

Summary
Quantitative disease resistance, often influenced by environmental factors, is thought to be the result of DNA sequence variants segregating at multiple loci. However, heritable differences in DNA methylation, so‐called transgenerational epigenetic variants, also could contribute to quantitative traits. Here, we tested this possibility using the well‐characterized quantitative resistance of Arabidopsis to clubroot, a Brassica major disease caused by Plasmodiophora brassicae.
For that, we used the epigenetic recombinant inbred lines (epiRIL) derived from the cross ddm1‐2 × Col‐0, which show extensive epigenetic variation but limited DNA sequence variation. Quantitative loci under epigenetic control (QTL epi) mapping was carried out on 123 epiRIL infected with P. brassicae and using various disease‐related traits.
EpiRIL displayed a wide range of continuous phenotypic responses. Twenty QTL epi were detected across the five chromosomes, with a bona fide epigenetic origin for 16 of them. The effect of five QTL epi was dependent on temperature conditions. Six QTL epi co‐localized with previously identified clubroot resistance genes and QTL in Arabidopsis.
Co‐localization of clubroot resistance QTL epi with previously detected DNA‐based QTL reveals a complex model in which a combination of allelic and epiallelic variations interacts with the environment to lead to variation in clubroot quantitative resistance.
Keywords: Arabidopsis, clubroot, epigenetics, EpiRIL, methylome, Plasmodiophora brassicae, quantitative resistance, transgenerational
Introduction
Clubroot caused by the protist Plasmodiophora brassicae is a major disease of Brassicaceae including the three most economically important Brassica species, B. napus, B. rapa and B. oleracea (Dixon, 2009), and the model plant Arabidopsis thaliana (Koch et al., 1991). Infection with P. brassicae leads to tumorous club formation on roots resulting from cell hyperplasia and hypertrophy (Ingram & Tommerup, 1972). Cropping practices and crop protection products have limited success in controlling clubroot (Dixon, 2009). Currently, one of the most effective ways to limit the impact of this disease is to use resistant varieties (Diederichsen et al., 2009). To date, both qualitative and quantitative trait loci (QTL) for clubroot resistance have been identified in different Brassicaceae species (Manzanares‐Dauleux et al., 2000a; Rocherieux et al., 2004; Alix et al., 2007; Jubault et al., 2008; Piao et al., 2009; Lee et al., 2016). Current approaches to generating resistant varieties rely mainly on a few loci controlling qualitative resistance, with the inevitable outcome of rapid adaptation of the pathogen populations (Diederichsen et al., 2009). In this context, diversification and access to other sources of clubroot resistance variability is becoming necessary.
Numerous studies (Dowen et al., 2012; Luna et al., 2012; Zhang et al., 2013; Liu et al., 2015; Aoun et al., 2017; Zheng et al., 2017) have reported that plant responses to abiotic (temperature, drought) and biotic stresses could be associated with epigenetic variation in addition to nucleotidic variation. For instance, Dowen et al. (2012) and Yu et al. (2013) have shown that Arabidopsis mutants altered in the maintenance of DNA methylation in the CG, CHG and CHH contexts, showed strong resistance to Pseudomonas syringae pv tomato strain DC3000. The role of epigenetics in the expression of adaptive plant traits thus suggests that epigenetic variability could be used for generating stress‐tolerant or resistant plants. Furthermore, the occurrence of natural DNA methylation variants (epialleles) in plants (Becker et al., 2011; Schmitz et al., 2011) and their implication in evolution (Weigel & Colot, 2012) suggest that epialleles could be considered as a source of variability in plant breeding. In this ‘epigenetic breeding’ approach (Gallusci et al., 2017), two conditions are needed: transgenerational inheritance of epialleles and a clear connection between epigenotype and observed phenotype. Previous studies demonstrated that epialleles could be stably transmitted across at least eight generations (Johannes et al., 2009; Teixeira et al., 2009), and that such heritable differences in DNA methylation could be associated with heritable phenotypic variation for several complex traits (Johannes et al., 2009; Reinders et al., 2009). However, linking heritable phenotypic variation to epigenetic variation remains challenging because of the difficulty in teasing apart its effects to that of DNA sequence variation in natural settings (Johannes et al., 2008; Quadrana & Colot, 2016). This problem can, however, be greatly alleviated in Arabidopsis by using experimental populations of so‐called epigenetic recombinant inbred lines (epiRIL), which show extensive epigenetic variation but limited DNA sequence variation (Johannes et al., 2009; Reinders et al., 2009). One such population (Johannes et al., 2009) was indeed used to build a genetic map based solely on heritable differences in DNA methylation (differentially methylated regions, DMR) (Colomé‐Tatché et al., 2012) and to identify epigenetic QTL (QTLepi) for several complex traits (Cortijo et al., 2014; Kooke et al., 2015; Aller et al., 2018).
In the present study, we used this same epiRIL population to determine the impact of heritable differences in DNA methylation on the response of Arabidopsis to clubroot. We first showed that ddm1 mutants were less susceptible to clubroot than the wild‐type Col‐0 and that the assessed subset of 123 epiRIL displayed a wide range of continuous phenotypic responses to clubroot. Twenty QTLepi were detected across the five chromosomes, with a bona fide epigenetic origin for 16 of them. We have thus demonstrated that heritable differences in DNA methylation also could contribute to quantitative resistance to clubroot. Six QTLepi co‐localized with previously identified clubroot resistance genes and QTL in Arabidopsis, revealing that quantitative resistance to clubroot in natural accessions could be controlled by both nucleotidic and epigenetic variations.
Materials and Methods
Plant material
Plant stocks
Mutant plants altered in genes encoding chromatin modifiers were ordered from the NASC. All plants were obtained from T‐DNA insertion in the Columbia (Col‐0) genetic background and showed a homozygous insertion in the plant genome. The mutant lines used were drm2‐2 (SALK_150863), hda 15 (SALK_004027C), atxr5 (SALK_130607C), hac1 (SALK_082118C), srt2 (SALK_149295C) and ddm1 (SALK_000590C). T‐DNA insertions and homozygosity were confirmed by PCR using the set of appropriate primers designed with the T‐DNA Primer Design interface (http://signal.salk.edu/tdnaprimers.2.html, Supporting Information Table S1). The epigenetic recombinant inbred lines (epiRIL) population is that derived from a cross between the Arabidopsis thaliana ecotype Col‐0 and a Col‐0 line carrying the ddm1‐2 mutant allele (Johannes et al., 2009). Note that the ddm1‐2 allele was obtained by EMS mutagenesis, not T‐DNA transformation (Vongs et al., 1993). EpiRIL population seeds were obtained from the Versailles Arabidopsis Stock Center (http://publiclines.versailles.inra.fr/) at the Institute Jean‐Pierre Bourgin.
Clubroot pathogen
All clubroot tests were performed with the Plasmodiophora brassicae eH isolate described by Fahling et al. (2003). Isolate eH belongs to the P1 pathotype according to the classification using the differential host set of Some et al. (1996). One millilitre of resting spore suspension (107 spores ml−1) prepared according to Manzanares‐Dauleux et al. (2000b) was used for pathogen inoculation 10 d after germination (stage 1.04; Boyes et al., 2001). The inoculum was applied to the bottom of the stem base of each seedling.
Growth conditions
The Arabidopsis accession Col‐0 and the six mutant lines (Col‐0 background) were evaluated in a randomized complete block design (with three blocks) against the eH P. brassicae isolate. For QTLepi detection, the 123 epiRIL that have been epigenotyped previously (Colomé‐Tatché et al., 2012), together with the two parental lines, were phenotyped in four biological replicates, split in two growth rooms, in a randomized complete block design (with two blocks per replicate and six plants per epigenotype per block). In growth room‐2, four temperature sensors per block were placed at the height of plants. For each pathological test, seed germination was synchronized by placing seeds on wet blotting paper in Petri dishes for 2 d at 4°C. Seeds were sown individually in pots (4 cm diameter) containing a sterilized mix (by autoclaving) composed of 2/3 compost and 1/3 vermiculite. Per block, six plants per genotype (T‐DNA mutants, Col‐0, ddm1‐2 and epiRIL) were grown in controlled conditions of 16 h light (110 μmol m−2 s−1) at 21°C and 8 h dark at 18°C.
