

Abstract
In BOLERO‐2, adding everolimus to exemestane resulted in a twofold increase in median progression‐free survival (PFS) vs exemestane in postmenopausal women with hormone receptor‐positive (HR+), human epidermal growth factor receptor 2‐negative (HER2−) advanced breast cancer (aBC) after progression on a non‐steroidal aromatase inhibitor (NSAI). Here, we report on the open‐label, single‐arm, phase IIIB 4EVER trial (NCT01626222). This trial evaluated the clinical effectiveness of everolimus plus exemestane in postmenopausal women with HR+, HER2− aBC who had progressed on or after an NSAI, but with no restrictions on the time of progression after NSAI, prior chemotherapy for advanced disease or previous exemestane. The primary endpoint was overall response rate (ORR; i.e. the percentage of patients with a best overall response of complete or partial response per RECIST 1.1) within the first 24 weeks of treatment. Secondary endpoints included PFS, overall survival, safety and health‐related quality of life. Between June 2012 and November 2013, 299 patients were enrolled at 82 German centers: 281 patients were evaluable for efficacy and 299 for safety. The ORR was 8.9% (95% confidence interval [CI]: 5.8–12.9%). Median PFS was 5.6 months (95% CI: 5.4–6.0 months). The most frequent grade 3/4 adverse events were stomatitis (8.4%), general physical health deterioration (6.7%), dyspnea (4.7%) and anemia (4.3%). The ORR in 4EVER was lower than in BOLERO‐2, likely due to inclusion of patients with more advanced disease and extensive pretreatment. These data confirm the clinical benefits and known safety profile of everolimus plus exemestane in postmenopausal women with HR+, HER2− aBC.
Keywords: estrogen receptor‐positive, HER2‐negative, advanced breast cancer, everolimus, exemestane
Short abstract
What's new?
Current treatment guidelines for HR+, HER2– advanced breast cancer support continued endocrine therapy after progression on first‐line treatment, including the use of everolimus and exemestane combined. Here, the authors report on the phase IIIB 4EVER trial, which evaluated the efficacy, safety and quality of life effects of everolimus plus exemestane in postmenopausal women with pretreated, HR+, HER2– advanced breast cancer. The patient population was broader than that evaluated in previous major trials, and thus more reflective of real‐world practice. Overall, the results confirm the clinical benefits and known safety profile of everolimus plus exemestane in this patient population.
Abbreviations
- aBC
advanced breast cancer
- AE
adverse event
- CDK
cyclin‐dependent kinase
- CI
confidence interval
- CNS
central nervous system
- CR
complete response
- CT
computed tomography
- CTCAE
Common Terminology Criteria for Adverse Events
- ECOG PS
Eastern Cooperative Oncology Group performance status
- EORTC QLQ‐BR32
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Breast Cancer Module
- EORTC QLQ‐C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
- EOT
end of treatment
- ER
estrogen receptor
- FAS
full analysis set
- GCP
Good Clinical Practice
- HER2
human epidermal growth factor receptor 2
- HR
hormone receptor
- HRQoL
health‐related quality of life
- mTOR
mammalian target of rapamycin
- NE
not estimable
- NSAI
non‐steroidal aromatase inhibitor
- ORR
overall response rate
- OS
overall survival
- PD
progressive disease
- PFS
progression‐free survival
- PgR
progesterone receptor
- PR
partial response
- QoL
quality of life
- RECIST
Response Evaluation Criteria In Solid Tumors
- SD
stable disease
Endocrine therapy is the preferred first‐line treatment for postmenopausal women with hormone receptor‐positive (HR+), human epidermal growth factor receptor 2‐negative (HER2−) advanced breast cancer (aBC), with aromatase inhibitors generally recommended over other options.1, 2, 3 In time, however, advanced breast cancers inevitably progress due to primary or secondary endocrine resistance. Treatment guidelines for HR+, HER2– aBC support continued endocrine therapy after progression on first‐line treatment.1, 2, 3 Second‐line treatment options for aBC are guided by the agents that were used in the (neo)adjuvant and first‐line aBC settings, as well as tolerability profiles.2 Available options for second‐line treatment include: aromatase inhibitors; the mammalian target of rapamycin (mTOR) inhibitor everolimus in combination with exemestane; tamoxifen; and, more recently, fulvestrant in combination with the cyclin‐dependent kinase (CDK) 4/6 inhibitors palbociclib or abemaciclib.1, 2, 3, 4
For patients who progress on aromatase inhibitors, the duration of response with second‐line endocrine monotherapy (i.e. exemestane or fulvestrant) is ~3.2–6.5 months.5, 6 The randomized, placebo‐controlled, phase III BOLERO‐2 trial evaluated the addition of everolimus to exemestane after failure of a nonsteroidal aromatase inhibitor (NSAI) in postmenopausal women with HR+, HER2– aBC.6, 7 Median progression‐free survival (PFS) by local assessment was increased by more than twofold in patients receiving everolimus plus exemestane vs those receiving placebo plus exemestane (7.8 vs 3.2 months; hazard ratio = 0.45; 95% confidence interval [CI]: 0.38–0.54; p < 0.0001).6 With a median 18‐month follow‐up, overall response rates (ORRs) were 12.6% (95% CI: 9.8–15.9%) and 1.7% (95% CI: 0.5–4.2%) for everolimus plus exemestane and placebo plus exemestane, respectively.6 These data led to the regulatory approval of everolimus plus exemestane for use in this setting, and recommendations in breast cancer treatment guidelines.1, 2, 3
Here, we report the final efficacy, safety and quality of life (QoL) results from the phase IIIB 4EVER trial, which evaluated the use of everolimus plus exemestane in a broader population compared with BOLERO‐2 and is thus more reflective of real‐world practice. Patients in 4EVER had to have progressed on or after an NSAI; but were not limited by the time of recurrence or progression after NSAI therapy, the number of previous chemotherapy lines for advanced disease or the use of previous exemestane therapy.
