) |
| Skin | 5(6%) | 3(8%) | 2(6%) | 0 |
| Soft tissue | 7(8%) | 3(8%) | 2(6%) | 0 |
| Lymph node | 5(6%) | 2(5%) | 2(6%) | 0 |
| Liver | 7(8%) | 3(8%) | 2(6%) | 0 |
| Spleen | 3(2%) | 3(8%) | 0 | 0 |
| Lung | 4(4%) | 1(3%) | 3(9%) | 0 |
| Pancreas | 1(1%) | 0 | 1(3%) | 0 |
| Pituitarium | 1(1%) | 0 | 0 | 0 |
| GI tract | 1(1%) | 0 | 0 | 0 |
| HS classification, n (%) | ||||
| SS LCH | 76(85%) | 31(84%) | 29(85%) | 1(100%) |
| MS R0– LCH | 6(7%) | 2(5%) | 3(9%) | 0 |
| MS R0+ LCH | 7(8%) | 4(11%) | 2(6%) | 0 |
| Outcome, n (%) | ||||
| Alive | 76(87%) | 32(86%) | 29(85%) | 1(100%) |
| Died | 0 | 0 | 0 | 0 |
| Unknown | 13(13%) | 5(14%) | 5(15%) | 0 |
Median age at diagnosis, years
Sanger sequencing
Sanger sequencing was performed for assessment of mutations in exons 2 and exons 3 of the MAP2K1 gene and the hotspot mutation BRAF V600E in exon 15 of the BRAF. DNA was extracted from primary paraffin‐embedded tissues. The sample was dewaxed in xylene and rehydrated in a series of ethanol washes and then placed in proteinase K buffer overnight at 37°C, DNA was extracted using phenol‐chloroform, air‐dried, and reconstituted in water. Then the DNA was subjected to conventional Polymerase chain reaction (PCR). A forward primer, 5′‐ GCTCTGATAGGAAAATGAGATC‐3′, and a reverse primer 5′‐ ACTGATGGGACCCACTCCATC ‐3′, were used to amplify a 139–base pair (bp) amplicon of BRAF exon 15. For MAP2K1 gene, A forward primer, 5′‐ CTCTAGCCTCCCACTTTGAT‐3′, and a reverse primer 5′‐ CTCACCTTTCTGGCCATGAC‐3′, were used to amplify a 342 bp amplicon of exon 2; and a forward primer, 5′‐ CTCCCTCTACCTTAAAGAGC‐3′, and a reverse primer 5′‐ TGTCACATACCATGTGCTCC‐3′, were used to amplify a 260 bp amplicon of exon 3.
PCR amplification was performed using Taq Hot Start Version Polymerase (TaKaRa Bio, Osaka, Japan) under the following conditions: 94°C for 2 min followed by 35 cycles of 94°C for 30 sec, 59°C for 30 sec, 72°C for 30 sec, and then final extension at 72°C for 5 min. The PCR products were sequenced using the same primer sets as shown above.
Whole‐genome sequencing
Library preparation
DNA concentrations were measured with the NanoDrop 2000 (Thermo Fisher Scientific, USA), and sheared with Covaris S220 Sonicator (Covaris) to target of 350 bp average size. Fragmented DNA was purified using Sample Purification Beads (Illumina, USA). Adapter‐ligated libraries were prepared with the TruSeq Nano DNA Sample Prep Kits (Illumina, USA) according to Illumina‐provided protocol.
Illumina sequencing and analysis
DNA concentration of the enriched sequencing libraries was measured with the Qubit 2.0 fluorometer dsDNA HS Assay (Thermo Fisher Scientific, USA). Size distribution of the resulting sequencing libraries was analyzed using Agilent BioAnalyzer 2100 (Agilent). The libraries were used in cluster formation on an Illumina cBOT cluster generation system with HiSeq PE Cluster Kits (illumina, USA). Paired‐end sequencing is performed using an Illumina HiSeq system following Illumina‐provided protocols for 2 × 150 paired‐end sequencing.
Somatic SNVs, indels, and structural variants were analyzed by comparing paired tumor and normal genomes using ANNOVAR and Manta software.
