

Abstract
Objective
Baricitinib is an orally administered inhibitor of JAK1 and JAK2 that has been shown to be effective in treating rheumatoid arthritis (RA). This study was undertaken to analyze changes in lymphocyte cell subsets during baricitinib treatment and to correlate these changes with clinical outcomes.
Methods
An integrated analysis was conducted by pooling data from 3 completed phase III trials comparing placebo with baricitinib treatment (RA‐BEAM, RA‐BUILD, and RA‐BEACON) and 1 ongoing long‐term extension study (RA‐BEYOND) in patients with active RA (n = 2,186).
Results
Baricitinib treatment was associated with an early transient increase in total lymphocyte count at week 4, which returned to baseline by week 12. Transient changes within normal reference ranges in T cells and subsets were observed with baricitinib treatment, up to week 104. B cells and relevant subpopulations increased after 4 weeks of baricitinib treatment, with no further increases noted through 104 weeks of treatment. Natural killer (NK) cells temporarily increased after 4 weeks of baricitinib treatment, before decreasing below baseline levels and then stabilizing over time. With baricitinib treatment, few correlations were observed between changes in lymphocyte subsets and clinical end points, and most correlations were also observed within the placebo group. A modest potential association between low NK cell numbers and treatment‐emergent infections was observed in the baricitinib 4 mg/day treatment group, but not for serious infections or herpes zoster.
Conclusion
Overall, these findings demonstrate that changes in lymphocyte subsets were largely within normal reference ranges across the baricitinib phase III RA clinical program and were not associated with increased risk of serious infections.
Targeted synthetic disease‐modifying antirheumatic drugs (DMARDs) are orally available low molecular weight molecules developed for the treatment of rheumatoid arthritis (RA). Recent additions to this drug class include JAK inhibitors, which target cytokine signaling pathways implicated in RA pathogenesis 1, 2, 3, 4, 5.
Baricitinib is a selective JAK1/JAK2 inhibitor 6 approved for the treatment of moderate‐to‐severe RA in adults in >40 countries including multiple European countries, Japan, and the United States. Phase III trials showed significant clinical efficacy of baricitinib in patients with active RA who were naive to conventional synthetic DMARDs (csDMARDs) or had inadequate responses to csDMARDs or biologic DMARDs 7, 8, 9, 10. Baricitinib was associated with significant clinical improvement compared to placebo (which typically was given along with stable background csDMARDs), and compared to the tumor necrosis factor (TNF) inhibitor adalimumab in the RA‐BEAM trial 7, 8, 10.
Baricitinib, along with other JAK inhibitors, has been shown to change circulating lymphocyte subset populations, largely within normal ranges 11, 12, 13. However, detailed analyses of effects on subsets are lacking, and their relevance to efficacy and safety is unclear 14. This study demonstrated changes in lymphocyte numbers, including subpopulations, during treatment in patients with active RA (n = 2,186). It comprised an integrated analysis of 3 completed phase III RA trials comparing placebo treatment to baricitinib treatment (RA‐BEAM, RA‐BUILD, and RA‐BEACON), and 1 ongoing phase III long‐term extension study (RA‐BEYOND) 7, 8, 10. This study also tested whether changes in lymphocyte subsets were associated with efficacy or adverse events.
Patients and methods
Patients and study designs. We examined data from 3 previously reported randomized, double‐blind, multicenter phase III trials (RA‐BEAM, RA‐BUILD, and RA‐BEACON) and 1 ongoing phase III long‐term extension study (RA‐BEYOND) with data up to January 1, 2016. Patients were ≥18 years of age with moderate‐to‐severe active RA. Descriptions of the patient cohorts for each of the trials are shown in Supplementary Table 1 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40680/abstract), and detailed methods were reported previously 7, 8, 10. Criteria for exclusion and permanent discontinuation included an abnormal lymphocyte count of <750 cells/μl and <200 cells/μl, respectively.
After completion of 1 of the 3 randomized trials, patients could enter the RA‐BEYOND study and receive either baricitinib 2 mg/day or 4 mg/day for up to 60 months. Patients entering RA‐BEYOND after 52 weeks in RA‐BEAM continued baricitinib 4 mg/day or switched to baricitinib 4 mg/day from adalimumab. Patients entering RA‐BEYOND after 24 weeks of treatment in RA‐BUILD or RA‐BEACON continued baricitinib 2 mg/day or 4 mg/day, or switched to baricitinib 4 mg/day from placebo. In RA‐BEYOND, patients who received baricitinib 4 mg/day for ≥15 months (including in the preceding originator trial) without rescue and achieved sustained low disease activity or remission were re‐randomized (under blinded conditions) to either continue baricitinib 4 mg/day or step down to baricitinib 2 mg/day. Studies were conducted in accordance with the ethical principles of the Declaration of Helsinki and Good Clinical Practice guidelines and were approved by the ethics committee or institutional review board of each center. All patients provided written informed consent.
