

Abstract
Autofluorescence from the intracellular metabolite, NAD(P)H, is a biomarker that is widely used and known to reliably screen and report metabolic activity as well as metabolic fluctuations within cells. As a ubiquitous endogenous fluorophore, NAD(P)H has a unique rate of fluorescence decay that is altered when bound to coenzymes. In this work we measure the shift in the fluorescence decay, or average fluorescence lifetime (1–3 ns), of NAD(P)H and correlate this shift to changes in metabolism that cells undergo during apoptosis. Our measurements are made with a flow cytometer designed specifically for fluorescence lifetime acquisition within the ultraviolet to violet spectrum. Our methods involved culture, treatment, and preparation of cells for cytometry and microscopy measurements. The evaluation we performed included observations and quantification of the changes in endogenous emission owing to the induction of apoptosis as well as changes in the decay kinetics of the emission measured by flow cytometry. Shifts in NAD(P)H fluorescence lifetime were observed as early as 15 min post‐treatment with an apoptosis inducing agent. Results also include a phasor analysis to evaluate free to bound ratios of NAD(P)H at different time points. We defined the free to bound ratios as the ratio of ‘short‐to‐long’ (S/L) fluorescence lifetime, where S/L was found to consistently decrease with an increase in apoptosis. With a quantitative framework such as phasor analysis, the short and long lifetime components of NAD(P)H can be used to map the cycling of free and bound NAD(P)H during the early‐to‐late stages of apoptosis. The combination of lifetime screening and phasor analyses provides the first step in high throughput metabolic profiling of single cells and can be leveraged for screening and sorting for a range of applications in biomedicine. © 2018 The Authors. Cytometry Part A published by Wiley Periodicals, Inc. on behalf of International Society for Advancement of Cytometry.
Keywords: fluorescence lifetime, metabolic mapping, endogenous fluorescence, flow cytometry, apoptosis, phasor, metabolism, NAD(P)H, label‐free
Short abstract

When endogenous fluorophores are purposefully excited, the emission signals that result are reliable, substantially bright, and correlate to the level of intrinsic species that fluoresce. For this reason, autofluorescence has been exploited and used as a beacon of intracellular function for purposes such as cancer diagnostics 1, 2, understanding effects of drugs, and therapies on cells 3, 4, 5, as well as for fundamental cell biology research when cells are perturbed for a myriad of reasons 6, 7. Among the numerous compounds that excite are, proteins, amino acids, lipids, flavins, and other coenzymes. All are ubiquitous to cells and emit across ultraviolet to red wavelength ranges. Owing to their ease of excitation, natural fluorophores are frequently used to draw conclusions about changes occurring in viable cells in a label‐free fashion.
Flow cytometry is a common method of evaluating cellular autofluorescence 8, 9 because intrinsic emission is measurable by many standard cytometry detector channels. Therefore, autofluorescence is naturally part of the collected cytometric signal. Whereas, the amount of autofluorescence detected might differ, cell‐to‐cell, depending on the color observed, cell type, fixative, cell viability, or level of extrinsic fluorescence. The unique ability of flow cytometry to collect multiparametric data has fostered the use of autofluorescence for compensation and to compare unlabeled cells from dimly labeled cells. Additionally, beyond the notion of autofluorescence as a ‘background’ signal, 10, 11, 12, 13, 14, 15 cytometry can involve autofluorescence for the discrimination of heterogeneous cell types (e.g., lymphocytes, cancer cells) 16, 17 or to measure single‐cell metabolic fluctuations at a higher throughput 18, 19. The use of autofluorescence as a biomarker is fairly common within the flow cytometry community overall.
In fact, considerable attention has been focused on the measurement of autofluorescence from flavins and pyridine nucleotides, these being: reduced nicotinamide adenine dinucleotide (NADH), reduced nicotinamide adenine dinucleotide phosphate (NADPH), and flavin adenine dinucleotide (FAD). These endogenous fluorophores are metabolic coenzymes/cofactors important to all cells for the production of ATP, and have been optically characterized for decades 1, 20, 21. FAD and NAD(P)H cycle from reduced to oxidized forms, thus facilitating redox reactions during energy production (e.g., oxidative phosphorylation and glycolysis). Therefore, when FAD and NAD(P)H are optically tracked (where NAD(P)H refers to both NAD cofactors), their fluorescence is correlated to changes in cellular respiration. The signals that result vary depending on the NAD(P)H reduced or oxidized form 20, 22, location inside of the cell (e.g., mitochondria, cytosol) 23, and binding status with enzymes (e.g., malate, lactate, isocitrate, and succinate dehydrogenases) 24, 25. It is this optical fluctuation during metabolic changes that makes them favorable for research in applications related to cancer 26, 27, cell proliferation 28, 29, 30, apoptosis 31, and cell differentiation 32, 33. In some cases, the bound enzyme is known or has been validated by additional biochemical analysis. Yet for many studies, the bound enzyme might depend on the variation in the metabolism and is of lesser importance compared to the purpose for detecting changes in the NAD(P)H autofluorescence. Examples of this include diagnostic imaging to reveal boundaries of demarcation between normal and cancerous tissues or measurement of optical redox ratios 29, 32, 34, 35, 36, 37, 38, 39, 40, 41.
What we find appealing and focus on herein, is the fact that NAD(P)H and FAD each have two‐component fluorescence lifetimes and an average emission decay that shifts depending on their enzyme‐bound state 21, 42. Moreover, their bound state cycles depending on activity during mitochondrial respiration through oxidative phosphorylation (bound NAD(P)H and FAD) or cytosolic energy production via glycolysis (unbound NAD(P)H and FAD) 43. The bound‐to‐unbound status is important from a fluorescence lifetime perspective because in the bound state (i.e., oxphos metabolic profile), NAD(P)H fluorescence lifetime increases from 1‐ns to up to 7‐ns depending on the binding partner 44, 45, 46, 47. Conversely, FAD lifetime is shortened by ~1 ns when bound 48, 49. These decay kinetic properties have been measured in prior studies including those described above, which demonstrate the utility of the lifetime as a metric of metabolism.
The bulk of work on lifetime shifts of NAD(P)H are accomplished with fluorescence lifetime imaging microscopy (FLIM). Many FLIM studies including intravital 28, 29, 50, 51 or in vitro approaches 37, 52, 53, 54 have established the significance of NAD(P)H lifetime as a reliable metric of metabolic fluctuations. FLIM collects data from clusters of cells and generally compares small numbers of cells (<50) with a high degree of intracellular resolution and sensitivity 55. The drawback of FLIM measurements is that cells are neither measured at high throughputs nor sorted 56. FLIM is highly sensitive in the numbers of photons it detects yet at the cost of slower integration and computational times (i.e., complex lifetime regression and deconvolution of the instrument response function) 57. Although the sensitivity of FLIM permits a highly accurate intracellular metabolic profile, the cell‐to‐cell variation might be lost as well as the ability to make rapid measurements of the metabolic changes when cells are screened against compounds that modulate metabolism.
In order to address the throughput issue, we have developed fluorescence lifetime flow cytometry (FLFC) 58, 59, 60 as a method of measuring metabolic fluctuations of individual cells at typical cytometry scan rates (i.e., ~1000 cells/sec). Our measurements with FLFC are aimed at rapidly scanning NAD(P)H fluorescence lifetime shifts in order to establish methods to profile the metabolism of large cell populations. We present a metabolic tracking study in which cultured mammalian cells are metabolically perturbed by induction of apoptosis. Apoptosis, as a well characterized programmed cell death mechanism, is known to cause a marked effect on metabolism owing to the changing energy demands of the cell as it prepares for degradation of intracellular constituents 61. While many parallels have been reported with respect to the biochemical events during apoptosis that affect metabolism, the measurements are either studied on few cells or on bulk populations 31, 33, 62. With FLFC we can rapidly screen cells at various times after apoptosis is initiated and then use NAD(P)H autofluorescence lifetime shifts to make correlations between the bound or unbound state of the metabolite during apoptosis. Therefore, we have optimized a FLFC system for NAD(P)H lifetime analysis and demonstrate that reliable NAD(P)H lifetime shifts during early and late apoptosis can be measured and used as the first step toward mapping metabolism dynamically in a screening assay that can also be combined with cell sorting.