Gene expression of the fusion gene
To assess the mRNA expression of the fusion gene PLEKHA6‐NTRK3, we extracted total RNA from the primary paraffin‐embedded tissues using RNeasy FFPE kit (Qiagen, Germany). Polymerase chain reaction amplification was performed using Taq Hot Start Version Polymerase (TaKaRa Bio, Osaka, Japan) under the following conditions: 94°C for 2 min followed by 35 cycles of 94°C for 30 sec, 59°C for 30 sec, 72°C for 30 sec, and then final extension at 72°C for 5 min. PCR was performed using with primers as follows. PLEKHA6‐NTRK3 primers: a forward primer, 5’‐ACCACCAACAGTGACATACC‐3′, and a reverse primer 5′‐ AGTCCTCCTCACCACTGAT‐3′.
Immunohistochemical
All the work related to human tissues were performed under the Institutional Review Board approved protocols approved at Shanghai Jiao Tong University, according to the Declaration of Helsinki, and the investigators obtained informed written consent from the subjects (wherever necessary). Human tissues specimens were collected from Shanghai Children's Medical Center, School of Medicine, Shanghai Jiao Tong University (Shanghai, China). The tissue sections from paraffin‐embedded deidentified human LCH specimens were stained with antibodies against CD1a (Abcam, ab201337), CD207 (EPR15863), S100 (ab868). IHC staining was scored as 0–3 according to the percentage of positive cells.
Cell culture
HEK293T cells and murine fibroblast NIH 3T3 cells were cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin G and 100 g/ml streptomycin. The cells were incubated at 37°C in 5% CO2.
Constructs and infection
We utilized a real‐time polymerase chain reaction (RT‐PCR) approach to cloning the entire coding sequence of the PLEKHA6‐NTRK3 fusion transcript using RNA isolated from primary paraffin‐embedded tissues into LVX‐puro Vector (Shanghai GeneChem, Shanghai, China) and confirmed by DNA sequencing. And we also cloned NTRK3 domain, PLEKHA6 domain and entire NTRK3 from human CDNA library into the same vector.
We transfected the lentiviral constructs with packaging plasmids PSPAX2 and PMD2G into HEK293T cells using the lipofection transfection method to produce replication‐defective virus. Supernatant was harvested 48 hr later and concentrated by 100 kd column (Amicon purification system, Millipore, USA), and the virus were transduced into NIH 3T3 cells supplemented with 8 μg/ml polybrene (Sigma). The medium was changed 24 hr after infection, and puro‐positive cells were selected using 1 μg/ml puromycin.
Western blot
Cells were harvested using lysis buffer. Cell lysates were subjected to SDS‐PAGE, transferred to nitrocellulose membranes and immunoblotted with the following antibodies: Erk (Cell Signaling#4695), phospho‐Erk (Cell Signaling#4370), Actin (HuaAn M1210–2). Immunoblots were analyzed using the Odyssey system (LI‐COR Biosciences).
Growth curve
Cell growth curves were compared among the cell lines with PLEKHA6‐NTRK3, NTRK3 domain, PLEKHA6 domain, entire NTRK3, and transduced puro‐expressing control cells according to the method. Briefly, 5E4 cells were seeded in each well of a 12‐well plate, and the growth curves were plotted by counting cells every 24 hr over a 6 day period with excel software.
Statistical analyses
Data are expressed as mean±SD. All analyses were two‐tailed and considered statistically significant when p values were less than 0.05.
Results
A novel fusion gene PLEKHA6‐NTRK3 was detected in a pediatric LCH patient
We performed Sanger sequencing for specifically assessment of the mutations BRAF V600E and MAP2K1 genes in the 89 pediatric LCH patients. Among them, 37 of 89 cases (41.6%) harbored the BRAF V600E mutations, and MAP2K1 mutations that were mutually exclusive with BRAF mutations were detected in 34 of 89 cases (38.2%) (Figs. 1 a–1c). The remaining 18 pediatric LCH patients were BRAF V600E‐negative and MAP2K1‐negative, we performed Whole‐Genome sequencing (WGS) of which had both blood samples and paraffin‐embedded tissues to search additional abnormal genomic alterations. A novel fusion gene PLEKHA6‐NTRK3 (1 of 89 cases, 1.1%) was identified in a pediatric patient (Figs. 1 d, and 1e).
Figure 1.