Analytical methodology. Whole blood samples were collected at week 0 (for placebo, n = 856–892; for baricitinib 2 mg/day, n = 392–403; for baricitinib 4 mg/day, n = 869–891), week 4 (placebo, n = 747–825; 2 mg/day, n = 358–384; 4 mg/day, n = 765–846), week 12 (placebo, n = 706–790; 2 mg/day, n = 348–361; 4 mg/day, n = 755–835), weeks 24–32 (placebo, n = 481–530; 2 mg/day, n = 272–295; 4 mg/day, n = 637–706), weeks 48–52 (2 mg/day, n = 182–196; 4 mg/day, n = 364–582), and weeks 96–104 (2 mg/day, n = 79–91; 4 mg/day, n = 120–135). Total lymphocyte count was analyzed using a Beckman Coulter cell counter. Lymphocyte subsets were analyzed using a BD FACSCanto II flow cytometer (BD Biosciences) and a previously optimized panel 15 (see Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40680/abstract). Data on Th1 and Th17 cells at the 96–104‐week time point were not acquired, and data on these subsets in the baricitinib 2 mg/day group were not available for the 48–52‐week time point due to an insufficient sample size. T cells, including CD4 and CD8 subpopulations, B cells, and natural killer (NK) cells, were identified using BD Multitest 6‐color TBNK reagent (BD Biosciences). A Human FoxP3 Buffer Set (BD Biosciences) was used to identify Treg cells. Additional flow cytometry methods and representative gating for cell subsets in the primary flow cytometry data are included in Supplementary Figures 1–4 and Supplementary Flow Cytometry Methods (http://onlinelibrary.wiley.com/doi/10.1002/art.40680/abstract).
Statistical analysis. An integrated data set was generated by pooling patient data from RA‐BEAM, RA‐BUILD, RA‐BEACON, and RA‐BEYOND. The placebo group came from RA‐BEAM, RA‐BUILD, and RA‐BEACON; data were censored at rescue or the end of the 24‐week placebo treatment period. The baricitinib 2 mg/day group came from RA‐BUILD and RA‐BEACON, with associated data from RA‐BEYOND; data were censored at rescue. The baricitinib 4 mg/day group came from RA‐BEAM, RA‐BUILD, and RA‐BEACON, with associated data from RA‐BEYOND; data were censored at rescue or dose step‐down.
For changes in lymphocyte subsets, a restricted maximum‐likelihood–based mixed model for repeated measures was used, which included treatment, visit, and treatment‐by‐visit interaction as fixed categorical effects and baseline as fixed continuous effects. Change from baseline was estimated by modeling 4 covariance structures (autoregressive, heterogeneous autoregressive, compound symmetry, and Toeplitz matrix) and selecting the variance–covariance structure with the smallest Akaike information criterion. Fisher's exact test was used for comparisons of incidence of treatment‐emergent (TE) infections between treatment groups by NK cell count category.
For rheumatoid factor (RF) and anti–citrullinated protein antibody (ACPA), mean changes from baseline were compared between treatment groups by analysis of covariance, with treatment and baseline values as covariates. Type III sums of squares were used for comparisons of each baricitinib dose to placebo, and least squares means, SEM, and P values were reported.
The weighted Pearson's correlation coefficient and its associated P value were used to test whether correlations between change from baseline in lymphocyte subset and Disease Activity Score in 28 joints 16 based on the high‐sensitivity C‐reactive protein level (DAS28‐hsCRP) were significant by treatment group.
Results
Changes in lymphocyte subsets. Total lymphocyte count increased transiently at week 4 with baricitinib 2 mg/day and 4 mg/day compared to placebo, returning to baseline by week 12 and remaining stable thereafter (Figure 1). At baseline, low lymphocyte counts were observed in 8.7%, 6.2%, and 6.6% of patients receiving placebo, baricitinib 2 mg/day, and baricitinib 4 mg/day, respectively, and high lymphocyte counts were observed in 4.3%, 4.7%, and 4.0% of patients. Based on 24 weeks of data up to rescue, the proportions of patients with ≥1 TE low lymphocyte count were 10.7%, 8.2%, and 8.9% with placebo, baricitinib 2 mg/day, and baricitinib 4 mg/day, respectively, and the proportions of patients with ≥1 TE high lymphocyte count were 4.4%, 11.2%, and 11.9%, respectively (Figure 1).
Figure 1.
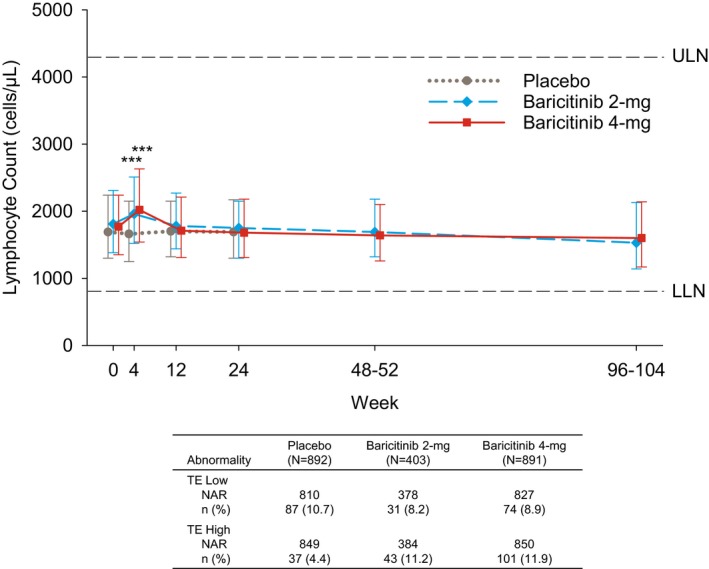
Top, Total lymphocyte counts during treatment. Values are the median and 25th–75th percentiles. Bottom, Patients with treatment‐emergent low lymphocyte count (TE low) and with treatment‐emergent high lymphocyte count (TE high). Values are the number (%) of the number at risk (NAR). *** = P ≤ 0.001 versus placebo, based on the least squares mean difference change from baseline. ULN = upper limit of normal; LLN = lower limit of normal.