Phenotyping
Phenotyping was performed 3 wk after inoculation (21 d post‐inoculation (dpi)). Plants were thoroughly rinsed with water and photographed. Infected roots were removed and frozen in liquid nitrogen. For the epiRIL and their parents, four disease‐related traits were assessed: longest leaf length (Lfi), disease index (DI), root biomass (Rbi) and pathogen DNA quantity (Pb), whereas for mutants, only DI was evaluated. DI was calculated according to Manzanares‐Dauleux et al. (2000b) and pathogen DNA quantification was determined by quantitative polymerase chain reaction (qPCR). These phenotypic traits were chosen due to their relevance for the study of the quantitative response to eH isolate on Arabidopsis (Jubault et al., 2008; Gravot et al., 2011; Lemarié et al., 2015a,b). The longest leaf length (Lfni) and the root biomass (Rbni) were also measured at 28 d post sowing on noninfected plants cultivated in growth room‐2 in a randomized complete block design with 24 plants per (epi)genotype. Two new variables were then obtained by calculating the difference between these ‘control’ values with those obtained on infected plants; these new variables were termed ΔLf and ΔRb.
Pathogen DNA quantification
Total genomic DNA (plant and pathogen) was extracted from lyophilized infected roots with the NucleoSpin Plant II kit according to manufacturer's instructions. After the evaluation of DNA concentration by Nanodrop ND‐1000, 5–10 ng of total genomic DNA extract and specific primers were used to quantify by qPCR pathogen the DNA quantity in each epiRIL and the parental lines. The pathogen‐specific primers designed on the 18S rRNA of P. brassicae used were PbF‐K1 5′ TTGGGTAATTTGCGCGCCTG 3′; PbR‐K1 5′ CAGCGGCAGGTCATTCAACA 3′. qPCR reactions were carried out in a thermocycler (LightCycler 480 II Roche), with Syber green (LightCycler® 480 SYBR Green I Master). The qPCR conditions used were: 50 cycles of denaturation at 95°C for 15 s, annealing/extension at 63°C for 30 s and 72°C for 30 s. Absolute quantification of pathogen DNA was carried out using a standard curve obtained based on a series of known amounts of pathogen DNA. The average of the pathogen quantity present in each epiRIL (ratio of pathogen DNA quantity to total DNA used for qPCR) was used in further statistical analyses and QTLepi detection.
Data analysis
Two generalized linear models (glm) were used to determine the effects of epigenome, temperature and epigenome × temperature interaction with R/glm2 (Marschner, 2011) in R v.3.2.2 (R Development Core Team, 2015). For each model, the family distribution option of the glm function was adapted according to the data distribution. The first glm (glm1) described in Eqn 1 was used to estimate the epigenotype effect of each epiRIL across biological replicates in each growth room:
| (Eqn 1) |
where μ, mean general effect; G i, differential effect between epigenotypes; R j, differential effect between replicates; B k (R j), interaction between blocks and replicates; and e ij, residual variance. Based on this model, broad sense heritability was estimated using the following equation:
| (Eqn 1.1) |
(, estimated epigenetic variance; , estimated environmental variance; and n, number of replicates per line).
The second generalized linear model (glm2) described in Eqn (Eqn 2) was used to estimate the epigenotype × temperature effect in growth room‐2 for each epiRIL across the biological replicates:
| (Eqn 2) |
where μ, mean general effect; G i, differential effect between epigenotypes; T j, differential temperature conditions; R k, differential effect between replicates; B l (R k), interaction between blocks and replicates; GT ij, interaction between epigenotype and temperature condition; TR jk, interaction between replicate and temperature condition; TB(R)jlk, interaction between block and temperature condition; and e ijkl, residual variance). Based on this model, broad sense heritability was estimated using the following equation:
| (Eqn 2.1) |
where , estimated epigenetic variance; , estimated epigenetic × temperature variance; , estimated environmental variance; and n, number of replicates per line.
For all traits, least square means on each effect according to the two models described above were estimated with the function lsmeans of the R/lsmeans package (,Lenth 2016) in R v.3.2.2 (R Core Team, 2015). The lsmeans function also was used to extract the epigenotype effect (G) and the interaction epigenotype × temperature (GT) of each trait according to the generalized model used. Differences in longest length leaf (ΔLf) and root biomass (ΔRb) between infected and control plants were calculated from G.
QTLepi detection
The G and GT of each trait were treated with the package R/qtl (Broman et al., 2003) in R v.3.2.2 (R Development Core Team, 2015). The package snow in R (Tierney et al., 2015) allowing the use of processor cores as cluster was used to reduce permutation calculation time. Simple Interval Mapping (SIM) was first carried out to identify potential QTLepi with the Haley–Knott (hk) method (Broman & Sen, 2009), using a step size of 2 cM and a window size of 10 cM. One thousand permutations with the hk method were carried out in order to determine SIM threshold levels for each condition and trait analysed. The significance level of threshold was fixed at α = 0.05. In order to integrate the possibility of the presence of multiple QTLepi on the same chromosome, a manual multiple QTL mapping (MQM) approach (Broman & Sen, 2009) was used based on the results of SIM analysis. For this, the stepwise function was used in order to select the QTLepi (forward and backward system, option ‘additive.only = FALSE’) based on the preliminary putative QTLepi identified by SIM. For each trait, a minimum of two potential QTLepi was used in the stepwise function even if only one potential QTLepi was detected in SIM. Logarithm of odds (LOD) thresholds were calculated using 1000 permutations with the function scantwo with a significance level of α = 0.05. Once the QTLepi selected, the model was fitted (fitqtl function), in order to calculate the LOD scores and the percentage of variation explained by each and all QTLepi (R 2). The confidence interval of each QTLepi was calculated with the lodint function with a LOD drop of one parameter. The epiallele effect was evaluated with the function effectplot. Putative interactions among the QTLepi incorporated in the model were tested with the function addint according to Broman & Sen (2009).
DNA sequence variation
Whole genome sequence data are available for the 123 epigenotyped epiRIL (Gilly et al., 2014). The joint identification of small‐scale variants (single‐base substitutions and indels < 100 bp) was performed using GATK HaplotypeCaller on the whole‐genome sequencing data available for 122 epiRIL. Raw variant calls were then filtered following GATK Best Practice suggestions and additional scripts. All variants were visually inspected using IGV and a subset of the detected single nucleotide polymorphism (SNP) was validated by PCR. For the analysis of transposition events (TE), TE‐Tracker (Gilly et al., 2014) was used to identify nonreference TE insertions in 102 epiRIL of sufficient genomic coverage. Raw calls were then filtered using in‐house scripts. All of the detected new TE insertions were visually inspected using IGV and a fraction of them were validated by PCR. Shared small‐scale and TE insertion variants are defined as present in at least 25% of the population. In order to evaluate the impact of shared sequence variants on the heritable variation observed in the epiRIL in response to clubroot, we compared various QTL models as described by Kooke et al. (2015). Three different models were tested: (1) using all QTL peak epigenetic markers (MM); (2) using each shared DNA sequence variant (SNP, indel, TE insertion) located in the confidence interval instead of the epigenetic marker at the peak; and (3) using both peak epigenetic markers and the shared DNA sequence variants included in the confidence interval. QTL detected using the three models were compared. If the DNA‐based markers had a more significant effect than the peak QTL epigenetic markers, then the model fitted was considered to be better or identical to the model with the peak epigenetic markers.
Results
ddm1 mutants have reduced susceptibility to P. brassicae
In order to determine if epigenetic variations could be associated with variations in response to clubroot infection, we assessed the response to infection of several Arabidopsis mutants affected in genes encoding chromatin modifiers. Infection of Col‐0 and six T‐DNA insertion mutants in genes encoding chromatin modifiers (atxr5, ddm1, drm2, hac1, hdc15 and srt2) with the P. brassicae eH isolate led to a range of phenotypic responses. A statistically significant DI effect (ANOVA: F = 4.99, P‐value = 0.005) (Table S2a) was identified 21 dpi suggesting that epigenetics was involved in plant response to infection by P. brassicae. A Dunnett's post hoc test (Table S2) revealed a significant difference between ddm1 and Col‐0 (t = −4.437, P‐value = 0.003) for the DI trait, ddm1 showing reduced symptoms compared to Col‐0 (Fig. 1).
Figure 1.

Clubroot symptoms shown by Col‐0 wild‐type and the ddm1 mutant (Col‐0 background). Arabidopsis Col‐0 and mutants were inoculated 10 d after germination and phenotyped 3 wk post‐inoculation. (a) Col‐0 ecotype; (b) ddm1 mutant. The ddm1 mutant showed a significant decrease in disease symptoms (Dunett's test, P < 0.02) compared to Col‐0. Plant individuals are representative of standard observations made in our experimental conditions. Bars, 1 cm.