Patients and Methods
Study design and participants
The 4EVER trial (NCT01626222) was a German multicenter, open‐label, single‐arm, phase IIIB study designed to evaluate the combination of everolimus and exemestane in postmenopausal women with HR+, HER2− locally advanced or metastatic breast cancer (Supporting Information Fig. S1). Eligible patients had recurrence while being on, or after completion of, adjuvant NSAI treatment (letrozole or anastrozole), or progression during, or following completion of, NSAI treatment for locally advanced or metastatic breast cancer. Letrozole or anastrozole did not have to be the last treatment prior to enrollment, but documentation of recurrence or progression during the most recent prior systemic therapy was required. Clinical evidence of recurrence or progression included at least one measurable lesion or, alternatively, evaluable bone lesions (lytic or mixed) in the absence of measurable disease. Additional primary inclusion criteria included confirmation of postmenopausal status, an Eastern Cooperative Oncology Group performance status (ECOG PS) ≤2 (on a scale from 0 to 5), and histologic or cytologic confirmation of estrogen receptor‐positive and/or progesterone receptor‐positive and HER2− breast cancer. Patients could have received prior exemestane, fulvestrant, and/or tamoxifen and any number of prior chemotherapy lines. Exclusion criteria included patients with only nonmeasurable lesions other than bone metastases, prior treatment with or known hypersensitivity to mTOR inhibitors, and HR− or HER2+ disease.
Patients received oral everolimus (10 mg) and exemestane (25 mg) daily for 48 weeks, or until disease progression, unacceptable toxicity, death or discontinuation for any other reason. Further treatment after 48 weeks was at the investigator's discretion. Study visits were performed at baseline and weeks 4, 12, 24, 36 and 48, or at the end of treatment (EOT) if occurring before 48 weeks. The end of study was planned for 1 year after recruitment of the last patient. After the last patient in the trial had taken the last dose of everolimus (i.e. after the last patient had completed the 48‐week treatment period or had discontinued treatment earlier), the current progression and survival status was updated for all patients. Patients were not followed until progression or death.
Patients were followed for safety for 28 days after the last dose of everolimus. In case of adverse events (AEs), everolimus dose interruptions or adjustments were permitted. In such cases, two reductions in the everolimus dose were allowed: initially to 5 mg daily, and a subsequent reduction to 5 mg every other day. Everolimus was resumed at the initial dose or a lower dose level per protocol requirements if recovery to grade 1 or lower was achieved in less than 4 weeks. Patients who interrupted everolimus for more than 4 weeks were discontinued from the study.
Tumor assessment and response were assessed by local radiology review according to Response Evaluation Criteria In Solid Tumors (RECIST) version 1.1. Patients had either at least one lesion that could be measured per RECIST v1.1 or bone lesions (lytic or mixed [lytic + sclerotic]) in the absence of measurable disease. For patients with measurable disease at baseline, tumor progression was evaluated at 24 and 48 weeks, or at EOT. All additional routine tumor evaluations were conducted and documented per RECIST v1.1. Positive areas on bone scans at baseline were further assessed by X‐ray or computed tomography (CT) scan, and monitored using the same modality at 24 and 48 weeks or at EOT. In the absence of measurable disease at baseline, the following were considered to indicate progression among patients with lytic or mixed (lytic + sclerotic) bone lesions: the appearance of one or more new lytic lesions in bone; the appearance of one or more new lesions outside of bone; or unequivocal progression of existing bone lesions.
All participants provided written informed consent prior to enrolment. The study was conducted in accordance with the International Conference on Harmonization Harmonized Tripartite Guidelines for Good Clinical Practice (GCP) and applicable local regulations, and with the ethical principles laid down in the Declaration of Helsinki. The protocol was reviewed and approved by the Institutional Review Board/Independent Ethics Committee/Research Ethics Board at each participating center.
Study endpoints
The primary study endpoint was overall response rate (ORR), which was defined as the proportion of patients with a best overall response of complete response (CR) or partial response (PR) within the first 24 weeks of treatment (ORR24w). No confirmatory CT assessments for CR or PR were required by the protocol as this would have placed additional radiation exposure on the patients. The best overall response for each patient was determined from the sequence of investigator‐assessed overall lesion responses per visit according to RECIST v1.1. Routine scans at week 4 and 12 were documented if performed as part of routine clinical practice. If a response (best overall response of CR or PR) was seen at these assessments, patients were considered to be “responders” per RECIST v1.1. Secondary endpoints included the ORR within the first 48 weeks of treatment (ORR48w); in addition, PFS, overall survival (OS), safety and health‐related quality of life (HRQoL) were assessed over the 48‐week treatment period (plus 28 days for safety). HRQoL was assessed using the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ‐C30) and the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Breast Cancer Module (EORTC QLQ‐BR32). Results of exploratory objectives will be reported separately.
Data analysis
In this single‐arm trial, the primary objective was to estimate the ORR24w; therefore, no comparative statistical hypothesis or model underlies the analysis. Patients who received at least one dose of study treatment, except for 18 patients from two study sites who were excluded because of GCP data validity non‐compliance issues, were included in the full analysis set (FAS) and included in the primary efficacy analysis. Sensitivity analyses were conducted to determine any impact of excluding these 18 patients by repeating the primary efficacy analyses on the full population of patients. The Clopper–Pearson method was used to determine the associated two‐sided exact 95% CIs for ORRs and individual response criteria. Preplanned subgroup analyses included prior treatment with chemotherapy or exemestane. PFS and OS were evaluated in the FAS and summarized using the Kaplan–Meier method. PFS and OS were measured from the start of treatment to the date of the first documented progression or death from any cause. If a patient had not progressed or was not known to have died, then PFS was censored at the date of last tumor assessment and survival was censored at the date of last contact. Descriptive statistics were used to summarize changes from baseline in individual HRQoL scores.
Post‐hoc Cox regression analyses were used to assess the impact of variables on patient outcome (PFS or overall survival). Hazard ratios were reported by patient subgroup according to the following variables: histologic grade 1, 2 or 3; time between initial diagnosis and first recurrence/metastasis; previous chemotherapy [yes/no]; prior exemestane [yes/no]; line of therapy [first line vs second line and higher or first line vs second line vs third line or higher]; visceral metastasis [lung, liver, central nervous system, abdomen/visceral; yes/no]; ECOG PS [0 vs ≥ 1]; age [35–54 years vs ≥ 55 years]; and weight).