CD3+, CD4+, and CD8+ T cell counts were increased at week 4 with baricitinib 2 mg/day and 4 mg/day compared to placebo, and returned to baseline by week 12. Inconsistent changes in these T cell subsets were observed at weeks 24–32 (Figures 2A–C) but were trending downward compared to baseline. At baseline, low CD3+ T cell counts were observed in 11.3%, 9.3%, and 9.1% of patients receiving placebo, baricitinib 2 mg/day, and baricitinib 4 mg/day, respectively; low CD4+ T cell counts were observed in 14.2%, 13.4%, and 13.5% of patients; low CD8+ T cell counts were observed in 7.9%, 7.6%, and 6.4% of patients. Based on 24 weeks of data up to rescue, the proportions of patients with ≥1 TE low CD3+, CD4+, or CD8+ T cell count did not differ between baricitinib and placebo treatments (Figures 2A–C).
Figure 2.
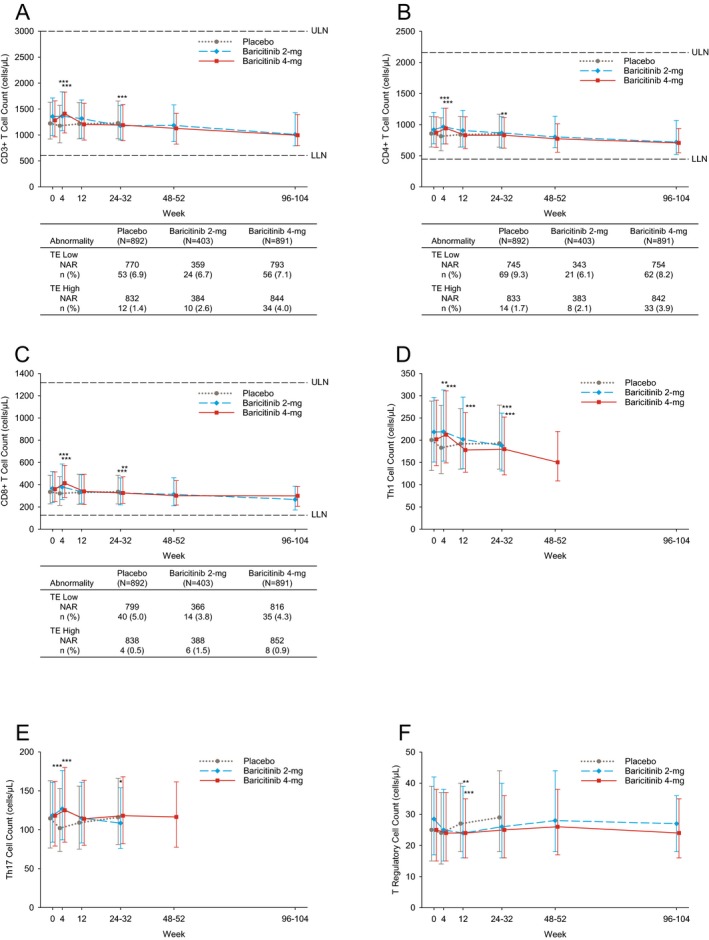
T cell counts during treatment. Counts of CD3+ cells (A), CD4+ cells (B), CD8+ cells (C), Th1 cells (D), Th17 cells (E), and Treg cells (F) are shown. Values are the median and 25th–75th percentiles. A–C include patients with TE high T cell count and TE low T cell count. Values are the number (%) of the NAR. * = P ≤ 0.05; ** = P ≤ 0.01; *** = P ≤ 0.001 versus placebo, based on the least squares mean difference change from baseline. See Figure 1 for definitions.
Th1 (CD4+CXCR3+CCR6−) cell counts were increased at week 4 with baricitinib treatment and were decreased below baseline through weeks 24–32 (Figure 2D). Th17 (CD4+CXCR3−CCR6+) cell counts increased at week 4 with baricitinib treatment, returned to baseline by week 12 with baricitinib 4 mg/day, and decreased at weeks 24–32 with baricitinib 2 mg/day (Figure 2E). Compared to the placebo group, which exhibited a modest increase at week 12, Treg cell counts were decreased at week 12 in both baricitinib groups and returned to near baseline by weeks 24–32, remaining stable thereafter (Figure 2F). For CD3+, CD4+, and CD8+ T cells, within‐group analysis demonstrated a significant decrease from baseline by weeks 96–104 with baricitinib 2 mg/day and 4 mg/day. For Th1 cells, a significant decrease from baseline by weeks 48–52 was observed with baricitinib 4 mg/day (Figures 2A–D).
CD19+ B cells and B cell subpopulations (including switched memory, non‐switched memory, mature naive, and immature transitional B cells) were significantly increased at weeks 4 through 24–32 in both baricitinib groups compared to placebo, and remained slightly above baseline or gradually returned to baseline over time (Figures 3A–E). At baseline, high CD19+ B cell counts were observed in 3.5%, 3.3%, and 3.6% of patients who received placebo, baricitinib 2 mg/day, and baricitinib 4 mg/day, respectively. Based on 24 weeks of data up to rescue, the proportions of patients with ≥1 TE high CD19+ B cell count were 1.2%, 3.7%, and 5.2% (Figure 3A).