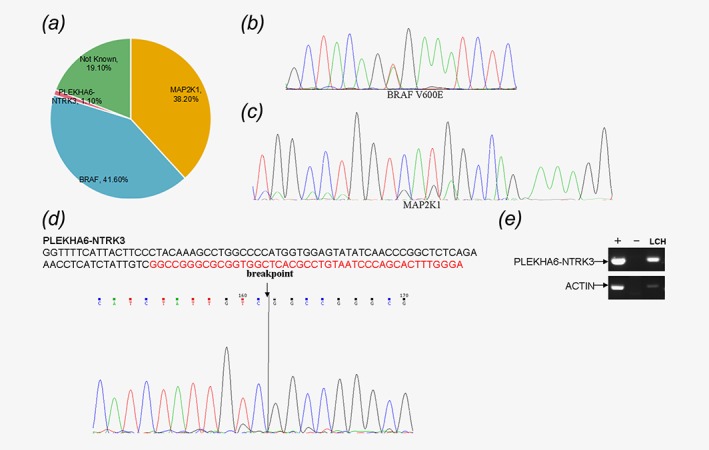
Specific sequencing data of the 89 patients with LCH. (a) The statistical charts of the sequencing information. (b) The mutation site of BRAF in exon 15. (c) The mutation site of MAP2K1 in exon 2 and 3. (d) New fusion gene PLEKHA6‐NTRK3. (e) PCR identifies the mRNA expression of the new fusion gene PLEKHA6‐NTRK3.
Clinical presentation
The patient with the fusion gene PLEKHA6‐NTRK3 was a 33‐month‐old male patient with a history of intermittent lower back pain more than 1 month, the physical examination showed spine appearance without deformity, and normal activity of the spine with no obvious limitation, while thoracolumbar local spinous process mild tenderness. The child had no other congenital anomalies and the family history was unremarkable. Whole‐body magnetic resonance imaging (MRI) revealed T12 vertebral wedges with soft tissue mass (Fig. 2 a), abdominal B‐ultra showed hepatic portal pancreatic lymph nodes were 1.5 × 0.7 cm, 1.7 × 0.11 cm.
Figure 2.
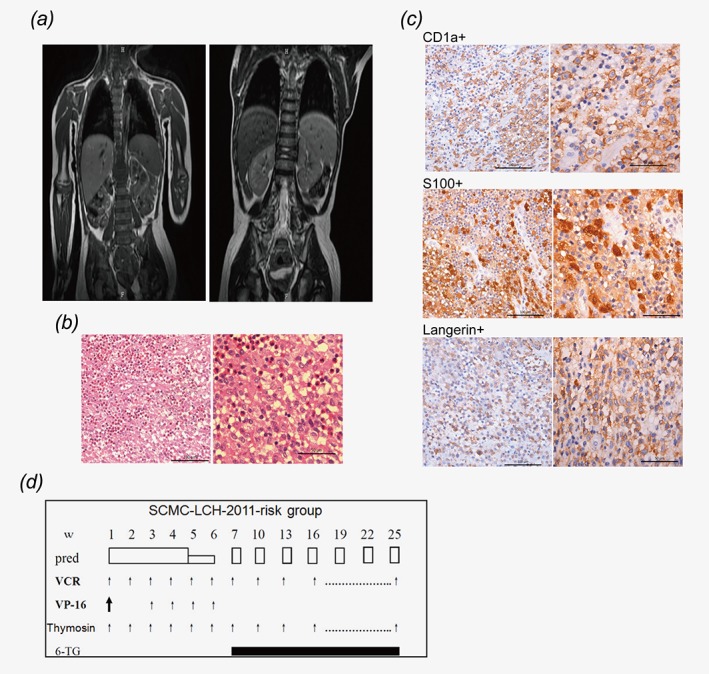
Clinical information of the patient with the fusion gene PLEKHA6‐NTRK3. (a) Left: Whole‐body magnetic resonance imaging (MRI) revealed T12 vertebral wedges with soft tissue mass during diagnosis. Right: After 1 year of treatment, MRI showed that the lesions disppered. (b) Morphology of typical abnormal histiocytic cells. (H&E) (c) Expression of CD1a+, S100+, Langerin+ on abnormal histiocytic cells of the patient. (d) The therapeutic regimen of the patient.
The patient underwent a surgical meanwhile process the biopsy of the soft tissue mass. Morphologically, the biopsy specimens were composed of abnormal histiocytic cells which were small oval cells with slight eosinophilic cytoplasm and grooved folded nuclei with minimal atypia, and the eosinophils infiltrating can be observed (Fig. 2 b). The patient was confirmed SS‐LCH for the histiocytes were positive for CD1a, langerin (CD207), and S100 on immunohistochemistry (Fig. 2 b).
