

Abstract
The concept of the high‐affinity state postulates that a certain subset of G‐protein‐coupled receptors is primarily responsible for receptor signaling in the living brain. Assessing the abundance of this subset is thus potentially highly relevant for studies concerning the responses of neurotransmission to pharmacological or physiological stimuli and the dysregulation of neurotransmission in neurological or psychiatric disorders. The high‐affinity state is preferentially recognized by agonists in vitro. For this reason, agonist tracers have been developed as tools for the noninvasive imaging of the high‐affinity state with positron emission tomography (PET). This review provides an overview of agonist tracers that have been developed for PET imaging of the brain, and the experimental paradigms that have been developed for the estimation of the relative abundance of receptors configured in the high‐affinity state. Agonist tracers appear to be more sensitive to endogenous neurotransmitter challenge than antagonists, as was originally expected. However, other expectations regarding agonist tracers have not been fulfilled. Potential reasons for difficulties in detecting the high‐affinity state in vivo are discussed.
Keywords: agonist high‐affinity state, experimental design, G‐protein‐coupled receptors, human brain, neurotransmitters, positron emission tomography
1. INTRODUCTION
Noninvasive imaging of neurotransmitter receptors with positron emission tomography (PET) provides insights into the number of receptors expressed in the brain and the functioning of brain networks. Analysis of the imaging data yields information about the role of particular neurotransmitters in the functioning of the brain in health as well as in neuropsychiatric disorders, including syndromes characterized by cognitive dysfunction.
Neurotransmitters bind to receptors that are either ligand‐gated ion channels (LGICs) or G‐protein‐coupled receptors (GPCRs). For some neurotransmitters, all receptors belong to a single receptor superfamily, for example, all known dopamine receptors are GPCRs. Other neurotransmitters bind to receptors from both superfamilies, for example, there are LGIC‐ and GPCR‐type receptors of the neurotransmitters acetylcholine and glutamate.
The signaling mechanism of LGICs is comparatively simple and quick—neurotransmitter binding opens the ionic channel that the receptor itself forms. A quick (millisecond time scale) and usually short‐lasting postsynaptic response is thus obtained. GPCRs, on the other hand, affect their downstream signaling pathways through the mediation of trimeric proteins called G‐proteins (Figure 1). The GPCR signaling mechanism is slower (second time scale) and more energy‐consuming than that of LGICs, but long lasting and much more versatile. Due to this versatility, GPCRs are the most popular targets for drugs in clinical use.1, 2 GPCR‐target drugs or ligands, can be described as agonists or antagonists depending on their effect on the receptor. Agonist drugs can be broadly defined as substances that act like the endogenous neurotransmitter (by binding to the receptor and inducing a physiological response), whereas antagonist drugs bind but do not induce a response and can block the action of the endogenous neurotransmitter.
Figure 1.
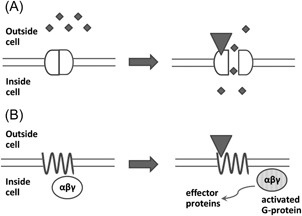
Simplified signaling mechanisms of ligand‐gated ion channels (A) and G‐protein‐coupled receptors (B). If an agonist (represented by a triangle) binds to a channel, the channel opens and ions (represented by small diamonds) can enter the cell. If the agonist binds to a receptor, the G‐protein (represented by an ellipse) dissociates from the receptor complex and activates specific effector proteins
In vitro studies in membrane homogenates from cultured cells or isolated tissues have shown that in a single population of GPCRs to which antagonist drugs have a single affinity, agonist drugs recognize two distinct receptor subpopulations: one for which they have high affinity and one for which they have low affinity. The existence of a receptor subpopulation that possesses high‐affinity toward the agonists (dubbed “high‐affinity state,” Rhigh) has been demonstrated for numerous neurotransmitter GPCRs including dopaminergic,3, 4 serotonergic,5, 6, 7 muscarinic,8 and opioid receptors.9 The high‐affinity state is commonly thought to be composed of receptor molecules bound to G‐proteins. A crystal structure of such an activated state of the receptor, in complex with the G‐protein, was first obtained in 2011.10
The relationship between G‐protein coupling and high‐affinity toward the agonist gave rise to a hypothesis that the relative abundance of Rhigh may characterize the responsiveness of the synaptic signaling machinery to agonist levels. Indeed, alterations of the fraction of receptors configured in the high‐affinity state, as measured in membrane homogenates, were found in pathological states associated with dysregulation of neurotransmission. For instance, the relative abundance of the high‐affinity state of µ‐opioid receptors was decreased in guinea pigs after chronic morphine treatment,11 while the high‐affinity state of muscarinic M1 receptors was downregulated in Alzheimer's disease.12, 13 Upregulation of the high‐affinity state of dopamine D2 receptors has been reported in several animal models of psychosis.14, 15
Assessing the availability of Rhigh may, therefore, provide more valuable information about the state of neurotransmission in vivo than assessing the availability of total receptors. Given that agonists preferentially bind to Rhigh, this hypothesis spurred the development of agonist PET tracers and their use for neuroreceptor imaging.
In this study we will review and discuss the molecular basis of the high‐affinity state, inherent advantages and shortcomings of agonist PET tracers stemming from their preferential binding to the high‐affinity state, agonist PET tracers currently available for receptor imaging, experimental methods used for the imaging of high‐affinity state in vivo, and evidence collected with these methods.
2. NATURE OF THE HIGH‐AFFINITY STATE OF GPCRS
2.1. G‐protein‐dependent high‐affinity state
The canonical view of the nature of the high‐affinity state is based on the so‐called ternary complex model of G‐protein signaling which originates from the studies of agonist binding to β‐adrenergic receptors in membrane homogenates.16, 17 This model claims that a “ternary” complex must form to launch the G‐protein signaling cascade, consisting of agonist, receptor, and G‐protein. Positive cooperativity between receptor‐agonist and receptor‐G‐protein binding creates the separation of the total receptor population into high‐ and low‐affinity states. For the agonist, receptors complexed with G‐proteins form the high‐affinity state, whereas free receptor molecules represent the low‐affinity state. Indeed, the preference of ligands for the G‐protein‐bound high‐affinity state was found to correlate with their intrinsic activity.18, 19, 20
Several newer and more sophisticated versions of the ternary complex model have been developed to account for pharmacological phenomena such as constitutive activity (presence of baseline signaling in the absence of agonists) and inverse agonism (existence of ligands that decrease rather than increase the level of signaling relative to baseline). These models imply the existence of more than two receptor species with different affinities for the agonist but the main premise remains the same: G‐protein binding is the main factor that determines the receptor's affinity toward the agonist.21, 22
An important feature of the G‐protein‐dependent high‐affinity state is its sensitivity to guanosine triphosphate (GTP). Indeed, the high‐affinity state of GPCRs detected in membrane homogenates usually disappears upon GTP addition.3, 4, 5, 6, 7, 8, 9 The reason for this is that the canonical G‐protein signaling cascade involves a so‐called GTP cycle (Figure 2). G‐proteins are heterotrimers and one of their subunits, Gα, has a binding site for guanosine nucleotide (this gives G‐proteins their name). In an inactive G‐protein, this site is occupied by guanosine diphosphate (GDP). Upon G‐protein activation by an agonist‐bound receptor, GDP is replaced by GTP from the cytoplasm, which leads to the dissociation of Gα‐subunit from the Gβ and Gγ subunits (together referred to as Gβγ). The G‐protein splits into two parts, which then activate downstream effectors. If the G‐protein uncouples from the receptor, the receptor quickly returns to its inactive “low‐affinity” state.23 Eventual hydrolysis of GTP to GDP in the Gα subunit lets the G‐protein reassemble and bind to the receptor again, which closes the cycle.24
Figure 2.
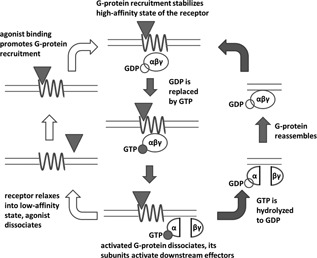
GPCR activation (left circuit, open arrows) and GTP cycle (right circuit, solid arrows). As in Figure 1, the agonist is represented by a triangle, the G‐protein by an ellipse and the receptor by a sinusoid line. The center of the figure shows the “ternary complex” consisting of agonist, receptor and G‐protein. GPCR, G‐protein‐coupled receptor; GTP, guanosine triphosphate
Therefore, the GTP cycle acts as a negative‐feedback loop, promoting G‐protein decoupling from the receptors and their (temporary) conversion into the low‐affinity state after agonist binding. Excess GTP shifts the equilibrium toward complete dissociation of G‐proteins from the receptors.
2.2. Oligomerization‐dependent high‐affinity state
The growing amount of evidence on GPCR oligomerization in cultured cells and living tissues 25 and on the pharmacological relevance of such oligomerization (see Ferre et al26 for review) has given rise to the concept of oligomerization‐dependent high‐affinity state. When the agonist interacts with a receptor oligomer, occupying and activating a single receptor unit within it, conformational changes in this receptor influence the conformation of other receptors within the same oligomer and decrease their affinity for other agonist molecules (Figure 3). In other words, separation into high‐ and low‐affinity states is caused by negative cooperativity effects of the agonist binding to oligomerized receptors.27
Figure 3.
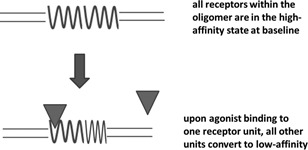
Oligomerization‐dependent high‐affinity state. In this schematic representation, the receptors are drawn as homodimers. Most higher order G‐protein‐coupled receptor complexes are homodimers, heterodimers or tetramers consisting of two different homodimers. The high‐affinity state of the receptor is pictured as a sinusoid, the low‐affinity state as a compressed sinusoid, and the agonist as a triangle is pictured as a compressed sinusoid and the agonist as a triangle
Receptor oligomerization is arguably mainly relevant for the explanation of the interplay between signaling pathways of different receptors26: interaction between oligomer subunits is conceptually simpler than interference of downstream cascades. However, among data from radioligand binding studies there are also some results that could be explained better by oligomerization than by G‐protein coupling, such as: (i) GTP‐insensitive high‐affinity agonist binding to dopamine D3 and serotonin 5‐HT2A receptors,28, 29 (ii) detection of high‐ and low‐affinity states of adenosine A2A receptors by antagonist ligands, 30 and (iii) detection of several (more than two) binding sites with different affinities to agonists in the muscarinic M2 receptor population.31
If there is cooperativity between receptor‐agonist and receptor‐receptor interaction, agonist binding might influence the degree of receptor oligomerization. Some studies indeed report such phenomena32, 33 but, in general, experimental data on the relationship between ligand binding and oligomerization are contradictory both in terms of whether ligand binding really promotes formation or dissociation of oligomers and whether this action is correlated with intrinsic activity (see Cottet et al34, 35 for review).
2.3. Influence of agonist binding on the high‐affinity state
In both G‐protein coupling and oligomerization‐dependent models of high‐affinity state, agonist binding to the receptor influences receptor interaction with other molecules and thus can alter the relative abundance of the high‐affinity state.
2.3.1. G‐protein‐dependent high‐affinity state
Under conditions where no feedback loops are present, as is the case with in vitro binding studies with nonliving material like membrane homogenates and tissue slices, the relationship between agonist concentration and percentage of receptors in the high‐affinity state at equilibrium is straightforward. In the absence of GTP, agonist binding can only increase G‐protein recruitment. Therefore, increasing agonist concentration will make the percentage of receptors in the “G‐protein‐dependent” high‐affinity state grow from some “floor” value (see Section 2.5) to the “ceiling” value determined by receptor‐G‐protein stoichiometry in the system (100% if the number of available G‐proteins is greater than or equal to the number of receptors). On the other hand, in the presence of excess GTP and negligible GTP hydrolysis, all G‐proteins activated by agonist‐bound receptors will be dissociated and uncoupled from the receptors, so at any agonist concentration, there will be no discernible high‐affinity state.
In living cells and tissues, however, the GTP cycle plays the role of a negative‐feedback loop, which counteracts excess high‐to‐low or low‐to‐high conversion of affinity states caused by the agonist. Depending on the combination of concentrations and kinetic rates, either G‐protein‐recruiting or G‐protein‐dissociating effects of an agonist can become dominant. Indeed, mathematical simulations of GPCR signaling have demonstrated the possibility of both agonist‐induced increase and decrease in the relative abundance of the G‐protein‐dependent high‐affinity state.22
2.3.2. Oligomerization‐dependent high‐affinity state
Negative cooperativity in agonist binding to oligomerized receptors implies that increasing agonist concentration will bring more and more receptors into “low‐affinity state.” The percentage of receptors in the high‐affinity state, equal to 100% in the absence of agonist, will decrease to 100%/N (N is the average number of receptors per oligomer) when the agonist occupies one receptor unit in each oligomer, converting all the other units to low‐affinity state. When agonist concentration raises so high that agonists start to occupy receptors in the low‐affinity state, the relative abundance of the high‐affinity state will fall even lower. There are no well described and widely accepted feedback loops for the oligomerization‐dependent model of the high‐affinity state.
2.4. Agonist‐induced receptor internalization
Activation of GPCRs by agonists promotes not only G‐protein binding to them but also their phosphorylation by G‐protein‐coupled receptor kinase (GRK) and internalization mediated by β‐arrestins.36 This provides an extra pathway through which the agonist can influence the relative abundance of the high‐affinity state. Internalized receptors are decoupled from G‐proteins (coupled to β‐arrestins instead) and removed from the cell surface to intracellular compartments, where the ionic environment and pH value can be different from extracellular conditions. This makes internalized receptors less accessible (especially for hydrophilic radioligands) and possibly also alters their affinity toward their ligands.
In vitro, β‐arrestin recruitment can happen within minutes.37, 38 Internalization of dopamine D2/3 receptors was observed within the same time frame in vivo and was shown to be dose‐dependent.39 Although it is not yet clear whether internalization mainly happens to receptors in the low‐ or high‐affinity state,40, 41 internalized dopamine D2/3 receptors on intact cells and µ‐opioid receptors incubated in a buffer imitating endosomal medium were shown to have decreased affinities toward their ligands.42, 43
Therefore, high concentrations of an agonist can promote receptor internalization and change the number and relative abundances of receptor subpopulations with different affinities toward imaging radioligands. On the other hand, in internalization‐deficient β‐arrestin knockout mice, baseline binding of dopamine D2/3 agonist and antagonist tracers was the same as in wild‐type controls.44 This may mean that at basal neurotransmitter levels there already is an equilibrium between neurotransmitter‐induced receptor internalization and recycling.
2.5. Relative abundance of high‐affinity state in the absence of the agonist
The oligomerization‐dependent model of the high‐affinity state implies that this is the state in which all receptors are configured in the absence of the agonist.
For the G‐protein‐dependent high‐affinity state, its baseline relative abundance, that is, the degree to which G‐proteins interact with the receptors in the absence of agonist is a matter of debate.45, 46 One extreme view, called collision coupling (Figure 4A), states that in living cells G‐proteins are not normally bound to the receptors but instead interact with them transiently when receptors become activated.47 Another extreme view (Figure 4B) states that G‐proteins are always bound (precoupled) to the receptors and do not decouple even after activation, which happens through structural rearrangement of the G‐protein rather than through dissociation.48, 49, 50
Figure 4.
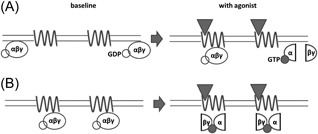
Two extreme modes of receptor‐G‐protein interaction. The agonist is represented by a triangle, the receptor by a sinusoid line and the G‐protein by an ellipse. A, In the collision coupling model, G‐proteins do not stably interact with receptors but agonist action on the receptor promotes G‐protein recruitment to and activation by the receptors, which results in the dissociation of G‐proteins. B, In the precoupling model, G‐proteins are stably bound to the receptors and rearrange their structures upon activation instead of dissociating. GDP, guanosine diphosphate; GTP, guanosine triphosphate
On the one hand, collision coupling provides a straightforward interpretation of differences in intrinsic activities of the agonists: agonist efficacy is related to the number of different G‐proteins that an agonist‐bound receptor can bind and activate per unit of time. Decoupling of G‐proteins from the receptors upon activation explains the disappearance of the high‐affinity state upon GTP addition in membrane homogenates. On the other hand, receptors and G‐proteins are known to be coisolated by immunoprecipitation and bioluminescence resonance energy transfer/fluorescence resonance energy transfer (BRET/FRET) experiments with mutated proteins incorporating fluorescent or bioluminescent probe demonstrate close contact between receptors and G‐proteins in the absence of agonists.45 Moreover, in BRET studies with α2 adrenergic and δ‐opioid receptors, these receptors were found to interact with G‐proteins both before and after activation by agonist.49, 50
A middle ground between the extreme views is, of course, possible, where some G‐proteins are bound to receptors at baseline but decoupled upon activation, or where G‐proteins are uncoupled at baseline but become bound to receptors upon activation. Moreover, BRET and FRET experiments image the whole population of the receptors, so constant presence of a RET signal, while showing that a fraction of receptors are engaged with G‐proteins, does not exclude the possibility of a rapid turnover of G‐proteins with which these receptors interact.
2.6. Summary
The existence of high‐ and low‐affinity states of GPCRs is commonly thought to be due to receptor interaction with G‐proteins. Being a part of the canonical GPCR signaling cascade, the receptor‐G‐protein coupling is directly related to the pharmacological activity of the agonists.
GPCR oligomerization (both homo and hetero), with negative cooperativity in agonist binding within the oligomer, can be an alternative mechanism leading to the formation of receptor subpopulations with different affinities for the agonist. It is plausible that at least for some GPCRs, oligomerization can contribute to the splitting of receptors into high‐ and low‐affinity states instead of, or in addition to, G‐protein coupling.
Both models of high‐affinity state imply that agonists preferentially bind to receptors that are almost ready to launch the signaling cascade, although in the oligomerization model it is so just because agonist binding makes unoccupied receptors “less ready.” Moreover, agonist binding can influence the relative abundance of the high‐affinity state, potentially promoting its formation or disintegration and launching receptor internalization in intact cells and living tissues. Such influence is most directly demonstrated for the G‐protein‐dependent model of the high‐affinity state.
3. EXPECTED ADVANTAGES AND DISADVANTAGES OF AGONIST TRACERS RELATIVE TO ANTAGONIST TRACERS
From the notion that agonists preferentially bind to a high‐affinity functional subset of receptors one can logically infer a number of applications in which agonist tracers should be superior, in theory, to antagonist tracers. Note that proposed advantages of agonist tracers mentioned below hold independently of whether the high‐affinity state is G‐protein‐dependent or oligomerization‐dependent.
3.1. Applications where agonist tracers have comparative advantage over antagonist tracers
3.1.1. Measurement of synaptic neurotransmission
An endogenous neurotransmitter is an agonist by definition, so it competes with the agonist tracer for the same subset of receptors—receptors configured in high‐affinity state—while an antagonist tracer also binds to receptors in the low‐affinity state that are “ignored” by the neurotransmitter except at very high concentrations. This means that a change in the concentration of neurotransmitter of a given magnitude will lead to greater change in agonist tracer binding compared with antagonist tracer binding (Figure 5).
Figure 5.
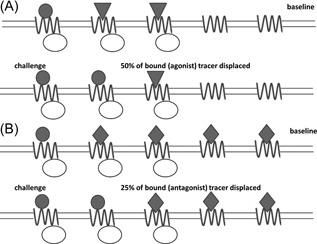
Greater sensitivity of agonist tracers to displacement (“challenge”) by neurotransmitter. Agonist tracers primarily bind to the receptors configured in the high‐affinity state (ie, coupled to G‐proteins), as do neurotransmitters. Therefore, the same change in receptor occupancy by the neurotransmitter displaces a greater fraction of bound agonist tracer (A) than of bound antagonist tracer (B). In this schematic diagram, the endogenous neurotransmitter is pictured as a circle, the agonist ligand as a triangle, the antagonist ligand as a diamond, the G‐protein as an ellipse, and the receptor as a sinusoid line
For some receptor families (eg, serotonin 5‐HT1A and 5‐HT2A receptors), all available antagonist tracers appear to be insensitive to alterations of endogenous neurotransmitter levels.51 As agonist tracers are supposed to be more sensitive than antagonist tracers to endogenous neurotransmitter competition, developing agonist ligands is considered a promising way to obtain a tool for the measurement of synaptic neurotransmission via these receptors.52
3.1.2. Studies of (pathological) alterations in receptor availability
In Section 1, a few examples were given of how alterations of the percentage of receptors configured in the high‐affinity state can accompany the disease. Since the high‐affinity state is the active form of the receptor involved in signaling and may be primarily affected by the disease, the abundance of the high‐affinity state could be a more meaningful biomarker than the total receptor density. Agonist tracers should then be a convenient tool for pinpointing alterations of the availability of receptors configured in the high‐affinity state in disease.
The results of some in vitro experiments with agonist and antagonist radioligands have supported the hypothesis that agonist tracers are superior to antagonists in detecting pathological changes in neuroreceptor availability. In vitro binding of the 5‐HT1A agonists [18F]F15599 and [18F]F13640 but not of the antagonist [18F]MPPF, in postmortem brain sections of Alzheimer's patients was decreased compared with control brains.53, 54 In unilateral 6‐hydroxydopamine‐induced lesions of the rat brain (exhibiting dopaminergic neurodegeneration similar to Parkinson's disease in humans, where upregulation of Rhigh is hypothesized), the ex vivo binding of dopamine D2/3 agonist [3H]NPA was changed to a greater extent than the in vitro binding of D2/3 antagonist [3H]raclopride.55
3.1.3. Measurement of agonist drug occupancy
Many drugs owe their effect to their agonist activity at one or more kinds of receptors. For instance, many antiparkinsonian drugs are D2/3 agonists56; muscarinic receptor agonists like milameline were tried as treatment of Alzheimer's disease57; the mechanism of action of antipsychotics may include not only D2/3 antagonism but also 5‐HT1A‐agonism58, 59; the active metabolite of clozapine (also an atypical antipsychotic) acts as an agonist at muscarinic M1 receptors60; opiate agonists are widely used as analgesics or antitussives and for treating diarrhea and opiate abuse.61
Increased sensitivity of agonist tracers to displacement by agonist drugs may be an advantage in occupancy studies: the opioid receptor antagonist [11C]diprenorphine failed to detect receptor occupancy by clinically relevant doses of opioid agonists.62, 63 However, no studies have so far been published, where the sensitivity of an agonist and an antagonist opioid receptor tracer with equal subtype‐selectivity to displacement by an agonist drug was compared head‐to‐head.
Agonist tracers can also complement antagonist tracers in the investigations of the affinity‐state preference of new drugs. The sensitivities of agonist and antagonist tracers to the displacement by the drug can be compared: drugs preferring the high‐affinity state will displace the agonist tracer more readily, while drugs not distinguishing between affinity states will show no difference in displacement efficacy. Two studies attempting this approach have been published64, 65 but both reported equal displacement of agonist and antagonist tracers by the drug, which can be interpreted in two ways: either the tested drugs were ideal antagonists or the hypothesis of greater agonist tracer displacement by agonist drug does not hold.
3.2. Intrinsic shortcomings of agonist tracers
Though the preference for the high‐affinity state makes agonist tracers potentially superior to antagonists in certain imaging applications, it also results in a number of specific difficulties associated with the development and use of agonist tracers.
3.2.1. Lower signal‐to‐noise ratios
The signal‐to‐noise ratio of a PET tracer is proportional to the density of receptors the tracer binds to in the brain (B avail) and to the tracer's affinity toward these receptors (1/K d). The density of receptors configured in the high‐affinity state (and thus recognized by agonist tracers) is by definition lower than the total receptor density.
Moreover, estimates of agonist affinity toward the high‐affinity state, acquired in membrane homogenates in vitro, may be systematically higher than the actual affinity in vivo. The reason why it may be so is the negative‐feedback between agonist‐receptor and receptor‐G‐protein binding in the GTP cycle (see Section 2.3), which is part of the G‐protein‐dependent model of the high‐affinity state. Indeed, GTP depletion was shown to increase the affinity of agonist but not antagonist ligands to opioid receptors in cultured cells.66 Therefore, affinity and nonspecific binding requirements for agonist tracers are stricter than for antagonists.
3.2.2. Greater likelihood of unwanted pharmacological effects
As agonist tracers preferentially bind to the functional subpopulation of the receptors, they may induce significant physiological responses at a rather low dose, which can distort the experimental results and cause discomfort to the patients.
Indeed, staying below the pharmacological dose range is a concern in opioid receptor imaging with agonist tracers.67, 68, 69 It was also reported as a potential concern in serotonin 5‐HT1A receptor imaging with the agonist [11C]CUMI‐101,70 even though first tests of the same compound in humans showed no adverse effects.71 Exceeding the pharmacological threshold is especially easy with tracers with low specific radioactivity and a related high injected mass of the radioligand. The risk of low specific radioactivity is increased when labeling chemistry is complex. For instance, the dopamine D2/3 receptor agonist [11C](+)PHNO was originally labeled via a four‐step route, resulting in a relatively low specific radioactivity.72 As a consequence, a high incidence of nausea (emesis is a typical effect of D2 agonism) was reported in patients injected with [11C](+)PHNO,73 and it was later found that [11C](+)PHNO human PET studies had frequently been performed under nontracer conditions.74
4. EXISTING PET AGONIST TRACERS FOR GPCR IMAGING IN THE CENTRAL NERVOUS SYSTEM
The greatest number of agonist PET tracers has been developed for the imaging of dopamine D2/3 receptors (see 75 for a review). Tracer development efforts in the last two decades have yielded a number of agonist radioligands for other receptors as well. The most promising agonist tracers developed for PET imaging of neuroreceptors are presented in Table 1, Figure 6 and Figure 7.
Table 1.
Agonist tracers developed for the imaging of high‐affinity state of neuroreceptors
| Receptors | Tracer names | In vitro evaluation | In vivo evaluation | Remarks | |||||
|---|---|---|---|---|---|---|---|---|---|
| Agonism proven by a , b | Preference for Rhigh proven by c | Rodents | Non‐human primates | Other animals | Humans | Sensitive to endogenous neurotransmitter levels d | |||
| Dopamine D1/5 | (S)(+)[11C]SKF82957 | ND | COMP+ GTPdis+76 | Rats77, 78, 79 | ND | ND | ND | Rats–77 | Lipophilic metabolites in brain tissue |
| (S)[11C]NNC 01‐0259 | MESS+80 | ND | ND | 81, 82, 83 | ND | ND | Primates–80 | Lipophilic metabolites in brain tissue | |
| Dopamine D2/3 | [11C]PHNO | ND | GTPdis+84 COMP+85 SAT−86 | Rats72, 87, 88 | 89 | Cats90 | 91 (first) | Rats+72, 87, 88 cats+90 primates+92 humans+93, 94 | Now primarily used as D3‐selective tracer95 derivation of 18F‐version unsuccessful96 |
| [11C]NPA | ND | COMP+56, 85 | Rats97 | 97, 98, 99 | Cats90 pigs100 | 101 (first) | Cats+90 primates+99 humans+102 | Relatively difficult radiosynthesis | |
| [11C]MNPA | ND | COMP+28 SAT−86 | Mice44, 103 rats104 | 105, 106, 107, 108, 109, 110 | ND | 111 (first) | Rats+104 mice+44 primates+109 | Lowest BPND (see Section 5.1.1) among D2/3 tracers used in humans | |
| [18F]MCL‐524 | ND | COMP+112 | ND | 113 | ND | ND | Primates+113 | Structurally related to NPA and MNPA | |
| Dopamine D2 | [11C]SV‐III‐130 | MESS+114 | ND | ND | 114 | ND | ND | Primates+114 | Possible 5‐HT1A binding |
| Dopamine D3 | [18F]LS‐3‐134 | MESS+115 | COMP−116 | ND | 115 | ND | ND | Primates+115 | Specific binding seen only after dopamine depletion |
| [18F]7‐OH‐FHXPAT | ND | GTPdis+117 | mice, rats117 | ND | ND | ND | ND | D3‐over‐D2 selectivity not fully characterized | |
| Serotonin 5‐HT1A | [11C]CUMI‐101 | GTPrec– 118 GTPrec±119 | ND | ND | 70, 120 | ND | 71, 121 | Rodents−122 primates+123 humans−124 humans±125 | Variable intrinsic activity 118, 119, 126 binds to adrenoceptors118, 126 derivation of 18F‐version successful127, 128 |
| [18F]F13714 | GTPrec+ MESS+129 | GTPdis+130 | Rats130 | 131 | Cats130 marmosets132 | ND | ND | Specific binding is irreversible | |
| [18F]F13640 | GTPrec+133 | ND | Rats134 | 134 | Cats134 | ND | Rats+134 | Slow, but reversible binding kinetics | |
| Serotonin 5‐HT2A | [11C]CIMBI‐36 | MESS+135 | ND | Rats, mice (only safety)136 | 137 | Pigs135 | 138, 139, 140 | Pigs+141 primates+142 humans−143 | Also binds to 5‐HT2C 137 alternative 11C‐labeling positions compared 140 derivation of 18F‐version unsuccessful144 |
| κ‐Opioid | [11C]GR103545 also known as (R)‐[11C]GR89696 | PHYS+ (for κ) | COMP±145 | Mice146 (race‐mate)147 (eutomer) | 68, 69, 148 | ND | 149, 150 | ND | Competition assay shows biphasic binding but this may reflect different affinities for κ |
| µ‐Opioid | [11C]carfentanil (mu‐OR) | PHYS+151 | ND | Mice152 rats43 | 153 | ND | 153(first) | Rats+43 humans+154, 155, 156 humans−157 | Derivation of 18F‐version successful, no follow‐up158 |
| µ/κ‐Opioid | [11C]PEO | GTPrec+159 | ND | Rats159 | ND | ND | ND | ND | Derivation of 18F‐version successful160, 161 |
| Muscarinic M1 | [11C]LSN3172176 | GTPrec+162 | COMP−162 | 163, 164 | ND | Imperfect subtype‐selectivity | |||
| [11C]AF‐150(S) | ND | COMP+20 | Rats165, 166 | ND | ND | ND | Rats±166 | Low signal‐to‐noise ratios | |
| Muscarinic M2 | [18F]FP‐TZTP | PHYS+167 | ND | Mice168 rats 169, 170, 171 | 170 | ND | 172(first) | Primates+173 | 11C‐version created, no follow‐up174 |
Abbreviations GDP, guanosine diphosphate; GTP, guanosine triphosphate; ND, no data available.
Coding of experimental paradigms aiming to confirm agonism: MESS monitoring secondary messenger levels in functional assays in vitro; GTPrec monitoring GTP recruitment to G‐proteins in vitro; PHYS monitoring physiological or behavioral effects of the compound in vivo or ex vivo.
Works confirming functional agonism are cited only if the preference for Rhigh has not been directly confirmed.
Coding of experimental paradigms aiming to confirm preferential binding to Rhigh: COMP obtaining a biphasic competition curve in vitro; SAT obtaining a biphasic saturation curve in vitro; GTPdis detecting the loss of specific binding upon GTP or GppNHp addition in vitro.
Coding of the outcomes of studies confirming sensitivity to endogenous neurotransmitter levels (and also agonism and Rhigh preference): +, positive outcome; −, negative outcome, ±, ambiguous results.
Figure 6.
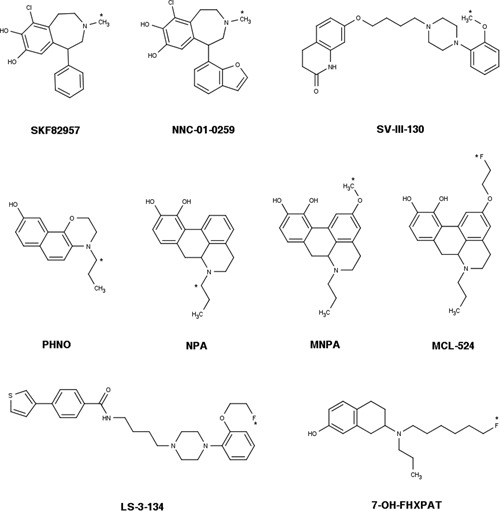
Chemical structures of agonist radioligands for dopaminergic receptors (see also Table 1). The position of the radionuclide in each molecule is indicated by an asterisk
Figure 7.
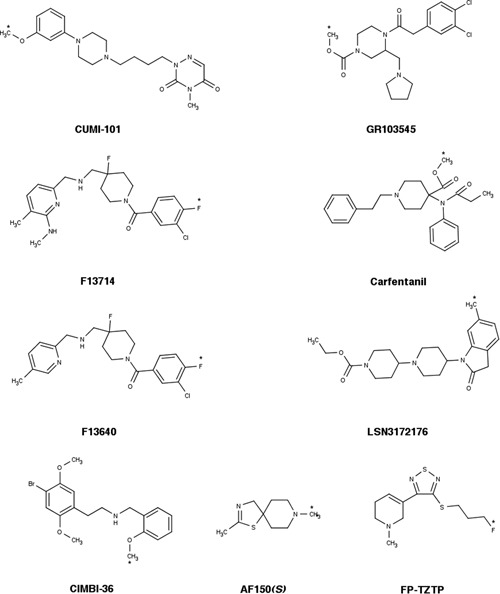
Chemical structures of agonist radioligands for serotonin, opioid, and muscarinic receptors (see also Table 1). [11C]PEO is not shown; its structure can be found in Van Waarde et al.175 The position of the radionuclide in each molecule is indicated by an asterisk
4.1. Definition and properties of an agonist tracer
An agonist tracer is usually defined as “a radiolabeled analog of a ligand with agonist activity.” There are many ways to confirm and measure the degree of agonist activity: behavioral or ex vivo studies examining the physiological effect of the drug, functional in vitro assays measuring the levels of certain secondary messengers, or the recruiting of proteins involved in signaling cascades to the receptors.
Because the intrinsic activity of a ligand is known to be correlated with the ratio of its affinities to the high‐ and low‐affinity receptor states,18, 19 it seems evident that agonists will preferentially bind to the high‐affinity state. However, agonism does not necessarily imply preferential binding to receptor‐G‐protein complexes, since noncanonical signaling pathways do exist. One example is cariprazine, a drug which was recently labeled with carbon‐11 and evaluated as a dopamine D2/3 receptor PET tracer. This compound showed partial D2/3 agonist activity in secondary messenger assays but did not recruit G‐proteins in vitro.176, 177, 178 Observations of G‐protein recruitment may also differ between in vitro setups. For instance, [11C]CUMI‐101, a tracer for serotonin 5‐HT1A receptors, was defined as an agonist based on the [35S]GTPyS assay (indirect measurement of G‐protein recruiting to receptors) in membrane homogenates from cell cultures expressing recombinant human receptors but was later found to act as an antagonist when the same assay was done in primate and rat brain homogenates.118, 119
Therefore, the most certain proof of the agonist radioligand's in vitro preference to the high‐affinity state is directly demonstrating that it recognizes high‐ and low‐affinity states of its receptor in natural tissue or in transfected cell culture. It is worth noting that for some agonist tracers, preferential in vitro binding to Rhigh was demonstrated only after the tracer had been evaluated in vivo (compare 105 and 28), while for some other radioligands the capability to discern affinity states in vitro was not assessed at all.80, 135
4.2. In vivo evaluation of a PET neuroreceptor tracer
Characteristics desirable for a PET tracer for brain imaging include the ability to pass the blood‐brain‐barrier, a low degree of metabolism, a high contrast between target (specific) and nontarget (nonspecific) binding, and pharmacokinetics that can be reliably quantified from a 60 to 90 minute‐long PET scan (see 179, 180, 181 for review). An important aim in PET imaging is the measurement of synaptic neurotransmission. For this reason, neuroreceptor tracers are tested for the sensitivity of their binding to changes of endogenous neurotransmitter levels, and agonists are supposed to be more sensitive than antagonists.
Neuroreceptor tracers are usually evaluated in rodents or non‐human primates before being moved to human studies. In non‐human primates, one can investigate the binding of the tracers with high spatial detail using clinical PET cameras. Evaluation in rodents is cheaper and enables the use of more invasive methods but interspecies differences in rodent, primate, and human physiology can be a confounding factor. In addition, the small size of rodents forces the researchers to use dedicated nonclinical “micro‐PET” cameras and does not permit to reliably image minor brain structures. To strike the right balance between controllability of the experimental conditions and image quality, tracers are sometimes evaluated in (relatively) large mammals, such as cats or pigs.
4.3. Availability of agonist PET neuroreceptor tracers
Agonist PET tracers can be divided into three categories (see Table 1). The first category includes radioligands that have reached the stage of in vivo evaluation in humans. The dopamine D2/3 ligands [11C](+)PHNO, [11C]NPA, and [11C]MNPA, the serotonin 5‐HT1A ligand [11C]CUMI‐101, the serotonin 5‐HT2A ligand [11C]CIMBI‐36, the κ‐opioid ligand [11C]GR103545, the µ‐opioid ligand [11C]carfentanil, and the muscarinic M2 receptor agonist [18F]FP‐TZTP fall into this category. The binding of most of these ligands has been shown to be sensitive to changes of endogenous neurotransmitter levels, although for [11C]CIMBI‐36 and [11C]CUMI‐101 the acquired results have been conflicting, and the sensitivity of [11C]GR103545 to endogenous opioids has not yet been evaluated. Many ligands from category one are used in PET imaging for other reasons than their agonist properties. [11C](+)PHNO, for example, is frequently selected as a tracer because it binds preferentially (though not selectively) to dopamine D3 receptors, and its preference for this subtype is higher than that of dopamine D2/3 antagonist ligands like [11C]raclopride.95 [11C]CUMI‐101 is used in clinical studies,182, 183 not because of its ability to image the high‐affinity state of serotonin 5‐HT1A receptors (which is actually under doubt—see below) but because of its high subtype‐selectivity and imaging contrast. [11C]carfentanil and [18F]TZTP are the only tracers available for µ‐opioid and muscarinic M1 receptors, so in their case, there are no antagonist tracers with which they could be compared head‐to‐head.