All patients who received at least one dose of study treatment and had at least one post‐baseline safety assessment were evaluated for safety. Safety was assessed using the Common Terminology Criteria for Adverse Events (CTCAE) version 4.03 and summarized by maximum severity.
Results
Patient disposition
The 4EVER study was conducted between June 25, 2012 (first patient first visit) and November 26, 2013 (last patient last visit). In total, 330 postmenopausal women with HR+, HER2− locally advanced or metastatic breast cancer whose disease was refractory to NSAI treatment were screened for eligibility. Of these, 299 women were enrolled at 82 study centers in Germany and received at least one dose of study treatment (everolimus plus exemestane). All 299 patients were included in the safety analyses. Eighteen patients from two study sites with GCP data validity non‐compliance issues were excluded; therefore, the FAS (used for efficacy analyses) comprised 281 patients.
Of the 299 patients enrolled and treated with everolimus plus exemestane, 36 (12.0%) patients completed 48 weeks of treatment. A total of 255 (85.3%) patients discontinued treatment before 48 weeks, most frequently due to progressive disease (38.8%), AEs (24.7%) or death (8.0%; Supporting Information Fig. S2). No information on treatment continuation or outcome was available for eight patients. Overall, 123 (41.1%) of patients completed the 24‐week visit. The median time on study (from screening to the last visit/progression and survival follow‐up; safety population) was 5.3 months (range: 0–14.6 months).
Patient characteristics
Baseline characteristics for patients included in the FAS are shown in Table 1. The 281 women had a median age of 67 years (range 35–85 years). Nearly all were white (99.3%). Most patients (60.9%) had an ECOG PS of 0 at baseline. All patients were postmenopausal, as required by the protocol.
Table 1.
Baseline patient demographics and disease characteristics (full analysis set)
| Baseline parameters | Full analysis set (N = 281) | |
|---|---|---|
| Patient demographics | ||
| Median age (range), years | 67 (35–85) | |
| Age group, n (%) | <65 years | 127 (45.2) |
| ≥65 years | 154 (54.8) | |
| Race, n (%) | White | 279 (99.3) |
| Asian | 2 (0.7) | |
| ECOG PS, n (%) 1 , 2 | 0 | 170 (60.9) |
| 1 | 98 (35.1) | |
| 2 | 11 (3.9) | |
| Missing | 2 (0.7) | |
| Disease characteristics, n (%) | ||
| Disease status | Metastatic | 270 (96.1) |
| Locally advanced | 5 (1.8) | |
| Metastatic and locally advanced | 6 (2.1) | |
| HR status 3 | ER+, PgR+ | 220 (78.3) |
| ER+, PgR− | 59 (21.0) | |
| ER−, PgR+ | 1 (0.4) | |
| ER−, PgR− | 1 (0.4) | |
| HER2 status 4 | HER2− | 277 (98.6) |
| HER2+ | 2 (0.7) | |
| Unknown/missing | 2 (0.7) | |
| Target lesion (in ≥10 patients) 5 | Liver | 145 (51.6) |
| Lymph nodes | 60 (21.4) | |
| Lung | 46 (16.4) | |
| Bone | 37 (13.2) | |
| Chest wall | 12 (4.3) | |
| Prior therapies, n (%) | ||
| Most recent prior therapy setting | Metastatic | 221 (78.6) |
| Adjuvant | 56 (19.9) | |
| Neoadjuvant | 1 (0.4) | |
| Missing | 3 (1.1) | |
| Prior therapy in the metastatic setting | Chemo‐ or endocrine therapy | 221 (78.6) |
| Chemotherapy | 151 (53.7) | |
| Endocrine therapy | 204 (72.6) | |
| Number of prior lines of therapy for metastatic disease 6 | 0 | 59 (21.0) |
| 1 | 52 (18.5) | |
| 2 | 45 (16.0) | |
| 3 | 34 (12.1) | |
| ≥4 | 87 (31.0) | |
| Unknown | 4 (1.4) | |
| NSAI as most recent therapy | 94 (33.5) | |
| Prior exemestane | 89 (31.7) | |
| Prior fulvestrant | 121 (43.1) | |
At screening.
No patient had an ECOG PS ≥3 at screening; however, one patient had an ECOG PS of 3 at baseline.
Assessed in primary tumor in 70.1% of patients and in metastatic sites in 29.9% of patients.
Assessed in primary tumor in 64.7% of patients, in metastatic sites in 34.8% of patients, and in unknown tumor stage for one patient.
Metastatic sites are not mutually exclusive (i.e. patients may have had metastasis at more than one site). Data on localization of target lesions were missing for 39 patients.
According to assessment by Study Steering Committee.
Abbreviations: ECOG PS: Eastern Cooperative Oncology Group performance status; ER: estrogen receptor; HER2: human epidermal growth factor receptor 2; HR: hormone receptor; NSAI: non‐steroidal aromatase inhibitor; PgR: progesterone receptor.
Most patients in the FAS had HR+ (n = 280, 99.6%), HER2– (n = 277, 98.6%) aBC. Five patients were enrolled and included in the FAS despite not meeting the inclusion criteria for receptor expression: one patient had estrogen receptor‐negative/progesterone receptor‐negative breast cancer; two patients had HER2+ breast cancer; and two patients had an undocumented HER2 status at screening.
Median time since initial diagnosis was 97.6 months (range: 4.3–415.3 months). Median time between the date of first recurrence/metastasis and the start of the study was 38 months (range: 0–327.4 months). All patients in the FAS had received prior NSAI therapy per inclusion requirements; 94 patients (33.5%) had received an NSAI as the most recent therapy, and 89 patients (31.7%) had received prior exemestane. Most patients (78.6%) had been previously treated in the metastatic setting, with more than 77% of patients having received one or more prior lines of therapy, and more than 59% having received more than two prior lines of therapy (Table 1).