The therapeutic regimen of the patient was chemotherapy of SCMC‐LCH‐2011‐risk group that adapted from the LCH‐II (arm B) study.18 He received an initial therapy of continuous prednisone (40 mg/m2 daily for 4 weeks, tapering over 2 weeks), vincristine (1.5 mg/m2 weekly for 7 weeks), and etoposide (100 mg/m2 daily, days 1–3, every 2 for 8 weeks). Continuation therapy was 6‐Thioguanine (40 mg /m2 daily) and a pulse of prednisone (40 mg /m2 daily, days 1–5, every 3 weeks) and vincristine (1.5 mg /m2/d, once every 3 weeks) (Fig. 2 d). After 1 year of treatment, reexamination of MRI showed the patient is disease free.
The functional analysis of the fusion gene PLEKHA6‐NTRK3
To further assess the effects of PLEKHA6‐NTRK3 expression, we cloned the sequence of PLEKHA6‐NTRK3, C‐terminal NTRK3 domain, N‐terminal PLEKHA6 domain and the entire NTRK3 from patient, and cDNA bank according to the WGS results and bind to the LVX‐puro plasmid vector, respectively (Fig. 3 a and 3b). Murine fibroblast NIH 3T3 cells were transduced with packaged lentivirus encoding PLEKHA6‐NTRK3, NTRK3 domain, PLEKHA6 domain, entire NTRK3, and empty vector separately.
Figure 3.
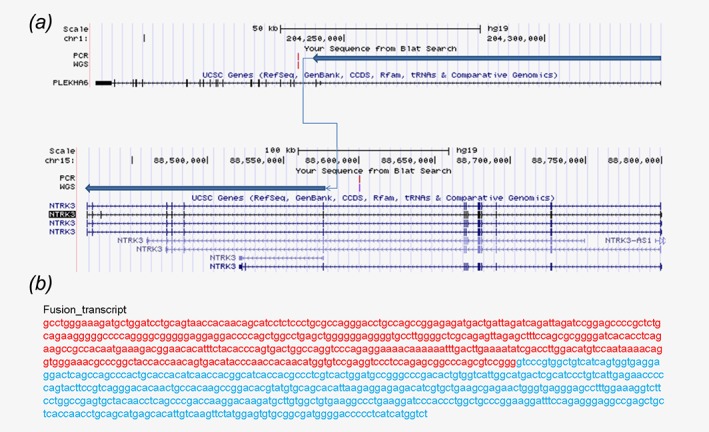
Plasmid design of the fusion gene PLEKHA6‐NTRK3. (a,b) Plasmid design of the fusion gene PLEKHA6‐NTRK3 and the gene sequence of it.
Compared to control NIH 3T3 cells, and the cell lines with empty vector, NTRK3 domain, PLEKHA6 domain and entire NTRK3, the p‐ERK of cell line with PLEKHA6‐NTRK3 was higher, it illustrated that MAPkinase pathway was specifically activated by fusion gene (Fig. 4 a). Activating of this pathway is related to many cellular functions, such as cell growth, proliferation.19, 20Indeed, our results showed that the ability of NIH 3T3 cells transduced with PLEKHA6‐NTRK3 to significantly promote the growth (Fig. 4 b). This result suggested that abnormal expression of NTRK3 can activate MAPkinase pathway to induce LCH (Fig. 4 c).
Figure 4.