The second category contains radioligands that have not (yet) been tested in humans but have been evaluated in non‐human primates. The dopamine D1/5 ligand (S)[11C]NNC 01‐0259, the dopamine D2/3 ligand [18F]MCL‐524, the dopamine D2 ligand [11C]SV‐III‐130, the dopamine D3 ligand [18F]LS‐3‐134, the serotonin 5‐HT1A ligands [18F]F13714 and [18F]F13640, and the muscarinic M1 ligand [11C]LSN3172176 belong to this group. The binding of the dopamine D2, D3, and D2/3 ligands from this category has been shown to be sensitive to changes of endogenous neurotransmitter levels but for the dopamine D1/5 the results were negative, and there are currently no D1/5 agonists or antagonists that would be sensitive to endogenous dopamine levels. Sensitivity of [18F]F13714, [18F]F13640, and [11C]LSN3172176 to endogenous serotonin/acetylcholine has not yet been evaluated in primates, although [18F]F13640 was sensitive to serotonin levels in rats. Some of these compounds were radiolabeled with 18F or 18F‐versions of previously developed 11C‐tracers because the longer half‐life of fluorine‐18 makes an imaging agent more suitable for wide‐scale clinical use. [18F]sufentanil has been created as a longer half‐life alternative to [11C]carfentanil but was not further developed.184
The third and last category contains tracers that have only been evaluated in rodents. The dopamine D1/5 ligand (S)(+)[11C]SKF82957, the dopamine D3 ligand [18F]7‐OH‐FHXPAT, the µ/κ‐opioid agonist [11C]PEO, and the muscarinic agonist [11C]AF‐150(S) fit in this category.
Binding of (S)(+)[11C]SKF82957 is known to be insensitive to changes of endogenous neurotransmitter levels. Available data for [11C]AF‐150(S) in this regard are inconclusive, and for other tracers from this category sensitivity to endogenous neurotransmitter levels has not yet been evaluated.
In the process of tracer development, some agonist tracers were found to have issues that hamper their routine application. Two radioligands for dopamine D1/5 receptors, (R)‐[11C]SKF82957 and (S)‐[11C]N‐methyl‐NNC 01‐0259,81, 185 are converted to lipophilic radioactive metabolites that penetrate into the brain and can confound the interpretation of imaging results, though this can be remedied by inhibiting the activity of the enzyme catechol‐O‐methyl transferase.77 Another D1/5 agonist, [11C]SKF75670, was originally developed together with [11C]SKF82957 but was abandoned because of inferior signal‐to‐noise ratios. Since it is structurally very similar to [11C]SKF82957, it may show the same unfavorable in vivo metabolism.78 The development of (S)‐[11C]N‐methyl‐NNC 01‐0259 was halted since tests examining its sensitivity to endogenous dopamine levels yielded a negative result.80 The D3‐selective agonist tracer [18F]LS‐3‐134 was shown to specifically bind to D3 receptors in monkey brain but specific binding was only measurable under dopamine depletion conditions.115 The muscarinic M1 receptor tracer [11C]AF‐150(S) showed both specific binding and sensitivity to endogenous acetylcholine levels in the rat brain but the low signal‐to‐noise ratios of this ligand cast doubt on its suitability for further research.165, 166
To demonstrate the preferential binding of agonist tracers to receptor high‐affinity state, a head‐to‐head comparison with reference tracers binding to all receptors (ie, antagonist tracers) is required. For D1/5, D2/3, 5‐HT1A, and 5‐HT2A agonist tracers counterpart antagonist tracers are available, which possess matching pharmacological selectivity (ie, bind with similar relative affinities to the same receptor subtypes within the relevant region of interest as their “corresponding” agonist tracers) and, in case of D2/3 and 5‐HT2A ligands, sensitivity to endogenous neurotransmitter levels.51, 186, 187, 188 The same is true for the κ‐opioid agonist [11C]GR103545, which was developed along with its antagonist counterpart [11C]LY2795050,189 and for the µ/κ‐opioid agonist [11C]PEO, which can be used in conjunction with the µ‐partial agonist and κ‐antagonist [11C]buprenorphine.159 For the µ‐opioid agonist [11C]carfentanil, the M2 agonist [18F]FP‐TZTP and M1 agonists [18F]AF‐150(S) and [11C]LSN3172176 there are no suitable antagonist counterparts.175, 190 In theory, non‐subtype‐selective radioactive antagonists might be used in head‐to‐head comparisons with radioactive agonists if receptors to which the antagonist, but not the agonist, binds are fully blocked with a nonradioactive drug, but the feasibility of that approach is questionable because of the possible pharmacological effects of such blockade.
In conclusion, agonist tracers for PET imaging of dopamine D2/3, serotonin 5‐HT1A and 5‐HT2A, µ and κ‐opioid, and M2 muscarinic receptors are available. Agonist tracers for dopamine D1/5 and muscarinic M1 receptors have issues that make their use for PET imaging problematic. Preference for the high‐affinity state in vitro has only been directly demonstrated for D1/5 and D2/3 tracers, some 5‐HT1A tracers and the M1 tracer [18F]AF‐150(S). Agonist tracers for D2/3, 5‐HT1A, 5‐HT2A, and κ‐opioid receptors can be matched with antagonist tracers binding to the same receptors for head‐to‐head comparisons.
5. PROVING THE EXISTENCE OF THE HIGH‐AFFINITY STATE IN VIVO
5.1. Outcome measures of in vivo experiments
In vivo experiments with neuroreceptor radioligands measure the availability of the receptors and changes in this availability in response to alterations of endogenous neurotransmitter levels or administration of exogenous drugs. Binding potentials (BPs) and target/nontarget ratios are “raw” measures representing receptor availability, which can later be recalculated to receptor densities or occupancies.
5.1.1. Binding potentials
The typical outcome measure of in vivo imaging experiments is the BP. This parameter is defined as the product of the density of binding sites (B max) and the affinity of the radioligand for these sites (inverse of the dissociation constant, 1/K d). BP is equal to the ratio of the concentrations of specifically bound and free ligand in the tissue of interest at equilibrium, provided that the administered dose of radioligand is sufficiently low (see the Appendix for more explanation). BP is estimated by fitting a kinetic model to measured time‐activity curves. Time‐dependent radioactivity in the region of interest and a reference region in the brain can be measured by PET imaging, and plasma radioactivity can be determined by blood sampling.191 Note that in vivo not all receptors may be available for binding, as they can be internalized, converted to low‐affinity state (for an agonist) or occupied by neurotransmitter, so in the in vivo context the term B avail is more suitable than B max.
Given the difficulty of determining the true concentration of the free ligand in the living tissue, other concentrations proportional to free ligand concentration in tissue are substituted in its place. Specifically, bound concentration is related to free plasma concentration (BPF), total plasma concentration (BPP), or “nondisplaceable” concentration (BPND), that is, the total concentration of free and nonspecifically bound ligand in the tissue.192 It is reasonable to assume that free ligand concentrations in the plasma and in the interstitial liquid of the brain tissue are equal at equilibrium, so BPF can be considered the “true” BP.
5.1.2. Target/nontarget ratios
When regions of interest are small relative to PET camera resolution (ie, subsections of rodent brain) it is often hard to obtain a reliable time‐activity curve with high temporal resolution. Also, in situations when a lot of experimental conditions have to be tested and compared, it is often infeasible to obtain time‐activity curves by PET or to sacrifice large groups of animals at different time points. In these situations, one can take advantage of the “pseudoequilibrium” state when the concentration ratios between receptor‐rich and receptor‐free tissues remain constant even as absolute concentrations are changing. For a typical neuroreceptor ligand, one can reasonably expect the pseudoequilibrium state to be reached within half an hour after injection. Once the time range in which the pseudoequilibrium exists is validated, specific binding can be estimated from tissue concentrations at a single time point within this time range. Such concentrations can be obtained by ex vivo dissection and radioactivity counting or from a static PET scan. Target/nontarget concentration ratios can be used as is or be recalculated to specific binding ratios (SBRs):
where T and NT are radioligand concentrations in receptor‐rich (“target”) and receptor‐poor (“nontarget”) regions of interest. In the absence of specific binding, SBR = 0, while T/NT = 1.
“Raw” specific binding, that is, the difference between radioligand concentrations in receptor‐poor and receptor‐rich regions can also be used as an outcome measure. However, when specific ligand binding is not normalized to the nonspecific binding at the same time point, its value is prone to intrasubject variations in pharmacokinetics. Therefore, the use of binding ratios is preferred.
5.1.3. Available receptor density
Available receptor density (B avail) can be estimated in a saturation experiment. Specific binding of a radioligand is determined by two parameters: the density of binding sites in the region of interest (B avail) and the affinity of the radioligand toward these receptors (1/K d). To independently estimate these two parameters, bound and free radioligand concentrations at equilibrium have to be estimated at least at two different injected doses (for radiotracers, injected dose is usually varied by changing molar radioactivity). Bound concentration can be estimated from a difference in equilibrium tracer concentrations between the target and nontarget regions, while free concentration can be back‐calculated from the binding potential (see Appendix). B avail can then be quantified by regression analysis. In PET imaging, the regression is often performed on “linearized” binding data: binding potential is plotted against absolute specific binding (Scatchard plot). Scatchard plot requires a simple linear regression and is therefore straightforward but also bias‐prone, as the X and Y axes are not independent of each other. Alternatively, B avail can be estimated by nonlinear regression of the binding curve (bound vs free concentration plot). For in vitro assays, where free and bound ligand concentrations can be independently estimated, nonlinear regression is the gold standard. However, for in vivo experiments the Scatchard plot has remained popular.
5.1.4. Receptor occupancy
Displacement of a tracer from its receptors by a competing ligand decreases the binding potential of the tracer. Receptor occupancy can be calculated as the change in binding potential or target‐nontarget ratio after drug administration, relative to baseline. The same holds for the occupancy of the receptors by endogenous neurotransmitter, when drugs stimulating neurotransmitter release or depletion are administered.
5.2. Experimental paradigms used to demonstrate the existence of high‐affinity state in vivo
To demonstrate that agonist tracers preferentially bind to a certain “high‐affinity” subset of receptors in vivo three approaches have been used (summarized in Table 2). One approach is to directly measure the available binding site densities for agonist and antagonist tracers and demonstrate that the binding site density available to the agonist is lower. Another is to infer the ratio of high‐affinity binding site density to total binding site density from the results of experiments where tracers compete for binding to the receptors with unlabeled ligands. A third approach is to demonstrate that agonist, but not antagonist, binding can be influenced by manipulations of receptor‐G‐protein coupling.
Table 2.
Experimental paradigms used for the detection of high‐affinity state in vivo
| Approach | Experimental paradigm | Minimum ligand set necessary | Minimum number of experimental conditions | Results confirming the presence of high‐affinity state In vivo | Shortcomings | Examples | |
|---|---|---|---|---|---|---|---|
| Risk of pharmacological effects | Other | ||||||
| Binding site density comparison | Saturation experiment | Labeled antagonist + la ‐ beled agonist | 4 Minimum 2 dose levels per radioligand | Lower B max value for agonist tracer than for antagonist tracer | Yes | 90, 122, 193 | |
| B avail = BPF × K d | Labeled antagonist + labeled agonist | 2 Single dose level per radioligand | Lower B max value for agonist tracer than for antagonist tracer | No | Requires arterial input with free fraction in plasma, in vitro K d is likely not equal to in vivo K d | 70 | |
| Correlation analysis | Labeled antagonist + labeled agonist | 2 Single dose level per radioligand | Presence of the main trend with upward or downward deviations from it for certain regions of interest | No | See Section 5.2.1, third subsection | 70, 132, 137, 139 | |
| Imaging in disorders | Labeled antagonist + labeled agonist | 4 Single dose level per radioligand, imaging in healthy and diseased state | Same specific binding for antagonist tracer in control and diseased state, different specific binding for agonist tracer | No | Upregulation or downregulation of high‐affinity state has to be demonstrated in vitro | 103, 194, 195 | |
| Competition studies | In vivo displacement curves | Labeled antagonist + unlabeled agonist | 5 Minimum number of dose levels to distinguish between mono‐ and biphasic curves | Displacement curve better explained by biphasic than by monophasic model | Yes | large number of dose levels to test | 86, 196, 197, 198 |
| Neurotransmitter challenge | Labeled antagonist + labeled agonist + stimulator of neurotransmitter release or depletion | 4 Single dose level per radioligand, imaging before and after challenge | Greater displacement of agonist tracer by the challenge of same magnitude | Yes | see Section 5.2.2 | 90, 98, 106 | |
| Exogenous drug challenge | Labeled antagonist + labeled agonist + unlabeled agonist | 4 Single dose level per radioligand, imaging before and after challenge | Greater displacement of agonist tracer by the challenge of same magnitude | Yes | 86, 107, 196, 199, 200 | ||
| Probing the nature of the high‐affinity state | Sensitivity to G‐protein coupling | Labeled antagonist + la ‐ beled agonist + agent for G‐protein decoupling | 4 Single dose level per radioligand, imaging before and after G‐protein decoupling | G‐protein decoupling decreases specific binding for the agonist but not for the antagonist radioligand | No | Requires intrathecal or intracerebral injections, does not look at what fraction of total receptors are in the high‐affinity state | Proof of concept presented in15 |
It is important to emphasize that to compare agonist and antagonist tracers with each other their pharmacological selectivity profiles (ie, relative binding affinities toward different receptor subtypes) should be identical within the region of interest used for comparison. Otherwise, any detected difference in binding behavior could be attributed to the relative preference of one of the tracers toward a certain receptor subtype.
In principle, all experiments described below can be performed not only with PET tracers labeled with short‐lived positron‐emitting isotopes but also with radioligands labeled with long‐lived isotopes such as tritium (3H). On the one hand, this makes the experimental paradigm nontranslatable to the clinical setup. On the other hand, this enables the use of elegant approaches like the double‐tracer study, where radioligands labeled with short‐lived (eg, 11C) and long‐lived (eg, 3H) isotopes are compared head‐to‐head in the same group of animals.194, 196
5.2.1. Approach 1: comparing binding site densities
Saturation study
As explained above, a minimum of two different radioligand doses needs to be tested to estimate binding site density (B avail). More doses will add precision and can reveal potential cooperativity effects or the presence of multiple binding sites with different affinities (eg, receptor affinity states), provided that radioligand binding to all these sites is distinguishable from nonspecific binding. However, published PET studies comparing binding site densities of agonist and antagonist tracers were restricted to two doses.90, 193 Another study used single time point SBRs as outcome measure and built saturation curves based on 9 to 10 data points.122
In two‐dose PET studies aimed at quantifying B avail, the low dose corresponds to the minimum amount of radioligand that can be injected, that is, the “tracer dose,” which should occupy less than or equal to 10% of the receptor population in the region of interest. The high‐dose is chosen to occupy about two‐thirds of that population.90, 193
Extracting density values from true binding potential measurements
It should be noted that performing a saturation assay with agonist radioligands can lead to unwanted and dangerous pharmacological effects, especially in the case of opioid ligands.68, 69
In a head‐to‐head comparison of 5‐HT1A agonist and antagonist tracers, Kumar et al70 attempted to circumvent this problem by comparing the “true” binding potentials (BPF) for the two tracers at low injected dose instead of performing a second high‐dose scan to independently measure B avail and K d. Given that BPF = B avail/K d, B avail can arguably be calculated from the BPF value using in vitro K d value for the corresponding tracer. However, there are two problems with this approach. First, to calculate BPF, one needs to obtain an arterial input curve and free fraction in plasma for the investigated radioligand, in addition to the time‐activity curve for the region of interest. Such a large amount of input data makes BPF prone to experimental error and bias. Second, the in vivo K d of the radioligand is not necessarily equal to the in vitro K d, especially if the latter is measured for receptors from a different animal species or in transfected cells.
Studying correlation between regional binding of agonist and antagonist
Binding potentials or target/nontarget ratios for an agonist tracer in various brain regions can be plotted against the corresponding measurements for an antagonist tracer, to examine their correlation. Such a plot may provide insight into the relationship between the densities of available binding sites for agonist and antagonist tracers while staying below the “tracer” threshold. If agonist binding in a certain brain region lies above the main trend on the correlation graph, it suggests that the relative abundance of receptors in the high‐affinity state in this region is higher than average, and vice versa.
This approach, however, has many limitations. If the relative abundance of the high‐affinity state is drastically different in each region, the correlation graph will be meaningless: there will be no main trend to pinpoint deviations from. If the relative abundance of the high‐affinity state is the same in all regions, the correlation graph will be a straight line, revealing no differences in agonist and antagonist binding and thus no evidence in favor of the existence of the high‐affinity state. Therefore, analysis of the correlation between agonist and antagonist binding cannot be the sole method of looking for the existence of high‐affinity state but can be an extra piece of data analysis in experiments based on other paradigms.
Studying agonist binding in disorders presumably caused by high‐affinity state dysregulation
In vitro experiments in membrane homogenates suggest that some neuropsychiatric disorders are accompanied by alterations in the relative abundance of the high‐affinity state in a given receptor population, while changes in overall receptor density relative to the healthy condition are either absent or much less pronounced. One example is animal models of psychosis where the high‐affinity state of dopamine D2/3 receptors is upregulated.14, 201
Therefore, another way to demonstrate the existence of a high‐affinity state in vivo is to show that its upregulation (or downregulation) can be noninvasively detected by agonist tracers. If the relative abundance of the high‐affinity state is altered but the overall receptor density remains (relatively) constant, the binding of the agonist but not of the antagonist tracer will be different in the diseased state relative to the healthy state. Ratios of BP or SBR values for agonist and antagonist tracers can be used as outcome measures to normalize for possible concomitant alterations in total receptor density.202
In this paradigm, the binding of each tracer only has to be assessed at a low and pharmacologically inactive dose. However, one has to demonstrate that the relative abundance of high‐affinity state really differs between healthy and diseased states, using experimental approaches other than PET (typically, in vitro assays). Moreover, in the diseased state, alteration of the relative abundance of the high‐affinity state may be accompanied by alterations in other parameters relevant for radioligand binding. For instance, changes in baseline neurotransmitter levels also differentially affect the binding of agonist and antagonist tracers (agonist binding is changed to a greater extent). Concomitant changes in several parameters pressing agonist tracer binding in different directions can offset each other, leading to little or no change in overall receptor availability to the agonist tracer compared with the healthy state.
5.2.2. Approach 2: studying tracer vulnerability to displacement by an unlabeled competitor
One important difference between tracer‐drug competition experiments in vitro and in vivo is that in the latter case the concentration of both tracer and drug at the receptors is not constant. While for the radioligand a true equilibrium between its concentrations in blood and brain tissue can be achieved by using bolus‐plus‐infusion injection scheme, the same is virtually infeasible for the unlabeled drug (tissue concentrations of which are much harder to monitor). Nevertheless, one can usually safely assume that the pharmacokinetics of the competing drug are dose‐linear within the investigated dose range, so the degree of displacement of the tracer by the drug is also dose‐linear.
Building in vivo displacement curves
In vitro, the high‐affinity state is detected by displacing an antagonist radioligand with ever increasing concentrations of unlabeled agonist drug. When the remaining specific binding of the radioligand is plotted against agonist concentrations, the displacement is shown to proceed in two phases: agonist first displaces the radioligand from high‐affinity sites then from low‐affinity sites. The same displacement curve can be built in vivo by plotting binding potentials or target/nontarget ratios for an antagonist tracer against an administered dose of unlabeled agonist drug.
The advantage of this paradigm is that it does not require an agonist radioligand. Antagonist radioligands are much more numerous than agonist radioligands, so the displacement curve paradigm is currently applicable to a wider range of receptors than other paradigms mentioned below.
The downside, however, is that the generation of a displacement curve is a laborious undertaking. The shape of the biphasic curve is determined by five parameters: maximum binding level (at no displacement), minimum binding level (full displacement), agonist affinities for high‐ and low‐affinity states, and the percentage of receptors in the investigated population configured in the high‐affinity state. This means that at least five different dose levels (including zero) have to be used to test whether the obtained curve is monophasic or biphasic.
Therefore, studies that used the displacement curve approach typically used SBRs obtained ex vivo at a single time point from large numbers of rodents,86, 196 though the use of PET scanning in primates has also been reported.197, 198 The actual number of dose levels tested was 6 to 9 in rodent and 9 in non‐human primates.
Comparing vulnerability to displacement by unlabeled agonist
An agonist tracer should be more vulnerable than an antagonist tracer to displacement (or “challenge”) by other agonists because it competes with them for the same subpopulation of receptors. Displacement can be elicited by administering an appropriate agonist drug or by stimulating endogenous neurotransmitter release.
The advantage of using exogenous agonist drugs for displacement is that these drugs can be selected to be subtype‐specific and to only occupy the receptor population that is being imaged (or even a defined subset of this population if the tracer binds to more than one receptor subtype).
On the other hand, manipulating neurotransmitter levels has the advantage of being “natural”: one looks at the competition of the tracer with the endogenous ligand, the action of which on the receptors is thought to govern the functioning of the brain [see Laruelle187 and Finnema51 for reviews]. One can also reasonably expect that the competition will only happen at receptors that are really situated in the synapses. Moreover, neurotransmitter levels can be both increased and decreased relative to the baseline. In the latter case, the expected result is greater increase, rather than greater decrease, of binding for the agonist tracer. However, manipulating neurotransmitter release has its downsides, too. First, the effect vs time relationship between the administration of the drug that stimulates a rise or fall in endogenous neurotransmitter level and synaptic receptor occupancy is more complex than when receptors are occupied with exogenous agonist. Second, the released neurotransmitter can act on other receptor subtypes beyond the one being imaged. Third, some drugs used to manipulate neurotransmitter levels are known to manipulate levels of several neurotransmitters at once (eg, amphetamine stimulates both dopamine and norepinephrine release). The lack of selectivity regarding what neurotransmitter is manipulated and which receptors are occupied can confound the interpretation of cause‐and‐effect relationships.
If the “tracer condition” is satisfied (radioligands occupy a negligible fraction of all receptors), the ratio of agonist and antagonist radioligand vulnerabilities to displacement by a challenge is a constant value as long as less than 100% of the high‐affinity state is occupied as a result of the challenge (Figure 8). Therefore, in theory, a single dose of agonist drug or neurotransmitter level manipulator should provide enough information to compare the vulnerability of agonist and antagonist tracers. In practice, because the actual percentage of receptors in the high‐affinity state is unknown, several doses are often tried, resulting in occupancies of up to 100%,86, 90, 107, 196 except in the human studies where the maximum challenge magnitude is limited by ethical considerations.93, 102
Figure 8.
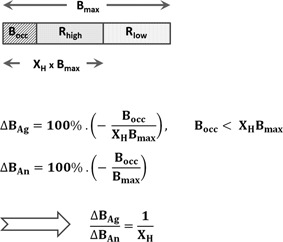
Relationship between agonist and antagonist tracer displacement (∆B Ag and ∆B An) and the fraction of receptors occupied by competing agonist drug or neurotransmitter. B max is the total receptor density available at baseline, X H is the fraction of receptors configured in the high‐affinity state, B occ is the amount of receptors occupied as a result of the challenge. If B occ < X H B max, that is, not all high‐affinity state receptors become occupied, the ratio of relative decreases of agonist and antagonist tracer binding is constant and equal to 1/X H
An important limitation of the vulnerability comparison paradigm is that preference for the high‐affinity state is not the only factor influencing the vulnerability of the radioligand to displacement by unlabeled drugs. For instance, many preclinical in vivo tracer binding experiments are performed in anesthetized animals, and isoflurane and ketamine anesthesia were found to increase the baseline binding of agonist D2/3 tracers, exaggerating the vulnerability of agonist tracers relative to antagonists.109, 203 The mechanism of such selective influence is unclear, although there are reports that anesthetics interfere with receptor‐G‐protein (un)coupling 204, 205 and alter endogenous neurotransmitter levels.206, 207
Furthermore, D2/3 antagonist tracers are known to differ between themselves in the sensitivity to changes in dopamine levels.187, 208 The underlying reasons can be more or less favorable binding kinetics (see Finnema et al51, 209 for discussion) or differences in affinity toward the surface and internalized receptors.210, 211 Although a head‐to‐head comparison of D2/3 agonist [11C]MNPA and D2/3 antagonist [18F]fallypride in internalization‐deficient mice demonstrated that competition with the neurotransmitter is sufficient to explain short‐term (though not long‐term) changes in binding for both tracers,44 this situation can be different for other receptor types.43
5.2.3. Studying vulnerability to G‐protein uncoupling in vivo
Addition of GTP or its analogs decreases specific binding of agonist but not antagonist ligands in vitro, so uncoupling of G‐proteins induced in vivo should lead to the same effects.
Seeman15 demonstrated that GTP addition to tissues extracted from an animal after D2/3 agonist radioligand injection accelerates radioligand dissociation from D2/3 receptors in the tissue and proposed the use of pertussis toxin to promote G‐protein decoupling from the receptors in vivo. Indeed, physiological effects of dopamine and opioid receptor agonists were inhibited by pertussis toxin injections.212, 213, 214
This approach probes the nature of the high‐affinity state, that is, it seeks an answer to the question “is G‐protein binding to the receptor significant for agonist binding to the receptor?” However, this question is not the same as “do agonists bind to a subset of all receptors?,” which is addressed in other paradigms. As discussed in Section 2, all receptors may be precoupled to G‐proteins. Moreover, G‐protein decoupling agents (pertussis toxin or anything else) will have to be introduced locally into the region of interest rather than systemically through intravenous, intraperitoneal, or subcutaneous injections. For brain imaging, that means that intrathecal and intracerebral injections will have to be used. Such injections are technically challenging and hardly (if anyhow) translatable to the clinic. Therefore, this paradigm has not yet been used for head‐to‐head comparisons of agonist and antagonist radioligands.
5.3. Current evidence on the existence of high‐affinity state in vivo
5.3.1. Dopamine receptors
The majority of studies attempting to demonstrate the existence of high‐affinity state in vivo has concerned dopamine D2/3 receptors. Agonists [11C](−)NPA, [11C]MNPA, and [11C](+)PHNO were compared with antagonists [11C]raclopride and [18F]fallypride. In some studies, these compounds were used in their unlabeled or 3H‐labeled forms. A large portion of these studies failed to demonstrate the existence in vivo of the subpopulation of D2/3 receptors configured in the high‐affinity state (see Skinbjerg et al215 for review).
Binding site densities for D2/3 agonist and antagonist tracers were found to be equal in one study,90 while in another study the average relative abundance of D2/3‐high was found to be 79%,193 which is close to the upper extreme of such percentages determined in vitro.216 Saturation of [11C](+)PHNO binding in monkey brain was found to be biphasic but the two binding sites most probably corresponded to D2 and D3 receptor subtypes rather than to high‐ and low‐affinity states.217
In rats with brain lesions induced by the dopaminergic neurotoxin 6‐hydroxydopamine, binding levels of D2/3 antagonist [11C]raclopride and of D2/3 agonist [3H]PHNO were increased to the same extent.194 No difference in baseline agonist binding relative to the healthy condition was found in dopamine β‐hydroxylase knockout (Dβh‐KO) mice,103 in rats withdrawn from chronic ethanol and in amphetamine‐sensitized rats.194 In these three animal models, an upregulation of D2/3‐high was previously demonstrated in vitro by the group of Seeman et al.201, 218, 219, 220 However, in vitro measurements of elevated striatal D2/3‐high in Dβh‐KO mice could not be replicated by the group that performed the in vivo imaging study.103 Moreover, most of the data on the elevation of D2/3‐high in ethanol‐withdrawn rats is based on comparison of B max values for antagonist radioligands in the presence and absence of GppNHp,201, 219 an indirect method of assessing D2/3‐high abundance, shortcomings of which are discussed in Van Wieringen et al.216
In clinical studies of diseases where alteration of D2/3‐high was suspected, binding potentials of agonist tracers in healthy and diseased subjects were similar,202, 221, 222, 223, 224 although some recent reports buck this trend.195, 225
The dopamine D2/3 antagonist radioligand raclopride (11C‐ or 3H‐labeled) was displaced by D2/3 agonist drugs in a monophasic manner.86, 196, 197 In a more recent study, up to 70% of [11C]raclopride binding was displaced by D2/3 agonist quinpirole without any evidence of biphasicity.39
The majority of studies comparing agonist and antagonist tracers’ vulnerability to displacement by agonist drugs found no difference in vulnerabilities,86, 107, 196 though some reports confirming greater vulnerability of agonist tracers do exist 199, 200 and the relative timing of tracer and drug administration were claimed to be important.200
Nevertheless, D2/3 agonists did prove to be more sensitive than antagonists to endogenous dopamine levels in anesthetized rodents,44 cats,90 primates,92, 98, 106 as well as in awake humans,93, 102 though not in awake rodents,86, 194, 196, 203 with a single exception.199 However, lack of consistency in preclinical data on neurotransmitter and agonist drug challenge raises a question whether this advantage of the agonists stems from their preference for the high‐affinity state or from other factors (see Section 5.2.2).
For dopamine D1/5 receptors, an in vivo displacement curve was built in baboons using the D1‐antagonist [11C]NNC‐112 and the D1‐agonist drug DAR‐0100A.198 Occupancies above 40% were not investigated, but the best‐fit curve was monophasic, not supporting the existence of a high‐affinity receptor subpopulation.
5.3.2. Serotonin receptors
A few head‐to‐head agonist‐antagonist comparisons done with serotonin receptor radioligands yielded ambiguous results.
Two studies found that about 80‐90% of the specific binding of antagonist 5‐HT1A ligand [11C]WAY‐100635 could be displaced by the 5‐HT1A agonist 8‐OH‐DPAT,226, 227 but in both cases only a single dose of the agonist was tried, so it is impossible to say whether 8‐OH‐DPAT displaces [11C]WAY‐100635 in a monophasic or a biphasic manner. In an ex vivo saturation study, the binding site density of 5‐HT1A agonist [3H]CUMI‐101 was 33% lower than that of the antagonist [3H]MPPF in rat frontal cortex, but 82% higher in rat hippocampus.122 BPF values for [11C]CUMI‐101 across the baboon brain were about 45% of BPF values for 5‐HT1A antagonist [11C]WAY‐100635,70 while in vitro K d values for the two tracers were comparable, but these findings should be interpreted with caution for reasons described in the subsection “Extracting density values from true binding potential measurements” of Section 5.2.1. Yet another study found considerable differences between the binding patterns of 5‐HT1A agonist [18F]F13174 and 5‐HT1A antagonist [18F]MPPF in marmosets: regional BPND values even did not correlate when animals were imaged without the use of anesthesia.132 However, the variability of data (especially for [18F]F13174) was high, so it is hard to draw any firm conclusions regarding the existence of separate 5‐HT1A receptor subpopulations recognized by [18F]F13174 and [18F]MPPF from the study.
Regional BPND values of the 5‐HT2A agonist [11C]CIMBI‐36 were compared with regional BPND values of the 5‐HT2A antagonist [11C]MDL100907 in monkeys 137 and regional BPND values of another 5‐HT2A antagonist [18F]altanserin in humans.139 Both analyses did yield deviations from linear correlation in the hippocampus and choroid plexus, but these could all be explained by the binding of [11C]CIMBI‐36 to 5‐HT2C receptors. In a recent non‐human primate study, [11C]CIMBI‐36 turned out to be more sensitive than [11C]MDL100907 to fenfluramine‐induced serotonin release,142 but, as discussed above for D2/3 radioligands, this does not per se prove that [11C]CIMBI‐36 preferentially binds to the high‐affinity state of 5‐HT2A receptors in vivo.
To sum up, the majority of data, both for dopamine and serotonin receptors, does not directly support the existence of a receptor subpopulation in vivo to which agonists preferentially bind. There are some undisputable differences in the behavior of agonist tracers and antagonist tracers, such as specific sensitivity of the former to anesthesia and greater sensitivity to synaptic neurotransmitter levels, but the reason for these differences is not clear.
5.4. Experimental data in light of the nature of the high‐affinity state
Some attempts to detect the high‐affinity state in vivo may have failed because the used radioligands were not sufficiently subtype‐selective or lacked sufficient intrinsic acitivity via the canonical GPCR pathway.118, 119, 228 In other cases, however, the reasons for failure were completely unclear. Therefore, the nature and functioning of the high‐affinity state in vivo has remained elusive.
Two explanations of the failure to detect high‐ and low‐affinity states in vivo have been put forward. One explanation proposes that all, or almost all, receptors are configured in the high‐affinity state in vivo 86 and permanently precoupled to G‐proteins, so that these G‐proteins, at least their Gα subunits, do not dissociate from the receptors after activation.215 Another explanation states that, whatever the baseline degree of receptor‐G‐protein precoupling, receptors can and do recruit new G‐proteins when occupied by agonists, so high agonist concentrations eventually make all receptors bind G‐proteins and thus convert to the high‐affinity state.196, 198 Indeed, receptors have been subjected to high agonist (drug or neurotransmitter) concentrations in virtually all experimental paradigms used for in vivo detection of the high‐affinity state (Table 2 ). In vivo imaging experiments last for tens of minutes, while time constants for receptor and G‐protein activation and for receptor‐ligand and receptor‐G‐protein binding vary from tens of milliseconds to a few seconds.37 Gradual G‐protein recruitment in response to agonist binding can thus confound experimental outcomes in currently used paradigms and can “inflate” the apparent relative abundance of the high‐affinity state in vivo. Indeed, in studies comparing the vulnerability of dopamine D2/3 agonist and antagonist tracers to drug challenge, the lowest doses of D2/3 agonist drugs and the dopamine release stimulator amphetamine (resulting in the lowest receptor occupancies and therefore minimal G‐protein recruitment) tended to produce the greatest relative difference between agonist and antagonist tracer displacement. In three such studies, the lowest dose of challenge drugs resulted in zero or negative displacement of the antagonist [11C]/[3H]raclopride but positive (though small and not statistically significant) displacement of agonist tracers [11C]/[3H]PHNO and [11C]/[3H]MNPA.86, 107, 196 Superior sensitivity of D2/3 agonists to amphetamine‐induced dopamine release in humans compared to the D2/3 antagonist [11C]raclopride was demonstrated at receptor occupancies of 22% or lower.93, 102 Finally, the D2/3 agonist [3H]PHNO was found to be more vulnerable than [3H]raclopride to displacement by the agonist (−)NPA when this agonist was co‐injected with the radioligands instead of being injected sometime before their administration, although this study used non‐normalized “raw” specific binding as outcome measure.200
Comparison of the in vivo imaging results with available in vitro data on detection of the high‐affinity state raises even more questions. There are no systematic reviews of in vitro studies assessing the percentage of receptors configured in the high‐affinity state for nondopaminergic receptors. However, recently published reviews dedicated to the affinity states of D2/3 receptors 216, 229 demonstrate not only the lack of agreement but also the absence of a trend in the assessments of the abundance of the high‐affinity state of D2/3 receptors in different setups:
-
(i)
Experiments in membrane homogenates from transfected cells and isolated tissues consistently showed that a significant portion of D2/3 receptors is configured in the low‐affinity state. Only about 20% of dopamine D2/3 receptors in the rodent brain and about 30% in the human brain are in the high‐affinity state.216
-
(ii)
No high‐affinity state of D2/3 receptors could be detected on intact cells. In bovine pituitary membranes, 55% of D2/3 receptors were in the high‐affinity state, but intact dispersed pituitary cells had only low‐affinity state.230 Similar results were demonstrated with transfected HEK293T and T‐REx‐293 cells expressing D2 receptors.28, 42 One study found 16% of D2 receptors on cultured rat pituitary adenoma cells configured in the high‐affinity state using [3H]domperidone as a radioligand,231 but these results have yet to be replicated.
-
(iii)
Autoradiographic studies where densities (Bmax) of D2/3 receptors in tissue slices were estimated, tended to produce lower Bmax values for agonist than for antagonist radioligands, but the variance between studies was too high to consider the difference significant.229 The majority of antagonist vs agonist drug displacement experiments demonstrated a single high‐affinity population of D2/3 receptors in tissue slices in vitro.229 This is in agreement with the data from in vivo imaging (see Section 5.3.1).
To explain the low relative abundance of the high‐affinity state in membrane homogenates within the precoupling model of receptor‐G‐protein interaction, one can assume that partial dissociation of receptor‐G‐protein complexes occurs during membrane preparation, but the exact mechanism is hard to define. Alternatively, one can assume the existence of a large intracellular reserve of receptors in the high‐affinity state. In transfected cells, the affinity of agonist ligands used as PET tracers toward internalized D2/3 receptors was shown to be about twofold lower than toward surface receptors,42 a change that would hardly be noticeable in saturation or competition curves. However, the total densities of D2/3 receptors measured in membrane homogenates by radioligand binding saturation and in vivo by PET are in good agreement,229 which does not support the existence of extra D2/3 receptors that are detectable in vivo but are not found in membrane homogenates.
The collision coupling model of receptor‐G‐protein interaction can be reconciled with measurements of the high‐affinity state in membrane homogenates if one assumes that in these homogenates only a fraction of the total G‐protein pool of the cell is available for recruitment to the receptors. Indeed, the number of G‐proteins in living cells is likely equal to or much greater than the number of their cognate receptors.45, 232, 233 However, given that G‐proteins are anchored to the lipid bilayer,234 it is not clear how such G‐protein reserve can both be easily accessible in the living tissue and become lost in membrane homogenates. Especially the findings of Seeman200 are puzzling in this regard. In that study, IC50 values for the displacement of [3H](+)PHNO and [3H]raclopride from striatal membranes by (−)NPA were measured. When radioligands and (−)NPA were added to the membranes simultaneously, the IC50 value observed for [3H](+)PHNO was sevenfold lower than for [3H]raclopride. However, when (−)NPA was added to the membranes 30 minutes earlier than the radioligands, the difference in IC50 values for [3H](+)PHNO and [3H]raclopride was only twofold. Similar vulnerabilities of [3H]raclopride and [3H](+)PHNO in the case of (−) NPA pre‐incubation could mean that (−)NPA can and does stimulate G‐protein recruitment to all or almost all receptors in the membranes, while greater vulnerability of [3H](+)PHNO in case of simultaneous addition may mean that in the absence of an agonist, only some receptors are G‐protein‐bound and therefore accessible to [3H](+)PHNO. However, in those experiments, radioligands were equilibrated with membrane homogenates for 2 hours before readout, which is much longer than the pre‐incubation step. Therefore, even in the case of simultaneous addition of radioligands and (−)NPA, there should have been enough time during the equilibration step for (−)NPA to elicit recruitment of “spare” G‐proteins to the receptors.
The absence of the high‐affinity state of D2/3 receptors in isolated intact cells 28,42,230 is even harder to reconcile with either of the two models mentioned above. Collision coupling at least provides a theoretical explanation of the disappearance of high‐affinity state upon agonist addition due to a high level of GTP in living cells. Still it remains puzzling why agonists force D2/3 receptors in dispersed cells from a natural tissue (bovine pituitary) to uncouple from G‐proteins (convert into the low‐affinity state), but promote G‐protein recruitment (conversion to the high‐affinity state) when they are acting on the same receptors in intact tissue.
Switching from the G‐protein‐dependent high‐affinity state model to the oligomerization‐dependent model leaves the same questions open: it is not clear how the degrees of receptor oligomerization can be different in membranes, dispersed cells and living tissues. Moreover, observation of almost all receptors configured in high‐affinity state in vivo is hard to reconcile with the oligomerization‐dependent model. At full oligomerization and full agonist occupancy, cooperativity‐induced high‐affinity state can have a relative abundance of no more than 50% (in the case of dimers), and higher values imply that few, if any, receptors are oligomerized.
6. CONCLUSION
The concept of the high‐affinity state postulates that a certain subset of receptors in the living brain is primarily responsible for signaling. Assessing the abundance of this subset is thus potentially very relevant for studies concerning the responses of neurotransmission to pharmacological or physical stimuli and the dysregulation of neurotransmission in neurological disorders.
A number of experimental paradigms have been developed for the estimation of the relative abundance of receptors configured in the high‐affinity state. The high‐affinity state is preferentially recognized by agonists in vitro, so the development of agonist PET tracers as tools for the noninvasive imaging of the high‐affinity state has become popular in recent decades.
The greatest number of agonist tracers has been developed for dopamine D2/3 receptors, but agonist tracers for dopamine D1, µ‐opioid, and muscarinic M2 receptors are also known, and in recent years, radiolabeled agonists for serotonin 5‐HT1A and 5‐HT2A, κ‐opioid and muscarinic M1 receptors have appeared. It should be noted, however, that for many of the nondopaminergic tracers the actual preference for the high‐affinity state has not been directly tested, because functional agonism is often assumed to imply preferential binding to the high‐affinity state.
For dopamine, serotonin and κ‐opioid receptors, head‐to‐head comparisons of agonist and antagonist tracers are now possible, while matching antagonist tracers for muscarinic M2 and µ‐opioid receptors have yet to be developed. Given that, beyond head‐to‐head agonist‐antagonist comparisons, antagonist tracers are also suitable for experiments like displacement curve generation (see Section 5.2.2), development of new antagonist tracers for muscarinic M2 and µ‐opioid receptors with a pharmacological selectivity matching that of existing agonist tracers will arguably be more useful for the assessment of the affinity states of these receptors than development of new agonist tracers.
Agonist tracers appear to be more sensitive to endogenous neurotransmitter challenge, as was originally expected. However, other expectations regarding agonist tracers have not been fulfilled. Agonist imaging did not reveal alterations in the relative abundance of the high‐affinity state in neurological disorders. The benefits of agonist tracers for the imaging of receptor occupancies by drugs have also not been proven.
Moreover, though the separation of GPCRs into subsets with high‐ and low‐affinity state is consistently observed in membrane homogenates in vitro, data from preclinical and clinical experiments do not support the existence of the high‐ and low‐affinity states in vivo. The majority of these data concerns dopamine D2/3 receptors but recent results on serotonin receptors paint the same picture.
The relative abundance of the high‐affinity state in vivo may simply be close to (or equal to) 100%, making the detection of low‐affinity state unfeasible. It is also possible that agonist drugs or tracers used for in vivo experiments may inflate the relative abundance of the high‐affinity state.
Critical revision of experimental approaches and collection of experimental evidence for nondopaminergic receptors will help clarify whether the high‐affinity state of GPCRs exists in vivo and whether agonist tracers really have advantages over antagonist tracers because of their preferential binding to the high‐affinity state.
ACKNOWLEDGMENTS
During the writing of this manuscript, VS was appointed as a PhD student at the Department of Nuclear Medicine and Molecular Imaging, University Medical Center Groningen. His PhD research was supported by a grant from the Dutch Technology Foundation (STW), No. 10127
Biographies
Vladimir Shalgunov performed PhD research at the University Medical Center Groningen (2009‐2013). His PhD project concerned the development of fluorine‐18‐labeled agonist radioligands for PET imaging of the high‐affinity state of dopamine D2/3 receptors. After returning to his native country, Russia, he worked as a research scientist in nanomedicine development and as a technician in radiopharmaceutical production (2014‐2017). In 2017, he was appointed as a postdoc at the Department of Drug Design and Pharmacology, University of Copenhagen (Denmark).
Aren van Waarde was trained as a biologist (animal physiology). He is a member of the permanent staff of the Department of Nuclear Medicine and Molecular Imaging at the University of Groningen, University Medical Center Groningen, The Netherlands. Since 1991, he is involved in the preclinical evaluation of novel radiopharmaceuticals, particularly ligands for receptors in the brain.
Jan Booij is Professor of Experimental Nuclear Medicine at the Amsterdam University Medical Centers, location Academic Medical Center, University of Amsterdam (The Netherlands). His research is mainly focused on the development and evaluation of novel radiopharmaceuticals for the dopaminergic system, which can be used to study the role of this system in neuropsychiatric disorders, including drug addiction as well as obesity.
Martin Michel is Professor of Pharmacology at the Johannes Gutenberg‐University in Mainz (Germany). His primary research interest is experimental and clinical receptor pharmacology in the autonomic nervous system.
Rudi Dierckx is Head of the Department of Nuclear Medicine and Molecular Imaging at University Medical Center Groningen in The Netherlands and Professor of Nuclear Medicine at the University Hospital in Ghent (Belgium). He is also Head of the Medical Imaging Center and Chairman ad interim of the Department of Radiology at UMCG. His primary research interests are PET studies of the human brain.
Philip Elsinga is Professor of Radiopharmaceutical Chemistry at the University of Groningen, President and Cofounder of the Dutch Society for Clinical Radiochemistry, member of the Board of Directors of the Society of Radiopharmaceutical Sciences, and Editor‐in‐Chief of EJNMMI Radiopharmacy and Chemistry.
APPENDIX A.
CALCULATION OF BINDING POTENTIALS
The ratio of the concentrations of specifically bound tracer (B) and free tracer (F) in the tissue at equilibrium is equal to the ratio of available receptor density and ligand‐receptor affinity (B max/K d) when the tracer only occupies a negligible fraction of all receptors (Figure A1).
Figure A1.
Relationship between the fraction of ligand‐occupied receptors and B/F ratio. At equilibrium, B/B max = F/(F + K d), therefore B/F = B max/(F + K d). If F ≪ K d, then B ≪ B max and B/F ~ B max/K d. Note that at low concentrations (F/K d ≪ 1), the ligand occupies only a small part of total receptor population, and the B/F ratio (binding potential) is stable (left part of the graph)
In practice, specifically bound tracer concentration B is often calculated as a difference between tracer concentrations in a receptor‐rich region of interest and a receptor‐poor reference region. Free tracer concentration, on the other hand, is replaced with directly measurable concentrations that are linearly dependent on “true” F at equilibrium.192 These concentrations can be:
-
–
Free tracer concentration in plasma (equal to F if the tracer is transferred from tissue to plasma by passive diffusion);
-
–
Total, that is, free and protein‐bound, tracer concentration in plasma;
-
–
Nondisplaceable, that is, free and nonspecifically bound, tracer concentration in tissue.
To determine tracer concentration in plasma (whether free or total) one needs to obtain arterial plasma samples, which is an invasive procedure. Therefore, the last option is the most popular: tracer concentration in receptor‐rich tissue is related to tracer concentration in receptor‐poor tissue.
Binding potentials are defined at equilibrium. Plasma and tissue concentrations of a tracer can be directly equilibrated with each other by administering the tracer in an infusion but such paradigms are hard to implement. Therefore, in most positron emission tomography (PET) and single‐photon emission computed tomography (SPECT) studies tracer is administered via a simple bolus injection. In that case, to calculate binding potentials, time‐activity curves for regions of interest are used as input data for so‐called kinetic modeling. A compartmental model is built up based on relevant theories. Tracer distribution is described in terms of its transfer between virtual compartments (like “free tracer,” “nonspecifically bound tracer,” and “specifically bound tracer”). The model is fitted to the time‐activity curves acquired by the PET camera. If the model can adequately describe the experimental curves, the values of individual kinetic parameters or their combinations are determined from the model fit. Fewer fitting parameters often mean a more robust fit, so many simplifying assumptions are often made. Graphical analyses, simplifying the fitting process to linear regression, are also widely used.235, 236 Fitted parameters are then used to obtain binding potentials, essentially by extrapolating the kinetics observed in the imaging experiments to the situation of equilibrium.
Shalgunov V, van Waarde A, Booij J, Michel MC, Dierckx RAJO, Elsinga PH. Hunting for the high‐affinity state of G‐protein‐coupled receptors with agonist tracers: Theoretical and practical considerations for positron emission tomography imaging. Med Res Rev. 2019;39:1014‐1052. 10.1002/med.21552
Present addressVladimir Shalgunov, Department of Drug Design and Pharmacology, University of Copenhagen, Copenhagen, Denmark.
References
REFERENCES
- 1. Overington JP, Al‐Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993‐996. [DOI] [PubMed] [Google Scholar]
- 2. Deupi X, Standfuss J. Structural insights into agonist‐induced activation of G‐protein‐coupled receptors. Curr Opin Struct Biol. 2011;21:541‐551. [DOI] [PubMed] [Google Scholar]
- 3. Sibley DR, De Lean A, Creese I. Anterior pituitary dopamine receptors. Demonstration of interconvertible high‐ and low‐affinity states of the D‐2 dopamine receptor. J Biol Chem. 1982;257:6351‐6361. [PubMed] [Google Scholar]
- 4. Leff SE, Hamblin MW, Creese I. Interactions of dopamine agonists with brain D1 receptors labeled by 3H‐antagonists. Evidence for the presence of high‐ and low‐affinity agonist‐binding states. Mol Pharmacol. 1985;27:171‐183. [PubMed] [Google Scholar]
- 5. Battaglia G, Shannon M, Titeler M. Guanyl nucleotide and divalent cation regulation of cortical S2 serotonin receptors. J Neurochem. 1984;43:1213‐1219. [DOI] [PubMed] [Google Scholar]
- 6. Gozlan H, Thibault S, Laporte AM, Lima L, Hamon M. The selective 5‐HT1A antagonist radioligand [3H]WAY 100635 labels both G‐protein‐coupled and free 5‐HT1A receptors in rat brain membranes. Eur J Pharmacol. 1995;288:173‐186. [DOI] [PubMed] [Google Scholar]
- 7. Watson J, Collin L, Ho M, et al. 5‐HT(1A) receptor agonist‐antagonist binding affinity difference as a measure of intrinsic activity in recombinant and native tissue systems. Br J Pharmacol. 2000;130:1108‐1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ehlert FJ. The relationship between muscarinic receptor occupancy and adenylate cyclase inhibition in the rabbit myocardium. Mol Pharmacol. 1985;28:410‐421. [PubMed] [Google Scholar]
- 9. Law PY, Hom DS, Loh HH. Multiple affinity states of opiate receptor in neuroblastoma x glioma NG108‐15 hybrid cells. Opiate agonist association rate is a function of receptor occupancy. J Biol Chem. 1985;260:3561‐3569. [PubMed] [Google Scholar]
- 10. Rasmussen SGF, Devree BT, Zou Y, et al. Crystal structure of the beta2 adrenergic receptor‐Gs protein complex. Nature. 2011;477:549‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Werling LL, McMahon PN, Cox BM. Selective changes in mu opioid receptor properties induced by chronic morphine exposure. Proc Natl Acad Sci USA. 1989;86:6393‐6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Flynn DD, Weinstein DA, Mash DC. Loss of high‐affinity agonist binding to M1 muscarinic receptors in Alzheimer's disease: implications for the failure of cholinergic replacement therapies. Ann Neurol. 1991;29:256‐262. [DOI] [PubMed] [Google Scholar]
- 13. Ladner CJ, Lee JM. Reduced high‐affinity agonist binding at the M(1) muscarinic receptor in Alzheimer's disease brain: differential sensitivity to agonists and divalent cations. Exp Neurol. 1999;158:451‐458. [DOI] [PubMed] [Google Scholar]
- 14. Seeman P, Schwarz J, Chen JF, et al. Psychosis pathways converge via D2High dopamine receptors. Synapse. 2006;60:319‐346. [DOI] [PubMed] [Google Scholar]
- 15. Seeman P. All roads to schizophrenia lead to dopamine supersensitivity and elevated dopamine D2(high) receptors. CNS Neurosci Ther. 2011;17:118‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist‐specific binding properties of the adenylate cyclase‐coupled beta‐adrenergic receptor. J Biol Chem. 1980;255:7108‐7117. [PubMed] [Google Scholar]
- 17. Kent RS, De Lean A, Lefkowitz RJ. A quantitative analysis of beta‐adrenergic receptor interactions: resolution of high‐ and low‐affinity states of the receptor by computer modeling of ligand binding data. Mol Pharmacol. 1980;17:14‐23. [PubMed] [Google Scholar]
- 18. Lahti RA, Figur LM, Piercey MF, Ruppel PL, Evans DL. Intrinsic activity determinations at the dopamine D2 guanine nucleotide‐binding protein‐coupled receptor: utilization of receptor state binding affinities. Mol Pharmacol. 1992;42:432‐438. [PubMed] [Google Scholar]
- 19. Fitzgerald LW, Conklin DS, Krause CM, et al. High‐affinity agonist binding correlates with efficacy (intrinsic activity) at the human serotonin 5‐HT2A and 5‐HT2C receptors: evidence favoring the ternary complex and two‐state models of agonist action. J Neurochem. 1999;72:2127‐2134. [DOI] [PubMed] [Google Scholar]
- 20. Fisher A, Heldman E, Gurwitz D, et al. New M1 agonists: selective signaling, neurotrophic‐like and cognitive effects—Implications in the treatment of Alzheimer's disease In: Hanin I, Yoshida M, Fisher A, eds. Alzheimer's and Parkinson's Diseases: Recent Developments. Boston, MA: Springer; 1995:pp. 449‐455. [Google Scholar]
- 21. Christopoulos A, Kenakin T. G‐protein‐coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323‐374. [DOI] [PubMed] [Google Scholar]
- 22. Stein RSL, Ehlert FJ. A kinetic model of GPCRs: analysis of G protein activity, occupancy, coupling and receptor‐state affinity constants. J Recept Signal Transduct Res. 2015;35:269‐283. [DOI] [PubMed] [Google Scholar]
- 23. Rosenbaum DM, Zhang C, Lyons JA, et al. Structure and function of an irreversible agonist‐beta(2) adrenoceptor complex. Nature. 2011;469:236‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bimbaumer L, Bimbaumer M. Signal transduction by G proteins: 1994 edition. J Recept Signal Transduct Res. 1995;15:213‐252. [DOI] [PubMed] [Google Scholar]
- 25. Khelashvili G, Dorff K, Shan J, et al. GPCR‐OKB: the G‐protein‐coupled receptor oligomer knowledge base. Bioinformatics. 2010;26:1804‐1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ferre S, Casado V, Devi LA, et al. G‐protein‐coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev. 2014;66:413‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Casadó V, Cortés A, Ciruela F, et al. Old and new ways to calculate the affinity of agonists and antagonists interacting with G‐protein‐coupled monomeric and dimeric receptors: the receptor‐dimer cooperativity index. Pharmacol Ther. 2007;116:343‐354. [DOI] [PubMed] [Google Scholar]
- 28. Skinbjerg M, Namkung Y, Halldin C, Innis RB, Sibley DR. Pharmacological characterization of 2‐methoxy‐N‐propylnorapomorphine's interactions with D2 and D3 dopamine receptors. Synapse. 2009;63:462‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. López‐Giménez JF, Villazón M, Brea J, et al. Multiple conformations of native and recombinant human 5‐hydroxytryptamine(2a) receptors are labeled by agonists and discriminated by antagonists. Mol Pharmacol. 2001;60:690‐699. [PubMed] [Google Scholar]
- 30. Orru M, Bakešová J, Brugarolas M, et al. Striatal pre‐ and post‐synaptic profile of adenosine A(2A) receptor antagonists. PLOS One. 2011;6:e16088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chidiac P, Green MA, Pawagi AB, Wells JW. Cardiac muscarinic receptors. Cooperativity as the basis for multiple states of affinity. Biochemistry. 1997;36:7361‐7379. [DOI] [PubMed] [Google Scholar]
- 32. Grant M, Collier B, Kumar U. Agonist‐dependent dissociation of human somatostatin receptor 2 dimers: a role in receptor trafficking. J Biol Chem. 2004;279:36179‐36183. [DOI] [PubMed] [Google Scholar]
- 33. Patel RC, Kumar U, Lamb DC, et al. Ligand binding to somatostatin receptors induces receptor‐specific oligomer formation in live cells. Proc Natl Acad Sci USA. 2002;99:3294‐3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cottet M, Faklaris O, Maurel D, et al. BRET and time‐resolved FRET strategy to study GPCR oligomerization: from cell lines toward native tissues. Front Endocrinol (Lausanne). 2012;3:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scarselli M, Annibale P, McCormick PJ, et al. Revealing G‐protein‐coupled receptor oligomerization at the single‐molecule level through a nanoscopic lens: methods, dynamics and biological function. FEBS J. 2016;283:1197‐1217. [DOI] [PubMed] [Google Scholar]
- 36. Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G‐protein‐coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107‐144. [DOI] [PubMed] [Google Scholar]
- 37. Lohse MJ, Nuber S, Hoffmann C. Fluorescence/bioluminescence resonance energy transfer techniques to study G‐protein‐coupled receptor activation and signaling. Pharmacol Rev. 2012;64:299‐336. [DOI] [PubMed] [Google Scholar]
- 38. Krasel C, Vilardaga JP, Bünemann M, Lohse MJ. Kinetics of G‐protein‐coupled receptor signalling and desensitization. Biochem Soc Trans. 2004;32:1029‐1031. [DOI] [PubMed] [Google Scholar]
- 39. Sander CY, Hooker JM, Catana C, Rosen BR, Mandeville JB. Imaging agonist‐induced D2/D3 receptor desensitization and internalization in vivo with PET/fMRI. Neuropsychopharmacology. 2016;41:1427‐1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ko F, Seeman P, Sun WS, Kapur S. Dopamine D2 receptors internalize in their low‐affinity state. Neuroreport. 2002;13:1017‐1020. [DOI] [PubMed] [Google Scholar]
- 41. Sun W, Ginovart N, Ko F, Seeman P, Kapur S. In vivo evidence for dopamine‐mediated internalization of D2‐receptors after amphetamine: differential findings with [3H]raclopride versus [3H]spiperone. Mol Pharmacol. 2003;63:456‐462. [DOI] [PubMed] [Google Scholar]
- 42. Guo N, Guo W, Kralikova M, et al. Impact of D2 receptor internalization on binding affinity of neuroimaging radiotracers. Neuropsychopharmacology. 2010;35:806‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Quelch DR, Katsouri L, Nutt DJ, Parker CA, Tyacke RJ. Imaging endogenous opioid peptide release with [11C]carfentanil and [3H]diprenorphine: influence of agonist‐induced internalization. J Cereb Blood Flow Metab. 2014;34:1604‐1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Skinbjerg M, Liow JS, Seneca N, et al. D2 dopamine receptor internalization prolongs the decrease of radioligand binding after amphetamine: a PET study in a receptor internalization‐deficient mouse model. Neuroimage. 2010;50:1402‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hein P, Bünemann M. Coupling mode of receptors and G proteins. Naunyn Schmiedebergs Arch Pharmacol. 2009;379:435‐443. [DOI] [PubMed] [Google Scholar]
- 46. Oldham WM, Hamm HE. Heterotrimeric G protein activation by G‐protein‐coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60‐71. [DOI] [PubMed] [Google Scholar]
- 47. Tolkovsky AM, Levitzki A. Mode of coupling between the beta‐adrenergic receptor and adenylate cyclase in turkey erythrocytes. Biochemistry. 1978;17:3795‐3810. [DOI] [PubMed] [Google Scholar]
- 48. Bunemann M, Frank M, Lohse MJ. Gi protein activation in intact cells involves subunit rearrangement rather than dissociation. Proc Natl Acad Sci USA. 2003;100:16077‐16082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Galés C, Van Durm JJJ, Schaak S, et al. Probing the activation‐promoted structural rearrangements in preassembled receptor‐G protein complexes. Nat Struct Mol Biol. 2006;13:778‐786. [DOI] [PubMed] [Google Scholar]
- 50. Audet N, Galés C, Archer‐Lahlou É, et al. Bioluminescence resonance energy transfer assays reveal ligand‐specific conformational changes within preformed signaling complexes containing delta‐opioid receptors and heterotrimeric G proteins. J Biol Chem. 2008;283:15078‐15088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Finnema SJ, Scheinin M, Shahid M, et al. Application of cross‐species PET imaging to assess neurotransmitter release in brain. Psychopharmacology (Berl). 2015;232:4129‐4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Paterson LM, Tyacke RJ, Nutt DJ, Knudsen GM. Measuring endogenous 5‐HT release by emission tomography: promises and pitfalls. J Cereb Blood Flow Metab. 2010;30:1682‐1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Becker G, Streichenberger N, Billard T, Newman‐Tancredi A, Zimmer L. A postmortem study to compare agonist and antagonist 5‐HT1A receptor‐binding sites in Alzheimer's disease. CNS Neurosci Ther. 2014;20:930‐934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vidal B, Sebti J, Verdurand M, et al. Agonist and antagonist bind differently to 5‐HT1A receptors during Alzheimer's disease: a post‐mortem study with PET radiopharmaceuticals. Neuropharmacology. 2016;109:88‐95. [DOI] [PubMed] [Google Scholar]
- 55. Palner M, Kjaerby C, Knudsen GM, Cumming P. Effects of unilateral 6‐OHDA lesions on [3H]‐N‐propylnorapomorphine binding in striatum ex vivo and vulnerability to amphetamine‐evoked dopamine release in rat. Neurochem Int. 2011;58:243‐247. [DOI] [PubMed] [Google Scholar]
- 56. Seeman P. Antiparkinson therapeutic potencies correlate with their affinities at dopamine D2(High) receptors. Synapse. 2007;61:1013‐1018. [DOI] [PubMed] [Google Scholar]
- 57. Heidrich A, Rösler M. Milameline: nonselective, partial muscarinic receptor agonist for the treatment of Alzheimer's disease? CNS Drug Rev. 2006;5:93‐104. [Google Scholar]
- 58. Bruins Slot LA, Kleven MS, Newman‐Tancredi A. Effects of novel antipsychotics with mixed D(2) antagonist/5‐HT(1A) agonist properties on PCP‐induced social interaction deficits in the rat. Neuropharmacology. 2005;49:996‐1006. [DOI] [PubMed] [Google Scholar]
- 59. Newman‐Tancredi A, Gavaudan S, Conte C, et al. Agonist and antagonist actions of antipsychotic agents at 5‐HT1A receptors: a [35S]GTPgammaS binding study. Eur J Pharmacol. 1998;355:245‐256. [DOI] [PubMed] [Google Scholar]
- 60. Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Toward a muscarinic hypothesis of schizophrenia. Mol Psychiatry. 2007;12:232‐246. [DOI] [PubMed] [Google Scholar]
- 61. Lever J. PET and SPECT imaging of the opioid system: receptors, radioligands and avenues for drug discovery and development. Curr Pharm Des. 2007;13:33‐49. [DOI] [PubMed] [Google Scholar]
- 62. Melichar JK, Hume SP, Williams TM, et al. Using [11C]diprenorphine to image opioid receptor occupancy by methadone in opioid addiction: clinical and preclinical studies. J Pharmacol Exp Ther. 2005;312:309‐315. [DOI] [PubMed] [Google Scholar]
- 63. Hume SP, Lingford‐Hughes AR, Nataf V, et al. Low sensitivity of the positron emission tomography ligand [11C]diprenorphine to agonist opiates. J Pharmacol Exp Ther. 2007;322:661‐667. [DOI] [PubMed] [Google Scholar]
- 64. Kodaka F, Ito H, Takano H, et al. Effect of risperidone on high‐affinity state of dopamine D2 receptors: a PET study with agonist ligand [11C](R)‐2‐CH3O‐N‐n‐propylnorapomorphine. Int J Neuropsychopharmacol. 2011;14:83‐89. [DOI] [PubMed] [Google Scholar]
- 65. Seneca N, Finnema SJ, Laszlovszky I, et al. Occupancy of dopamine D(2) and D(3) and serotonin 5‐HT(1)A receptors by the novel antipsychotic drug candidate, cariprazine (RGH‐188), in monkey brain measured using positron emission tomography. Psychopharmacology (Berl). 2011;218:579‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yabaluri N, Medzihradsky F. Reversible modulation of opioid receptor binding in intact neural cells by endogenous guanosine triphosphate. Mol Pharmacol. 1995;48:690‐695. [PubMed] [Google Scholar]
- 67. Zubieta JK, Smith YR, Bueller JA, et al. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293:311‐315. [DOI] [PubMed] [Google Scholar]
- 68. Talbot PS, Narendran R, Butelman ER, et al. 11C‐GR103545, a radiotracer for imaging kappa‐opioid receptors in vivo with PET: synthesis and evaluation in baboons. J Nucl Med. 2005;46:484‐494. [PubMed] [Google Scholar]
- 69. Tomasi G, Nabulsi N, Zheng MQ, et al. Determination of in vivo B max and K d for 11C‐GR103545, an agonist PET tracer for kappa‐opioid receptors: a study in non‐human primates. J Nucl Med. 2013;54:600‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kumar JSD, Milak MS, Majo VJ, et al. Comparison of high‐and low‐affinity serotonin 1A receptors by PET in vivo in non‐human primates. J Pharmacol Sci. 2012;120:254‐257. [DOI] [PubMed] [Google Scholar]
- 71. Hines CS, Liow JS, Zanotti‐Fregonara P, et al. Human biodistribution and dosimetry of (1)(1)C‐CUMI‐101, an agonist radioligand for serotonin‐1a receptors in brain. PLOS One. 2011;6:e25309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wilson AA, McCormick P, Kapur S, et al. Radiosynthesis and evaluation of [11C]‐(+)‐4‐propyl‐3,4,4a,5,6,10b‐hexahydro‐2H‐naphtho[1,2‐b][1,4]oxazin‐9‐ol as a potential radiotracer for in vivo imaging of the dopamine D2 high‐affinity state with positron emission tomography. J Med Chem. 2005;48:4153‐4160. [DOI] [PubMed] [Google Scholar]
- 73. Mizrahi R, Houle S, Vitcu I, Ng A, Wilson AA. Side effects profile in humans of (11)C‐(+)‐PHNO, a dopamine D(2/3) agonist ligand for PET. J Nucl Med. 2010;51:496‐497. [DOI] [PubMed] [Google Scholar]
- 74. Searle GE, Beaver JD, Tziortzi A, et al. Mathematical modelling of [11C]‐(+)‐PHNO human competition studies. Neuroimage. 2013;68:119‐132. [DOI] [PubMed] [Google Scholar]
- 75. Finnema SJ, Bang‐Andersen B, V. wikstrom H, Halldin C. Current state of agonist radioligands for imaging of brain dopamine D2/D3 receptors in vivo with positron emission tomography. Curr Top Med Chem. 2010;10:1477‐1498. [DOI] [PubMed] [Google Scholar]
- 76. Neumeyer JL, Baindur N, Niznik HB, Guan HC, Seeman P. (+/‐)‐3‐Allyl‐6‐bromo‐7,8‐dihydroxy‐1‐phenyl‐2,3,4,5‐tetrahydro‐1H‐3‐ benzazepin, a new high‐affinity D1 dopamine receptor ligand: synthesis and structure‐activity relationship. J Med Chem. 1991;34:3366‐3371. [DOI] [PubMed] [Google Scholar]
- 77. Palner M, McCormick P, Parkes J, Knudsen GM, Wilson AA. Systemic catechol‐O‐methyl transferase inhibition enables the D1 agonist radiotracer R‐[11C]SKF 82957. Nucl Med Biol. 2010;37:837‐843. [DOI] [PubMed] [Google Scholar]
- 78. DaSilva JN, Wilson AA, Valante CV, Hussey D, Wilson D, Houle S. In vivo binding of [11C]SKF 75670 and [11C]SKF 82957 in rat brain: two dopamine D‐1 receptor agonist ligands. Life Sci. 1996;58:1661‐1670. [DOI] [PubMed] [Google Scholar]
- 79. DaSilva JN, Schwartz RA, Greenwald ER, Lourenco CM, Wilson AA, Houle S. Dopamine D1 agonist R‐[11C]SKF 82957: synthesis and in vivo characterization in rats. Nucl Med Biol. 1999;26:537‐542. [DOI] [PubMed] [Google Scholar]
- 80. Finnema SJ, Bang‐Andersen B, Jørgensen M, et al. The dopamine D(1) receptor agonist (S)‐[(1)(1)C]N‐methyl‐NNC 01‐0259 is not sensitive to changes in dopamine concentration—a positron emission tomography examination in the monkey brain. Synapse. 2013;67:586‐595. [DOI] [PubMed] [Google Scholar]
- 81. Foged C, Halldin C, Lundkvist C, et al. Preparation of the dopamine D1 agonists [11C]SKF92857 and [11C]N‐methyl‐NNC‐01‐259 and PET stiudies in monkeys. J Label Compd Radiopharm. 1997;40:571‐573. [Google Scholar]
- 82. Finnema S, Bang‐Andersen B, Farde L, et al. PET evaluation of the partial dopamine D1 receptor agonist radioligands R‐ and S‐[11C]N‐methyl‐NNC01‐0259. Neuroimage. 2006;31:T114. [Google Scholar]
- 83. Finnema SJ, Bang‐Andersen B, Jørgensen M, et al. COMT inhibition prevents the formation of lipophilic radiometabolites of catechols—An example with (S)‐[11C]N‐methyl‐NNC01‐0259. Neuroimage. 2008;41:T91. [Google Scholar]
- 84. Seeman P, Ulpian C, Larsen RD, Anderson PS. Dopamine receptors labelled by PHNO. Synapse. 1993;14:254‐262. [DOI] [PubMed] [Google Scholar]
- 85. Seeman P, Ko F, Willeit M, McCormick P, Ginovart N. Antiparkinson concentrations of pramipexole and PHNO occupy dopamine D2(high) and D3(high) receptors. Synapse. 2005;58:122‐128. [DOI] [PubMed] [Google Scholar]
- 86. Peng T, Zysk J, Dorff P, et al. D2 receptor occupancy in conscious rat brain is not significantly distinguished with [3H]‐MNPA, [3H]‐(+)‐PHNO, and [3H]‐raclopride. Synapse. 2010;64:624‐633. [DOI] [PubMed] [Google Scholar]
- 87. Galineau L, Wilson AA, Garcia A, Houle S, Kapur S, Ginovart N. In vivo characterization of the pharmacokinetics and pharmacological properties of [11C]‐(+)‐PHNO in rats using an intracerebral beta‐sensitive system. Synapse. 2006;60:172‐183. [DOI] [PubMed] [Google Scholar]
- 88. Egerton A, Hirani E, Ahmad R, et al. Further evaluation of the carbon11‐labeled D2/3 agonist PET radiotracer PHNO: reproducibility in tracer characteristics and characterization of extrastriatal binding. Synapse. 2010;64:301‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Narendran R, Slifstein M, Guillin O, et al. Dopamine (D2/3) receptor agonist positron emission tomography radiotracer [11C]‐(+)‐PHNO is a D3 receptor preferring agonist in vivo. Synapse. 2006;60:485‐495. [DOI] [PubMed] [Google Scholar]
- 90. Ginovart N, Galineau L, Willeit M, et al. Binding characteristics and sensitivity to endogenous dopamine of [11C]‐(+)‐PHNO, a new agonist radiotracer for imaging the high‐affinity state of D2 receptors in vivo using positron emission tomography. J Neurochem. 2006;97:1089‐1103. [DOI] [PubMed] [Google Scholar]
- 91. Willeit M, Ginovart N, Kapur S, et al. High‐affinity states of human brain dopamine D2/3 receptors imaged by the agonist [11C]‐(+)‐PHNO. Biol Psychiatry. 2006;59:389‐394. [DOI] [PubMed] [Google Scholar]
- 92. Gallezot JD, Kloczynski T, Weinzimmer D, et al. Imaging nicotine‐ and amphetamine‐induced dopamine release in rhesus monkeys with [(11)C]PHNO vs [(11)C]raclopride PET. Neuropsychopharmacology. 2014;39:866‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shotbolt P, Tziortzi AC, Searle GE, et al. Within‐subject comparison of [(11)C]‐(+)‐PHNO and [(11)C]raclopride sensitivity to acute amphetamine challenge in healthy humans. J Cereb Blood Flow Metab. 2012;32:127‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Graff‐Guerrero A, Willeit M, Ginovart N, et al. Brain region binding of the D2/3 agonist [11C]‐(+)‐PHNO and the D2/3 antagonist [11C]raclopride in healthy humans. Hum Brain Mapp. 2008;29:400‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Boileau I, Nakajima S, Payer D. Imaging the D3 dopamine receptor across behavioral and drug addictions: Positron emission tomography studies with [11C]‐(+)‐PHNO. Eur Neuropsychopharmacol. 2015;25:1410‐1420. [DOI] [PubMed] [Google Scholar]
- 96. Vasdev N, Seeman P, Garcia A, et al. Syntheses and in vitro evaluation of fluorinated naphthoxazines as dopamine D2/D3 receptor agonists: radiosynthesis, ex vivo biodistribution and autoradiography of [(18)F]F‐PHNO. Nucl Med Biol. 2007;34:195‐203. [DOI] [PubMed] [Google Scholar]
- 97. Hwang DR, Kegeles LS, Laruelle M. (−)‐N‐[(11)C]propyl‐norapomorphine: a positron‐labeled dopamine agonist for PET imaging of D(2) receptors. Nucl Med Biol. 2000;27:533‐539. [DOI] [PubMed] [Google Scholar]
- 98. Narendran R, Hwang DR, Slifstein M, et al. In vivo vulnerability to competition by endogenous dopamine: comparison of the D2 receptor agonist radiotracer (−)‐N‐[11C]propyl‐norapomorphine ([11C]NPA) with the D2 receptor antagonist radiotracer [11C]‐raclopride. Synapse. 2004;52:188‐208. [DOI] [PubMed] [Google Scholar]
- 99. Narendran R, Slifstein M, Hwang DR, et al. Amphetamine‐induced dopamine release: duration of action as assessed with the D2/3 receptor agonist radiotracer (−)‐N‐[(11)C]propyl‐norapomorphine ([11C]NPA) in an anesthetized non‐human primate. Synapse. 2007;61:106‐109. [DOI] [PubMed] [Google Scholar]
- 100. Cumming P, Gillings NM, Jensen SB, Bjarkam C, Gjedde A. Kinetics of the uptake and distribution of the dopamine D(2,3) agonist (R)‐N‐[1‐(11)C]n‐propylnorapomorphine in brain of healthy and MPTP‐treated Gottingen miniature pigs. Nucl Med Biol. 2003;30:547‐553. [DOI] [PubMed] [Google Scholar]
- 101. Narendran R, Frankle WG, Mason NS, et al. Positron emission tomography imaging of D(2/3) agonist binding in healthy human subjects with the radiotracer [(11)C]‐N‐propyl‐norapomorphine: preliminary evaluation and reproducibility studies. Synapse. 2009;63:574‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Narendran R, Mason NS, Laymon CM, et al. A comparative evaluation of the dopamine D(2/3) agonist radiotracer [11C](−)‐N‐propyl‐norapomorphine and antagonist [11C]raclopride to measure amphetamine‐induced dopamine release in the human striatum. J Pharmacol Exp Ther. 2010;333:533‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Skinbjerg M, Seneca N, Liow JS, et al. Dopamine beta‐hydroxylase‐deficient mice have normal densities of D(2) dopamine receptors in the high‐affinity state based on in vivo PET imaging and in vitro radioligand binding. Synapse. 2010;64:699‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Seneca N, Zoghbi SS, Skinbjerg M, et al. Occupancy of dopamine D2/3 receptors in rat brain by endogenous dopamine measured with the agonist positron emission tomography radioligand [11C]MNPA. Synapse. 2008;62:756‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Finnema SJ, Seneca N, Farde L, et al. A preliminary PET evaluation of the new dopamine D2 receptor agonist [11C]MNPA in cynomolgus monkey. Nucl Med Biol. 2005;32:353‐360. [DOI] [PubMed] [Google Scholar]
- 106. Seneca N, Finnema SJ, Farde L, et al. Effect of amphetamine on dopamine D2 receptor binding in non‐human primate brain: a comparison of the agonist radioligand [11C]MNPA and antagonist [11C]raclopride. Synapse. 2006;59:260‐269. [DOI] [PubMed] [Google Scholar]
- 107. Finnema SJ, Halldin C, Bang‐Andersen B, et al. Dopamine D(2/3) receptor occupancy of apomorphine in the non‐human primate brain—a comparative PET study with [11C]raclopride and [11C]MNPA. Synapse. 2009;63:378‐389. [DOI] [PubMed] [Google Scholar]
- 108. Seneca N, Skinbjerg M, Zoghbi SS, et al. Kinetic brain analysis and whole‐body imaging in monkey of [11C]MNPA: a dopamine agonist radioligand. Synapse. 2008;62:700‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ohba H, Harada N, Nishiyama S, Kakiuchi T, Tsukada H. Ketamine/xylazine anesthesia alters [11C]MNPA binding to dopamine D2 receptors and response to methamphetamine challenge in monkey brain. Synapse. 2009;63:534‐537. [DOI] [PubMed] [Google Scholar]
- 110. Tsukada H, Ohba H, Nishiyama S, Kakiuchi T. Differential effects of stress on [(1)(1)C]raclopride and [(1)(1)C]MNPA binding to striatal D(2)/D(3) dopamine receptors: a PET study in conscious monkeys. Synapse. 2011;65:84‐89. [DOI] [PubMed] [Google Scholar]
- 111. Otsuka T, Ito H, Halldin C, et al. Quantitative PET analysis of the dopamine D2 receptor agonist radioligand 11C‐(R)‐2‐CH3O‐N‐n‐propylnorapomorphine in the human brain. J Nucl Med. 2009;50:703‐710. [DOI] [PubMed] [Google Scholar]
- 112. Sromek AW, Si YG, Zhang T, George SR, Seeman P, Neumeyer JL. Synthesis and evaluation of fluorinated aporphines: potential positron emission tomography ligands for D2 receptors. ACS Med Chem Lett. 2011;2:189‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Finnema SJ, Stepanov V, Nakao R, et al. (18)F‐MCL‐524, an (18)F‐labeled dopamine D2 and D3 receptor agonist sensitive to dopamine: a preliminary PET study. J Nucl Med. 2014;55:1164‐1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Xu J, Vangveravong S, Li S, et al. Positron emission tomography imaging of dopamine D2 receptors using a highly selective radiolabeled D2 receptor partial agonist. Neuroimage. 2013;71:168‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Mach RH, Tu Z, Xu J, et al. Endogenous dopamine (DA) competes with the binding of a radiolabeled D(3) receptor partial agonist in vivo: a positron emission tomography study. Synapse. 2011;65:724‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Rangel‐Barajas C, Malik M, Taylor M, Neve KA, Mach RH, Luedtke RR. Characterization of [(3)H]LS‐3‐134, a novel arylamide phenylpiperazine D3 dopamine receptor selective radioligand. J Neurochem. 2014;131:418‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Mukherjee J, Majji D, Kaur J, et al. PET radiotracer development for imaging high‐affinity state of dopamine D2 and D3 receptors: Binding studies of fluorine‐18 labeled aminotetralins in rodents. Synapse. 2017;71(3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Shrestha SS, Liow JS, Lu S, et al. (11)C‐CUMI‐101, a PET radioligand, behaves as a serotonin 1A receptor antagonist and also binds to alpha(1) adrenoceptors in brain. J Nucl Med. 2014;55:141‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Hendry N, Christie I, Rabiner EA, Laruelle M, Watson J. In vitro assessment of the agonist properties of the novel 5‐HT1A receptor ligand, CUMI‐101 (MMP), in rat brain tissue. Nucl Med Biol. 2011;38:273‐277. [DOI] [PubMed] [Google Scholar]
- 120. Kumar JSD, Prabhakaran J, Majo VJ, et al. Synthesis and in vivo evaluation of a novel 5‐HT1A receptor agonist radioligand [O‐methyl‐ 11C]2‐(4‐(4‐(2‐methoxyphenyl)piperazin‐1‐yl)butyl)‐4‐methyl‐1,2,4‐triazine‐3,5(2H,4H)dione in non‐human primates. Eur J Nucl Med Mol Imaging. 2007;34:1050‐1060. [DOI] [PubMed] [Google Scholar]
- 121. Milak MS, DeLorenzo C, Zanderigo F, et al. In vivo quantification of human serotonin 1A receptor using 11C‐CUMI‐101, an agonist PET radiotracer. J Nucl Med. 2010;51:1892‐1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Palner M, Underwood MD, Kumar DJS, et al. Ex vivo evaluation of the serotonin 1A receptor partial agonist [(3)H]CUMI‐101 in awake rats. Synapse. 2011;65:715‐723. [DOI] [PubMed] [Google Scholar]
- 123. Milak MS, Severance AJ, Prabhakaran J, et al. In vivo serotonin‐sensitive binding of [11C]CUMI‐101: a serotonin 1A receptor agonist positron emission tomography radiotracer. J Cereb Blood Flow Metab. 2011;31:243‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Pinborg LH, Feng L, Haahr ME, et al. No change in [(1)(1)C]CUMI‐101 binding to 5‐HT(1A) receptors after intravenous citalopram in human. Synapse. 2012;66:880‐884. [DOI] [PubMed] [Google Scholar]
- 125. Selvaraj S, Turkheimer F, Rosso L, et al. Measuring endogenous changes in serotonergic neurotransmission in humans: a [11C]CUMI‐101 PET challenge study. Mol Psychiatry. 2012;17:1254‐1260. [DOI] [PubMed] [Google Scholar]
- 126. Shrestha SS, Liow JS, Jenko K, Ikawa M, Zoghbi SS, Innis RB. The 5‐HT1A receptor PET radioligand 11C‐CUMI‐101 has significant binding to alpha1‐adrenoceptors in human cerebellum, limiting its use as a reference region. J Nucl Med. 2016;57:1945‐1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Majo VJ, Milak MS, Prabhakaran J, et al. Synthesis and in vivo evaluation of [(18)F]2‐(4‐(4‐(2‐(2‐fluoroethoxy)phenyl)piperazin‐1‐yl)butyl)‐4‐methyl‐1,2,4‐triazine‐3,5(2H,4H)‐dione ([(18)F]FECUMI‐101) as an imaging probe for 5‐HT1A receptor agonist in non‐human primates. Bioorg Med Chem. 2013;21:5598‐5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kumar JSD, Underwood MD, Simpson NR, et al. Autoradiographic evaluation of [(18)F]FECUMI‐101, a high affinity 5‐HT1AR ligand in human brain. ACS Med Chem Lett. 2016;7:482‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Koek W, Vacher B, Cosi C, et al. 5‐HT1A receptor activation and antidepressant‐like effects: F 13714 has high efficacy and marked antidepressant potential. Eur J Pharmacol. 2001;420:103‐112. [DOI] [PubMed] [Google Scholar]
- 130. Lemoine L, Becker G, Vacher B, et al. Radiosynthesis and preclinical evaluation of 18F‐F13714 as a fluorinated 5‐HT1A receptor agonist radioligand for PET neuroimaging. J Nucl Med. 2012;53:969‐976. [DOI] [PubMed] [Google Scholar]
- 131. Tavares A, Becker G, Barret O, et al. Initial evaluation of [18F]F13714, a novel 5‐HT1A receptor agonist in non‐human primates. Eur J Nucl Med Mol Imaging. 2013;40(Suppl.2):S56. [Google Scholar]
- 132. Yokoyama C, Mawatari A, Kawasaki A, et al. Marmoset serotonin 5‐HT1A receptor mapping with a biased agonist PET probe 18F‐F13714: comparison with an antagonist tracer 18F‐MPPF in awake and anesthetized states. Int J Neuropsychopharmacol. 2016;19:pyw079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Maurel JL, Autin JM, Funes P, Newman‐Tancredi A, Colpaert F, Vacher B. High‐efficacy 5‐HT1A agonists for antidepressant treatment: a renewed opportunity. J Med Chem. 2007;50:5024‐5033. [DOI] [PubMed] [Google Scholar]
- 134. Vidal B, Fieux S, Colom M, et al. (18)F‐F13640 preclinical evaluation in rodent, cat and primate as a 5‐HT1A receptor agonist for PET neuroimaging. Brain Struct Funct. 2018;223:2973‐2988. [DOI] [PubMed] [Google Scholar]
- 135. Ettrup A, Hansen M, Santini MA, et al. Radiosynthesis and in vivo evaluation of a series of substituted 11C‐phenethylamines as 5‐HT2A agonist PET tracers. Eur J Nucl Med Mol Imaging. 2011;38:681‐693. [DOI] [PubMed] [Google Scholar]
- 136. Ettrup A, Holm S, Hansen M, et al. Preclinical safety assessment of the 5‐HT2A receptor agonist PET radioligand [11C]Cimbi‐36. Mol Imaging Biol. 2013;15:376‐383. [DOI] [PubMed] [Google Scholar]
- 137. Finnema SJ, Stepanov V, Ettrup A, et al. Characterization of [(11)C]Cimbi‐36 as an agonist PET radioligand for the 5‐HT(2A) and 5‐HT(2C) receptors in the non‐human primate brain. Neuroimage. 2014;84:342‐353. [DOI] [PubMed] [Google Scholar]
- 138. Ettrup A, da Cunha‐Bang S, McMahon B, et al. Serotonin 2A receptor agonist binding in the human brain with [11C]Cimbi‐36. J Cereb Blood Flow Metab. 2014;34:1188‐1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Ettrup A, Svarer C, McMahon B, et al. Serotonin 2A receptor agonist binding in the human brain with [11C]Cimbi‐36: Test‐retest reproducibility and head‐to‐head comparison with the antagonist [18F]altanserin. Neuroimage. 2016;130:167‐174. [DOI] [PubMed] [Google Scholar]
- 140. Johansen A, Hansen HD, Svarer C, et al. The importance of small polar radiometabolites in molecular neuroimaging: A PET study with [(11)C]Cimbi‐36 labeled in two positions. J Cereb Blood Flow Metab. 2018;38:659‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Jørgensen LM, Weikop P, Villadsen J, et al. Cerebral 5‐HT release correlates with [(11)C]Cimbi36 PET measures of 5‐HT2A receptor occupancy in the pig brain. J Cereb Blood Flow Metab. 2017;37:425‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Yang KC, Stepanov V, Martinsson S, et al. Fenfluramine reduces [11C]Cimbi‐36 binding to the 5‐HT2A receptor in the non‐human primate brain. Int J Neuropsychopharmacol. 2017;20:683‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Ettrup A, McMahon B, Skibsted A, et al. Serotonin 2A receptor agonist binding with [11C]Cimbi‐36 in the human brain is unaltered by citalopram/pindolol and acute tryptophan depletion. Eur Neuropsychopharmacol. 2016;26:S307‐S308. [Google Scholar]
- 144. Herth MM, Petersen IN, Hansen HD, et al. Synthesis and evaluation of (18)F‐labeled 5‐HT2A receptor agonists as PET ligands. Nucl Med Biol. 2016;43:455‐462. [DOI] [PubMed] [Google Scholar]
- 145. Caudle RM, Mannes AJ, Iadarola MJ. GR89,696 is a kappa‐2 opioid receptor agonist and a kappa‐1 opioid receptor antagonist in the guinea pig hippocampus. J Pharmacol Exp Ther. 1997;283:1342‐1349. [PubMed] [Google Scholar]
- 146. Ravert HT, Mathews WB, Musachio JL, Scheffel U, Finley P, Dannals RF. [11C]‐methyl 4‐[(3,4‐dichlorophenyl)acetyl]‐3‐[(1‐pyrrolidinyl)‐methyl]‐1‐ piperazinecarboxylate ([11C]GR89696): synthesis and in vivo binding to kappa opiate receptors. Nucl Med Biol. 1999;26:737‐741. [DOI] [PubMed] [Google Scholar]
- 147. Ravert HT, Scheffel U, Mathews WB, Musachio JL, Dannals RF. [(11)C]‐GR89696, a potent kappa opiate receptor radioligand; in vivo binding of the R and S enantiomers. Nucl Med Biol. 2002;29:47‐53. [DOI] [PubMed] [Google Scholar]
- 148. Schoultz BW, Hjornevik T, Willoch F, et al. Evaluation of the kappa‐opioid receptor‐selective tracer [(11)C]GR103545 in awake rhesus macaques. Eur J Nucl Med Mol Imaging. 2010;37:1174‐1180. [DOI] [PubMed] [Google Scholar]
- 149. Tomasi G, Zheng MQ, Weinzimmer D, Lin S, Nabulsi N, Williams W. Kinetic modeling of the kappa agonist tracer [11C]GR103545 in humans. J Nucl Med. 2010;51:1293.20660376 [Google Scholar]
- 150. Naganawa M, Jacobsen LK, Zheng MQ, et al. Evaluation of the agonist PET radioligand [(1)(1)C]GR103545 to image kappa opioid receptor in humans: kinetic model selection, test‐retest reproducibility and receptor occupancy by the antagonist PF‐04455242. Neuroimage. 2014;99:69‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Leysen J, Tollenaere JP, Koch MHJ, Laduron P. Differentiation of opiate and neuroleptic receptor binding in rat brain. Eur J Pharmacol. 1977;43:253‐267. [DOI] [PubMed] [Google Scholar]
- 152. Saji H, Tsutsumi D, Magata Y, Iida Y, Konishi J, Yokoyama A. Preparation and biodistribution in mice of [11C]carfentanil: a radiopharmaceutical for studying brain mu‐opioid receptors by positron emission tomography. Ann Nucl Med. 1992;6:63‐67. [DOI] [PubMed] [Google Scholar]
- 153. Frost JJ, Wagner HN, Jr. , Dannals RF, et al. Imaging opiate receptors in the human brain by positron tomography. J Comput Assist Tomogr. 1985;9:231‐236. [DOI] [PubMed] [Google Scholar]
- 154. Henriksen G, Willoch F. Imaging of opioid receptors in the central nervous system. Brain. 2008;131:1171‐1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Colasanti A, Searle GE, Long CJ, et al. Endogenous opioid release in the human brain reward system induced by acute amphetamine administration. Biol Psychiatry. 2012;72:371‐377. [DOI] [PubMed] [Google Scholar]
- 156. Mick I, Myers J, Stokes PRA, et al. Amphetamine induced endogenous opioid release in the human brain detected with [(1)(1)C]carfentanil PET: replication in an independent cohort. Int J Neuropsychopharmacol. 2014;17:2069‐2074. [DOI] [PubMed] [Google Scholar]
- 157. Guterstam J, Jayaram‐Lindström N, Cervenka S, et al. Effects of amphetamine on the human brain opioid system—a positron emission tomography study. Int J Neuropsychopharmacol. 2013;16:763‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Henriksen G, Platzer S, Marton J, et al. Syntheses, biological evaluation, and molecular modeling of 18F‐labeled 4‐anilidopiperidines as mu‐opioid receptor imaging agents. J Med Chem. 2005;48:7720‐7732. [DOI] [PubMed] [Google Scholar]
- 159. Marton J, Schoultz BW, Hjo̷rnevik T, et al. Synthesis and evaluation of a full‐agonist orvinol for PET‐imaging of opioid receptors: [11C]PEO. J Med Chem. 2009;52:5586‐5589. [DOI] [PubMed] [Google Scholar]
- 160. Riss PJ, Hong YT, Marton J, et al. Synthesis and evaluation of 18F‐FE‐PEO in rodents: an 18F‐labeled full agonist for opioid receptor imaging. J Nucl Med. 2013;54:299‐305. [DOI] [PubMed] [Google Scholar]
- 161. Schoultz BW, Hjørnevik T, Reed BJ, et al. Synthesis and evaluation of three structurally related (1)(8)F‐labeled orvinols of different intrinsic activities: 6‐O‐[(1)(8)F]fluoroethyl‐diprenorphine ([(1)(8)F]FDPN), 6‐O‐[(1)(8)F]fluoroethyl‐buprenorphine ([(1)(8)F]FBPN), and 6‐O‐[(1)(8)F]fluoroethyl‐phenethyl‐orvinol ([(1)(8)F]FPEO). J Med Chem. 2014;57:5464‐5469. [DOI] [PubMed] [Google Scholar]
- 162. Mogg AJ, Eessalu T, Johnson M, et al. In vitro pharmacological characterization and in vivo validation of LSN3172176 a novel M1 selective muscarinic receptor agonist tracer molecule for positron emission tomography. J Pharmacol Exp Ther. 2018;365:602‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Jesudason C, Barth V, Goldsmith P, et al. Discovery of two novel, selective agonist radioligands as PET imaging agents for the M1 muscarinic acetylcholine receptor. J Nucl Med. 2017;58(Suppl.1):546. [Google Scholar]
- 164. Nabulsi N, Holden D, Zheng MQ, et al. Evaluation of a novel, selective M1 muscarinic acetylcholine receptor ligand [11C]‐LSN3172176 in non‐human primates. J Nucl Med. 2017;58(Suppl.1):275. [Google Scholar]
- 165. Buiter HJ, Leysen J, Fisher A, Huisman M, Knol D, Lammertsma AA. [11C]AF150(S): an agonist PET ligand for the M1 muscarinic acetylcholine receptor. J Nucl Med. 2010;51:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Buiter HJ, Windhorst AD, Huisman MC, et al. [11C]AF150(S), an agonist PET ligand for M1 muscarinic acetylcholine receptors. EJNMMI Res. 2013;3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Sauerberg P, Olesen PH, Nielsen S, et al. Novel functional M1 selective muscarinic agonists. Synthesis and structure‐activity relationships of 3‐(1,2,5‐thiadiazolyl)‐1,2,5,6‐tetrahydro‐1‐methylpyridines. J Med Chem. 1992;35:2274‐2283. [DOI] [PubMed] [Google Scholar]
- 168. Jagoda EM, Kiesewetter DO, Shimoji K, et al. Regional brain uptake of the muscarinic ligand, [18F]FP‐TZTP, is greatly decreased in M2 receptor knockout mice but not in M1, M3 and M4 receptor knockout mice. Neuropharmacology. 2003;44:653‐661. [DOI] [PubMed] [Google Scholar]
- 169. Kiesewetter DO, Lee J, Lang L, Park SG, Paik CH, Eckelman WC. Preparation of 18F‐labeled muscarinic agonist with M2 selectivity. J Med Chem. 1995;38:5‐8. [DOI] [PubMed] [Google Scholar]
- 170. Kiesewetter DO, Carson RE, Jagoda EM, Herscovitch P, Eckelman WC. In vivo muscarinic binding of 3‐(alkylthio)‐3‐thiadiazolyl tetrahydropyridines. Synapse. 1999;31:29‐40. [DOI] [PubMed] [Google Scholar]
- 171. Ravasi L, Tokugawa J, Nakayama T, et al. Imaging of the muscarinic acetylcholine neuroreceptor in rats with the M2 selective agonist [18F]FP‐TZTP. Nucl Med Biol. 2012;39:45‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Podruchny TA, Connolly C, Bokde A, et al. In vivo muscarinic 2 receptor imaging in cognitively normal young and older volunteers. Synapse. 2003;48:39‐44. [DOI] [PubMed] [Google Scholar]
- 173. Carson RE, Kiesewetter DO, Jagoda E, Der MG, Herscovitch P, Eckelman WC. Muscarinic cholinergic receptor measurements with [18F]FP‐TZTP: control and competition studies. J Cereb Blood Flow Metab. 1998;18:1130‐1142. [DOI] [PubMed] [Google Scholar]
- 174. Reid AE, Ding YS, Eckelman WC, et al. Comparison of the pharmacokinetics of different analogs of 11C‐labeled TZTP for imaging muscarinic M2 receptors with PET. Nucl Med Biol. 2008;35:287‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Van Waarde A, Absalom AR, Visser AK, Dierckx RA. Positron emission tomography (PET) imaging of opioid receptors In: Dierckx RA, Otte A, De Vries EFJ, van Waarde A, Luiten PGM, eds. PET and SPECT of Neurobiological Systems. Heidelberg: Springer; 2014:pp. 583‐623. [Google Scholar]
- 176. Kiss B, Horvath A, Nemethy Z, et al. Cariprazine (RGH‐188), a dopamine D(3) receptor‐preferring, D(3)/D(2) dopamine receptor antagonist‐partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333:328‐340. [DOI] [PubMed] [Google Scholar]
- 177. Tadori Y, Forbes RA, McQuade RD, Kikuchi T. In vitro pharmacology of aripiprazole, its metabolite and experimental dopamine partial agonists at human dopamine D2 and D3 receptors. Eur J Pharmacol. 2011;668:355‐365. [DOI] [PubMed] [Google Scholar]
- 178. Tóth M, Varrone A, Steiger C, et al. Brain uptake and distribution of the dopamine D3 /D2 receptor partial agonist [11C]cariprazine: an in vivo positron emission tomography study in non‐human primates. Synapse. 2013;67:258‐264. [DOI] [PubMed] [Google Scholar]
- 179. Wong DF, Pomper MG. Predicting the success of a radiopharmaceutical for in vivo imaging of central nervous system neuroreceptor systems. Mol Imaging Biol. 2003;5:350‐362. [DOI] [PubMed] [Google Scholar]
- 180. Pike VW. PET radiotracers: crossing the blood‐brain barrier and surviving metabolism. Trends Pharmacol Sci. 2009;30:431‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Van de Bittner GC, Ricq EL, Hooker JM. A philosophy for CNS radiotracer design. Acc Chem Res. 2014;47:3127‐3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Selvaraj S, Walker C, Arnone D, et al. Effect of citalopram on emotion processing in humans: a combined 5‐HT1A [(11)C]CUMI‐101 PET and functional MRI study. Neuropsychopharmacology. 2018;43:655‐664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Selvaraj S, Mouchlianitis E, Faulkner P, et al. Presynaptic serotoninergic regulation of emotional processing: a multimodal brain imaging study. Biol Psychiatry. 2015;78:563‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Henriksen G, Platzer S, Hauser A, et al. 18F‐labeled sufentanil for PET‐imaging of mu‐opioid receptors. Bioorg Med Chem Lett. 2005;15:1773‐1777. [DOI] [PubMed] [Google Scholar]
- 185. DaSilva JN, Wilson AA, Nobrega JN, Jiwa D, Houle S. Synthesis and autoradiographic localization of the dopamine D‐1 agonists [11C]SKF 75670 and [11C]SKF 82957 as potential PET radioligands. Appl Radiat Isot. 1996;47:279‐284. [DOI] [PubMed] [Google Scholar]
- 186. Hatano K, Ishiwata K, Elsinga P. PET tracers for imaging of the dopaminergic system. Curr Med Chem. 2006;13:2139‐2153. [DOI] [PubMed] [Google Scholar]
- 187. Laruelle M. Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab. 2000;20:423‐451. [DOI] [PubMed] [Google Scholar]
- 188. Paterson LM, Kornum BR, Nutt DJ, Pike VW, Knudsen GM. 5‐HT radioligands for human brain imaging with PET and SPECT. Med Res Rev. 2013;33:54‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Zheng MQ, Nabulsi N, Kim SJ, et al. Synthesis and evaluation of 11C‐LY2795050 as a kappa‐opioid receptor antagonist radiotracer for PET imaging. J Nucl Med. 2013;54:455‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Tsukada H. PET imaging of muscarinic receptors In: Dierckx RA, Otte A, De Vries EFJ, van Waarde A, Luiten PGM, eds. PET and SPECT of Neurobiological Systems. Heidelberg: Springer; 2014:pp. 445‐464. [Google Scholar]
- 191. Mintun MA, Raichle ME, Kilbourn MR, Wooten GF, Welch MJ. A quantitative model for the in vivo assessment of drug binding sites with positron emission tomography. Ann Neurol. 1984;15:217‐227. [DOI] [PubMed] [Google Scholar]
- 192. Innis RB, Cunningham VJ, Delforge J, et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27:1533‐1539. [DOI] [PubMed] [Google Scholar]
- 193. Narendran R, Hwang DR, Slifstein M, et al. Measurement of the proportion of D2 receptors configured in state of high affinity for agonists in vivo: a positron emission tomography study using [11C]N‐propyl‐norapomorphine and [11C]raclopride in baboons. J Pharmacol Exp Ther. 2005;315:80‐90. [DOI] [PubMed] [Google Scholar]
- 194. McCormick PN, Kapur S, Reckless G, Wilson AA. Ex vivo [11C]‐(+)‐PHNO binding is unchanged in animal models displaying increased high‐affinity states of the D2 receptor in vitro. Synapse. 2009;63:998‐1009. [DOI] [PubMed] [Google Scholar]
- 195. Kubota M, Nagashima T, Takano H, et al. Affinity states of striatal dopamine D2 receptors in antipsychotic‐free patients with schizophrenia. Int J Neuropsychopharmacol. 2017;20:928‐935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. McCormick PN, Kapur S, Seeman P, Wilson AA. Dopamine D2 receptor radiotracers [(11)C](+)‐PHNO and [(3)H]raclopride are indistinguishably inhibited by D2 agonists and antagonists ex vivo. Nucl Med Biol. 2008;35:11‐17. [DOI] [PubMed] [Google Scholar]
- 197. Kortekaas R, Maguire RP, Cremers TI, Dijkstra D, van Waarde A, Leenders KL. In vivo binding behavior of dopamine receptor agonist (+)‐PD 128907 and implications for the “ceiling effect” in endogenous competition studies with [(11)C]raclopride‐a positron emission tomography study in Macaca mulatta . J Cereb Blood Flow Metab. 2004;24:531‐535. [DOI] [PubMed] [Google Scholar]
- 198. Slifstein M, Suckow RF, Javitch JA, Cooper T, Lieberman J, Abi‐Dargham A. Characterization of in vivo pharmacokinetic properties of the dopamine D1 receptor agonist DAR‐0100A in non‐human primates using PET with [11C] NNC112 and [11C] raclopride. J Cereb Blood Flow Metab. 2011;31:293‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Cumming P, Wong DF, Gillings N, Hilton J, Scheffel U, Gjedde A. Specific binding of [11C]raclopride and N‐[3H]propyl‐norapomorphine to dopamine receptors in living mouse striatum: occupancy by endogenous dopamine and guanosine triphosphate‐free G protein. J Cereb Blood Flow Metab. 2002;22:596‐604. [DOI] [PubMed] [Google Scholar]
- 200. Seeman P. Dopamine DHigh receptors measured ex vivo are elevated in amphetamine‐sensitized animals. Synapse. 2009;63:186‐192. [DOI] [PubMed] [Google Scholar]
- 201. Seeman P, Weinshenker D, Quirion R, et al. Dopamine supersensitivity correlates with D2High states, implying many paths to psychosis. Proc Natl Acad Sci USA. 2005;102:3513‐3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Narendran R, Martinez D, Mason NS, et al. Imaging of dopamine D2/3 agonist binding in cocaine dependence: a [11C]NPA positron emission tomography study. Synapse. 2011;65:1344‐1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. McCormick PN, Ginovart N, Wilson AA. Isoflurane anaesthesia differentially affects the amphetamine sensitivity of agonist and antagonist D2/D3 positron emission tomography radiotracers: implications for in vivo imaging of dopamine release. Mol Imaging Biol. 2011;13:737‐746. [DOI] [PubMed] [Google Scholar]
- 204. Aronstam RS, Dennison RL, Jr . Anesthetic effects on muscarinic signal transduction. Int Anesthesiol Clin. 1989;27:265‐272. [DOI] [PubMed] [Google Scholar]
- 205. Ishizawa Y. Mechanisms of anesthetic actions and the brain. J Anesth. 2007;21:187‐199. [DOI] [PubMed] [Google Scholar]
- 206. Finnema SJ, Hughes ZA, Haaparanta‐Solin M, et al. Amphetamine decreases alpha2C‐adrenoceptor binding of [11C]ORM‐13070: a PET study in the primate brain. Int J Neuropsychopharmacol. 2014;18(3):pyu081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Whittington RA, Virag L. Isoflurane decreases extracellular serotonin in the mouse hippocampus. Anesth Analg. 2006;103:92‐98. [DOI] [PubMed] [Google Scholar]
- 208. Laruelle M. Measuring dopamine synaptic transmission with molecular imaging and pharmacological challenges: the state of the art. Neuromethods. 2012;2012:163‐203. [Google Scholar]
- 209. Morris ED, Yoder KK. Positron emission tomography displacement sensitivity: predicting binding potential change for positron emission tomography tracers based on their kinetic characteristics. J Cereb Blood Flow Metab. 2007;27:606‐617. [DOI] [PubMed] [Google Scholar]
- 210. Ginovart N. Imaging the dopamine system with in vivo [11C]raclopride displacement studies: understanding the true mechanism. Mol Imaging Biol. 2005;7:45‐52. [DOI] [PubMed] [Google Scholar]
- 211. Quelch DR, Withey SL, Nutt DJ, Tyacke RJ, Parker CA. The influence of different cellular environments on PET radioligand binding: an application to D2/3 dopamine receptor imaging. Neuropharmacology. 2014;85:305‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Wong CS, Su YF, Chang KJ, Watkins WD. Intrathecal pertussis toxin treatment attenuates opioid antinociception and reduces high‐affinity state of opioid receptors. Anesthesiology. 1992;77:691‐699. [DOI] [PubMed] [Google Scholar]
- 213. Self D, Terwilliger R, Nestler E, Stein L. Inactivation of Gi and G(o) proteins in nucleus accumbens reduces both cocaine and heroin reinforcement. J Neurosci. 1994;14:6239‐6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Fujita N, Nakahiro M, Fukuchi I, Saito K, Yoshida H. Effects of pertussis toxin on D2‐dopamine receptor in rat striatum: evidence for coupling of Ni regulatory protein with D2‐receptor. Brain Res. 1985;333:231‐236. [DOI] [PubMed] [Google Scholar]
- 215. Skinbjerg M, Sibley DR, Javitch JA, Abi‐Dargham A. Imaging the high‐affinity state of the dopamine D2 receptor in vivo: fact or fiction? Biochem Pharmacol. 2012;83:193‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Van Wieringen JP, Booij J, Shalgunov V, Elsinga P, Michel MC. Agonist high‐ and low‐affinity states of dopamine D(2) receptors: methods of detection and clinical implications. Naunyn Schmiedebergs Arch Pharmacol. 2013;386:135‐154. [DOI] [PubMed] [Google Scholar]
- 217. Gallezot JD, Beaver JD, Gunn RN, et al. Affinity and selectivity of [(1)(1)C]‐(+)‐PHNO for the D3 and D2 receptors in the rhesus monkey brain in vivo. Synapse. 2012;66:489‐500. [DOI] [PubMed] [Google Scholar]
- 218. Seeman P, Tallerico T, Ko F, Tenn C, Kapur S. Amphetamine‐sensitized animals show a marked increase in dopamine D2 high receptors occupied by endogenous dopamine, even in the absence of acute challenges. Synapse. 2002;46:235‐239. [DOI] [PubMed] [Google Scholar]
- 219. Seeman P, Tallerico T, Ko F. Alcohol‐withdrawn animals have a prolonged increase in dopamine D2High receptors, reversed by general anesthesia: relation to relapse? Synapse. 2004;52:77‐83. [DOI] [PubMed] [Google Scholar]
- 220. Seeman P, McCormick PN, Kapur S. Increased dopamine D2(High) receptors in amphetamine‐sensitized rats, measured by the agonist [(3)H](+)PHNO. Synapse. 2007;61:263‐267. [DOI] [PubMed] [Google Scholar]
- 221. Graff‐Guerrero A, Mizrahi R, Agid O, et al. The dopamine D2 receptors in high‐affinity state and D3 receptors in schizophrenia: a clinical [11C]‐(+)‐PHNO PET study. Neuropsychopharmacology. 2009;34:1078‐1086. [DOI] [PubMed] [Google Scholar]
- 222. Suridjan I, Rusjan P, Addington J, Wilson A, Houle S, Mizrahi R. Dopamine D2 and D3 binding in people at clinical high risk for schizophrenia, antipsychotic‐naive patients and healthy controls while performing a cognitive task. J Psychiatry Neurosci. 2013;38:98‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Boileau I, Payer D, Chugani B, et al. The D2/3 dopamine receptor in pathological gambling: a positron emission tomography study with [11C]‐(+)‐propyl‐hexahydro‐naphtho‐oxazin and [11C]raclopride. Addiction. 2013;108:953‐963. [DOI] [PubMed] [Google Scholar]
- 224. Mizrahi R, Suridjan I, Kenk M, et al. Dopamine response to psychosocial stress in chronic cannabis users: a PET study with [11C](+)PHNO. Neuropsychopharmacology. 2013;38:673‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Frankle WG, Paris J, Himes M, Mason NS, Mathis CA, Narendran R. Amphetamine‐induced striatal dopamine release measured with an agonist radiotracer in schizophrenia. Biol Psychiatry. 2018;83:707‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Farde L, Ginovart N, Ito H, et al. PET‐characterization of [carbonyl‐11C]WAY‐100635 binding to 5‐HT1A receptors in the primate brain. Psychopharmacology (Berl). 1997;133:196‐202. [DOI] [PubMed] [Google Scholar]
- 227. Mathis CA, Simpson NR, Mahmood K, Kinahan PE, Mintun MA. [11C]WAY 100635: a radioligand for imaging 5‐HT1A receptors with positron emission tomography. Life Sci. 1994;55:L403‐L407. [DOI] [PubMed] [Google Scholar]
- 228. Seeman P. Dopamine agonist radioligand binds to both D2High and D2Low receptors, explaining why alterations in D2High are not detected in human brain scans. Synapse. 2012;66:88‐93. [DOI] [PubMed] [Google Scholar]
- 229. Cumming P. Absolute abundances and affinity states of dopamine receptors in mammalian brain: a review. Synapse. 2011;65:892‐909. [DOI] [PubMed] [Google Scholar]
- 230. Sibley DR, Mahan LC, Creese I. Dopamine receptor binding on intact cells. Absence of a high‐affinity agonist‐receptor binding state. Mol Pharmacol. 1983;23:295‐302. [PubMed] [Google Scholar]
- 231. Seeman P. Dopamine D2(High) receptors on intact cells. Synapse. 2008;62:314‐318. [DOI] [PubMed] [Google Scholar]
- 232. Ostrom RS, Post SR, Insel PA. Stoichiometry and compartmentation in G‐protein‐coupled receptor signaling: implications for therapeutic interventions involving G(s). J Pharmacol Exp Ther. 2000;294:407‐412. [PubMed] [Google Scholar]
- 233. González‐Maeso J, Rodríguez‐Puertas R, Meana JJ. Quantitative stoichiometry of G‐proteins activated by mu‐opioid receptors in postmortem human brain. Eur J Pharmacol. 2002;452:21‐33. [DOI] [PubMed] [Google Scholar]
- 234. Gilman AG. G proteins: transducers of receptor‐generated signals. Annu Rev Biochem. 1987;56:615‐649. [DOI] [PubMed] [Google Scholar]
- 235. Logan J, Fowler JS, Volkow ND, et al. Graphical analysis of reversible radioligand binding from time‐activity measurements applied to [N‐11C‐methyl]‐(−)‐cocaine PET studies in human subjects. J Cereb Blood Flow Metab. 1990;10:740‐747. [DOI] [PubMed] [Google Scholar]
- 236. Ichise M, Liow JS, Lu JQ, et al. Linearized reference tissue parametric imaging methods: application to [11C]DASB positron emission tomography studies of the serotonin transporter in human brain. J Cereb Blood Flow Metab. 2003;23:1096‐1112. [DOI] [PubMed] [Google Scholar]