Efficacy
Overall response rate
The ORR24w was 8.9% (95% CI: 5.8–12.9%; Table 2). Among the 25 responders, one patient had a CR and 24 (8.5%) patients had a PR. The 82 (29.2%) patients with an unknown response status at 24 weeks were not excluded from the ORR calculation (i.e. stayed in the denominator). The reasons for an unknown response status were: no tumor assessment until discontinuation of study treatment (74 patients [90.2%]), missing RECIST data at week 24 (7 patients [8.5%]), or lost to follow‐up (1 patient [1.2%]). The disease control rate (CR + PR + stable disease) within the first 24 weeks of treatment was 33.5% (95% CI: 28.0–39.3%). ORR48w was 10.3% (95% CI: 7.0–14.5%) in the FAS, with one patient experiencing a CR and 28 (10.0%) patients a PR. The disease control rate up to 48 weeks was 35.9% (95% CI: 30.3–41.9%).
Table 2.
Responses to everolimus plus exemestane in the full analysis set and by prior therapy
| All patients in the FAS (N = 281) | Prior exemestane | Prior chemotherapy | |||
|---|---|---|---|---|---|
| No (n = 190) | Yes (n = 91) | No (n = 130) | Yes (n = 151) | ||
| Week 24 | |||||
| ORR24w, 1 n (%) [95% CI] | 25 (8.9) [5.8–12.9] | 17 (8.9) [5.3–13.9] | 8 (8.8) [3.9–16.6] | 15 (11.5) [6.6–18.3] | 10 (6.6) [3.2–11.8] |
| CR | 1 (0.4) | 1 (0.5) | 0 | 0 | 1 (0.7) |
| PR | 24 (8.5) | 16 (8.4) | 8 (8.8) | 15 (11.5) | 9 (6.0) |
| SD | 69 (24.6) | 47 (24.7) | 22 (24.2) | 40 (30.8) | 29 (19.2) |
| PD | 105 (37.4) | 72 (37.9) | 33 (36.3) | 41 (31.5) | 64 (42.4) |
| Unknown 2 | 82 (29.2) | 54 (28.4) | 28 (30.8) | 34 (26.2) | 48 (31.8) |
| Week 48 | |||||
| ORR48w, 1 n (%) [95% CI] | 29 (10.3) [7.0–14.5] | 21 (11.1) [7.0–16.4] | 8 (8.8) [3.9–16.6] | 17 (13.1) [7.8–20.1] | 12 (7.9) [4.2–13.5] |
| CR | 1 (0.4) | 1 (0.5) | 0 | 0 | 1 (0.7) |
| PR | 28 (10.0) | 20 (10.5) | 8 (8.8) | 17 (13.1) | 11 (7.3) |
| SD | 72 (25.6) | 47 (24.7) | 25 (27.5) | 40 (30.8) | 32 (21.2) |
| PD | 112 (39.9) | 77 (40.5) | 35 (38.5) | 44 (33.8) | 68 (45.0) |
| Unknown 2 | 68 (24.2) | 45 (23.7) | 23 (25.3) | 29 (22.3) | 39 (25.8) |
Responses were evaluated using Response Evaluation Criteria In Solid Tumors version 1.1, assessed by local radiology review.
Rate of patients with best overall response of CR or PR.
Included in calculation of ORR (not regarded as responders).
Abbreviations: CI: confidence interval; CR: complete response; FAS: full analysis set; ORR: overall response rate; PD: progressive disease; PR; partial response; SD: stable disease.
ORR24w and ORR48w were numerically higher for patients who had not received prior chemotherapy in the metastatic setting vs those who had (ORR24w: 11.5% [95% CI: 6.6–18.3%] vs 6.6% [95% CI: 3.2–11.8%]; ORR48w: 13.1% [95% CI: 7.8–20.1%] vs 7.9% [95% CI: 4.2–13.5%]). No differences in ORR were observed in subgroup analyses based on prior exemestane (Table 2).
Progression‐free survival
Median PFS in the FAS was 5.6 months (95% CI: 5.4–6.0 months; Fig. 1 a). The Kaplan–Meier‐estimated PFS rate was 50.0% at 24 weeks and 19.3% at 48 weeks of treatment.
Figure 1.
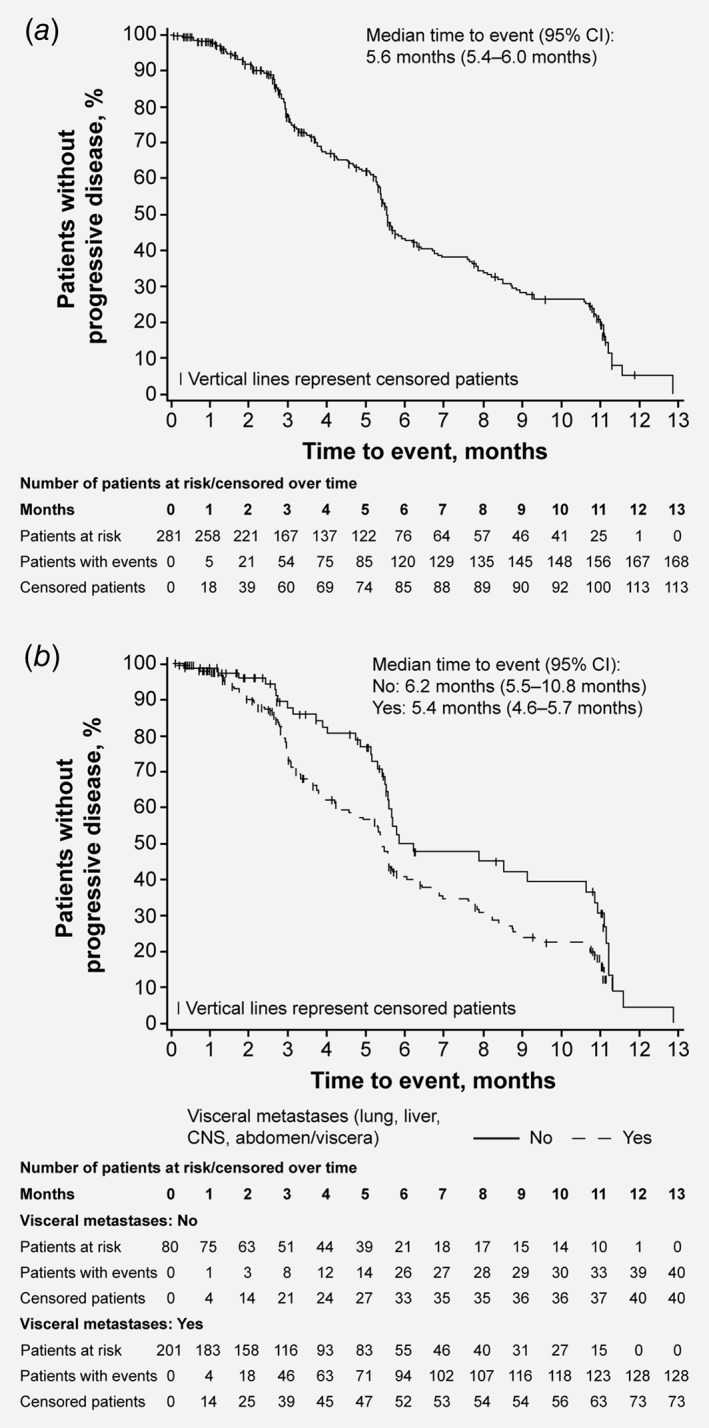
Progression‐free survival. Kaplan–Meier estimates and associated 95% CIs are shown for: (a) all patients in the full analysis set; (b) patients with or without visceral metastases.