Materials and Methods
Cell Culture and Staurosporine Treatment
HeLa cells (ATCC®, Manassas, VA) were cultured in Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific Inc., Waltham, MA) and incubated in 5% CO2 at 37°C in a humidified incubator. Cells were seeded in a 6‐well plate and allowed to reach 85–90% confluency prior to treatment with an apoptosis inducing agent, staurosporine (STS) (Cell Signaling Technology, Boston, MA). A fresh STS stock solution was made for each experiment by dissolving in dimethyl sulfoxide (DMSO) to a final concentration of 1 mM and diluting to a final concentration of 1 μM in DMEM (STS‐media). 2 ml of the STS‐media was added to each well of a 6‐well plate and cells were harvested after 15 min, 30 min, 60 min, and 180 min of treatment using Trypsin‐EDTA (5%) (Thermo Fisher Scientific Inc.). A cell pellet was obtained with centrifugation at 500 rcf for 5 min and then resuspended in 1X phosphate buffer saline (PBS).
Annexin V PI Apoptosis Assay
Apoptosis induction was verified after treatment with STS using an annexin V propidium iodide (PI) apoptosis detection kit (BD Biosciences, Franklin Lakes, NJ). Cells were analyzed for early and late apoptosis using a BD Accuri™ C6 (BD Biosciences, San Jose, CA) flow cytometer with 488 nm excitation. Fluorescence signals from cells stained with FITC and PI were collected in 530/30 nm and 585/40 nm emission channels, respectively. Cytometry results were processed with FCS Express V4 software (De Novo Software, Glendale, CA).
Fluorescence Microscopy
Epifluorescence microscopy images were collected to confirm autofluorescence measurements. Preparation included culturing cells as a monolayer in 35 mm glass bottom micro well dishes (MatTek Corporation, Ashland, MA) following the previously mentioned growth protocol. Cells were washed with PBS before imaging. In order to capture endogenous fluorescence of NAD(P)H and morphological cell features, an Axio Observer Z1/7 scope with Plan‐Apochromat 20×/0.8 M27 objective was used (ZEISS, Oberkochen, Germany). Bright field images were obtained using the Axiocam 506‐3 V TL lamp. Autofluorescence of NAD(P)H was captured using a 335–383 nm excitation filter and 420–470 nm emission filter corresponding to the NAD(P)H fluorescence spectrum 63. NAD(P)H fluorescence images (2000 ms) for untreated HeLa cells as well treated cells after 15 min, 30 min, 60 min, and 180 min of treatment were captured. Images were processed for brightness and contrast (Fiji, www.fiji.sc). Standard workflow for image‐wide cell segmentation and calculation of mean intensity from objects was followed using CellProfiler (www.cellprofiler.org) to count number of cells and quantify autofluorescence intensities.
Time Domain Spectrophotometer Measurements
A time‐correlated single photon counting (TCSPC) spectrograph (HORIBA Scientific, Edison, NJ) was used to measure fluorescence decay kinetic characteristics of bulk solutions of fluorescent microspheres and cultured cells. Glacial Blue fluorescence microspheres (5‐μm, Bangs Laboratories Inc., Fishers, IN) were first measured in order to prepare for use as calibration standards during NAD(P)H FLFC measurements. The TCSPC provided the a priori knowledge of the fluorescence lifetimes of the microspheres (measured at ~1‐ns) for use as a standard with FLFC. Subsequent to the microsphere measurements, suspensions of HeLa cells untreated and treated for apoptosis induction using STS (15 min, 30 min, 60 min, and 180 min) were also measured. The TCSPC measurement of cells provided data to confirm the lifetime shifts of NAD(P)H measurable with flow cytometry. Cell preparation included suspension in PBS (~105 cells/ml) and use of a mini magnetic stirrer during the TCSPC integration. The TCSPC system operated with a pulsed light‐emitting diode at 393 nm (DeltaDiode™ DD‐390) for excitation, and at 90o from the excitation onto the cuvette, fluorescence was collected. A photodetector collected light centered at 448 nm +/−10 nm. Data were acquired using DataStation v2.4, analysis software accompanying the instrument.
Fluorescence Lifetime Flow Cytometry
A benchtop two color channel flow cytometer was optimized for our NAD(P)H fluorescence lifetime measurements (Fig. 3). We first took the backbone of a simple flow cytometer that we commissioned and outsourced for assembly (Kinetic River Corp., San Jose, CA) with the goal of procuring a sensitive instrument capable of detecting autofluorescence. We then modified this flow cytometer by adding frequency‐domain components 58, 59, 60 and signal processing algorithms necessary for lifetime acquisition. Briefly, our frequency‐domain approach includes fluorescence lifetime measurements through the radio‐frequency modulation of an excitation source and the collection of the phase‐shifted and amplitude‐attenuated modulated emission signal. When the emission signal is compared to the excitation source, the phase shift and amplitude demodulation can be measured and used to calculate the fluorescence lifetime. Therefore, we designed this system by adding and aligning a 378 nm 60 mW solid state laser (Vortran Laser Technology Inc., Sacramento, CA). Appropriate mirrors and lenses were used in the path of the laser at orthogonal positions to obtain a collimated and focused beam. A digitally generated sinusoidal radio frequency signal was sent to the laser for modulation (Tektronik AFG‐3102 digital function generator, Beaverton, OR). The laser modulation frequency was set at 6.25 MHz for all experiments. Fluorescence emission was captured using a 448/20 nm band pass filter and a photomultiplier tube (Hamamatsu, Bridgewater, NJ). Side scattered light was captured using a 375/6 nm band pass filter and an identical PMT detector. High frequency preamplifiers and PMT voltage control (DarklingX LLC.) were added for frequency‐domain capabilities. When lifetime measurements were conducted, the PMT voltage was held constant for each experiment; intensity fluctuations were controlled with the addition of neutral density filters. A Chemyx Fusion 200 dual syringe drive injection pump (Chemyx Inc., Stafford, TX) was used to inject the samples at volumetric flow rates ranging from 0.05 to 0.5 μl/min. Phosphate buffer saline sheath fluid pressure was maintained at 25 psig. Signals after amplification were directed to a customized data acquisition system (DarklingX LLC.) digitizing the detected optical signals at 50 million samples per second 64. Digitized cytometric waveforms from fluorescence and side scatter channels were saved and used for secondary analyses with MATLAB (The MathWorks®, Natick, MA). Histograms and bivariate scatter plots were generated after collecting the peak value from each cell based on the peak of the modulated emission and side scatter signals. Fluorescence lifetime histograms were generated by calculating the average fluorescence lifetime measured per‐cell. The computational steps are described in detail elsewhere 65 and involve taking the correlated sets of fluorescence and side scatter waveforms per‐cell and processing with a fast Fourier transform (FFT). The FFT output leads to our ability to identify a phase angle difference between the fluorescence emission signal and excitation signal (scattered light collected at 90°) as well as a demodulation depth value per‐cell. Using the phase angle and demodulation depth, the average fluorescence lifetime is calculated assuming single exponential decay kinetics.
Figure 3.
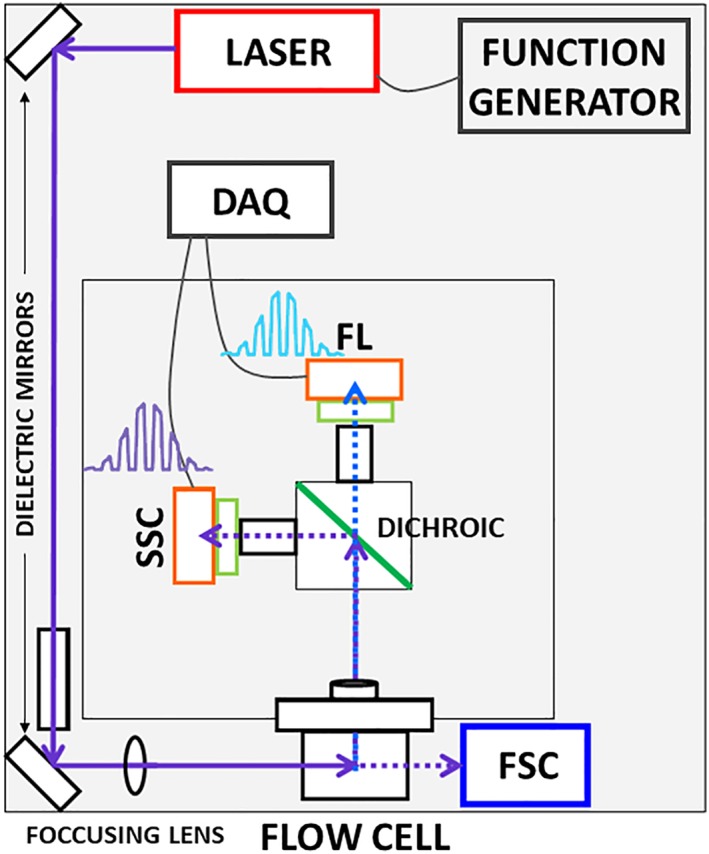
Schematic of fluorescence lifetime flow cytometer (FLFC) built for improved sensitivity to capture dim autofluorescence signals. A 375 nm excitation laser is modulated at RF frequency to excite the cells passing through the flow cell. Light after passing through different focusing lenses and dichroic mirrors is collected as side scattered light, captured at 90° angle using 376/6 nm band pass filter and fluorescence is captured using 448/20 nm band pass filter. Both signals are amplified and directed to a data acquisition system which digitizes the signal at 50 MSPS. Digitized signals are saved as comma separated files to be processed in MATLAB®. [Color figure can be viewed at wileyonlinelibrary.com]
Phasor Analyses
Moving beyond average fluorescence lifetimes, we used phasor graphs to gain insight on contributions of multiple fluorescence lifetimes per‐cell 59. Taking the phase angle and demodulation depth we developed phasor plots where a phase angle and demodulation polar vector is computed. Cartesian transformed values result in the product of demodulation and the cosine of the phase angle (x‐axis) as well as the product of demodulation and sine of the phase angle (y‐axis). All phasor plots were generated in R (r-project.org) using ggplot2 package. The universal semicircle with the equation x2 + y2 = x centered at 0.5 with a radius of 0.5 was drawn to represent the boundary where events with single exponential decay kinetic profiles reside. Therefore the (0,0) origin is interpreted as the longest lifetime possible (infinite) attributable to a zero‐demodulation point (e.g., noisiest signal).