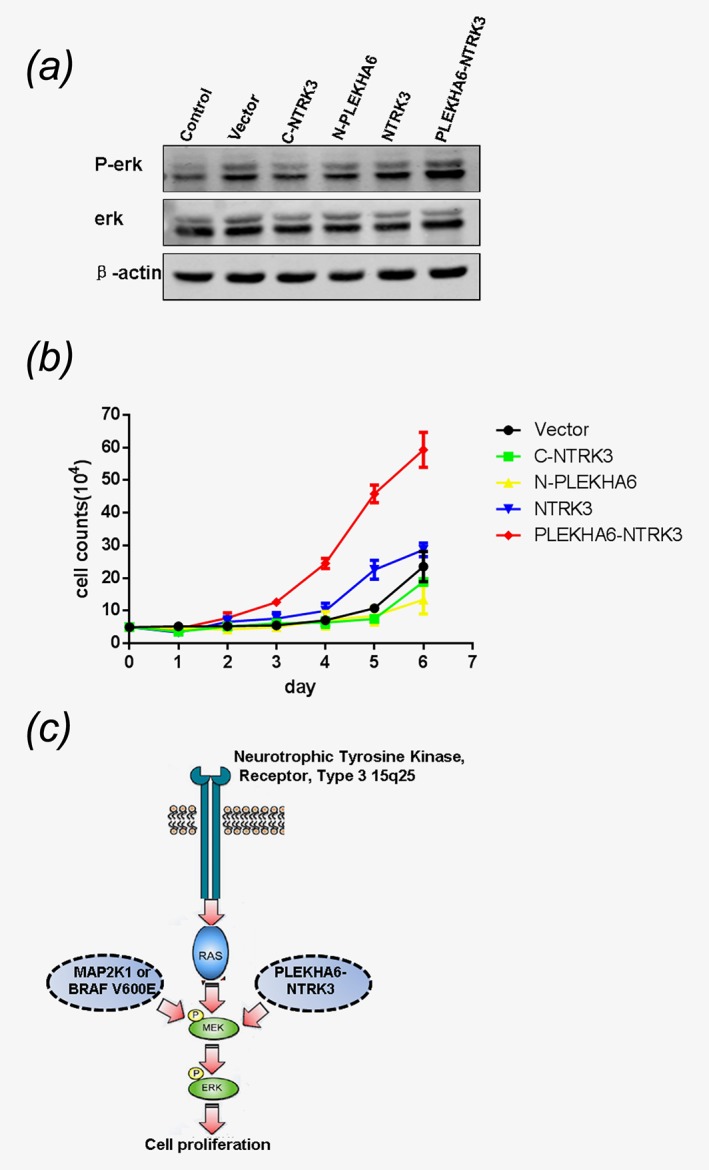
The fusion gene PLEKHA6‐NTRK3 activate the ERK pathway and promote the cell growth. (a) The fusion gene PLEKHA6‐NTRK3 activates the RAS–RAF–MEK–ERK pathway in NIH 3 T3 cells. (b) The fusion gene PLEKHA6‐NTRK3 promotes the cell growth. (c) Pattern diagram of the activition of the RAS–RAF–MEK–ERK pathway.
Discussion
We reported here a new fusion gene PLEKHA6‐NTRK3 in LCH, this mutation constitutively activated the the MAPKinase RAS–RAF–MEK–ERK cell signaling pathway, to promote the cell growth. So it shrinked the unknown part of the molecular spectrum of LCH and needed be included in LCH molecular screening panel to better define its prevalence in LCH.
In our study, the BRAF V600E and MAP2K1 mutation rates were 41.6% and 38.2% (Fig. 1 a and 1c), respectively, which were higher than the previous reported in Asian patients.13, 21 BRAF V600E mutations were the most important and highest frequency genomic alterations in LCH, followed by MAP2K1 mutations.4, 10, 11 Both mutations can constitutive activated of the MAPKinase cell signaling pathway, which is a key regulator of many cellular functions involving cell growth, proliferation, and differentiation. Except LCH, these two mutations have been implicated in several human cancers including melanomas,22, 23 hairy cell leukemia24, 25 and precancerous lesions26 as well as some non‐cancerous lesions such as benign nevus cell nevi,27, 28, 29 hyperplastic polyp of the colon,30 and so on. BRAF inhibitor Vemurafenib has been used for the treatment of the LCH, and some research showed that combination of Vemurafenib and MEK kinase inhibitors is effective in the treatment of melanomas.5, 31 All these manifested that RAS–RAF–MEK–ERK pathway play an important role in cancer generation.
The new fusion gene PLEKHA6‐NTRK3 which was detected in a BRAF V600E‐negative and MAP2K1‐negative patient is associated with RAS–RAF–MEK–ERK pathway, for it specifically activates it (Fig. 4 a). Recurrent kinase fusion involving BRAF, ALK, and NTRK1 has been found to be associated with the pathogenesis of LCH,14 and the fusion genes that involve NTRK3 have been detected in many other tumors. For example, fusion gene ETV6‐NTRK3 was associated with congenital mesoblastic nephroma, and reported as a primary event in human secretory breast carcinoma,32, 33, 34 and multiple fusion partners targeting NTRK3 such as EML4‐NTRK3 could contribute to the development of congenital fibrosarcoma.35 We predict that this translocation may cause a novel fusion mutation PLEKHA6‐NTRK3, which might be a driving event of LCH.