In predefined univariate analyses, no difference in median PFS was observed in patients treated with or without prior exemestane (5.6 months [95% CI: 4.2–6.9 months] vs 5.5 months [95% CI: 5.3–6.3 months], respectively). Patients who had received prior chemotherapy in the metastatic setting had a numerically shorter median PFS than patients who had not received prior chemotherapy (5.2 months [95% CI: 4.2–5.5 months] vs 6.2 months [95% CI: 5.6–7.7 months], respectively). Median PFS was also numerically shorter in patients with visceral metastases vs those without per a post‐hoc analysis (5.4 months [95% CI: 4.6–5.7 months] vs 6.2 months [95% CI: 5.5–10.8 months], respectively; Fig. 1 b). A post‐hoc multivariate analysis supported the finding that prior exemestane had no influence on PFS but, in contrast to the univariate results, also found prior chemotherapy to have no impact. However, the multivariate analysis suggested that the independent variable of visceral metastases appeared to influence PFS (p = 0.0414).
Overall survival
The median OS time was not yet reached due to the short duration of the study (Fig. 2). The Kaplan–Meier‐estimated OS rates at 24 and 48 weeks of treatment were 79.4% and 66.9%, respectively.
Figure 2.

Overall survival. Kaplan–Meier estimates of overall survival are shown for all patients in the full analysis set.
Exploratory evaluations of patient subgroups showed no substantial differences in OS at 24 or 48 weeks in patients with or without prior exemestane. OS rates were, however, numerically higher for patients who had not received prior chemotherapy in the metastatic setting compared with patients who had received prior chemotherapy, both at 24 weeks (87.1% vs 72.8%) and 48 weeks (80.3% vs 55.3%). Post‐hoc multivariate analyses indicated a possible association between visceral metastases and prior chemotherapy with OS (data not shown).
Quality of life
The EORTC QLQ‐C30 mean global health status score (mean ±standard deviation) did not improve between baseline and week 48/EOT (−8.8 ±18.8 points; Fig. 3). For the EORTC QLQ‐C30 functional scales, mean ±standard deviation decreases (i.e. deterioration) of ≥5 points between baseline and EOT were observed for social functioning (−11.5 ±29.6 points), role functioning (−6.7 ±28.4 points) and physical functioning (−6.2 ±19.6 points). For the EORTC QLQ‐C30 symptom scales, mean ±standard deviation increases (i.e. deterioration) of ≥5 points between baseline and week 48/EOT were observed for loss of appetite (+16.2 ±37.0 points), insomnia (+11.2 ±32.4 points), fatigue (+10.9 ±31.2 points), diarrhea (+10.7 ±28.8 points), dyspnea (+7.9 ±28.8 points) and nausea/vomiting (+5.3 ±26.0 points). Increases in symptom scores and the observed decrease in the global health status/QoL score were more pronounced at earlier post‐baseline visits than at later visits. The estimated change (±standard error) in the global health status/QoL score from baseline was 5.1 ±1.5 and 5.5 ±3.3 points at weeks 12 and 48, respectively, and 3.5 ±2.0 points and 1.6 ±2.7 points at weeks 24 and 36, respectively.
Figure 3.
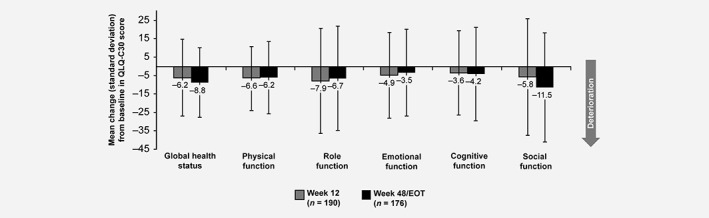
Health‐related quality of life (full analysis set). Mean (SD) changes from baseline to Weeks 12 and EOT/48 weeks in health‐related quality of life assessed using the EORTC QLQ‐C30, including global health status (an indicator of quality of life) and functional subscales (indicators of performance in functions of daily living). A decrease in EORTC QLQ‐C30 scores indicates deterioration.
The median time to ≥5% deterioration in EORTC QLQ‐C30 global health status/QoL score from baseline was 4.2 months (95% CI: 3.0–5.5 months). ECOG PS (grades 0–4) did not change appreciably over the course of the first 36 weeks of the study (Supporting Information Fig. S3). The proportion of patients with an ECOG PS of 0 (fully active, able to carry on all predisease performance without restriction) decreased from 61.7% to 45.8% between study weeks 36 and 48. In line with this change, the proportion of patients with an ECOG PS of 1 increased from 33.3% to 42.5%, respectively, and the proportion of patients with an ECOG PS of 2 increased from 3.3% to 10.5%.