Phasor plots constructed with FLFC data are used to unmix multiple lifetime components using a single frequency of modulation. Distinct differences in decay kinetics between free and bound NAD(P)H are thus revealed on a single phasor graph. The median phasor point for a data set is considered as a linear combination of three fluorescence species: free NAD(P)H, bound NAD(P)H, and a third long lifetime species, given by the maximum lifetime possible. Equations (1) and (2) represent the median phasor coordinates for i'th fluorescence species. The fractional contribution αi can be solved using matrix calculations using Eq. (3).
| (1) |
| (2) |
| (3) |
This permits facile calculations for relative contributions of free NAD(P)H and bound NAD(P)H thereby allowing us to quantify a free‐to‐bound NAD(P)H ratio for a large cell population with single cell resolution.
Results
Apoptosis Induction
Apoptosis was induced in HeLa cells by treatment with 1 μM staurosporine (STS). The bivariate histogram of PI‐positive cells versus annexin V‐positive cells is shown for unlabeled HeLa cells (Fig. 1A, top left), cells labeled before treatment with STS (Fig. 1A, top right), cells labeled after 1‐h STS treatment (Fig. 1A, bottom left), and cells labeled after 3 h of STS treatment (Fig. 1A, bottom right).The percentage of annexin‐V positive cells increases (from 0% to 27% then 48%) over time after treatment and at the 3 h time point the percentage of PI positive cells is the highest (9%). We also confirmed apoptosis induction with imaging by observing morphology (Fig. 1B). It is evident in the basic epifluorescence microscopy and bright field images that cells are becoming irregularly shaped and appear detached and smaller. Additionally, autofluorescence [NAD(P)H channel] was measured to evaluate changes in the autofluorescence brightness. The mean intensity values (a.u., normalized, 0 < n < 1) from the segmented objects (cell boundaries) in the fluorescence images increased from 0.01 to 0.016, 0.023, and 0.026 comparing untreated cells to cells treated after 15 min, 1 h, and 3 h, respectively (supplementary information). Mean intensity is defined as the average pixel intensities within segmented regions of interest.
Figure 1.
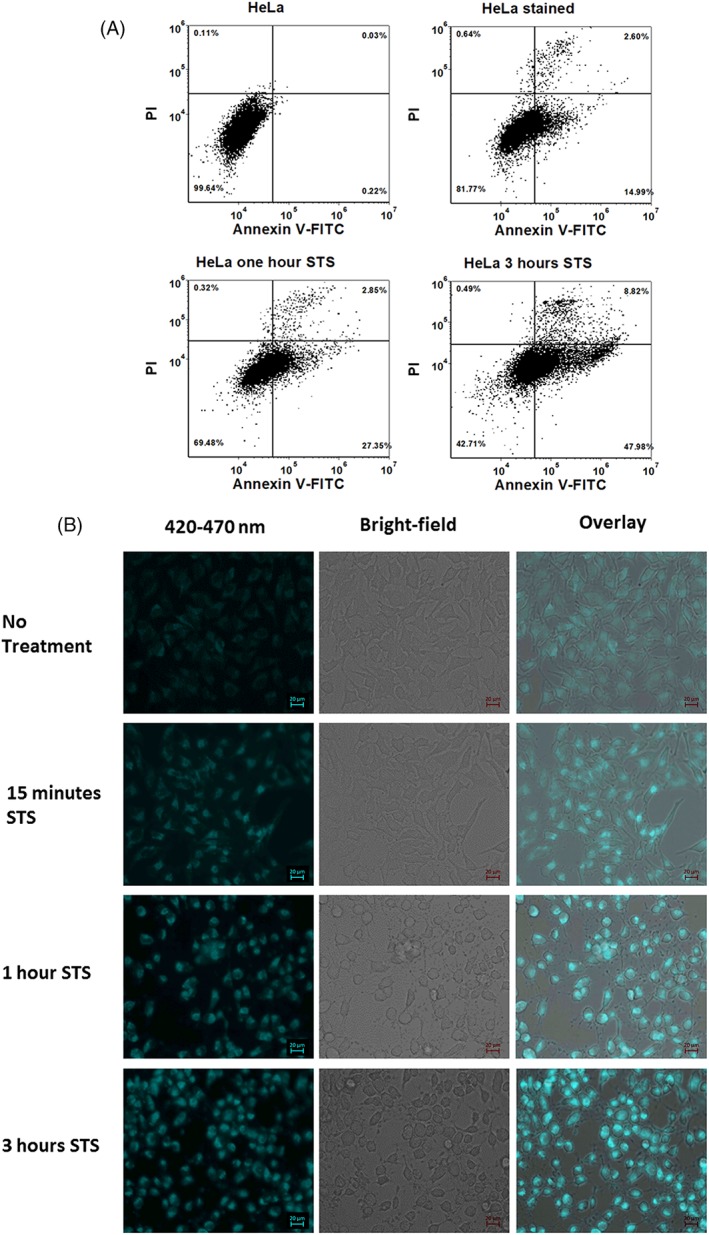
(A) Annexin V‐FITC Propidium Iodide (PI) binding assay for evaluation of apoptosis. 1 μM staurosporine (STS) was used to induce apoptosis in HeLa cells. Dot plot graphs generated from flow cytometric analysis (488 nm excitation) show percentage apoptotic cells in stained cells with no STS treatment (control), cells treated with STS for 1 h and cells treated with STS for 3 h. X‐axis represents fluorescence collected from channel with a 530/30 nm bandpass filter corresponding to Annexin V‐FITC. Y‐axis represents fluorescence collected from channel with a 585/40 nm bandpass filter corresponding to PI. Quadrant gates were generated using unstained HeLa cells as universal negative control. (B) Fluorescence microscopy of HeLa cells. Fluorescence images were obtained from unstained cells treated with STS to capture NADH fluorescence intensity. A 335–383 nm excitation filter and 420–470 nm emission filter were used. Bright‐field and fluorescence channels are overlaid. The scale bar for micrographs is 20 μm. [Color figure can be viewed at wileyonlinelibrary.com]
TCSPC Measurements
The TCSPC results include evaluation of treated cells as well as fluorescence microspheres. The Glacial blue microspheres measured for system calibration were found to have an average fluorescence lifetime of approximately 1 ns (data not shown). The TCSPC data for treated and untreated cells measured in bulk are plotted in Figure 2. A representative timed photon emission count (i.e., exponential decay curve) shown in Figure 2 is collected to confirm the direction that the fluorescence lifetime shifts with a time‐domain instrument. Using a qualitative assessment (i.e., graphical trend), the STS treated cell suspension, when measured for the bulk fluorescence decay kinetics, exhibited a longer decay than the nontreated cells. As described later, the fluorescence lifetime measured with FLFC shifted to longer decay times after 15 min and longer STS treatments. Therefore, the TCSPC measurements trend similarly.
Figure 2.
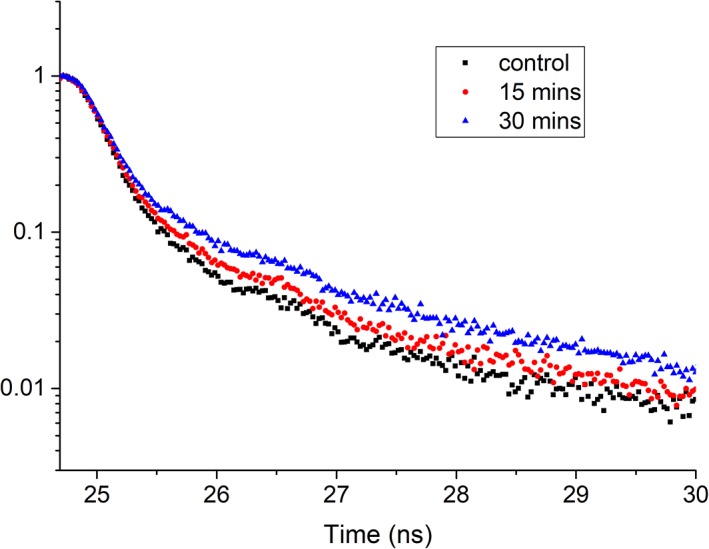
Fluorescence decay curves from cuvette measurements of HeLa cells. Untreated cell sample (control, black squares) along with cells treated with STS measured after 15 min (red dots) and 30 min (blue triangles) show a shift in the decay kinetics. A 393‐nm peak wavelength laser diode was used for excitation and emission was captured using a 448/20 nm bandpass filter. The time calibration for this instrument was equivalent to 2.64078e‐2 ns/channel. [Color figure can be viewed at wileyonlinelibrary.com]
FLFC Measurements and Phasor Analyses
The FLFC results are provided in Figures 4 and 5. Figure 4 includes representative data of the fluorescence microsphere population measured with the FLFC system. Included is a fluorescence peak histogram (Fig. 4A) and a bivariate plot of the side scatter versus fluorescence values for ~2000 microspheres measured. Figure 4A also includes statistics of the microsphere population; the mean fluorescence detected with our FLFC instrument was 2998 (a.u.) and the coefficient of variation was 13%. Figure 4B is a histogram of the average fluorescence lifetime distribution of the fluorescence microspheres used for calibration. The mean fluorescence lifetime is 0.98 ns and the standard deviation is 0.3 ns.
Figure 4.
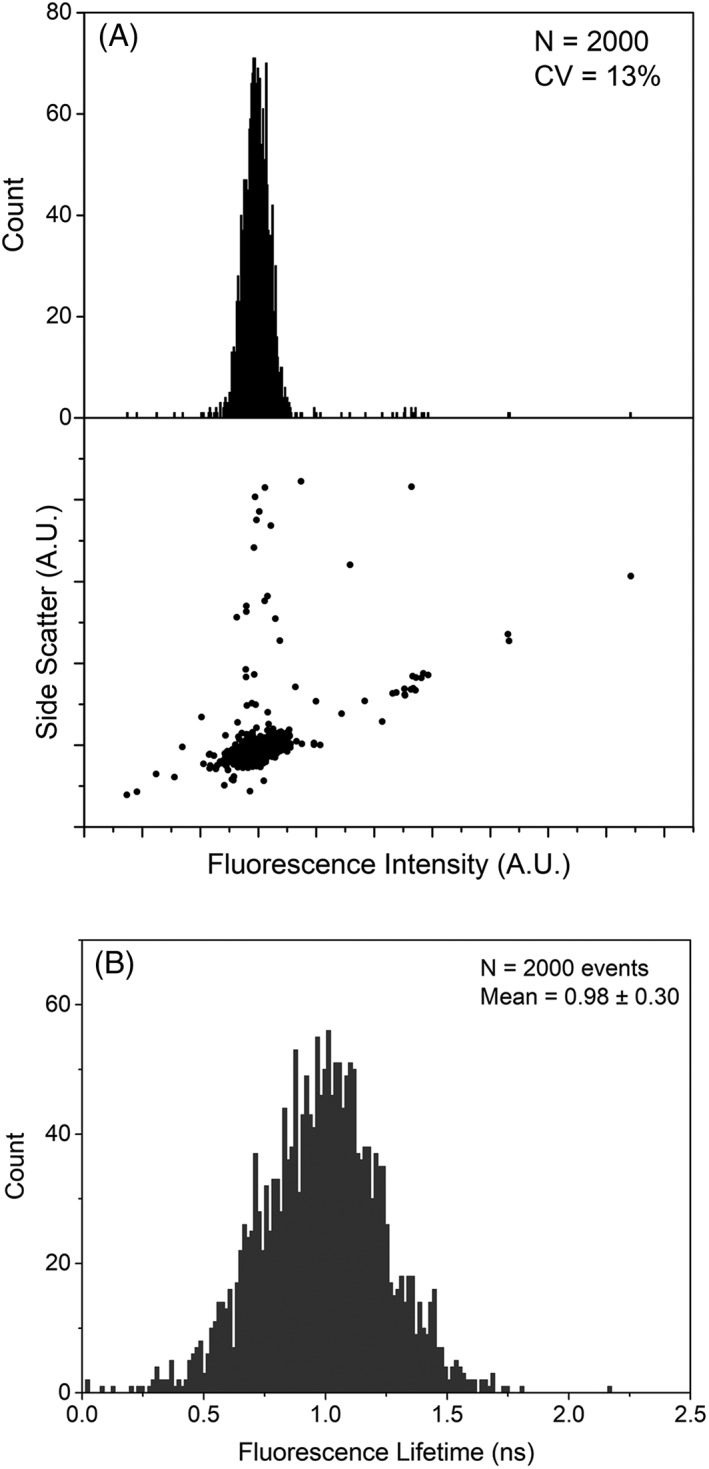
Fluorescence intensity and fluorescence lifetime histograms for glacial blue fluorescent microspheres. (A) 1‐D fluorescence histogram and dot plot (scatter vs. fluorescence). The side scatter and fluorescence are the amplitude of the modulated signals after Fourier transformation. The glacial blue amplitude measured with the FLFC resulted in a CV of 13% indicating optimal alignment and performance of the flow cytometer. (B) Fluorescence lifetime histogram for 2,000 events of glacial blue microspheres. The mean fluorescence lifetime is 0.98 ns with standard deviation of 0.3 ns.
Figure 5.
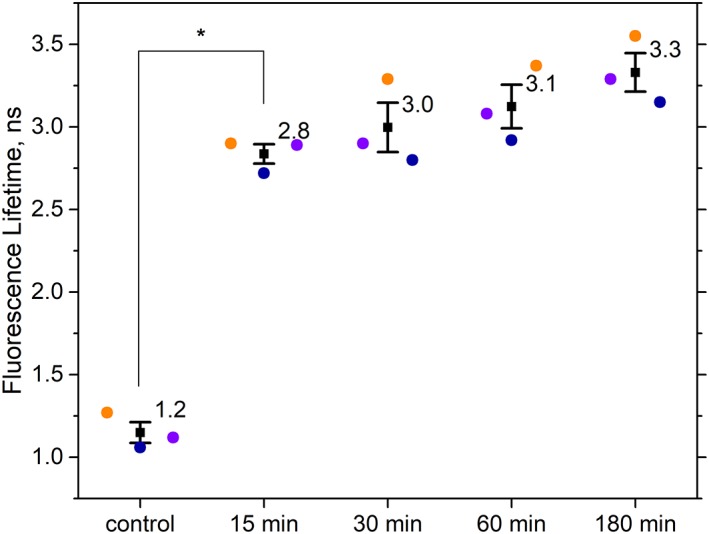
Flow cytometric fluorescence lifetime measurements of HeLa cells before and after induction of apoptosis. Mean fluorescence lifetimes of cell samples (n ~3000) are plotted for three independent experimental repeats (purple, blue, and orange circles). Fluorescence lifetimes were calculated using the phase difference between a correlated side scatter and fluorescence modulated waveform signal for each cell. Data points shown are untreated HeLa cells (control), HeLa cells treated with 1 μM STS for 15 min, 30 min, 60 min, and 180 min. Mean of the three mean fluorescence lifetime values are plotted as a square with error bars showing standard error of means. Tukey post hoc test showed a significant difference in the fluorescence lifetime between control and cells treated with STS for 15 min, with P‐value <10e‐5. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 5 provides data with results of the NAD(P)H autofluorescence lifetime measurements made before and after STS treatment. In Figure 5, are final data showing the mean and standard deviation of the resulting fluorescence lifetimes for each treatment. FLFC measurements were made for cell populations (n = 3000) where an average fluorescence lifetime was calculated per‐cell. Then an average fluorescence lifetime for each population was computed. Finally, the mean and standard deviation values provided in Figure 5 were calculated based on the three independent experimental repeats for this study. The results include averages of the untreated cell populations (i.e., zero‐time point) as well as cells measured after 15 min, 30 min, 60 min, and 180 min of STS treatment. The average fluorescence lifetime (detected for NAD(P)H emission) when HeLa cells were not treated is ~1 ns. The fluorescence lifetime then shifted to 2.8 ns at 15 min after STS treatment. At longer time points, the fluorescence lifetime gradually increased to a maximum of 3.3 ns at 3 h after treatment. Although the data are not shown, an increase in the autofluorescence intensity was also observed with the FLFC system as apoptosis proceeded. The autofluorescence intensity changes in cytometry are similar to that detected by microscopy (see Fig. 1B).
Figure 6A is a representative phasor plot of all cell populations where the individual phase and modulation are plotted per‐cell (viewed as smoothed distributions, i.e., density plot). In Figure 6A are data representative of the overall cell‐to‐cell distributions within the universal semicircle. A magnified region is inset for optimal visualization of these data. In flow cytometry, a phasor graph includes population statistics per‐cell as opposed to per‐pixel as in FLIM. From this graph the population of cells that are not undergoing apoptosis are distinct and have the lowest excited state decay time, separate from the cells undergoing treatment. When cells are treated, the population shifts, as indicated on the phasor plot with the populations higher in phase and modulation. Figure 6A also includes a centroid (dark dot), a visual guide of the location of the median of the cell population. It is obvious with the phasor plot that the longer the cells are exposed to an apoptosis‐inducing agent, the fluorescence lifetime shifts to a longer time. The data were found to be highly consistent when observing the NAD(P)H emission with our FLFC system. Figure 6B was also annotated to make a prediction about the contribution of free versus bound NAD(P)H on a per‐cell basis for a large cell population. Figure 6B shows how relative contributions of three different fluorescence species in a mixture can be visualized and calculated.
Figure 6.
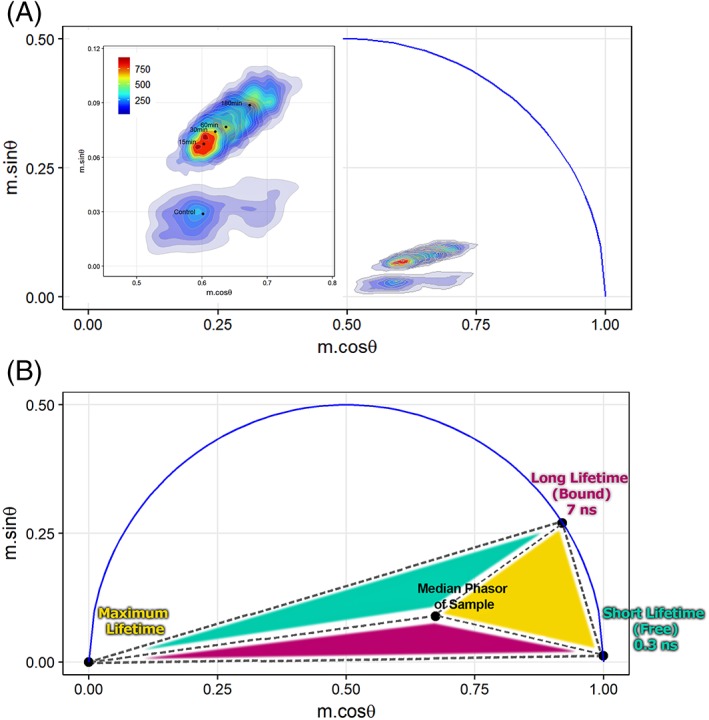
FLFC‐derived phasor plots. (A) Phasor graphs showing populations of untreated cells and cells treated with STS to trigger apoptosis. A MATLAB routine permitted extraction of the phase angle and demodulation depth from each set of correlated cytometric waveforms at 6.25 MHz. Plots were generated in R (r‐project.org) and represent individual cells by density regions where the regions of more cells are represented by warmer colors (red). Inset shows magnified view of overlaid cell populations with the median phasor points (black dots) for each sample. (B) Phasor plot with highlighted (colored) areas used to evaluate the fractional contributions. Short lifetime (S) of 0.3 ns represents the free NAD(P)H lifetime. Long lifetime (L) of 7 ns represents the bound NAD(P)H lifetime. Maximum lifetime component is chosen as the third vertex of the triangle which encompasses the cell populations collected herein. Fractional contribution of free and bound NAD(P)H can be estimated using vector algebra in phasor space as well as visualized by the total area of the triangle opposite of the vertex that specifies a short, long, or maximum lifetime (e.g., the size of the cyan colored region—opposite triangle to short lifetime vertex—is proportional to the total contribution of a short lifetime component). [Color figure can be viewed at wileyonlinelibrary.com]
The proportion of fractional contributions obtained for short lifetimes (0.3 ns) and long lifetimes (7 ns) were calculated as S/L ratios and are plotted in Figure 7. This ratio is calculated for three independent repeats for control (i.e., untreated cell sample), and cells measured after treating with STS after 15 min, 30 min, 60 min, and 180 min. The higher S/L ratio corresponds to larger free‐to‐bound NAD(P)H which is observed for the control (untreated) HeLa cell samples. Conversely, the low S/L ratio represents an increase in the bound fraction of NAD(P)H. This trend is again consistent with fluorescence lifetime results showing an increase in the average fluorescence lifetime due to an increase in bound NAD(P)H.
Figure 7.
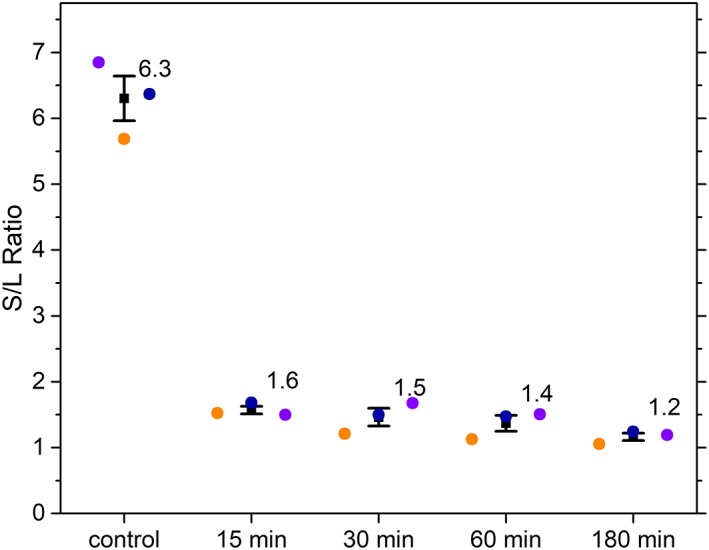
Ratio of fractional contribution of short and long lifetime components (S/L ratio) for the control cell and each time point post‐apoptosis induction. S/L ratios of each sample calculated using phasor analysis, are plotted for three independent experimental repeats (colored circles). Error bars represents standard error of the mean. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
A FLFC system was optimized 58, 59, 60, 66 and used to demonstrate how screening of individual cells for the autofluorescence lifetime shifts of NAD(P)H is a possible method of counting, and eventually sorting, cells based on metabolic shifts that occur during cell death. Our overall results include an increase in both the autofluorescence intensity and lifetime when apoptosis is induced by using STS. STS is a microbial alkaloid and nonselective inhibitor of protein kinase C (PKC) and cyclin‐dependent kinases (CDKs) 67. Based on numerous studies, STS is known to induce mitochondrial apoptosis, causing mitochondrial membrane potential disruption, cytochrome c release, Caspase 3 activation, and production of reactive oxygen species 68, 69, 70. In this study we use 1 μM of STS for treatment based on prior work showing Caspase 3 activation at 2 h after application of this dose 31. We did not evaluate for the specific NAD(P)H binding partner activity during apoptosis, which will be the focus of future work.
Our ability to screen for the fluorescence lifetime shifts with flow cytometry is important for a wide variety of metabolic studies and applications. Our FLFC system can detect autofluorescence lifetime increases at shifts resolvable based on the range of lifetimes reported for NAD(P)H. The 3‐ns value we measured confirms results of other studies in which the induction of apoptosis causes an increase in the fluorescence lifetime of NAD(P)H during STS treatment 62. The FLIM studies of these changes determined the subcellular location of NAD(P)H during apoptosis 23 and confirmed the presence of mitochondrial respiration in cultured cells. Similar trends have been observed in different cell types, in tissues, under various growth conditions, and with a range of apoptosis inducing agents 31, 33, 62, 71. In apoptosis, oxidative phosphorylation is enhanced and thus mitochondrial reactive oxygen species levels increase, which activates inflammatory responses leading to mitochondria dysfunction. In prior FLIM studies relating apoptosis to NAD(P)H fluorescence lifetimes, 23, 31, 62, 72, 73 the changes were found to occur before mitochondrial membrane potential depletion, ATP depletion, and Caspase3 activation 62. The increases we detect were also at short time points, which we selected to compare cells before and shortly after apoptosis induction.
Phasor plots in this study enable visualization of cell populations with changing fluorescence decay kinetics. Often, endogenous fluorophores exhibit two or more exponential decay profiles owing to the distribution of bound and free NAD(P)H inside the cell. Therefore the possibility of evaluating the fractional contributions of bound and unbound NAD(P)H (i.e., S/L ratio) 74, 75 is possible using the phasor analysis. Phasor graphical analyses that are derived from FLIM data utilize the reciprocal property of phasors; “gated” pixels yield a lifetime image map of a handful of cells providing an evaluation of the multiple decay kinetics. These are valuable graphical evaluations in that they provide simple ways to quantify redox ratios 54, evaluate glycolytic rates, and find absolute concentrations of NAD(P)H 76.
When the free‐to‐bound ratio (F/B) of NAD(P)H is measured, it is an important metabolic metric because it can be easily compared across samples as well as across treatments causing metabolic perturbations. Using a phasor graph to establish short‐to‐long fluorescence lifetimes of NAD(P)H we predict the actual F/B ratio at a single cell level. With a simple analysis we make this interpretation using the calculated fractional contributions (α1, α2) as well as visually, based on area of the triangles formed by the vertices and sample point (i.e., point of linear superposition falling inside the triangle). In FLIM, F/B ratios have been correlated to coefficients of the exponential decay fit (i.e., fractional contributions), which are parameters obtained after a bi‐exponential fit 50. FLIM‐phasor permit the estimation of F/B ratios by “windowing” the pixels which are closer to single exponential lifetime points corresponding to the bound and free states of NAD(P)H 45, 47. The S/L ratios that we evaluated using linear unmixing of the FLFC phasor data (Fig. 7) are comparable to the fractional coefficients reported previously in a cell death 2‐p FLIM study 51, 62.
Our calculations of the bound fraction (long lifetime) of NADH using only one lifetime (i.e., reported highest bound state lifetime: 7 ns) is limited because the bound state of NAD(P)H depends on the binding to different substrates. Additionally, we use a maximum lifetime value (i.e., origin of the phasor, see Fig. 6B) for the S/L analysis for simplicity as this is the extreme upper limit of all lifetimes possible. One would expect that measurements of NAD(P)H when bound to specific and distinct substrates, multiple distinct lifetimes results, thus more can be revealed about unique F/B ratios for a given NAD(P)H‐to‐substrate interaction 50, 77. Nonetheless, our use of a singular NAD(P)H‐bound value as well as the chosen maximum lifetime, results in an agreement between our reported S/L ratios and those reported in the literature. These data therefore represent the effect of apoptosis on free‐to‐bound ratios for large censuses of cells using the law of linear combinations within the phasor space 45.
Conclusion
As NAD(P)H emits brightly and has unique decay kinetic properties, it is quite valuable for quantitative cell science experiments. In this study, we use FLFC to collect fluorescence lifetime measurements of NAD(P)H and evaluate autofluorescence lifetimes under different cell conditions in a high throughput manner. We show how simple cytometry measurements and analyses with phasor plots provide an adequate spatial dimension for visualizing and mapping cell populations that are biochemically perturbed. The two‐fold significance of this study is that 1) it establishes a method for reliable quantitation of intracellular metabolite behavior and 2) accurately demonstrates the ability of the established method to evaluate apoptosis at early stages in a label‐free manner.
Author Contributions Statement
K.N. and F.A. wrote the paper, prepared the samples, performed measurements with the flow cytometer and made the calculations for the fluorescence lifetime values. K.D.H. led interpretation of cell experiments. J.P.H. directed the work and its interpretation and guarantee the integrity of its results.
Supporting information
Figure S1: Fluorescence images of HeLa cells before and post Staurosporine (STS) treatment. Left panels show images captured on Axio Observer Z1/7 scope with Plan‐Apochromat 20×/0.8 M27 objective (ZEISS, Oberkochen, Germany), using a 335–383 nm excitation filter and 420–470 nm emission filter. Left panel images were used as input for CellProfiler (www.cellprofiler.org) image analysis pipeline for automatic ROI segmentation. Right panels show segmented cell boundaries as output from CellProfiler analysis workflow.
Table S1: Summary of mean pixel intensity data after quantification from images in Figure S1.
Supporting information MIFlowCyt checklist
Acknowledgments
This work was supported in part by the Cowboys for Cancer Research Foundation and the by NSF CAREER award DBI‐1150202 to JPH.
Literature Cited
- 1. Heikal AA. Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies. Biomark Med 2010;4:241–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miranda‐Lorenzo I, Dorado J, Lonardo E, Alcala S, Serrano AG, Clausell‐Tormos J, Cioffi M, Megias D, Zagorac S, Balic A. Intracellular autofluorescence: a biomarker for epithelial cancer stem cells. Nat Methods 2014;11:1161–1169. [DOI] [PubMed] [Google Scholar]
- 3. Walsh AJ, Cook RS, Manning HC, Hicks DJ, Lafontant A, Arteaga CL, Skala MC. Optical metabolic imaging identifies glycolytic levels, subtypes, and early‐treatment response in breast cancer. Cancer Res 2013;73:6164–6174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Walsh AJ, Cook RS, Sanders ME, Aurisicchio L, Ciliberto G, Arteaga CL, Skala MC. Quantitative optical imaging of primary tumor organoid metabolism predicts drug response in breast cancer. Cancer Res 2014;74:5184–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cannon T, Shah A, Skala M. Autofluorescence imaging captures heterogeneous drug response differences between 2D and 3D breast cancer cultures. Biomed Opt Express 2017;8:1911–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Debreczeny MP, Bates R, Fitch RM, Galen KP, Ge J, Dorshow RB. Human skin auto‐fluorescence decay as a function of irradiance and skin type. In Proceedings of the International Society for Optics and Photonics. p 78971T.
- 7. Croce AC, Ferrigno A, Santin G, Piccolini VM, Bottiroli G, Vairetti M. Autofluorescence of liver tissue and bile: organ functionality monitoring during ischemia and reoxygenation. Lasers Surg Med 2014;46:412–421. [DOI] [PubMed] [Google Scholar]
- 8. Thorell B. Flow cytometric analysis of cellular endogenous fluorescence simultaneously with emission from exogenous fluorochromes, light scatter and absorption. Cytometry A 1981;2:39–43. [DOI] [PubMed] [Google Scholar]
- 9. Thorell B. Flow‐cytometric monitoring of intracellular flavins simultaneously with NAD (P) H levels. Cytometry A 1983;4:61–65. [DOI] [PubMed] [Google Scholar]
- 10. Sebestyén Z, Nagy P, Horváth G, Vámosi G, Debets R, Gratama JW, Alexander DR, Szöllősi J. Long wavelength fluorophores and cell‐by‐cell correction for autofluorescence significantly improves the accuracy of flow cytometric energy transfer measurements on a dual‐laser benchtop flow cytometer. Cytometry A 2002;48:124–135. [DOI] [PubMed] [Google Scholar]
- 11. Hulspas R, O'Gorman MR, Wood BL, Gratama JW, Sutherland DR. Considerations for the control of background fluorescence in clinical flow cytometry. Cytometry B Clin Cytom 2009;76:355–364. [DOI] [PubMed] [Google Scholar]
- 12. Li F, Yang M, Wang L, Williamson I, Tian F, Qin M, Shah PK, Sharifi BG. Autofluorescence contributes to false‐positive intracellular Foxp3 staining in macrophages: a lesson learned from flow cytometry. J Immunol Methods 2012;386:101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roederer M, Murphy RF. Cell‐by‐cell autofluorescence correction for low signal‐to‐noise systems: Application to epidermal growth factor endocytosis by 3T3 fibroblasts. Cytometry A 1986;7:558–565. [DOI] [PubMed] [Google Scholar]
- 14. Steinkamp JA, Stewart CC. Dual‐laser, differential fluorescence correction method for reducing cellular background autofluorescence. Cytometry A 1986;7:566–574. [DOI] [PubMed] [Google Scholar]
- 15. Corsetti JP, Sotirchos SV, Cox C, Cowles JW, Leary JF, Blumburg N. Correction of cellular autofluorescence in flow cytometry by mathematical modeling of cellular fluorescence. Cytometry A 1988;9:539–547. [DOI] [PubMed] [Google Scholar]
- 16. Shah AT, Cannon TM, Higginbotham JN, Coffey RJ, Skala MC. Autofluorescence flow sorting of breast cancer cell metabolism. J Biophotonics 2017;10:1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pantanelli SM, Li Z, Fariss R, Mahesh SP, Liu B, Nussenblatt RB. Differentiation of malignant B‐lymphoma cells from normal and activated T‐cell populations by their intrinsic autofluorescence. Cancer Res 2009;69:4911–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robinson JP, Bruner LH, Bassoe CF, Hudson JL, Ward PA, Phan SH. Measurement of intracellular fluorescence of human monocytes relative to oxidative metabolism. J Leukoc Biol 1988;43:304–310. [DOI] [PubMed] [Google Scholar]
- 19. Buschke D, Squirrell J, Vivekanandan A, Rueden C, Eliceiri K, Ogle B. Noninvasive sorting of stem cell aggregates based on intrinsic markers. Cytometry A 2014;85:353–358. [DOI] [PubMed] [Google Scholar]
- 20. Chance B, Cohen P, Jobsis F, Schoener B. Intracellular Oxidation‐Reduction States in Vivo: The microfluorometry of pyridine nucleotide gives a continuous measurement of the oxidation state. Science 1962;137:499–508. [DOI] [PubMed] [Google Scholar]
- 21. Lakowicz JR, Szmacinski H, Nowaczyk K, Johnson ML. Fluorescence lifetime imaging of free and protein‐bound NADH. Proc Natl Acad Sci 1992;89:1271–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blacker TS, Mann ZF, Gale JE, Ziegler M, Bain AJ, Szabadkai G, Duchen MR. Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. Nat Commun 2014;5:3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Levitt JM, Baldwin A, Papadakis A, Puri S, Xylas J, Münger K, Georgakoudi I. Intrinsic fluorescence and redox changes associated with apoptosis of primary human epithelial cells. J Biomed Opt 2006;11:064012. [DOI] [PubMed] [Google Scholar]
- 24. Wakita M, Nishimura G, Tamura M. Some characteristics of the fluorescence lifetime of reduced pyridine nucleotides in isolated mitochondria, isolated hepatocytes, and perfused rat liver in situ. J Biochem 1995;118:1151–1160. [DOI] [PubMed] [Google Scholar]
- 25. Piersma SR, Visser AJ, de Vries S, Duine JA. Optical spectroscopy of nicotinoprotein alcohol dehydrogenase from Amycolatopsis methanolica: a comparison with horse liver alcohol dehydrogenase and UDP‐galactose epimerase. Biochemistry 1998;37:3068–3077. [DOI] [PubMed] [Google Scholar]
- 26. Bird DK, Yan L, Vrotsos KM, Eliceiri KW, Vaughan EM, Keely PJ, White JG, Ramanujam N. Metabolic mapping of MCF10A human breast cells via multiphoton fluorescence lifetime imaging of the coenzyme NADH. Cancer Res 2005;65:8766–8773. [DOI] [PubMed] [Google Scholar]
- 27. Skala MC, Squirrell JM, Vrotsos KM, Eickhoff JC, Gendron‐Fitzpatrick A, Eliceiri KW, Ramanujam N. Multiphoton microscopy of endogenous fluorescence differentiates normal, precancerous, and cancerous squamous epithelial tissues. Cancer Res 2005;65:1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Skala MC, Riching KM, Bird DK, Gendron‐Fitzpatrick A, Eickhoff J, Eliceiri KW, Keely PJ, Ramanujam N. In vivo multiphoton fluorescence lifetime imaging of protein‐bound and free nicotinamide adenine dinucleotide in normal and precancerous epithelia. J Biomed Opt 2007;12:024014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Skala MC, Riching KM, Gendron‐Fitzpatrick A, Eickhoff J, Eliceiri KW, White JG, Ramanujam N. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc Natl Acad Sci 2007;104:19494–19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heaster TM, Walsh AJ, Zhao Y, Hiebert SW, Skala MC. Autofluorescence imaging identifies tumor cell‐cycle status on a single‐cell level. J Biophotonics 2018. Jan;11. doi: 10.1002/jbio.201600276. Epub 2017 May 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang H‐W, Ghukasyan V, Chen C‐T, Wei Y‐H, Guo H‐W, Yu J‐S, Kao F‐J. Differentiation of apoptosis from necrosis by dynamic changes of reduced nicotinamide adenine dinucleotide fluorescence lifetime in live cells. J Biomed Opt 2008;13:054011. [DOI] [PubMed] [Google Scholar]
- 32. Wallrabe H, Svindrych Z, Alam SR, Siller KH, Wang T, Kashatus D, Hu S, Periasamy A. Segmented cell analyses to measure redox states of autofluorescent NAD (P) H, FAD & Trp in cancer cells by FLIM. Sci Rep 2018;8:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buschke DG, Squirrell JM, Fong JJ, Eliceiri KW, Ogle BM. Cell death, non‐invasively assessed by intrinsic fluorescence intensity of NADH, is a predictive indicator of functional differentiation of embryonic stem cells. Biol Cell 2012;104:352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wright BK, Andrews LM, Jones MR, Stringari C, Digman MA, Gratton E. Phasor‐FLIM analysis of NADH distribution and localization in the nucleus of live progenitor myoblast cells. Microsc Res Tech 2012;75:1717–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Plotegher N, Stringari C, Jahid S, Veronesi M, Girotto S, Gratton E, Bubacco L. NADH fluorescence lifetime is an endogenous reporter of α‐synuclein aggregation in live cells. FASEB J 2015;29:2484–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alam SR, Wallrabe H, Svindrych Z, Chaudhary AK, Christopher KG, Chandra D, Periasamy A. Investigation of Mitochondrial Metabolic Response to Doxorubicin in Prostate Cancer Cells: An NADH, FAD and Tryptophan FLIM Assay. Sci Rep 2017;7:10451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stringari C, Cinquin A, Cinquin O, Digman MA, Donovan PJ, Gratton E. Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proc Natl Acad Sci U S A 2011;108:13582–13587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pate KT, Stringari C, Sprowl‐Tanio S, Wang K, TeSlaa T, Hoverter NP, McQuade MM, Garner C, Digman MA, Teitell MA, et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J 2014;33:1454–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van De Winkel M, Pipeleers D. Autofluorescence‐activated cell sorting of pancreatic islet cells: purification of insulin‐containing B‐cells according to glucose‐induced changes in cellular redox state. Biochem Biophys Res Commun 1983;114:835–842. [DOI] [PubMed] [Google Scholar]
- 40. Hanson MS, Steffen A, Danobeitia JS, Ludwig B, Fernandez LA. Flow Cytometric Quantification of Glucose‐Stimulated β‐Cell Metabolic Flux Can Reveal Impaired Islet Functional Potency. Cell Transplant 2008;17:1337–1347. [DOI] [PubMed] [Google Scholar]
- 41. Smelt M, Faas M, De Haan B, De Vos P. Pancreatic beta‐cell purification by altering FAD and NAD (P) H metabolism. Exp Diabetes Res 2008;2008:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van den Berg PA, Feenstra KA, Mark AE, Berendsen HJ, Visser AJ. Dynamic conformations of flavin adenine dinucleotide: simulated molecular dynamics of the flavin cofactor related to the time‐resolved fluorescence characteristics. J Phys Chem B 2002;106:8858–8869. [Google Scholar]
- 43. Ghukasyan VV, Kao F‐J. Monitoring cellular metabolism with fluorescence lifetime of reduced nicotinamide adenine dinucleotide. J Phys Chem C 2009;113:11532–11540. [Google Scholar]
- 44. Mossakowski AA, Pohlan J, Bremer D, Lindquist R, Millward JM, Bock M, Pollok K, Mothes R, Viohl L, Radbruch M. Tracking CNS and systemic sources of oxidative stress during the course of chronic neuroinflammation. Acta Neuropathol 2015;130:799–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Datta R, Alfonso‐García A, Cinco R, Gratton E. Fluorescence lifetime imaging of endogenous biomarker of oxidative stress. Sci Rep 2015;5:9848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leben R, Ostendorf L, van Koppen S, Rakhymzhan A, Hauser AE, Radbruch H, Niesner RA. Phasor‐Based Endogenous NAD (P) H Fluorescence Lifetime Imaging Unravels Specific Enzymatic Activity of Neutrophil Granulocytes Preceding NETosis. Int J Mol Sci 2018;19:1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Datta R, Heylman C, George SC, Gratton E. Label‐free imaging of metabolism and oxidative stress in human induced pluripotent stem cell‐derived cardiomyocytes. Biomed Opt Express 2016;7:1690–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nakashima N, Yoshihara K, Tanaka F, Yagi K. Picosecond fluorescence lifetime of the coenzyme of D‐amino acid oxidase. J Biol Chem 1980;255:5261–5263. [PubMed] [Google Scholar]
- 49. Lakowicz JR. Principles of Fluorescence Spectroscopy. New York, NY: Springer, 2006. [Google Scholar]
- 50. Sharick JT, Favreau PF, Gillette AA, Sdao SM, Merrins MJ, Skala MC. Protein‐bound NAD (P) H Lifetime is Sensitive to Multiple Fates of Glucose Carbon. Sci Rep 2018;8:5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bower AJ, Marjanovic M, Zhao Y, Li J, Chaney EJ, Boppart SA. Label‐free in vivo cellular‐level detection and imaging of apoptosis. J Biophotonics 2017;10:143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stringari C, Sierra R, Donovan PJ, Gratton E. Label‐free separation of human embryonic stem cells and their differentiating progenies by phasor fluorescence lifetime microscopy. J Biomed Opt 2012;17:046012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stringari C, Edwards RA, Pate KT, Waterman ML, Donovan PJ, Gratton E. Metabolic trajectory of cellular differentiation in small intestine by Phasor Fluorescence Lifetime Microscopy of NADH. Sci Rep 2012;2:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stringari C, Nourse JL, Flanagan LA, Gratton E. Phasor fluorescence lifetime microscopy of free and protein‐bound NADH reveals neural stem cell differentiation potential. PLoS One 2012;7:e48014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Suhling K, Hirvonen LM, Levitt JA, Chung P‐H, Tregidgo C, Le Marois A, Rusakov DA, Zheng K, Ameer‐Beg S, Poland S. Fluorescence lifetime imaging (FLIM): Basic concepts and some recent developments. Med Photon 2015;27:3–40. [Google Scholar]
- 56. Lee D‐H, Li X, Ma N, Digman MA, Lee AP. Rapid and label‐free identification of single leukemia cells from blood in a high‐density microfluidic trapping array by fluorescence lifetime imaging microscopy. Lab Chip 2018;18:1349–1358. [DOI] [PubMed] [Google Scholar]
- 57. Becker W. Fluorescence lifetime imaging–techniques and applications. J Microsc 2012;247:119–136. [DOI] [PubMed] [Google Scholar]
- 58. Cao R, Jenkins P, Peria W, Sands B, Naivar M, Brent R, Houston JP. Phasor plotting with frequency‐domain flow cytometry. Opt Express 2016;24:14596–14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sands B, Jenkins P, Peria WJ, Naivar M, Houston JP, Brent R. Measuring and sorting cell populations expressing isospectral fluorescent proteins with different fluorescence lifetimes. PLoS One 2014;9:e109940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jenkins P, Naivar MA, Houston JP. Toward the measurement of multiple fluorescence lifetimes in flow cytometry: maximizing multi‐harmonic content from cells and microspheres. J Biophotonics 2015;8:908–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Andersen JL, Kornbluth S. The tangled circuitry of metabolism and apoptosis. Mol Cell 2013;49:399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yu J‐S, Guo H‐W, Wang H‐W, Wang C‐H, Wei Y‐H. Increase of reduced nicotinamide adenine dinucleotide fluorescence lifetime precedes mitochondrial dysfunction in staurosporine‐induced apoptosis of HeLa cells. J Biomed Opt 2011;16:036008. [DOI] [PubMed] [Google Scholar]
- 63. Kirkpatrick ND, Zou C, Brewer MA, Brands WR, Drezek RA, Utzinger U. Endogenous fluorescence spectroscopy of cell suspensions for chemopreventive drug monitoring. Photochem Photobiol 2005;81:125–134. [DOI] [PubMed] [Google Scholar]
- 64. Naivar MA, Parson JD, Wilder ME, Habbersett RC, Edwards BS, Sklar L, Nolan JP, Graves SW, Martin JC, Jett JH. Open, reconfigurable cytometric acquisition system: ORCAS. Cytometry A 2007;71:915–924. [DOI] [PubMed] [Google Scholar]
- 65. Houston JP, Naivar MA, Freyer JP. Digital analysis and sorting of fluorescence lifetime by flow cytometry. Cytometry A 2010;77:861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Houston JP, Naivar MA, Jenkins P, Freyer JP. Capture of fluorescence decay times by flow cytometry. Curr Protoc Cytom 2012;59:1.25.1–1.25.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Senderowicz AM. Inhibitors of cyclin‐dependent kinase modulators for cancer therapy. Prog Drug Res 2005;63:183–206. [DOI] [PubMed] [Google Scholar]
- 68. Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng T‐I, Jones DP, Wang X. Prevention of apoptosis by Bcl‐2: release of cytochrome c from mitochondria blocked. Science 1997;275:1129–1132. [DOI] [PubMed] [Google Scholar]
- 69. Cai J, Jones DP. Superoxide in apoptosis Mitochondrial generation triggered by cytochromec loss. J Biol Chem 1998;273:11401–11404. [DOI] [PubMed] [Google Scholar]
- 70. Kruman I, Guo Q, Mattson MP. Calcium and reactive oxygen species mediate staurosporine‐induced mitochondrial dysfunction and apoptosis in PC12 cells. J Neurosci Res 1998;51:293–308. [DOI] [PubMed] [Google Scholar]
- 71. Zhao Y, Marjanovic M, Chaney EJ, Graf BW, Mahmassani Z, Boppart MD, Boppart SA. Longitudinal label‐free tracking of cell death dynamics in living engineered human skin tissue with a multimodal microscope. Biomed Opt Express 2014;5:3699–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang H‐W, Wei Y‐H, Guo H‐W. Reduced nicotinamide adenine dinucleotide (NADH) fluorescence for the detection of cell death. Anticancer Agents Med Chem 2009;9:1012–1017. [DOI] [PubMed] [Google Scholar]
- 73. Ghukasyan V, Buryakina T, Kao F‐J. Metabolic mapping of cell culture growth by NADH fluorescence lifetime imaging. Proceedings of SPIE ‐ The International Society for Optical Engineering. Communications and Photonics Conference and Exhibition (ACP), 2009 AsiaVolume: 2009‐Supplement. IEEE 2009;7634:1–8. [Google Scholar]
- 74. Lakner PH, Monaghan MG, Möller Y, Olayioye MA, Schenke‐Layland K. Applying phasor approach analysis of multiphoton FLIM measurements to probe the metabolic activity of three‐dimensional in vitro cell culture models. Sci Rep 2017;7:42730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Stringari C, Cinquin A, Cinquin O, Digman MA, Donovan PJ, Gratton E. Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proc Natl Acad Sci 2011;108:13582–13587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ma N, Digman MA, Malacrida L, Gratton E. Measurements of absolute concentrations of NADH in cells using the phasor FLIM method. Biomed Opt Express 2016;7:2441–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Radbruch H, Mothes R, Bremer D, Seifert S, Köhler R, Pohlan J, Ostendorf L, Günther R, Leben R, Stenzel W. Analyzing nicotinamide adenine dinucleotide phosphate oxidase activation in aging and vascular amyloid pathology. Front Immunol 2017;8:844. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Fluorescence images of HeLa cells before and post Staurosporine (STS) treatment. Left panels show images captured on Axio Observer Z1/7 scope with Plan‐Apochromat 20×/0.8 M27 objective (ZEISS, Oberkochen, Germany), using a 335–383 nm excitation filter and 420–470 nm emission filter. Left panel images were used as input for CellProfiler (www.cellprofiler.org) image analysis pipeline for automatic ROI segmentation. Right panels show segmented cell boundaries as output from CellProfiler analysis workflow.
Table S1: Summary of mean pixel intensity data after quantification from images in Figure S1.
Supporting information MIFlowCyt checklist