Figure 3.
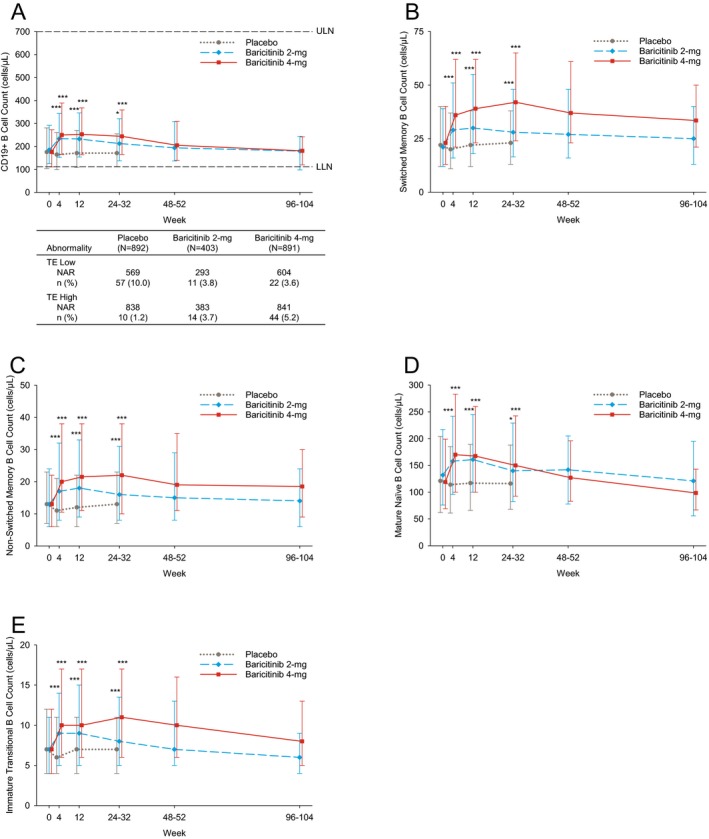
B cell counts during treatment. Counts of CD19+ B cells (A), switched memory B cells (B), non‐switched memory B cells (C), mature naive B cells (D), and immature transitional B cells (E) are shown. Values are the median and 25th–75th percentiles. A includes patients with TE high B cell count and TE low B cell count. Values are the number (%) of the NAR. * = P ≤ 0.05; *** = P ≤ 0.001 versus placebo, based on the least squares mean difference change from baseline. See Figure 1 for definitions.
Finally, NK cell counts were increased significantly by week 4 with baricitinib compared to placebo; changes were transient, and NK cell counts were decreased by weeks 12 through 24–32 in both baricitinib groups (Figure 4). In within‐group analysis, NK cell counts in patients treated with baricitinib 2 mg/day and 4 mg/day were below baseline at the week 12 through weeks 48–52 assessment (P ≤ 0.05). While NK cell counts in patients treated with baricitinib 4 mg/day were below baseline by the weeks 96–104 assessment, the change from baseline for patients receiving 2 mg/day (P > 0.05) was no longer significant by this point (Figure 4). At baseline, low NK cell counts were observed in 15.1%, 11.1%, and 14.9% of patients receiving placebo, baricitinib 2 mg/day, and baricitinib 4 mg/day, respectively. The proportions of patients with ≥1 TE low NK cell count were 9.9%, 14.8%, and 20.4% (Figure 4). Among patients with a TE low NK cell count at their last visit (while receiving baricitinib) who had a follow‐up value available (n = 16), the count normalized during the posttreatment follow‐up period in 11 patients (69%).
Figure 4.
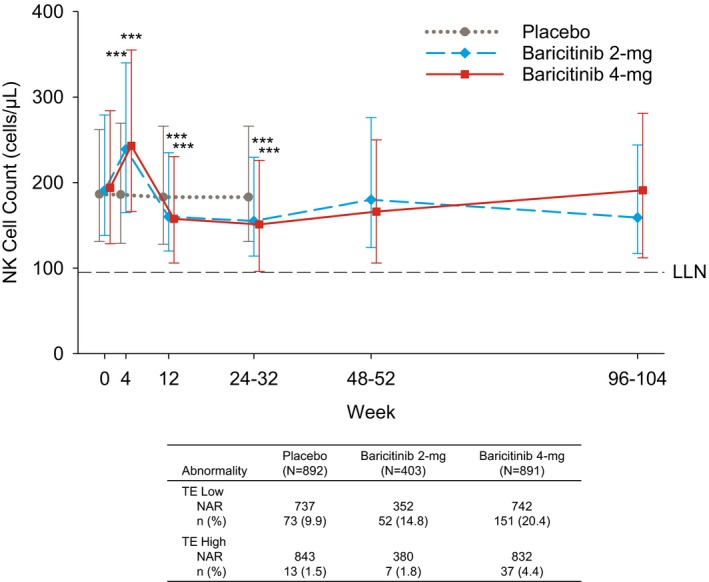
Top, Natural killer (NK) cell count during treatment. Values are the median and 25th–75th percentiles. Bottom, Patients with TE low NK cell count and with TE high NK cell count. Values are the number (%) of the NAR. *** = P ≤ 0.001 versus placebo, based on the least squares mean difference change from baseline. See Figure 1 for other definitions.