LCH has been known as a neoplastic disorder, it is also considered as an immune disorder, while the pathogenesis of it remains obscure.36, 37 Discovery of new pathogenic genes are conducive to elucidate the pathogenesis of this disease.
Acknowledgements
This work was supported in part by National Natural Science Foundation of China (No.81470315, 81772663 to Y.L.; No.81670136 to J.T.; and No.30670883, 81270623 to S.S); Shanghai Jiao Tong University School of Medical Engineering Cross Fund (No. YG2017MS32), Collaborative Innovation Center for Translation Medicine at Shanghai Jiao Tong University School of Medicine (TM201502) to Y.L.; The Science and Technology Commission of Shanghai Municipality (14411950600) to Jing Chen.
Conflict of interest: All authors declare no conflict of interest.
J.C, X.H, and M.Y contributed equally to this work.
Contributor Information
Yanxin Li, Email: liyanxin@scmc.com.cn.
Shuhong Shen, Email: shenshuhong@scmc.com.cn.
References
- 1. Collin M, Bigley V, McClain KL, et al. Cell(s) of origin of langerhans cell histiocytosis. Hematol Oncol Clin N Am 2015;29:825–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Geissmann F, Lepelletier Y, Fraitag S, et al. Differentiation of langerhans cells in Langerhans cell histiocytosis. Blood 2001;97:1241–8. [DOI] [PubMed] [Google Scholar]
- 3. Hogstad B, Berres ML, Chakraborty R, et al. RAF/MEK/extracellular signal‐related kinase pathway suppresses dendritic cell migration and traps dendritic cells in Langerhans cell histiocytosis lesions, J Exp Med 2018;215:319–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Badalian‐Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010;116:1919–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Varadi Z, Banusz R, Csomor J, et al. Effective BRAF inhibitor vemurafenib therapy in a 2‐year‐old patient with sequentially diagnosed Langerhans cell histiocytosis and Erdheim‐Chester disease. OncoTargets Ther 2017;10:521–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Diamond EL, Subbiah V, Lockhart AC, et al. Vemurafenib for BRAF V600‐mutant Erdheim‐Chester disease and Langerhans cell histiocytosis: analysis of data from the histology‐independent, Phase 2, open‐label VE‐BASKET study. JAMA Oncol 2017;4(3):384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haroche J, Cohen‐Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim‐Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013;121:1495–500. [DOI] [PubMed] [Google Scholar]
- 8. Nelson DS, Quispel W, Badalian‐Very G, et al. Somatic activating ARAF mutations in Langerhans cell histiocytosis. Blood 2014;123:3152–5. [DOI] [PubMed] [Google Scholar]
- 9. Berres ML, Lim KP, Peters T, et al. BRAF‐V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 2015;212:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chakraborty R, Hampton OA, Shen X, et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood 2014;124:3007–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brown NA, Furtado LV, Betz BL, et al. High prevalence of somatic MAP2K1 mutations in BRAF V600E‐negative Langerhans cell histiocytosis. Blood 2014;124:1655–8. [DOI] [PubMed] [Google Scholar]
- 12. Alayed K, Medeiros LJ, Patel KP, et al. BRAF and MAP2K1 mutations in Langerhans cell histiocytosis: a study of 50 cases. Hum Pathol 2016;52:61–7. [DOI] [PubMed] [Google Scholar]
- 13. Zeng K, Ohshima K, Liu Y, et al. BRAFV600E and MAP2K1 mutations in Langerhans cell histiocytosis occur predominantly in children. Hematol Oncol 2017;35:845–51. [DOI] [PubMed] [Google Scholar]
- 14. Chakraborty R, Burke TM, Hampton OA, et al. Alternative genetic mechanisms of BRAF activation in Langerhans cell histiocytosis. Blood 2016;128:2533–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nelson DS, van Halteren A, Quispel WT, et al. MAP2K1 and MAP3K1 mutations in Langerhans cell histiocytosis. Genes Chromosomes Cancer 2015;54:361–8. [DOI] [PubMed] [Google Scholar]
- 16. Diamond EL, Durham BH, Haroche J, et al. Diverse and Targetable Kinase Alterations Drive Histiocytic Neoplasms. Cancer Discov 2016;6:154–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morimoto A, Oh Y, Shioda Y, et al. Recent advances in Langerhans cell histiocytosis. Pediatr Int 2014;56:451–61. [DOI] [PubMed] [Google Scholar]