Heritable differences in DNA methylation are associated with differential susceptibility to P. brassicae
EpiRIL response to P. brassicae is quantitative
In order to identify epialleles involved in clubroot response, QTLepi detection was carried out on the subset of 123 lines of the epiRIL population used previously in Colomé‐Tatché et al. (2012). In total, each genotype was assessed against P. brassicae isolate eH in four biological replicates, split in two growth rooms, each biological replicate being composed of two blocks. Distribution of the four disease‐related traits assessed on the 123 epiRIL showed continuous distribution suggesting polygenic control of these traits (Fig. 2). However, significant differences were observed for all traits between the biological replicates (Mann–Whitney U‐test) set up in the two growth rooms (DI: W = 3685.5, P‐value < 2.2e‐16; Lfi: W = 233 530, P‐value < 2.2e‐16; Pb: W = 4826, P‐value < 2.2e‐16), suggesting an influence of the growth conditions on the epiRIL response to clubroot (Table S3). Indeed, higher levels of disease symptoms were observed on the Col‐0 parent line (growth room‐1: DI = 53.25 ± 2.36; growth room‐2: DI = 90.75 ± 9.64) and on the epiRIL population (growth room‐1: DI = 51.17 ± 11.42; growth room‐2: DI = 86.27 ± 10.74) growing in growth room‐2 compared to growth room‐1. Consequently, we decided to analyse data from the two growth rooms independently. Analysis of biological replicates grown in each growth room showed a significant epigenotype effect (glm1, Eqn 1) for nearly all phenotypic traits measured (P‐value ranged from 0.02 to < 2.2e‐16 in growth room‐1, and from 0.35 to < 2.2e‐16 in growth room‐2; Table S4). Broad‐sense heritability (H 2) was estimated for each trait using Eqn 1.1 and ranged from 0.46 to 0.76 in the growth room‐1 and from 0.44 to 0.65 in the growth room‐2 depending on the trait studied (Table 1).
Figure 2.
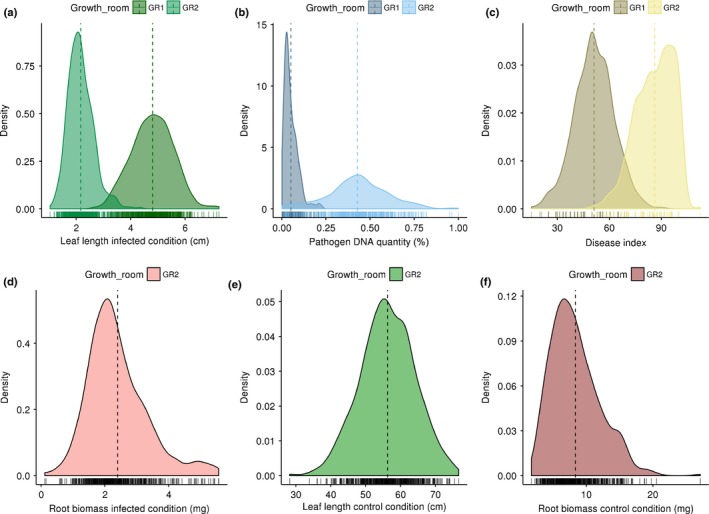
Distribution of phenotypic data measured in the two growth rooms used for pathological tests. For each trait studied, data are coloured to show in which growth chamber the Arabidopsis plants were grown. Vertical coloured dashed lines indicate means of traits in each growth room. Data distribution is shown for leaf length (a), pathogen DNA quantity (b), disease index (c) and root biomass (d) in infected condition, and for leaf length (e) and root biomass (f) in noninfected condition. GR1 corresponds to growth room‐1. GR2 corresponds to growth room‐2.
Table 1.
Heritability estimates for each trait in the Arabidopsis epigenetic recombinant inbred lines population
| Trait | Heritability model | H 2 Growth room‐1 | H 2 Growth room‐2 |
|---|---|---|---|
| DI | glm1 | 0.55 | 0.50 |
| Pb | glm1 | 0.46 | 0.44 |
| Lfi | glm1 | 0.76 | 0.65 |
| Rbi | glm1 | NA | 0.62 |
| DI | glm2 | NA | 0.50 |
| Pb | glm2 | NA | 0.50 |
| Lfi | glm2 | NA | 0.67 |
| Rbi | glm2 | NA | 0.50 |
H 2 is the broad sense heritability calculated with the formula (Eqn 1): and 2: including the variance of the temperature × epigenotype interaction. , the estimated epigenetic variance; , the estimated temperature × epigenetic interaction variance; , the estimated environmental variance; n, the number of replicates per line. DI, disease index; Pb, pathogen DNA quantity; Lfi, leaf length in infected condition; Rbi, root biomass in infected condition. NA, not available.
QTL analysis of data obtained in growth room‐1
Phenotypic data measured on the two biological replicates set up in growth room‐1 were used in glm1 (Eqn 1) to extract the epigenotype effect (G) with the lsmean function. The epigenotype effect G identified for each trait was then used for the QTLepi analysis. In total, five QTLepi were detected (Fig. 3). Two QTLepi were identified for Pb on chromosomes 1 and 4 (Pb1epi‐At1, Pb1epi‐At4) explaining 14.64% and 9.65% of the phenotypic variability, respectively. Three QTLepi were detected for Lfi on chromosomes 1, 3 and 5 (Lfi1epi‐At1, Lfi1epi‐At3, Lfi1epi‐At5) explaining 15.49%, 15.01% and 8.11% of phenotypic variation, respectively (Table 2). The variance explained by each fitted QTL model was of 43.82% and 19.59% for Pb and Lfi, respectively. Surprisingly, no QTLepi was identified for DI despite a significant effect of epigenotype on this trait. Confidence intervals of the QTLepi detected ranged from 6.23 to 36 cM (Table 2). No epistatic interaction was found between QTLepi for either trait. For the three QTLepi detected for Lfi (markers nearest of the peak LOD score: MM52, MM427 and MM728), wild‐type (WT) epialleles were associated with an increase in the trait values. The ddm1‐derived epiallele was associated with an increase in pathogen quantity at QTLepi on chromosome 1 (peak marker: MM123) whereas it was associated with a decrease in the Pb value on chromosome 4 (peak marker MM550) (Fig. S1; Table 2).
Figure 3.
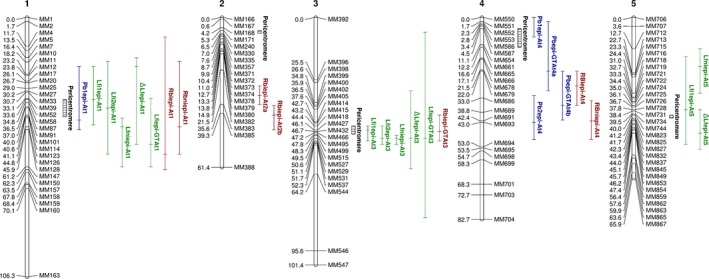
Epigenetic quantitative trait loci (QTL) identified in the epigenetic recombinant inbred lines (epiRIL) population in response to Plasmodiophora brassicae infection. Numbers above chromosomes indicate Arabidopsis chromosomes. MM indicates markers. For each marker, position in cM is indicated. The vertical bar length is equal to the one‐ logarithm of odds (LOD) likelihood confidence interval. The dash in the bar indicates the peak position. QTL name indicates the trait associated and chromosomal localization.
Table 2.