Changes in lymphocyte counts and clinical effects of baricitinib treatment. Negative correlations between changes from baseline in the DAS28‐hsCRP and counts of CD19+ B cell and several B cell subpopulations were observed with placebo and baricitinib treatment, indicating larger improvements in the DAS28‐hsCRP with larger increases in B cell counts (see Supplementary Table 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40680/abstract). A negative correlation was also observed between changes in the DAS28‐hsCRP and counts of Th17 and Treg cells with baricitinib 4 mg/day (Supplementary Table 3). The increase in CD19+ B cells observed with baricitinib (Figure 3A) occurred while RF titers had significantly decreased from baseline at week 24 with baricitinib 2 mg/day and 4 mg/day compared to placebo, and ACPA titers were significantly decreased from baseline at week 24 with baricitinib 4 mg/day only (see Supplementary Table 4 at http://onlinelibrary.wiley.com/doi/10.1002/art.40680/abstract).
Additionally, we tested whether changes in NK cell numbers were associated with increased infection rates in baricitinib‐treated RA patients, based on 24 weeks of data up to rescue. Compared to patients who never experienced a low NK cell count during placebo treatment, increases in frequency of overall infections were seen in both baricitinib groups, and an increase in frequency of herpes zoster infection was seen in the baricitinib 4 mg/day group. However, the rate of serious infection was similar among all 3 groups (Table 1). Patients who experienced ≥1 low NK cell value during the treatment period exhibited a dose‐related increase in overall infections with baricitinib versus placebo; herpes zoster and serious infection rates were similar among all 3 groups (Table 1). Similar associations were observed between changes in CD4+ T cell counts and infection rates (see Supplementary Table 5 at http://onlinelibrary.wiley.com/doi/10.1002/art.40680/abstract). Among patients who experienced a low NK cell count prior to their first TE infection, the majority experienced a low NK cell count <3 months before the infection, across treatment groups (Supplementary Table 6).
Table 1.
Incidence of TE infection by NK cell count, weeks 0–24a
| Placebo (n = 860) | Baricitinib 2 mg/day (n = 393) | Baricitinib 4 mg/day (n = 868) | |
|---|---|---|---|
| Never‐low NK cell count, no. (%)b | 718 (83.5) | 316 (80.4) | 618 (71.2)c |
| Patients with ≥1 TE infection | 213 (29.7) | 114 (36.1)d | 217 (35.1)d |
| Patients with ≥1 TE herpes zoster | 3 (0.4) | 3 (0.9) | 13 (2.1)e |
| Patients with ≥1 serious infection | 13 (1.8) | 4 (1.3) | 10 (1.6) |
| Low NK cell count, no. (%)f | 142 (16.5) | 77 (19.6) | 250 (28.8)c |
| Patients with ≥1 TE infection | 47 (33.1) | 29 (37.7) | 112 (44.8)d |
| Patients with ≥1 TE herpes zoster | 0 | 1 (1.3) | 2 (0.8) |
| Patients with ≥1 serious infection | 1 (0.7) | 1 (1.3) | 3 (1.2) |
Data are from the time of treatment initiation through 24 weeks, with data censored at rescue. TE = treatment‐emergent.
Patients who never experienced low natural killer (NK) cell values (defined as values <95 cells/μl) at any time postbaseline up to time of rescue.
P ≤ 0.001 versus baricitinib 2 mg/day and versus placebo, by Fisher's exact test.
P ≤ 0.05 versus placebo.
P ≤ 0.01 versus placebo.
Patients who experienced ≥1 low NK cell value at any time postbaseline up to the time of rescue.
Discussion
In this study, we characterized changes in lymphocyte subsets in RA patients (n = 2,186) from phase III and long‐term extension trials in the baricitinib clinical program and assessed whether observed lymphocyte changes correlated with clinical effects of baricitinib treatment. Changes in lymphocyte subsets were generally within the normal range and were largely consistent across the baricitinib phase III RA clinical trials, which included patients with different responsiveness to prior DMARD therapies 11, 12, 13. In this integrated analysis, a transient increase in total lymphocyte count was observed after 4 weeks of treatment with baricitinib, returning to baseline by week 12. Overall changes in lymphocyte subsets from a phase III study of baricitinib in the treatment of DMARD‐naive RA patients (RA‐BEGIN) 13 generally reflected similar patterns of change as those in this analysis. An early transient increase in lymphocyte count was also observed in patients with plaque psoriasis who were treated with baricitinib 17.
These findings may in part relate to the timing of baricitinib administration. In previous phase I studies, transient increases in total lymphocyte counts were observed within hours of baricitinib administration, returning to baseline prior to the next daily dose 6. Transient changes in T cells and subsets were observed with baricitinib treatment, with cell counts remaining largely within normal reference ranges. While CD3+, CD4+, and CD8+ T cells decreased below baseline by weeks 96–104, longer‐term data will be required to determine whether changes continue or a new baseline is established in patients.