Safety
In the safety population, the median time to treatment discontinuation was 3.9 months (95% CI: 3.4–4.8 months) and the median duration of exposure to the combination of everolimus plus exemestane was 133 days (range 2–393 days; equivalent to 4.4 months [range 0.1–12.9 months]; Supporting Information Table S1). The overall incidence of all‐cause, all‐grade AEs was 98.7% (Table 3). The most frequent all‐cause, all‐grade AEs were stomatitis (49.2%), fatigue (36.1%), diarrhea (26.4%), nausea (26.1%), decreased appetite (25.4%) and dyspnea (24.7%). The most common grade 3/4 AEs were stomatitis (8.4%), general physical health deterioration (6.7%), dyspnea (4.7%) and anemia (4.3%). Overall, 74 (24.7%) patients permanently discontinued study treatment due to AEs. The most common AEs leading to study treatment discontinuation were stomatitis (4.3%), dyspnea (2.3%), nausea (2.0%), pneumonitis (2.0%) and vomiting (2.0%). Of the 281 patients in the FAS, 151 (53.7%) required at least one dose reduction of everolimus, most commonly due to AEs (85.7%; Supporting Information Table S2). Similarly, AEs were the most common reasons for dose reduction (72.2%) in the 40 (14.2%) patients who required ≥1 dose reduction of exemestane. Median relative dose intensity of everolimus in the safety population was 98.3% (range: 4.8–100%).
Table 3.
Most common adverse events (occurring in ≥5% of patients) regardless of study drug relationship (safety population)
| Safety population (N = 299) | ||
|---|---|---|
| Most common adverse events, n (%) | Any grade | Grade 3/4 |
| Any adverse event | 295 (98.7) | 176 (58.9) |
| Stomatitis | 147 (49.2) | 25 (8.4) |
| Fatigue | 108 (36.1) | 10 (3.3) |
| Diarrhea | 79 (26.4) | 6 (2.0) 1 |
| Nausea | 78 (26.1) | 9 (3.0) |
| Decreased appetite | 76 (25.4) | 9 (3.0) 1 |
| Dyspnea | 74 (24.7) | 14 (4.7) |
| Rash | 68 (22.7) | 3 (1.0) 1 |
| Dysgeusia | 55 (18.4) | 2 (0.7) 1 |
| Anemia | 53 (17.7) | 13 (4.3) |
| Cough | 53 (17.7) | 2 (0.7) 1 |
| Peripheral edema | 50 (16.7) | 3 (1.0) 1 |
| Weight decrease | 45 (15.1) | 2 (0.7) |
| Vomiting | 43 (14.4) | 10 (3.3) |
| Epistaxis | 43 (14.4) | 0 |
| Headache | 37 (12.4) | 0 |
| General physical health deterioration | 36 (12.0) | 20 (6.7) |
| Arthralgia | 32 (10.7) | 1 (0.3) 1 |
| Pyrexia | 30 (10.0) | 2 (0.7) 1 |
| Pruritus | 29 (9.7) | 1 (0.3) 1 |
| Aphthous stomatitis | 27 (9.0) | 2 (0.7) 1 |
| Aspartate aminotransferase increase | 27 (9.0) | 6 (2.0) 1 |
| Bone pain | 27 (9.0) | 0 |
| Constipation | 26 (8.7) | 1 (0.3) 1 |
| Alanine aminotransferase increase | 26 (8.7) | 6 (2.0) 1 |
| Insomnia | 26 (8.7) | 4 (1.3) 1 |
| Alopecia | 26 (8.7) | 0 |
| Nasopharyngitis | 24 (8.0) | 1 (0.3) 1 |
| Thrombocytopenia | 23 (7.7) | 5 (1.7) |
| Back pain | 23 (7.7) | 2 (0.7) 1 |
| Dry skin | 23 (7.7) | 2 (0.7) 1 |
| Leukopenia | 22 (7.4) | 2 (0.7) 1 |
| Pneumonitis | 22 (7.4) | 7 (2.3) |
| Abdominal pain, upper | 20 (6.7) | 2 (0.7) 1 |
| Dry mouth | 19 (6.4) | 0 |
| Gamma‐glutamyltransferase increase | 19 (6.4) | 8 (2.6) |
| Malignant neoplasm progression | 19 (6.4) | 11 (3.7) |
| Pneumonia | 19 (6.4) | 10 (3.3) |
| Pleural effusion | 19 (6.4) | 9 (3.0) |
| Nail disorder | 16 (5.4) | 0 |
| Urinary tract infection | 15 (5.0) | 6 (2.0) |
| Hyperglycemia | 15 (5.0) | 4 (1.3) 1 |
| Polyneuropathy | 15 (5.0) | 1 (0.3) 1 |
Only grade 3 events; no grade 4 events were reported.
Adverse events were assessed according to the Common Terminology Criteria for Adverse Events, version 4.03.
A total of 36 (12%) patients died during the study or during the safety follow‐up. Of these, five deaths were due to serious AEs suspected by investigators to be related to everolimus. Disease progression was cited as a contributory cause of death in three of the five cases (one patient died from multiorgan failure, one from poor general condition and acute liver failure, and one from acute renal failure). General physical health deterioration was recorded as the cause of death in the other two patients (cause of death was pneumonitis in one patient, and bilateral pneumonia in the other).
Discussion
BOLERO‐2 was the first phase III trial to show a significant improvement in PFS among postmenopausal women with NSAI‐resistant HR+, HER2− aBC treated with everolimus plus exemestane vs exemestane alone.6, 7 A subsequent non‐interventional study, BRAWO, collected data on the routine clinical use of everolimus and exemestane in postmenopausal women with HR+, HER2− aBC progressing on or after NSAI therapy.8 Although studies with different designs and study populations should be compared with caution, the current 4EVER trial results support the previously reported clinical benefits and known safety profile of everolimus plus exemestane.
Relative to BOLERO‐2, the 4EVER trial expanded patient eligibility through broader inclusion criteria; patients were required to have progressed on or after NSAI therapy; but were not limited by the time of recurrence or progression, number of previous chemotherapy lines for advanced disease, or previous exemestane therapy. Patients in 4EVER were typically older than those in the experimental arm (n = 485) of BOLERO‐2 (median 67 vs 62 years, respectively), and were of a similar age to patients in BRAWO (median 66 years).6, 8 In line with the broader inclusion criteria, patients in 4EVER were more heavily pretreated than those in BOLERO‐2: 31.7% (4EVER) vs 0% (BOLERO‐2) had received prior exemestane; 43.1% (4EVER) vs 17.0% (BOLERO‐2) had received prior fulvestrant; and 53.7% (4EVER) vs 26.0% (BOLERO‐2) had received prior chemotherapy in the metastatic setting.6
As might be expected based on the more heavily pretreated patient population, the ORR with everolimus plus exemestane in 4EVER (8.9% [95% CI: 5.8–12.9] within the first 24 weeks of treatment) was lower than in other trials with the combination in this setting (BOLERO‐2: ORR for the everolimus plus exemestane arm was 12.6% [95% CI: 9.8–15.9] at median 18‐month follow‐up;6 BRAWO: ORR for everolimus plus exemestane was 19.4% at the second interim analysis [12‐month follow‐up]).9 In contrast to BOLERO‐2 and BRAWO, tumor assessments as per the study protocol were only performed at 24 and 48 weeks/EOT in 4EVER. Furthermore, patients without known tumor assessment up to 24 weeks were defined as having an “unknown” response status and were included in ORR calculation. This leads to a rather conservative calculation of ORR24w. It is also notable that in BRAWO, patients were generally treated with everolimus plus exemestane as an earlier line of therapy than in 4EVER (first‐ or second‐line therapy: 55.0% vs 39.5%; third‐ or fourth‐line therapy: 31.6% vs 28.1%; ≥fifth‐line: 13.4% vs 31.0%, respectively).8 In addition, a lower starting dose of everolimus was used in some patients in BRAWO (5 vs 10 mg in 4EVER), potentially reducing the number of patients who required treatment interruptions.8
PFS results followed the same trend (median PFS for everolimus plus exemestane [95% CI]: 4EVER, 5.6 months [5.4–6.0 months]; BOLERO‐2, 7.8 months; BRAWO, 8.0 months [6.7–9.1 months]).6, 8 Importantly, the ORR and median PFS in 4EVER were considerably longer than in the placebo plus exemestane arm of BOLERO‐2 (ORR: 1.7% [95% CI: 0.5–4.2]); PFS: 3.2 months, local assessment).6 Median OS was not yet reached in the present study due to the short study duration (median 5.3 months from study start to last visit/progression and survival follow‐up).
In the current study, exploratory analyses indicated that prior exemestane use and prior chemotherapy use did not affect PFS achieved with everolimus plus exemestane. In contrast, visceral metastases had an impact on PFS, consistent with the known role of visceral metastases in influencing aBC prognosis.10 However, these findings should be interpreted with caution due to the post‐hoc nature of the analyses and the potential influence of the patient population. Further evaluation of the role of visceral metastases in prognosis could be explored in future trials.
Overall, the safety results of the 4EVER study were consistent with the established safety profile of everolimus plus exemestane in postmenopausal women with aBC.6, 7 This is particularly encouraging given the older, advanced, and heavily pretreated population of patients in the 4EVER trial, with many patients having seen one or even multiple lines of prior chemotherapies in the metastatic setting. In a subanalysis of BOLERO‐2, no new safety concerns were identified among elderly patients (≥65 years and ≥70 years) treated with everolimus plus exemestane, but rates of grade 3/4 adverse events were higher than younger patients (with the notable exception of stomatitis, rash, diarrhea and headache).11 Although we cannot compare between trials, it is encouraging that rates of grade 3/4 adverse events in 4EVER were more consistent with the younger population of patients (<70 years) in BOLERO‐2.11 Over half of patients in 4EVER had received prior chemotherapy for metastatic breast cancer vs 26% in the experimental arm of BOLERO‐2.6 Despite these differences, rates of adverse events indicative of bone marrow suppression were unremarkable in 4EVER, and appeared consistent with BOLERO‐2.6, 7 In 4EVER, AEs leading to permanent discontinuation of study treatment were reported for 24.7% of patients, and AEs that required at least one dose reduction of everolimus were reported for 53.7% of patients. These rates are consistent with those from BOLERO‐2 and BRAWO.8 Of note, this study did not employ standardized methods for stomatitis prevention. Prophylactic use of dexamethasone oral solution has been shown to substantially reduce the incidence and severity of stomatitis in patients receiving everolimus and exemestane and thus could help patients remain on therapy for longer.12
There were some weaknesses of the present study. Foremost, this was a single‐arm trial, which can impact the interpretation of the results, especially findings from subgroup analyses. Eighteen patients were excluded from the FAS due to concerns with data validity following GCP non‐compliance issues. However, a sensitivity analysis of the primary endpoint showed no evident difference in ORR24w of treatment (primary endpoint) regardless of whether these 18 patients were included. The relatively high number of patients with an “unknown” best overall response by week 24 was due to early study discontinuations without prior RECIST evaluation. These “unknown” responses were treated conservatively and were analyzed in the same way as progressive disease for the calculation of the ORR, as required by RECIST v1.1.
Study treatment was accompanied by deterioration in most of the tested HRQoL scores, which could be expected in this patient population and late therapy setting. Evidence‐based guidelines interpret the observed mean scores for deterioration between baseline and week 48/EOT as “medium” impact for social functioning, “small” impact for physical functioning and “trivial” impact for role functioning (EORTC QLQ‐C30 functional scale) and “medium” impact for fatigue, insomnia and loss of appetite, and “small” impact for nausea/vomiting, dyspnea and diarrhea (EORTC QLQ‐C30 symptom scale).13 Deterioration in a global health status/QoL score and in specific symptom scores was highest at the beginning of treatment and leveled off thereafter, and the affected symptom scores may have resulted from the side effects of study treatment. Efforts to proactively manage side effects, for example with stomatitis prevention strategies, may minimize the impact of treatment on quality of life in clinical practice.
In conclusion, the 4EVER study demonstrates the efficacy of everolimus plus exemestane treatment in a more advanced and heavily pretreated patient population than that included in the phase III BOLERO‐2 trial and the noninterventional BRAWO study. Safety results were in line with the known safety profile of everolimus. Future evaluation of the efficacy of everolimus plus exemestane following treatment with CDK4/6 inhibitors such as palbociclib, ribociclib or abemaciclib will be of interest.
Supporting information
Table S1. Duration of study drug exposure by treatment (safety population)
Table S2. Dose reductions (full analysis set)
Supporting Information Figure S1.
Supporting Information Figure S2.
Supporting Information Figure S3.
Acknowledgements
The authors thank the patients and their families who participated in this trial. They also acknowledge the contribution of Professor Diethelm Wallwiener of University Hospital Tübingen, Germany, who contributed to the trial and to early drafts of this manuscript. Medical editorial assistance was provided by Alison Lovibond PhD of Articulate Science LLC and was funded by Novartis Pharmaceuticals Corporation.
Conflicts of Interest: Hans Tesch received honoraria and travel expenses from Novartis Pharmaceuticals Corporation. Michael P. Lux received honoraria for lectures and advisory boards from Novartis Pharmaceuticals Corporation, Pfizer, MSD, Hexal, Roche, AstraZeneca, Eisai and Medac. Julia Kreuzeder, Mathias Muth and Claudia Quiering are employees of Novartis Pharma GmbH, Nuremberg. Diana Lüftner has been a member of a speakers’ bureau and participant in advisory boards for Novartis Pharmaceuticals Corporation. Christian M. Kurbacher reports receiving honoraria from and acting in an advisory role for Novartis Pharmaceuticals Corporation. Thomas Decker has received honoraria for advisory boards from Novartis Pharmaceuticals Corporation. Peter A. Fasching has received honoraria from Amgen, Roche, Pfizer, Novartis Pharmaceuticals Corporation, Teva and Celgene. His institution received research grants from Novartis Pharmaceuticals Corporation. Frederik Marmé reports receiving honoraria from Roche, Pfizer, Novartis Pharmaceuticals Corporation, Amgen, AstraZeneca, Genomic Health, Tesaro, Celgene and Eisai. Wolfgang Janni reports receiving research grants from Novartis Pharmaceuticals Corporation. Andrea Distelrath, Christoph Mundhenke, Oliver Stoetzer, Peyman Hadji, Andreas Schneeweiss and Florin‐Andrei Taran have no conflicts of interest to disclose.
References
- 1. National Comprehensive Cancer Network, (NCCN) . Breast Cancer. v2, 2017.
- 2. Cardoso F, Costa A, Senkus E, et al. 3rd ESO‐ESMO international consensus guidelines for Advanced Breast Cancer (ABC 3). Breast 2017;31:244–259. [DOI] [PubMed] [Google Scholar]
- 3. Rugo HS, Rumble BR, Macrae E, et al. Endocrine therapy for hormone receptor–positive metastatic breast cancer: American Society of Clinical Oncology guideline. J Clin Oncol 2016;34:3069–3103. [DOI] [PubMed] [Google Scholar]
- 4. U.S. Food and Drug Administration . FDA approves abemaciclib for HR‐positive, HER2‐negative breast cancer. Available at: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm578081.htm.
- 5. Di Leo A, Jerusalem G, Petruzelka L, et al. Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor‐positive advanced breast cancer. J Clin Oncol 2010;28:4594–4600. [DOI] [PubMed] [Google Scholar]
- 6. Yardley DA, Noguchi S, Pritchard KI, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO‐2 final progression‐free survival analysis. Adv Ther 2013;30:870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone‐receptor‐positive advanced breast cancer. N Engl J Med 2012;366:520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fasching PA, Decker T, Schneeweiss A, et al. Breast cancer treatment with everolimus and exemestane for ER+ women—results of the 2nd interim analysis of the non‐interventional trial BRAWO. Ann Oncol 2014;25:v1–v41. abstract LBA9. [Google Scholar]
- 9. Jackisch C, Grischke E, Schneeweiss A, et al. Subgroup analysis on efficacy in the routine treatment: results of the 2nd interim analysis of BRAWO, the non‐interventional trial “Breast Cancer Treatment with Everolimus and Exemestane for HR+ Women”. Cancer Res 2014;75(9, Suppl.): P5–19‐12. [Google Scholar]
- 10. Campone M, Bachelot T, Gnant M, et al. Effect of visceral metastases on the efficacy and safety of everolimus in postmenopausal women with advanced breast cancer: subgroup analysis from the BOLERO‐2 study. Eur J Cancer 2013;49:2621–2632. [DOI] [PubMed] [Google Scholar]
- 11. Prichard KI, Burris HA, Ito Y, et al. Safety and efficacy of everolimus with exemestane vs. exemestane alone in elderly patients with HER2‐negative, hormone receptor positive breast cancer in BOLERO‐2. Clin Breast Cancer 2013;13:421–432. [DOI] [PubMed] [Google Scholar]
- 12. Rugo HS, Seneviratne L, Beck JT, et al. Prevention of everolimus‐related stomatitis in women with hormone receptor‐positive, HER2‐negative metastatic breast cancer using dexamethasone mouthwash (SWISH): a single‐arm, phase 2 trial. Lancet Oncol 2017;18:654–662. [DOI] [PubMed] [Google Scholar]
- 13. Cocks K, King MT, Velikova G, et al. Evidence‐based guidelines for interpreting change scores for the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30. Eur J Cancer 2012;48:1713–1721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Duration of study drug exposure by treatment (safety population)
Table S2. Dose reductions (full analysis set)
Supporting Information Figure S1.
Supporting Information Figure S2.
Supporting Information Figure S3.