Summary of epigenetic quantitative trait loci (QTLepi) detected in the Arabidopsis epigenetic recombinant inbred lines (epiRIL) population by multiple QTL mapping (MQM)
| Trait | Condition | Test location | QTL | Chr | Pos | Lod | Closest peak marker | Confidence interval (cM) | R 2 (%) | R 2 for all QTL by trait | Favourable allele | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| G‐Rbi | Infected | Growth room‐2 | Rbiepi‐At1 | 1 | 56.00 | 3.04 | MM147 | 8.00 | 62.33 | 6.67 | 44.59 | WT |
| Growth room‐2 | Rbiepi‐At2a | 2 | 32.00 | 7.48 | MM383 | 28.00 | 35.62 | 17.91 | WT | |||
| Growth room‐2 | Rbiepi‐At2b | 2 | 40.00 | 6.18 | MM385 | 36.00 | 46.00 | 14.42 | ddm1‐2 | |||
| Growth room‐2 | Rbiepi‐At4 | 4 | 28.00 | 4.79 | MM686 | 22.02 | 36.00 | 10.88 | WT | |||
| G‐Lfi | Infected | Growth room‐1 | Lfi1epi‐At1 | 1 | 33.60 | 6.51 | MM 52 | 20.00 | 44.00 | 15.50 | 43.82 | WT |
| Growth room‐1 | Lfi1epi‐At3 | 3 | 46.10 | 6.32 | MM427 | 44.36 | 50.59 | 15.01 | WT | |||
| Growth room‐1 | Lfi1epi‐At5 | 5 | 37.80 | 3.60 | MM728 | 16.00 | 52.00 | 8.11 | WT | |||
| Growth room‐2 | Lfi2epi‐At1 | 1 | 37.05 | 5.14 | MM91 | 18.00 | 56.00 | 15.65 | 26.16 | WT | ||
| Growth room‐2 | Lfi2epi‐At3 | 3 | 49.45 | 4.04 | MM515 | 44.37 | 52.00 | 12.06 | WT | |||
| G‐Pb | Infected | Growth room‐1 | Pb1epi‐At1 | 1 | 42.00 | 4.36 | MM123 | 20.00 | 45.93 | 14.64 | 19.59 | WT |
| Growth room‐1 | Pb1epi‐At4 | 4 | 0.00 | 2.95 | MM550 | 0.00 | 8.00 | 9.65 | ddm1‐2 | |||
| Growth room‐2 | Pb2epi‐At4 | 4 | 42.99 | 3.05 | MM693 | 32.00 | 50.00 | 10.86 | 10.86 | ddm1‐2 | ||
| GT‐Lfi | Infected | Growth room‐2 | LfiepiGT‐At1 | 1 | 52.00 | 3.32 | MM128 | 33.07 | 61.20 | 10.67 | 19.34 | WT |
| Growth room‐2 | LfiepiGT‐At3 | 3 | 49.45 | 2.47 | MM547 | 6.00 | 82.00 | 7.81 | WT | |||
| GT‐Rbi | Infected | Growth room‐2 | RbiepiGT‐At3 | 3 | 46.65 | 2.52 | MM432 | 40.00 | 50.59 | 9.06 | 9.06 | WT |
| GT‐Pb | Infected | Growth room‐2 | PbepiGT‐At4a | 4 | 22.00 | 2.38 | MM679 | 1.73 | 30.00 | 8.04 | 15.89 | ddm1‐2 |
| Growth room‐2 | PbepiGT‐At4b | 4 | 36.00 | 4.50 | MM689 | 22.02 | 42.00 | 15.88 | ddm1‐2 | |||
| G‐Lfni | Noninfected | Growth room‐2 | Lfniepi‐At1 | 1 | 44.80 | 8.32 | MM126 | 41.15 | 61.20 | 19.66 | 46.20 | WT |
| Growth room‐2 | Lfniepi‐At3 | 3 | 49.46 | 5.75 | MM515 | 48.32 | 52.00 | 12.93 | WT | |||
| Growth room‐2 | Lfniepi‐At5 | 5 | 20.00 | 4.76 | MM713 | 12.73 | 31.00 | 10.49 | WT | |||
| G‐Rbni | Noninfected | Growth room‐2 | Rbniepi‐At1 | 1 | 44.80 | 3.39 | MM126 | 18.00 | 56.00 | 10.44 | 22.93 | WT |
| Growth room‐2 | Rbniepi‐At4 | 4 | 42.43 | 3.72 | MM691 | 34.00 | 50.00 | 11.51 | WT | |||
| G‐ΔLf | Noninfected – Infected | Growth room‐2 | ΔLfepi‐At1 | 1 | 20.00 | 3.79 | MM10 | 16.45 | 52.00 | 10.44 | 31.55 | ddm1‐2 |
| Growth room‐2 | ΔLfepi‐At3 | 3 | 49.46 | 3.68 | MM515 | 30.00 | 62.00 | 10.11 | ddm1‐2 | |||
| Growth room‐2 | ΔLfepi‐At5 | 5 | 47.36 | 3.08 | MM854 | 37.80 | 54.00 | 8.38 | ddm1‐2 | |||
Confidence intervals are in cM. Peak positions are indicated in cM and with the marker nearest to the logarithm of odds (LOD) score peak. R², phenotypic variation explained by the QTLepi. Chr, chromosome. For Lfi and Rbi, favourable alleles are associated with an increase in the value. For Pb, favourable alleles are associated with a decrease in the value. G‐Rbi, G‐Lfi and G‐Pb represent epigenetic QTL for each trait in infected condition. G‐Lfni and G‐Rbni represent epigenetic QTL for each trait in control condition. G‐ΔLf represents epigenetic QTL obtained by difference between leaf length in infected condition and leaf length in control condition. GT‐Rbi, GT‐Lfi and GT‐Pb represent epigenetic × temperature QTL for each trait.WT, wild‐type; ddm1‐2, mutant allele.DI, disease index; Pb, pathogen DNA quantity; Lfi, leaf length in infected condition; Rbi, root biomass in infected condition; Lfni and Rbni, respectively, leaf length and root biomass in control (noninfected) condition; ΔLf, change in leaf length.
QTL analysis of data from growth room‐2
As above, phenotypic data measured on the two biological replicates in growth room‐2 were used in glm1 to extract the epigenotype effect with the lsmean function. Once again, the epigenotype effect G identified for each trait was then used for the QTLepi analysis. In total, seven QTLepi were detected (Fig. 3). One QTLepi was detected on chromosome 4 for Pb (Pb2epi‐At4) and two QTLepi on chromosomes 1 and 3 were detected for Lfi (Lfi2epi‐At1, Lfi2epi‐At3). QTL mapping also revealed four QTLepi controlling Rbi, which was measured only in this growth room: one on chromosome 1, two on chromosome 2 and one on chromosome 4 (Rbiepi‐At1, Rbiepi‐At2a, Rbiepi‐At2b, Rbiepi‐At4). Again, no QTLepi was identified for DI despite a significant effect of the epigenotype. The variance explained by each fitted QTL model was of 10.86%, 26.16% and 44.59% for Pb, Lfi and Rbi, respectively. The QTLepi identified for Pb on chromosome 4 explained 10.86% of the variability. The two QTLepi detected on chromosomes 1 and 3 for Lfi explained (respectively) 15.65% and 12.06% of the phenotypic variation (Table 2). Confidence intervals of the detected QTLepi ranged from 7.62 to 54.33 cM (Table 2). No epistatic interaction was found between QTLepi for Lfi and Rbi traits. The additive allele effect of the two QTLepi detected for Lfi (peak markers: MM91 and MM515) and of three of the four QTLepi detected for Rbi (peak markers: MM147, MM383, MM686) was in the same direction: WT‐derived epialleles were associated with an increase in trait value. For the Pb QTL (peak marker: MM693) and one of the four QTLepi of Rbi (peak marker: MM385), the ddm1‐derived epiallele was associated with a decrease in trait value (Fig. S1; Table 2).
For Lfi, QTLepi detected on chromosomes 1 and 3 in growth room‐1 (Lfi1epi‐At1, Lfi1epi‐At3) co‐localized with the QTLepi detected on chromosomes 1 and 3 in growth room‐2 (Lfi2epi‐At1, Lfi2epi‐At3). The QTLepi on chromosome 5 was detected only in growth room‐1. The Pb QTLepi detected in the two growth rooms were different. These results indicated that the QTL detection was possibly dependent on the growth room conditions (Fig. 3; Table 2).
As ddm1 mutants displayed smaller roots and leaf lengths than WT in control conditions (i.e. noninoculated; Kakutani et al., 1996; Cortijo et al., 2014; Table S3), a similar experiment was carried out also in growth room‐2 without clubroot infection to determine the impact of developmental alteration due to the mutation on clubroot symptoms. The Lfni and Rbni data assessed on the 123 epiRIL in control condition showed continuous distribution, suggesting polygenic control of these traits (Fig. 2). Data analysis showed a significant epigenotype effect (glm1) on both phenotypic traits (Lfni: χ² = 15 771.8, P‐value < 2.2e‐16; Rbni: χ² = 33.09, P‐value = 3.48e‐10). In total, five QTLepi were detected (Fig. 3): three QTLepi were identified for Lfni on chromosomes 1, 3 and 5 (Lfniepi‐At1, Lfniepi‐At3 and Lfniepi‐At5) explaining 19.66%, 12.93% and 10.49% of the phenotypic variability, respectively. Two QTLepi were detected for Rbni on chromosomes 1 and 4 (Rbniepi‐At1, Rbniepi‐At4) explaining 10.44% and 11.51% of phenotypic variation, respectively (Table 2). The additive allele effect of all the QTLepi detected for Lfni (peak markers: MM126, MM515 and MM713) and Rbni (peak markers: MM126 and MM691) was in the same direction: WT‐derived epialleles were associated with an increase in trait value (Fig. S1). Three QTLepi were detected with ΔLf on chromosomes 1, 3 and 5 (ΔLfepi‐At1, ΔLfepi‐At3 and ΔLfepi‐At5) explaining 10.44%, 10.11% and 8.38% of the phenotypic variability, respectively (Fig. 3). No QTLepi was identified for ΔRb. The additive allele effect of all the QTLepi detected for ΔLf (peak markers: MM10, MM515 and MM854) was also in the same direction but in this case the ddm1‐2 epialleles were associated with the decrease of the value (Fig. S1).
Temperature affects the plant response to P. brassicae in the epiRIL population
In order to explain the differences observed between the two growth rooms, we paid specific attention to the temperature conditions, as this parameter was shown to be critical for the development of clubroot disease (Siemens et al., 2002; Sharma et al., 2011). Although similar values of global mean temperature were noted in both growth rooms (growth room‐1 = 19.95°C, growth room‐2 = 20.06°C), significant differences (F = 2.67, P‐value = 0.002) in global temperature variances were registered between the two rooms (Fig. S2a). We then analysed mean and variance temperature values for each photoperiod (day and night). Day and night mean temperatures were similar in both chambers (growth room‐1: day temperature = 20.98°C, night temperature = 17.83°C; growth room‐2: day temperature = 20.85°C, night temperature = 18.51°C) and were very close to the required values (day temperature = 21°C, night temperature = 18°C). Conversely, the temperature variance in the two growth rooms and for both periods was significantly different (Fisher permutation test; day period: F = 9.05, P‐value = 0.002; night period: F = 3.67, P‐value = 0.002). The temperature range in growth room‐1 was 10.83°C for the day period and 4.98°C for the night period; the temperature range in growth room‐2 was 3.51°C for the day period and 1.88°C for the night period (Fig. S2b,c).
Effect of temperature variation on plant response to clubroot
Based on these observations, a more detailed analysis was carried out in growth room‐2 to evaluate the impact of the temperature variability on epiRIL response to clubroot. For this, we used data from temperature sensors placed at the height of the plants in growth room‐2. Significant differences (Kruskal–Wallis test) in the median temperatures were observed (χ² = 4115.3, df = 15, P‐value < 2.2e‐16) according to the location of the sensors. Pairwise analysis (Dunnett's test) of the differences between temperatures measured by the sensors showed significant temperature variations at several positions in the growth room (Table S5) and a temperature gradient from 21.4 to 24.1°C (Table S6).
A significant temperature effect (glm2) was shown for two of the phenotypic traits measured (P‐value ranged from 0.54 to < 2.2e‐16; Table S4). Moreover, a significant interaction between temperature and epigenotype effects (GT interaction) on the phenotypic traits measured was identified for nearly all traits (P‐value ranged from 0.44 to < 2.2e‐16; Table S4). Heritability (H²) in the case of temperature and epigenotype interaction was estimated for each trait using the formula (2.1). Heritability ranged from 0.50 to 0.67 according to the dataset used (Table 1).
Detection of epigenotype × temperature QTLepi
In order to determine whether temperature variation could influence plant response to clubroot in the epiRIL population, we associated each epiRIL with the mean temperature data measured with the temperature sensor placed in the block where the epiRIL was grown. These data were used in glm2 (Eqn (Eqn 2)) to extract the GT interaction using the lsmean function. The GT interaction values identified for each trait were then used for the QTLepi analysis. Two QTLepiGT (epigenotype × temperature QTLepi) were detected on the chromosomes 1 and 3 for Lfi (LfiepiGT‐At1, LfiepiGT‐At3), and one QTLepiGT on chromosome 3 was detected for Rbi (RbiepiGT‐At3). No QTLepi was detected for Pb at a 5% significance level using the SIM by stepwise approach but two QTLepi were detected on the chromosome 4 using a 10% significance level (PbepiGT‐At4a, PbepiGT‐At4b). Again, no QTLepi could be detected for DI (Fig. 3). The variance explained by the fitted QTL model was 19.34% for Lfi, 9.06% for Rbi and 15.89% for Pb. The QTLepi found for Lfi on chromosomes 1 and 3 explained 10.67% and 7.81% of the variability. The QTLepi found for Rbi explained 9.06% of phenotypic variation. The two QTLepi found for Pb on chromosome 4 explained 8.04% and 15.88% of the variation (Table 2). Confidence intervals of the detected QTLepi ranged from 10.59 to 76 cM (Table 2). No epistatic interaction was found between QTLepi for Lfi and Pb traits. For all QTLepi detected, the WT epialleles were associated with an increase in the values (Fig. S1; Table 2). Comparison of the QTLepi and QTLepiGT (taking into account the interaction with temperature) showed that all QTLepiGT co‐localized totally or partially with QTLepi detected in at least one growth room (Fig. 3). Indeed, the comparison of the confidence intervals of QTLepi detected using the leaf length trait showed that the QTLepi Lfi1epi‐At1 and Lfi2epi‐At1 overlapped with the QTLepiGT LfiepiGT‐At1. Likewise, Lfi1epi‐At3 and Lfi2epi‐At3 confidence intervals co‐localized with the confidence interval of LfiepiGT‐At3 (Fig. 3; Table 2). For the QTLepi detected using the quantification of the pathogen DNA (Pb) two overlaps were identified on chromosome 4, one between Pb1epi‐At4 and PbepiGT‐At4a in the region extending from the short arm to the pericentromeric region and another between Pb2epi‐At4, PbepiGT‐At4a and PbepiGT‐At4b on the long arm of chromosome 4 (Fig. 3; Table 2).
Impact of DNA sequence variation within QTLepi confidence intervals
Although the epiRIL population was designed to minimize as much as possible DNA sequence variation, the presence of a small number of segregating nucleotidic variants cannot be avoided, notably as a result of the known effect of loss of DNA methylation on TE activity. In order to investigate the potential contribution of parental DNA sequence variants to the differential susceptibility to P. brassicae associated with the 20 QTLepi, we identified using whole genome sequencing data all sequence variants shared by more 25% of the epiRIL. In total, 63 small‐scale and 11 TE insertion variants were located within the 20 QTLepi, respectively. Eleven shared insertions of TE were detected in the QTLepi confidence intervals, with 18 QTLepi showing at least one insertion and two QTLepi showing no insertion. All QTLepi detected included at least one small‐scale sequence polymorphism in their confidence interval (Dataset S1).
Effects of the shared TEs and sequence variants were tested as described in Kooke et al. (2015). For 16 of 20 QTLepi, the effect of the epigenetic marker at the QTL peak was more significant than the effect of the TE or the small‐scale sequence variant (Dataset S2). However, for LfiepiGT‐At1, LfiepiGT‐At3, Lfi1epi‐At5 and ΔLfepi‐At3, the significant effect observed for a SNP or a TE was greater than for the epigenetic marker at the QTL peak (Dataset S2). In this case, we considered that these four QTLepi were actually caused by DNA sequence variants. However, a linkage disequilibrium between the significant genetic markers and a causal epiallele could also be possible.
Discussion
The aim of the present study was to investigate the role of epigenetic modifications in the Arabidopsis response to Plasmodiophora brassicae. To determine whether epigenetic regulation does play a role in clubroot quantitative resistance, a reverse genetics approach, using six T‐DNA insertional mutants (Col‐0 background) in genes involved in epigenetic pathways, was first carried out. For the six mutants tested, the disease index (DI) was only significantly reduced in the ddm1 mutant compared to Col‐0. This finding suggests that ddm1 confers decreased susceptibility of Arabidopsis to P. brassicae. This observation is in agreement with previous results obtained by Kellenberger et al. (2016) and Sharma et al. (2017) in the Brassica genus, which linked plant responses to biotic and abiotic stress to epigenetic regulations. Moreover, the involvement of ddm1 in the Arabidopsis–P. brassicae interaction further supports the hypothesis that DNA methylation plays a role in plant pathogen infections (Dowen et al., 2012; López Sánchez et al., 2016; Hewezi et al., 2017). To decipher the epigenetic architecture of the clubroot resistance in Arabidopsis, we then carried out a epigenetic QTL (QTLepi) detection experiment using the epigenetic recombinant inbred lines (epiRIL) population. Four disease‐related traits (DI, pathogen DNA quantity (Pb), leaf length (Lfi) and root biomass (Rbi)), mostly used previously in Arabidopsis (Jubault et al., 2008; Gravot et al., 2011) to evaluate plant response to clubroot, were monitored. These traits were chosen in order to characterize disease development (disease index and pathogen DNA quantity) and the consequences of P. brassicae infection on plant development (Rbi and Lfi).
Epigenetic QTL control Arabidopsis clubroot resistance
From our experiments using four biological replicates, we have shown a significant epigenetic effect on the response to P. brassicae infection. Thus, heritable plant responses induced by DNA methylation appear to be involved in the plant response to P. brassicae. The moderate to high heritability values (from 0.33 to 0.76) observed were similar to those described in control and abiotic stress conditions for this population (Johannes et al., 2009; Colomé‐Tatché et al., 2012; Kooke et al., 2015). Moreover, these heritability values also were similar to those described by Jubault et al. (2008) on a RIL population infected with P. brassicae. The moderate heritability levels calculated for the traits mean that this epigenetic variability could be considered for use in breeding. As the plant response to P. brassicae varied depending on the growth room used, we carried out the QTLepi detection experiments using data from each growth room. In total, sixteen additive QTLepi grouped in six genomic regions were identified distributed throughout four A. thaliana chromosomes. Among the 16 QTLepi, five QTLepi were involved in pathogen multiplication (Pb) and 11 were involved in plant (foliar and root) development in response to P. brassicae infection. The identification of three QTLepi for differences in longest length leaf (ΔLf) highlighted the modulation by P. brassicae infection of the foliar development variation already present in the epiRIL population (illustrated in Fig. S3). Clubroot resistance response is therefore composed of factors reducing the impact of the clubroot infection on foliar development as well as factors reducing pathogen development. For four of the 20 QTLepi initially detected (LfiepiGT‐At1, LfiepiGT‐At3, Lfiepi‐At5 and ΔLfepi‐At3), analysis of shared transposition events (TE) and sequence variants included in their confidence intervals showed that the effect of DNA‐based markers was greater than the effect of epigenetic markers, suggesting that these QTL are not bona fide epigenetic. Among the four phenotypic traits evaluated, no QTLepi was detected for DI despite a significant epigenotype effect in the two growth rooms (P = 1.57e‐06 and 1.65e‐04 for growth room‐1 and ‐2, respectively). Results of the reverse genetics approach showed that ddm1 was less susceptible to P. brassicae compared to Col‐0 suggesting that ddm1 epialleles confer a reduction in symptoms. In this context, the absence of QTLepi detected for the DI trait may be explained by a sampling effect, because only a subset (123 of 505 lines) of the epiRIL population was phenotyped in this study, and/or a low proportion of ddm1 epi‐haplotypes in the population subset (27%), which may have been insufficient to detect small QTL effects (Holland, 2007). Most of the QTLepi detected in this study co‐localized with the pericentromeric regions. This may be explained by a more stable loss of methylation in those regions due to the loss of methylation maintenance on repeats and TE in ddm1 (Kooke et al., 2015). Concerning the effect of the epialleles, the majority of the wild‐type (WT) epialleles were associated with an increase in the morphological trait values (length of leaves and root biomass). However, the ddm1‐derived epiallele led to a decrease in the amount of pathogen DNA for two of the three QTLepi detected on chromosomes 1 and 4. This finding highlighted the positive effect of this epiallele on plant resistance, in agreement with the results obtained in the mutant test of this study and by Dowen et al. (2012) who showed a modest increase in ddm1 resistance to P. syringae.
Stability and pleiotropy of clubroot epigenetic QTL
Several QTL detected were stable across growth rooms. Indeed, despite the temperature variations between the two growth rooms, two overlapping leaf length QTLepi (growth room‐1: Lfi1epi‐At1 and Lfi1epi‐At3; growth room‐2: Lfi2epi‐At1 and Lfi2epi‐At3) were detected in each growth room. Furthermore, for Pb, the QTLepi Pb1epi‐At4, detected in the growth room‐1 overlapped with the QTLepi PbepiGT‐At4a detected in the growth room‐2 (Fig. 3; Table 2). The co‐localization on chromosome 1 of four QTLepi controlling three different traits (Lfi, Pb and Rbi) and on chromosome 4 of three QTLepi controlling two traits (Pb and Rbi) may suggest the presence of pleiotropic QTLepi (Fig. 3; Table 2). However, analysis of the correlation (Spearman rho correlation) between traits showed only a moderate correlation (ρ = 0.52, P < 2.2e‐16) between Lfi and Rbi. Fine mapping is necessary to overcome the bias imposed by the large size of the QTLepi confidence intervals and make further conclusions on the possibility of a pleiotropic gene or an effect due to linked genes.
Temperature modulates Arabidopsis clubroot responses
Taking into consideration the temperature variations in growth room‐2, we tested the hypothesis that temperature plays a potential role in epiRIL and Col‐0 clubroot symptom variations. These observations are in agreement with the variations in clubroot severity observed on Brassica rapa subsp. chinensis and B. napus according to the temperature used for growing the plants (Sharma et al., 2011; Gossen et al., 2014). Siemens et al. (2002) had already evoked a possible link between temperature, clubroot response and epigenetics when studying the Arabidopsis mutant tu8 (mutant in the LIKE HETEROCHROMATIN PROTEIN 1 LHP1) which presented different levels of response to clubroot depending on the temperature conditions. Similar environmental effects on the modulation of QTL controlling clubroot response also have been shown with nitrogen supply variations in B. napus (Laperche et al., 2017; Aigu et al., 2018) and with flooding in Arabidopsis (Gravot et al., 2016). Here, our analyses suggested that the temperature effect was partly triggered by the interaction with the plant epigenome for the traits Rbi and Pb. The identification of temperature‐dependent QTLepi controlling pathogen quantity (PbepiGT‐At4a and PbepiGT‐At4b) suggests the presence of QTLepi involved in the control of the pathogen development under temperature dependence. Similar observation concerning the detection of QTL temperature dependance was carried out by Aoun et al. (2017) using the model Arabidopsis–Ralstonia solanacearum. In their study, Aoun et al. (2017) showed that an increase of the temperature of 3°C during the interaction between Arabidopsis and R. solanacearum led to an increase of the sensitivity of most accessions and the loss of detection of one major QTL associated with the resistance. The influence of the temperature on the epiRIL response to P. brassicae could be explained by the increase of the environmental sensitivity triggered by the DNA hypomethylation suggested by Kooke et al. (2015).
Clubroot resistance, a sophisticated system of regulation involving genetics and epigenetics
Interestingly, the comparison of the clubroot genetic QTL identified previously (Jubault et al., 2008) in Arabidopsis and the clubroot epigenetic QTL identified in this study highlighted overlapping of some confidence intervals. Indeed, the confidence intervals of six QTLepi (Rbiepi‐At1, ΔLfepi‐ At1and Pb1epi‐At1, Lfi1epi‐At1, Lfi2epi‐At1, ΔLfepi‐At1) overlapped with two QTL (Pb‐At1 and Pb‐At4) found in the Bur‐0 × Col‐0 RIL population by linkage analysis (Jubault et al., 2008) and the major gene RPB1 described by Arbeiter et al. (2002), respectively. These co‐localizations suggest that quantitative resistance to clubroot is modulated by a system involving both nucleotidic and epigenetic variations. These results illustrate the fact that in classical populations used for QTL detection, dissociation between causal genetic and epigenetic variations, both in linkage disequilibrium with markers, is nearly impossible (Schmitz et al., 2013a,b). In addition, our findings are consistent with the suggestion that during plant–pathogen interactions, plant transgenerational changes in genome structure and in DNA methylation patterns are possible (Boyko & Kovalchuk, 2011). The identification of two QTLepi (Rbiepi‐At2 and Pb1epi‐At4) that did not show any overlap with previously reported QTL for these traits suggests that at these loci only epigenetic variation may be responsible for plant response variation. However, an absence of co‐localization could also be due to the absence of nucleotidic and/or epigenetic variations at these loci in the previously studied populations but does not exclude their existence in other genotypes.
This first study on the role of epialleles in the Arabidopsis–P. brassicae interaction brings to light the possibility of a complex model of quantitative resistance where alleles and epialleles act in concert. Furthermore, our study has shown that the temperature variations could influence epiRIL response to P. brassicae. In order to confirm whether epialleles are involved in plant response to clubroot infection, the QTLepi confidence intervals must be reduced to find causal epialleles, notably through a fine‐mapping approach. The assessment of epiRIL in pathological tests in contrasting controlled temperature conditions is also needed to validate the possible role of temperature in modulating the epigenetic plant response to clubroot infection.
Author contributions
BL, EJ, AE, MJ, AG, JL and CL carried out the experiments and collected the data; BL, JL and CL carried out the molecular biology work; BL and VB carried out the genetic analyses; BL and AE carried out the bioinformatics analyses; VB, ME and VC provided DNA sequence information about the epiRIL used in this study; BL, VB, MJM‐D and MJ wrote the article, assisted by AG and VC for manuscript editing; and BL, MJM‐D and MJ designed and coordinated the study.
Supporting information
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Dataset S1 Location of segregating DNA sequence variants (SNP, indel, TE, SV) among the clubroot QTL intervals.
Dataset S2 Statistic analysis of TE and sequence variant effect compared to methylated markers on QTLepi detection for each trait study.
Fig. S1 Epiallele effects at the closest QTLepi peak marker.
Fig. S2 Boxplot of temperature data recorded in each growth room.
Fig. S3 Comparison of the epiallele effects at the closest QTLepi peak marker between Lfi and Lfni in growth chamber‐2.
Table S1 List of primer sets used to confirm homozygosity of the T‐DNA insertion in mutants.
Table S2 Disease index for Col‐0 and mutants after infection by P. brassicae, and Dunnett's post hoc test results.
Table S3 Phenotypic responses of epiRIL and their parent lines to infection by P. brassicae.
Table S4 Analysis of epigenotype, temperature and interaction between temperature and epigenotype effect for each trait in infected condition.
Table S5 Dunnett's test comparison of temperatures recorded by each temperature sensor in growth room‐2.
Table S6 Median temperature monitored by each temperature sensor in growth room‐2.
Acknowledgements
We acknowledge our IGEPP Colleagues for their technical support and the Biological Resource Centers BrACySol and Versailles Arabidopsis Stock Center for providing Brassica and epiRIL seeds, respectively. This research was supported by AGROCAMPUS OUEST, INRA and Université de Rennes. Benjamin Liégard was a PhD student co‐funded by INRA BAP department and Brittany Region.
References
- Aigu Y, Laperche A, Mendes J, Lariagon C, Guichard S, Gravot A, Manzanares‐Dauleux MJ. 2018. Nitrogen supply exerts a major/minor switch between two QTLs controlling Plasmodiophora brassicae spore content in rapeseed. Plant Pathology 67: 1574–1581. [Google Scholar]
- Alix K, Lariagon C, Delourme R, Manzanares‐Dauleux MJ. 2007. Exploiting natural genetic diversity and mutant resources of Arabidopsis thaliana to study the A. thaliana–Plasmodiophora brassicae interaction. Plant Breed 126: 218–221. [Google Scholar]
- Aller EST, Jagd LM, Kliebenstein DJ, Burow M. 2018. Comparison of the relative potential for epigenetic and genetic variation to contribute to trait stability. G3 8: 1733–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoun N, Tauleigne L, Lonjon F, Deslandes L, Vailleau F, Roux F, Berthomé R. 2017. Quantitative disease resistance under elevated temperature: genetic basis of new resistance mechanisms to Ralstonia solanacearum . Frontiers in Plant Science 8: 1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbeiter A, Fahling M, Graf H, Sacristán MD, Siemens J. 2002. Resistance of Arabidopsis thaliana to the obligate biotrophic parasite Plasmodiophora brassicae . Plant Protection Science ‐ Prague 38: 519–522. [Google Scholar]
- Becker C, Hagmann J, Müller J, Koenig D, Stegle O, Borgwardt K, Weigel D. 2011. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480: 245–249. [DOI] [PubMed] [Google Scholar]
- Boyes DC, Zayed AM, Ascenzi R, McCaskill AJ, Hoffman NE, Davis KR, Görlach J. 2001. Growth stage‐based phenotypic analysis of Arabidopsis: a model for high throughput functional genomics in plants. Plant Cell 13: 1499–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyko A, Kovalchuk I. 2011. Genetic and epigenetic effects of plant–pathogen interactions: an evolutionary perspective. Molecular Plant 4: 1014–1023. [DOI] [PubMed] [Google Scholar]
- Broman KW, Sen Ś. 2009. Fit and exploration of multiple‐QTL models In: A guide to QTL mapping with R/qtl. Statistics for biology and health. New York, NY, USA: Springer, 241–282. [Google Scholar]
- Broman KW, Wu H, Sen Ś, Churchill GA. 2003. R/qtl: QTL mapping in experimental crosses. Bioinformatics 19: 889–890. [DOI] [PubMed] [Google Scholar]
- Colomé‐Tatché M, Cortijo S, Wardenaar R, Morgado L, Lahouze B, Sarazin A, Etcheverry M, Martin A, Feng S, Duvernois‐Berthet E et al 2012. Features of the Arabidopsis recombination landscape resulting from the combined loss of sequence variation and DNA methylation. Proceedings of the National Academy of Sciences, USA 109: 16240–16245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortijo S, Wardenaar R, Colomé‐Tatché M, Gilly A, Etcheverry M, Labadie K, Caillieux E, Hospital F, Aury J‐M, Wincker P et al 2014. Mapping the epigenetic basis of complex traits. Science 343: 1145–1148. [DOI] [PubMed] [Google Scholar]
- Diederichsen E, Frauen M, Linders EGA, Hatakeyama K, Hirai M. 2009. Status and perspectives of clubroot resistance breeding in Crucifer crops. Journal of Plant Growth Regulation 28: 265–281. [Google Scholar]
- Dixon GR. 2009. The occurrence and economic impact of Plasmodiophora brassicae and clubroot disease. Journal of Plant Growth Regulation 28: 194–202. [Google Scholar]
- Dowen RH, Pelizzola M, Schmitz RJ, Lister R, Dowen JM, Nery JR, Dixon JE, Ecker JR. 2012. Widespread dynamic DNA methylation in response to biotic stress. Proceedings of the National Academy of Sciences, USA 109: E2183–E2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahling M, Graf H, Siemens J. 2003. Pathotype separation of Plasmodiophora brassicae by the host plant. Journal of Phytopathology 151: 425–430. [Google Scholar]
- Gallusci P, Dai Z, Génard M, Gauffretau A, Leblanc‐Fournier N, Richard‐Molard C, Vile D, Brunel‐Muguet S. 2017. Epigenetics for plant improvement: current knowledge and modeling avenues. Trends in Plant Science 22: 610–623. [DOI] [PubMed] [Google Scholar]
- Gilly A, Etcheverry M, Madoui M‐A, Guy J, Quadrana L, Alberti A, Martin A, Heitkam T, Engelen S, Labadie K et al 2014. TE‐Tracker: systematic identification of transposition events through whole‐genome resequencing. BMC Bioinformatics 15: 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen BD, Deora A, Peng G, Hwang S‐F, McDonald MR. 2014. Effect of environmental parameters on clubroot development and the risk of pathogen spread. Canadian Journal of Plant Pathology 36: 37‐48. [Google Scholar]
- Gravot A, Grillet L, Wagner G, Jubault M, Lariagon C, Baron C, Deleu C, Delourme R, Bouchereau A, Manzanares‐Dauleux MJ. 2011. Genetic and physiological analysis of the relationship between partial resistance to clubroot and tolerance to trehalose in Arabidopsis thaliana . New Phytologist 191: 1083–1094. [DOI] [PubMed] [Google Scholar]
- Gravot A, Richard G, Lime T, Lemarié S, Jubault M, Lariagon C, Lemoine J, Vicente J, Robert‐Seilaniantz A, Holdsworth MJ et al 2016. Hypoxia response in Arabidopsis roots infected by Plasmodiophora brassicae supports the development of clubroot. BMC Plant Biology 16: 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewezi T, Lane T, Piya S, Rambani A, Rice JH, Staton M. 2017. Cyst nematode parasitism induces dynamic changes in the root epigenome. Plant Physiology 174: 405–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland J. 2007. Genetic architecture of complex traits in plants. Current Opinion in Plant Biology 10: 156–161. [DOI] [PubMed] [Google Scholar]
- Ingram DS, Tommerup IC. 1972. The life history of Plasmodiophora brassicae Woron . Proceedings of the Royal Society of London. Series B: Biological Sciences 180: 103–112. [Google Scholar]
- Johannes F, Colot V, Jansen RC. 2008. Epigenome dynamics: a quantitative genetics perspective. Nature Reviews. Genetics 9: 883–890. [DOI] [PubMed] [Google Scholar]
- Johannes F, Porcher E, Teixeira FK, Saliba‐Colombani V, Simon M, Agier N, Bulski A, Albuisson J, Heredia F, Audigier P et al 2009. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genetics 5: e1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jubault M, Lariagon C, Simon M, Delourme R, Manzanares‐Dauleux MJ. 2008. Identification of quantitative trait loci controlling partial clubroot resistance in new mapping populations of Arabidopsis thaliana . TAG. Theoretical and Applied Genetics 117: 191–202. [DOI] [PubMed] [Google Scholar]
- Kakutani T, Jeddeloh JA, Flowers SK, Munakata K, Richards EJ. 1996. Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proceedings of the National Academy of Sciences, USA 93: 12406–12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger RT, Schlüter PM, Schiestl FP. 2016. Herbivore‐induced DNA demethylation changes floral signalling and attractiveness to pollinators in Brassica rapa . PLoS ONE 11: e0166646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch E, Cox R, Williams PH. 1991. Infection of Arabidopsis thaliana by Plasmodiophora brassicae . Journal of Phytopathology 132: 99–104. [Google Scholar]
- Kooke R, Johannes F, Wardenaar R, Becker F, Etcheverry M, Colot V, Vreugdenhil D, Keurentjes JJB. 2015. Epigenetic basis of morphological variation and phenotypic plasticity in Arabidopsis thaliana . Plant Cell 27: 337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laperche A, Aigu Y, Jubault M, Ollier M, Guichard S, Glory P, Strelkov SE, Gravot A, Manzanares‐Dauleux MJ. 2017. Clubroot resistance QTL are modulated by nitrogen input in Brassica napus . TAG. Theoretical and Applied Genetics 130: 669–684. [DOI] [PubMed] [Google Scholar]
- Lee J, Izzah NK, Choi B‐S, Joh HJ, Lee S‐C, Perumal S, Seo J, Ahn K, Jo EJ, Choi GJ et al 2016. Genotyping‐by‐sequencing map permits identification of clubroot resistance QTLs and revision of the reference genome assembly in cabbage (Brassica oleracea L.). DNA Research: An International Journal for Rapid Publication of Reports on Genes and Genomes 23: 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemarié S, Robert‐Seilaniantz A, Lariagon C, Lemoine J, Marnet N, Jubault M, Manzanares‐Dauleux MJ, Gravot A. 2015a. Both the jasmonic acid and the salicylic acid pathways contribute to resistance to the biotrophic clubroot agent Plasmodiophora brassicae in Arabidopsis. Plant & Cell Physiology 56: 2158–2168. [DOI] [PubMed] [Google Scholar]
- Lemarié S, Robert‐Seilaniantz A, Lariagon C, Lemoine J, Marnet N, Levrel A, Jubault M, Manzanares‐Dauleux M, Gravot A. 2015b. Camalexin contributes to the partial resistance of Arabidopsis thaliana to the biotrophic soilborne protist Plasmodiophora brassicae . Frontiers in Plant Science 6: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenth RV. 2016. Least‐squares means: the R Package lsmeans. Journal of Statistical Software 69: 1–33. [Google Scholar]
- Liu J, Feng L, Li J, He Z. 2015. Genetic and epigenetic control of plant heat responses. Frontiers in Plant Science 6: 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López Sánchez A, Stassen JHM, Furci L, Smith LM, Ton J. 2016. The role of DNA (de)methylation in immune responsiveness of Arabidopsis . The Plant Journal: for Cell and Molecular Biology 88: 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna E, Bruce TJA, Roberts MR, Flors V, Ton J. 2012. Next‐generation systemic acquired resistance. Plant Physiology 158: 844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzanares‐Dauleux MJ, Delourme R, Baron F, Thomas G. 2000a. Mapping of one major gene and of QTLs involved in resistance to clubroot in Brassica napus . TAG. Theoretical and Applied Genetics 101: 885–891. [Google Scholar]
- Manzanares‐Dauleux MJ, Divaret I, Baron F, Thomas G. 2000b. Evaluation of French Brassica oleracea landraces for resistance to Plasmodiophora brassicae . Euphytica 113: 211–218. [Google Scholar]
- Marschner IC. 2011. glm2: Fitting generalized linear models with convergence problems. The R Journal 3: 12–15. [Google Scholar]
- Piao Z, Ramchiary N, Lim YP. 2009. Genetics of clubroot resistance in Brassica species. Journal of Plant Growth Regulation 28: 252–264. [Google Scholar]
- Quadrana L, Colot V. 2016. Plant transgenerational epigenetics. Annual Review of Genetics 50: 467–491. [DOI] [PubMed] [Google Scholar]
- R Development Core Team . 2015. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; [WWW document] URL https://www.R-project.org/. [Google Scholar]
- Reinders J, Wulff BBH, Mirouze M, Marí‐Ordóñez A, Dapp M, Rozhon W, Bucher E, Theiler G, Paszkowski J. 2009. Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes & Development 23: 939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocherieux J, Glory P, Giboulot A, Boury S, Barbeyron G, Thomas G, Manzanares‐Dauleux MJ. 2004. Isolate‐specific and broad‐spectrum QTLs are involved in the control of clubroot in Brassica oleracea . TAG. Theoretical and applied genetics 108: 1555–1563. [DOI] [PubMed] [Google Scholar]
- Schmitz RJ, He Y, Valdes‐Lopez O, Khan SM, Joshi T, Urich MA, Nery JR, Diers B, Xu D, Stacey G et al 2013a. Epigenome‐wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Research 23: 1663–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RJ, Schultz MD, Lewsey MG, O'Malley RC, Urich MA, Libiger O, Schork NJ, Ecker JR. 2011. Transgenerational epigenetic instability is a source of novel methylation variants. Science 334: 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RJ, Schultz MD, Urich MA, Nery JR, Pelizzola M, Libiger O, Alix A, McCosh RB, Chen H, Schork NJ et al 2013b. Patterns of population epigenomic diversity. Nature 495: 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Gossen BD, McDonald MR. 2011. Effect of temperature on cortical infection by Plasmodiophora brassicae and clubroot severity. Phytopathology 101: 1424–1432. [DOI] [PubMed] [Google Scholar]
- Sharma R, Vishal P, Kaul S, Dhar MK. 2017. Epiallelic changes in known stress‐responsive genes under extreme drought conditions in Brassica juncea (L.) Czern. Plant Cell Reports 36: 203–217. [DOI] [PubMed] [Google Scholar]
- Siemens J, Nagel M, Ludwig‐Muller J, Sacristan MD. 2002. The interaction of Plasmodiophora brassicae and Arabidopsis thaliana: parameters for disease quantification and screening of mutant lines. Journal of Phytopathology 150: 592–605. [Google Scholar]
- Some A, Manzanares MJ, Laurens F, Baron F, Thomas G, Rouxel F. 1996. Variation for virulence on Brassica napus L. amongst Plasmodiophora brassicae collections from France and derived single‐spore isolates. Plant Pathology 45: 432–439. [Google Scholar]
- Teixeira FK, Heredia F, Sarazin A, Roudier F, Boccara M, Ciaudo C, Cruaud C, Poulain J, Berdasco M, Fraga MF et al 2009. A role for RNAi in the selective correction of DNA methylation defects. Science 323: 1600–1604. [DOI] [PubMed] [Google Scholar]
- Tierney L, Rossini AJ, Li N. 2009. Snow: a parallel computing framework for the R system. International Journal of Parallel Programming 37: 78‐90. [Google Scholar]
- Vongs A, Kakutani T, Martienssen RA, Richards EJ. 1993. Arabidopsis thaliana DNA methylation mutants. Science 260: 1926–1928. [DOI] [PubMed] [Google Scholar]
- Weigel D, Colot V. 2012. Epialleles in plant evolution. Genome Biology 13: 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A, Lepère G, Jay F, Wang J, Bapaume L, Wang Y, Abraham A‐L, Penterman J, Fischer RL, Voinnet O et al 2013. Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proceedings of the National Academy of Sciences, USA 110: 2389–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y‐Y, Fischer M, Colot V, Bossdorf O. 2013. Epigenetic variation creates potential for evolution of plant phenotypic plasticity. New Phytologist 197: 314–322. [DOI] [PubMed] [Google Scholar]
- Zheng X, Chen L, Xia H, Wei H, Lou Q, Li M, Li T, Luo L. 2017. Transgenerational epimutations induced by multi‐generation drought imposition mediate rice plant's adaptation to drought condition. Scientific Reports 7: 39843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Dataset S1 Location of segregating DNA sequence variants (SNP, indel, TE, SV) among the clubroot QTL intervals.
Dataset S2 Statistic analysis of TE and sequence variant effect compared to methylated markers on QTLepi detection for each trait study.
Fig. S1 Epiallele effects at the closest QTLepi peak marker.
Fig. S2 Boxplot of temperature data recorded in each growth room.
Fig. S3 Comparison of the epiallele effects at the closest QTLepi peak marker between Lfi and Lfni in growth chamber‐2.
Table S1 List of primer sets used to confirm homozygosity of the T‐DNA insertion in mutants.
Table S2 Disease index for Col‐0 and mutants after infection by P. brassicae, and Dunnett's post hoc test results.
Table S3 Phenotypic responses of epiRIL and their parent lines to infection by P. brassicae.
Table S4 Analysis of epigenotype, temperature and interaction between temperature and epigenotype effect for each trait in infected condition.
Table S5 Dunnett's test comparison of temperatures recorded by each temperature sensor in growth room‐2.
Table S6 Median temperature monitored by each temperature sensor in growth room‐2.