We observed that B cells and subpopulations increased after 4 weeks of baricitinib treatment and remained above baseline or stabilized over time. JAK inhibition modulates cytokine signaling, and therefore, possible downstream alterations in adhesion molecule expression and chemokine production may affect B cell trafficking with baricitinib treatment, resulting in elevated peripheral B cell numbers. Furthermore, differentiation of the B cell lineage may be induced in compensation for JAK1‐ and JAK2‐mediated inhibition of cytokine production, such as type I and type II interferons. Importantly, baricitinib treatment did not result in increased autoantibody titers, suggesting that the increase in B cells may not be pathologic. It is possible that some of the class‐switched memory B cells, increased by baricitinib in a dose‐dependent manner, may be regulatory B cells, which inhibit disease activity. Further studies would be needed to clarify these possibilities.
NK cells transiently increased after 4 weeks of baricitinib treatment, before decreasing below baseline levels and then gradually stabilizing over time. Notably, baseline low NK cell counts were observed in up to 15.1% of patients in this analysis. The overall effect of JAK1 and JAK2 inhibition on peripheral blood mononuclear cell subsets may potentially reflect the combined effects of a number of different biologic processes. These include demargination resulting from endothelial deactivation and altered marrow production of cellular subtypes. Identifying the predominant process that results in the observed change in peripheral blood may depend on the half‐life of the circulating cell.
Changes in lymphocyte subsets have also been characterized with tofacitinib, a JAK inhibitor that preferentially inhibits JAK1 and JAK3 18. Phase II and phase III clinical trials involving patients with RA treated with tofacitinib showed a transient increase in total lymphocytes early in treatment, with a gradual decrease over time 19, 20, 21. In phase II RA clinical trials, variable changes in T cells were observed with short‐term tofacitinib treatment, while B cells and NK cells increased and decreased from baseline, respectively 19, 20. To our knowledge, robust data from systematic follow‐up of tofacitinib‐treated RA patients from baseline through long‐term treatment are unavailable.
Changes in lymphocyte subsets have been characterized in clinical trials with other JAK inhibitors. Phase II clinical trials involving patients with RA treated with the selective JAK1 inhibitor filgotinib showed no clear dose‐dependent changes in the rates of TE low lymphocyte or NK cell counts during short‐term treatment, while similar studies with upadacitinib showed decreased lymphocyte and NK counts following administration of that agent 22, 23, 24, 25. These data suggest the possibility that different JAK inhibitors may be associated with slightly different lymphocyte changes over time, and in vitro JAK selectivity is not an easy predictor of these changes. It should be noted that lymphocyte subpopulations were monitored in 2,186 patients with active RA who participated in the baricitinib phase III program, including long‐term treatment, making it the most extensive analysis of lymphocyte subset changes with a JAK inhibitor described to date.
Changes in lymphocyte levels have also been reported in patients with RA who were treated with compounds that target TNF or interleukin‐6 (IL‐6) signaling. The phase III RA‐BEAM trial, as well as other studies, showed that the TNF inhibitor adalimumab produced a sustained increase in total lymphocyte counts over time in patients with RA 12, 21, 26. Consistent with an increase in total lymphocyte levels over time, the RA‐BEAM trial demonstrated that adalimumab treatment also resulted in increased T cells, B cells, and NK cells 12. Several studies with small patient populations have analyzed changes in lymphocyte subsets in RA patients who were treated with tocilizumab, a monoclonal antibody that inhibits the IL‐6 receptor. Two studies showed that tocilizumab treatment resulted in increased Treg cells, with one study showing decreased Th17 cells with treatment 27, 28. Conflicting changes in B cell numbers have been demonstrated with tocilizumab treatment; one study showed increased total B cells, with no significant changes in B cell subsets, while another showed decreased levels of memory B cell subsets 28, 29. Overall, these findings demonstrate that RA therapies that target different biologic pathways are associated with distinct changes in lymphocyte subsets.
Taking into consideration these changes, we attempted to determine, in an integrated analysis, if there are clinical correlations between changes in lymphocyte subsets and the DAS28‐hsCRP. Negative correlations between changes in the DAS28‐hsCRP and B cell subpopulations were observed with placebo and baricitinib treatment. In addition, changes in the DAS28‐hsCRP were negatively correlated with Th17 and Treg cell numbers in the baricitinib 4 mg/day group. Though statistically significant, the strength of these associations was modest. The increase in B cells with baricitinib treatment was not associated with an increase in autoantibody titers. Finally, patients who never experienced low NK cell counts as well as patients who experienced ≥1 low NK cell value with baricitinib 4 mg/day experienced a small increase in the rate of overall infection (though not serious infection). Low NK cell counts did not correlate with the rate of herpes zoster infection, a known adverse reaction with baricitinib, suggesting that NK modulation is not a dominant mechanism in this association.
The present findings do not mechanistically explain the observed changes in lymphocyte subsets. Several mechanisms could contribute to the changes, operating either in isolation or in combination. Thus, cell number may indicate transiently increased synthesis (perhaps reflecting an immediate effect of drug on marrow homeostasis), effects on cellular mobilization from lymphoid or inflamed tissues, effects on cellular egress from the circulation, or effects on cellular survival. Moreover, the impact of JAK inhibition is distinct in different subsets, leading, most notably in the B cell compartment, to a new homeostatic circulatory level. Whether these effects are mediated directly on lymphocyte subsets via JAK‐sensitive receptors or via altered chemokine or chemokine receptor expression (dependent on JAK‐driven pathways) is unclear and warrants further investigation.
A significant limitation of this study is that the follow‐up period, while considerable, does not span many years, though RA can require lifelong treatment. Longer‐term lymphocyte subset data continue to be collected during the ongoing long‐term extension study RA‐BEYOND, which currently provides up to 5 years of treatment with baricitinib after patients complete an originating study; evaluation of these longer‐term data, when available, will assist in addressing this limitation.