- 18. Gao YJ, Su M, Tang JY, et al. Treatment outcome of children with multisystem langerhans cell histiocytosis: the experience of a single children's hospital in Shanghai, China. J Pediatr Hematol Oncol 2018;40:e9–e12. [DOI] [PubMed] [Google Scholar]
- 19. Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol 2004;5:875–85. [DOI] [PubMed] [Google Scholar]
- 20. Montagut C, Settleman J. Targeting the RAF‐MEK‐ERK pathway in cancer therapy. Cancer Lett 2009;283:125–34. [DOI] [PubMed] [Google Scholar]
- 21. Sasaki Y, Guo Y, Arakawa F, et al. Analysis of the BRAFV600E mutation in 19 cases of Langerhans cell histiocytosis in Japan. Hematol Oncol 2017;35:329–34. [DOI] [PubMed] [Google Scholar]
- 22. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54. [DOI] [PubMed] [Google Scholar]
- 23. Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol 2011;29:3085–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dietrich S, Glimm H, Andrulis M, et al. BRAF inhibition in refractory hairy‐cell leukemia. N Engl J Med 2012;366:2038–40. [DOI] [PubMed] [Google Scholar]
- 25. Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4‐34‐expressing hairy‐cell leukemias. Nat Genet 2014;46:8–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rosenberg DW, Yang S, Pleau DC, et al. Mutations in BRAF and KRAS differentially distinguish serrated versus non‐serrated hyperplastic aberrant crypt foci in humans. Cancer Res 2007;67:3551–4. [DOI] [PubMed] [Google Scholar]
- 27. Taube JM, Begum S, Shi C, et al. Benign nodal nevi frequently harbor the activating V600E BRAF mutation. Am J Surg Pathol 2009;33:568–71. [DOI] [PubMed] [Google Scholar]
- 28. Tan JM, Tom LN, Jagirdar K, et al. The BRAF and NRAS mutation prevalence in dermoscopic subtypes of acquired naevi reveals constitutive mitogen‐activated protein kinase pathway activation. Br J Dermatol 2018;178:191–7. [DOI] [PubMed] [Google Scholar]
- 29. Kiuru M, Tartar DM, Qi L, et al. Improving classification of melanocytic nevi: BRAF V600E expression associated with distinct histomorphologic features. J Am Acad Dermatol 2018;79:221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Agaimy A, Stoehr R, Vieth M, et al. Benign serrated colorectal fibroblastic polyps/intramucosal perineuriomas are true mixed epithelial‐stromal polyps (hybrid hyperplastic polyp/mucosal perineurioma) with frequent BRAF mutations. Am J Surg Pathol 2010;34:1663–71. [DOI] [PubMed] [Google Scholar]
- 31. Grob JJ, Amonkar MM, Karaszewska B, et al. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health‐related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600‐mutation‐positive melanoma (COMBI‐v): results of a phase 3, open‐label, randomised trial. Lancet Oncol 2015;16:1389–98. [DOI] [PubMed] [Google Scholar]
- 32. Knezevich SR, McFadden DE, Tao W, et al. A novel ETV6‐NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet 1998;18:184–7. [DOI] [PubMed] [Google Scholar]
- 33. Bourgeois JM, Knezevich SR, Mathers JA, et al. Molecular detection of the ETV6‐NTRK3 gene fusion differentiates congenital fibrosarcoma from other childhood spindle cell tumors. Am J Surg Pathol 2000;24:937–46. [DOI] [PubMed] [Google Scholar]
- 34. Tognon C, Knezevich SR, Huntsman D, et al. Expression of the ETV6‐NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2002;2:367–76. [DOI] [PubMed] [Google Scholar]
- 35. Tannenbaum‐Dvir S, Glade Bender JL, Church AJ, et al. Characterization of a novel fusion gene EML4‐NTRK3 in a case of recurrent congenital fibrosarcoma. Cold Spring Harb Mol Case Stud 2015;1:a000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jezierska M, Stefanowicz J, Romanowicz G, et al. Langerhans cell histiocytosis in children —a disease with many faces. Recent advances in pathogenesis, diagnostic examinations and treatment. Postepy Dermatol Alergol 2018;35:6–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weiss LM, Jaffe R, Facchetti F. In: Swerdlow SH, Campo E, Harris NL, et al. eds Tumours derived from Langerhans cells. In: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues Revised, 4th edn. Lyon, France: International Agency for Research on Cancer (IARC), 2017. 470–3 Ref 3988241. [Google Scholar]