In conclusion, this integrated analysis of lymphocyte subsets during the course of treatment with baricitinib in a large phase III RA data set has shown distinct changes in different lymphocyte populations, including B cell increases, variable changes in T cell counts, and transient reduction in NK cells, without strong association with efficacy or safety end points. The later kinetics of these analytes during longer‐term baricitinib treatment, and any clinical correlations, continue to be evaluated.
Author contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Tanaka had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Tanaka, McInnes, Taylor, Chen, Issa, Zuckerman, Emery.
Acquisition of data
de Bono, Issa, Rooney.
Analysis and interpretation of data
Tanaka, McInnes, Taylor, Byers, Chen, de Bono, Issa, Macias, Rogai, Rooney, Schlichting, Zuckerman, Emery.
Role of the study sponsor
The study was designed by Eli Lilly and Company in consultation with an academic advisory board and Incyte Corporation. Eli Lilly and Company provided data analysis, laboratory, and site?monitoring services and was involved in data interpretation. Writing assistance was provided by Eli Lilly and Company. All authors and Eli Lilly and Company reviewed and approved the manuscript. The authors maintained control over the final content.
Supporting information
Acknowledgments
We acknowledge Julie Sherman (Eli Lilly and Company) for figure assistance. We also thank all patients and study investigators.
ClinicalTrials.gov identifiers: NCT01710358, NCT01721057, NCT01721044, NCT01885078.
Supported by Eli Lilly and Company and Incyte Corporation.
Dr. Tanaka has received speaking fees and/or honoraria from AbbVie, Asahi‐kasei, Astellas, Chugai, Daiichi‐Sankyo, Eisai, Janssen, Mitsubishi‐Tanabe, Novartis, Sanofi, Takeda, and UCB (less than $10,000 each) and from Eli Lilly and Company, Bristol‐Myers Squibb, GlaxoSmithKline, Pfizer, and YL Biologics (more than $10,000 each) and has received grant support from AbbVie, Astellas, Bristol‐Myers Squibb, Chugai, Daiichi‐Sankyo, Eisai, Mitsubishi‐Tanabe, MSD, Ono, Taisho‐Toyama, and Takeda. Dr. McInnes has received consulting fees from AbbVie, Eli Lilly and Company, Janssen, Novartis, Pfizer, and Roche (less than $10,000 each) and grant support from those companies. Dr. Taylor has received consulting fees from AbbVie, Galapagos, Pfizer, and UCB (less than $10,000 each) and from Eli Lilly and Company (more than $10,000) and has received grant support from Celgene, Eli Lilly and Company, Galapagos, and UCB. Drs. Byers, Chen, de Bono, Macias, Rogai, Rooney, Schlichting, and Zuckerman and Mr. Issa own stock or stock options in Eli Lilly and Company. Dr. Emery has served as an expert witness on behalf of AbbVie, Eli Lilly and Company, Bristol‐Myers Squibb, MSD, Novartis, Pfizer, Roche, Samsung, Sandoz, and UCB.
References
- 1. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med 2011;365:2205–19. [DOI] [PubMed] [Google Scholar]
- 2. Tanaka Y. Current concepts in the management of rheumatoid arthritis. Korean J Intern Med 2016;31:210–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Furst DE, Emery P. Rheumatoid arthritis pathophysiology: update on emerging cytokine and cytokine‐associated cell targets. Rheumatology (Oxford) 2014;53:1560–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schwartz DM, Bonelli M, Gadina M, O'Shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol 2016;12:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smolen JS, Landewé R, Bijlsma J, Burmester G, Chatzidionysiou K, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease‐modifying antirheumatic drugs: 2016 update. Ann Rheum Dis 2017;76:960–77. [DOI] [PubMed] [Google Scholar]
- 6. Shi JG, Chen X, Lee F, Emm T, Scherle PA, Lo Y, et al. The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. J Clin Pharmacol 2014;54:1354–61. [DOI] [PubMed] [Google Scholar]
- 7. Genovese MC, Kremer J, Zamani O, Ludivico C, Krogulec M, Xie L, et al. Baricitinib in patients with refractory rheumatoid arthritis. N Engl J Med 2016;374:1243–52. [DOI] [PubMed] [Google Scholar]
- 8. Dougados M, van der Heijde D, Chen YC, Greenwald M, Drescher E, Liu J, et al. Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: results from the RA‐BUILD study. Ann Rheum Dis 2017;76:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fleischmann R, Schiff M, van der Heijde D, Ramos‐Remus C, Spindler A, Stanislav M, et al. Baricitinib, methotrexate, or combination in patients with rheumatoid arthritis and no or limited prior disease‐modifying antirheumatic drug treatment. Arthritis Rheumatol 2017;69:506–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Taylor PC, Keystone EC, van der Heijde D, Weinblatt ME, Del Carmen Morales L, Reyes Gonzaga J, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med 2017;376:652–62. [DOI] [PubMed] [Google Scholar]
- 11. Emery P, McInnes I, Genovese M, Smolen J, Kremer J, Dougados M, et al. Characterization of changes in lymphocyte subsets in baricitinib‐treated patients with rheumatoid arthritis in two Phase 3 studies [abstract]. Arthritis Rheumatol 2015;67 Suppl 10 URL: https://acrabstracts.org/abstract/characterization-of-changes-in-lymphocyte-subsets-in-baricitinib-treated-patients-with-rheumatoid-arthritis-in-two-phase-3-studies/. [Google Scholar]
- 12. Tanaka Y, Fleischmann R, Schiff M, Takeuchi T, Keystone E, Weinblatt M, et al. Characterization of changes in lymphocyte subsets in baricitinib‐treated patients with rheumatoid arthritis in a phase 3 study (RA‐BEAM) [abstract]. Ann Rheum Dis 2016;75 Suppl 2:262–3. [Google Scholar]
- 13. Takeuchi T, Fleischmann R, Schiff M, Issa M, Macias W, Rooney T, et al. Characterization of changes in lymphocyte subsets in baricitinib‐treated patients with early, DMARD naïve, rheumatoid arthritis in a phase 3 study [abstract]. Ann Rheum Dis 2017;76 Suppl 2:1147. [Google Scholar]
- 14. Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol 2017;13:320. [DOI] [PubMed] [Google Scholar]
- 15. Mujtaba MA, Komocsar WJ, Nantz E, Samaniego MD, Henson SL, Hague JA, et al. Effect of treatment with tabalumab, a B cell–activating factor inhibitor, on highly sensitized patients with end‐stage renal disease awaiting transplantation. Am J Transplant 2016;16:1266–75. [DOI] [PubMed] [Google Scholar]
- 16. Prevoo ML, van ‘t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty‐eight–joint counts: development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 1995;38:44–8. [DOI] [PubMed] [Google Scholar]
- 17. Papp KA, Menter MA, Raman M, Disch D, Schlichting DE, Gaich C, et al. A randomized phase 2b trial of baricitinib, an oral Janus kinase (JAK) 1/JAK2 inhibitor, in patients with moderate‐to‐severe psoriasis. Br J Dermatol 2016;174:1266–76. [DOI] [PubMed] [Google Scholar]
- 18. Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J Med Chem 2014;57:5023–38. [DOI] [PubMed] [Google Scholar]
- 19. Hodge JA, Kawabata TT, Krishnaswami S, Clark JD, Telliez JB, Dowty ME, et al. The mechanism of action of tofacitinib–an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol 2016;34:318–28. [PubMed] [Google Scholar]
- 20. Van Vollenhoven R, Choy E, Lee EB, Hazra A, Anisfeld A, Lazariciu I, et al. Tofacitinib, an oral Janus kinase inhibitor, in the treatment of rheumatoid arthritis: changes in lymphocytes and lymphocyte subset counts and reversibility after up to 8 years of tofacitinib treatment [abstract]. Ann Rheum Dis 2016;75 Suppl 2:258. [Google Scholar]
- 21. Schulze‐Koops H, Strand V, Nduaka C, DeMasi R, Wallenstein G, Kwok K, et al. Analysis of haematological changes in tofacitinib‐treated patients with rheumatoid arthritis across phase 3 and long‐term extension studies. Rheumatology (Oxford) 2017;56:46–57. [DOI] [PubMed] [Google Scholar]
- 22. Westhovens R, Taylor PC, Alten R, Pavlova D, Enríquez‐Sosa F, Mazur M, et al. Filgotinib (GLPG0634/GS‐6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (MTX) in patients with active rheumatoid arthritis and insufficient response to MTX: results from a randomised, dose‐finding study (DARWIN 1). Ann Rheum Dis 2017;76:998–1008. [DOI] [PubMed] [Google Scholar]
- 23. Kavanaugh A, Kremer J, Ponce L, Cseuz R, Reshetko OV, Stanislavchuk M, et al. Filgotinib (GLPG0634/GS‐6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: results from a randomised, dose‐finding study (DARWIN 2). Ann Rheum Dis 2017;76:1009–19. [DOI] [PubMed] [Google Scholar]
- 24. Kremer JM, Emery P, Camp HS, Friedman A, Wang L, Othman AA, et al. A phase IIb study of ABT‐494, a selective JAK‐1 inhibitor, in patients with rheumatoid arthritis and an inadequate response to anti‐tumor necrosis factor therapy. Arthritis Rheumatol 2016;68:2867–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Genovese MC, Smolen JS, Weinblatt ME, Burmester GR, Meerwein S, Camp HS, et al. Efficacy and safety of ABT‐494, a selective JAK‐1 inhibitor, in a phase IIb study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol 2016;68:2857–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti–tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo‐controlled, 52‐week trial. Arthritis Rheum 2004;50:1400–11. [DOI] [PubMed] [Google Scholar]
- 27. Samson M, Audia S, Janikashvili N, Ciudad M, Trad M, Fraszczak J, et al. Inhibition of interleukin‐6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum 2012;64:2499–503. [DOI] [PubMed] [Google Scholar]
- 28. Daïen CI, Gailhac S, Audo R, Mura T, Hahne M, Combe B, et al. High levels of natural killer cells are associated with response to tocilizumab in patients with severe rheumatoid arthritis. Rheumatology (Oxford) 2015;54:601–8. [DOI] [PubMed] [Google Scholar]
- 29. Roll P, Muhammad K, Schumann M, Kleinert S, Einsele H, Dörner T, et al. In vivo effects of the anti–interleukin‐6 receptor inhibitor tocilizumab on the B cell compartment. Arthritis Rheum 2011;63:1255–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials