

Summary
We consider the problem of Bayesian sample size determination for a clinical trial in the presence of historical data that inform the treatment effect. Our broadly applicable, simulation-based methodology provides a framework for calibrating the informativeness of a prior while simultaneously identifying the minimum sample size required for a new trial such that the overall design has appropriate power to detect a non-null treatment effect and reasonable type I error control. We develop a comprehensive strategy for eliciting null and alternative sampling prior distributions which are used to define Bayesian generalizations of the traditional notions of type I error control and power. Bayesian type I error control requires that a weighted-average type I error rate not exceed a prespecified threshold. We develop a procedure for generating an appropriately sized Bayesian hypothesis test using a simple partial-borrowing power prior which summarizes the fraction of information borrowed from the historical trial. We present results from simulation studies that demonstrate that a hypothesis test procedure based on this simple power prior is as efficient as those based on more complicated meta-analytic priors, such as normalized power priors or robust mixture priors, when all are held to precise type I error control requirements. We demonstrate our methodology using a real data set to design a follow-up clinical trial with time-to-event endpoint for an investigational treatment in high-risk melanoma.
Keywords: Bayesian power, Clinical trial design, Power prior, Sample size determination, Sampling prior, Type I error rate
1. Introduction
We consider the problem of Bayesian sample size determination (SSD) for a clinical trial in the presence of historical data that inform the treatment effect. The purpose of our methodology is to provide a principled framework for calibrating the informativeness of a prior while simultaneously identifying the minimum sample size required for a new trial such that the overall design has adequate power and reasonable type I error control. Our design focuses on Bayesian versions of the type I error rate and power. These Bayesian operating characteristics are shown to be weighted averages of the type I error rate and power for fixed parameter values with weights determined by the posterior distribution of the parameters given the historical data after conditioning on the relevant hypothesis being true. Bayesian type I error control requires that a weighted-average type I error rate not exceed a prespecified threshold.
There are many instances where it may be of interest to borrow information on a treatment effect parameter. For example, for rare diseases it may be difficult or impossible to recruit large numbers of patients, and if trials have previously been conducted evaluating similar treatments (e.g., pharmacological products in the same drug class) in the disease population, using historical trial data to inform a prior for the effect of a similar investigational treatment may be appealing. A second example is when a trial comparing an investigational treatment to a standard of care (SOC) has an inconclusive outcome and a second trial is subsequently planned to further explore efficacy. A third example may arise when designing a trial in a geographic region under the purview of one regulatory body when similar trials were previously conducted in geographic regions under the purview of other regulatory bodies. For such scenarios, using existing information to inform a prior on the treatment effect may be reasonable, but the pertinence of the prior information will likely never be indisputable.
Pocock (1976) proposed a set of criteria to be evaluated when determining whether borrowing information on historical control groups is acceptable. These criteria may be interpreted to mean that the characteristics of the enrolled subjects, of the reference treatment, and of the effectiveness of the reference treatment as delivered to the enrolled subjects are all comparable between the historical and new trials. With these requirements met, one may also conclude that information on the effect of an investigational treatment studied in both trials might be combined without concern. However, in cases where the reference treatment corresponds to a potentially evolving SOC, when the investigational treatment is being studied in a new population (e.g., a different geographic locale), or when the investigational treatment is not identical across trials, these criteria will not be fully met. If one desires to borrow the prior information in such cases, it is important to balance that desire with the need to control type I errors in a principled way.
One of the challenges practitioners face when incorporating prior information on a treatment effect is that it is not possible to do so if one requires classical frequentist type I error control. For a design to exhibit frequentist type I error control, the type I error rate cannot exceed some prespecified threshold for any possible null value of the parameter. If one wishes to control the type I error rate in the traditional frequentist sense, all prior information must be disregarded in the analysis. This property is provable for simple normal models (see Appendix A of the supplementary material available at Biostatistics online) which inform large sample behavior for a wide class of realistic data models that are commonly used in practice. To address this challenge, we propose controlling a weighted-average (or Bayesian) type I error rate where the weights are assigned through elicitation of a null sampling prior distribution defined using the historical trial posterior distribution after conditioning on the null hypothesis. Several authors have developed methodology using notions of an average type I error rate. Examples include the work of Spiegelhalter and Freedman (1986), Brown and others (1987), Rubin and Stern (1998), Chen and others (2011, 2014a,b), and Ibrahim and others (2012). Designs based on a Bayesian type I error rate are considered by the Center for Devices and Radiological Health (CDRH) of the U.S. Food and Drug Administration (FDA), provided the historical data is of sufficient quality (Pennello and Thompson, 2007). One of the challenges with using Bayesian type I error rates is that such constructs are non-unique by nature. A principled approach is needed for eliciting the null sampling prior that defines the weights. The work of Ibrahim and others (2012) and Chen and others (2014a,b) focus on controlling a Bayesian type I error rate based on a null sampling prior that places all mass on a zero-value for the treatment effect (i.e., similar to classical frequentist type I error control). Necessarily, their approach is extremely conservative with respect to use of the prior information. Our work in this area is novel in that we formalize a procedure for translating historical data into a default null (DN) sampling prior that, in the authors’ opinion, more reasonably balances the use of the prior information with the need to control type I errors. We also provide a framework for customizing the default sampling prior weights that can be used as a mechanism for compromise between regulatory and non-regulatory stakeholders regarding the stringency of type I error control for a given historical data set.
Though our discussion thus far has focused exclusively on the control of type I error rates, ensuring adequate power is equally important. Although historical data may suggest a meaningful treatment effect, uncertainty in its magnitude is not often reflected in power calculations. Our design procedure provides a framework for more conservatively powering a trial by ensuring high weighted-average (or Bayesian) power where the weights are assigned through elicitation of an alternative sampling prior distribution defined using the historical trial posterior distribution after conditioning on the alternative hypothesis. We are not the first authors to advocate the need for more conservatively powering clinical trials or for the use of Bayesian power as a favorable construct for that purpose. Many of the works cited above explore some version of average power, as does the work of O’Hagan and Stevens (2001) who refer to this quantity as the assurance of the design. The simulation-based SSD methodology we develop herein is the first comprehensive framework that facilitates clinical trial design using these Bayesian operating characteristics that is applicable broadly to designs based on normal, binary, count, and time-to-event endpoints, including regression models.
Choosing null and alternative sampling priors is a key component of Bayesian design, as is choosing an analysis (or fitting) prior. Our method uses a partial-borrowing power prior (Ibrahim and Chen, 2000; Ibrahim and others, 2012) where the amount of borrowing is fixed a priori to ensure desired operating characteristics are met. Our formulation of the power prior is quite basic and therefore straightforward to implement with any data model for which the observed data likelihood exists in closed-form. Moreover, by exploiting an asymptotic connection between Bayesian analysis with the power prior and maximum likelihood analysis using case weights, one is able to perform design simulations with minimal use of Markov Chain Monte Carlo (MCMC) methods, greatly reducing the computational burden of the methodology.
Multiple authors have proposed more complex meta-analytic priors that attempt to let the new trial data influence how much of the prior information is used. Recent developments include the normalized power prior (NPP) (Duan and others, 2006), commensurate priors (Hobbs and others, 2011), robust meta-analytic-predictive priors (MAP) (Schmidli and others, 2014), and supervised methods (Pan and others, 2016) that manually adjust the informativeness of the prior based on measures of conflict between the prior information and the new trial data, assessed at the time of the analysis. We present simulation studies which demonstrate that, when a specific type I error constraint is placed on the design, these types of priors offer no efficiency gains over the simple partial-borrowing power prior in the sense that priors can be found resulting in designs with identical Bayesian power from each family of priors we considered.
The aforementioned simulation studies and companion case study using a data set from a real cancer clinical trial demonstrate that when one permits our proposed Bayesian type I error control, some non-zero fraction of the prior information can be incorporated into the design and analysis of the new trial. However, borrowing the prior information is not free. When the historical data posterior distribution is highly informative, the size of the future trial must be large to allow borrowing a significant amount of the available information. Furthermore, when one designs a trial to have high Bayesian power, the sample size required will generally be much larger than the sample size required for a similarly designed trial that is powered to detect the most likely treatment effect suggested by the historical data. Hence, our Bayesian design methodology is not simply a mechanism for reducing the sample size of a future trial, but rather a procedure for utilizing the information from the historical data to inform all aspects of the new trial’s design.
The rest of the article is organized as follows: in Section 2, we formally define the Bayesian type I error rate and power, develop DN and default alternative (DA) sampling priors, and provide suggestions for how these default sampling priors can be further customized. In Section 3, we develop a procedure for constructing an appropriately sized hypothesis test based on the Bayesian type I error rate. In Section 4, we present results from a simulation study that explores the properties of designs based on the Bayesian type I error rate and power and compare several choices for the analysis prior. In Section 5, we provide a detailed application of our methodology to design a follow-up trial using a time-to-event endpoint. We close the article with some discussion in Section 6.
2. A Bayesian formulation of the type I error rate and power
To formally define the Bayesian type I error rate and Bayesian power, we first need to introduce the concept of a sampling prior as described by Wang and Gelfand (2002) and extended by Chen and others (2011) to investigate the type I error rate and power. Sampling priors are also referred to as design priors (O’Hagan and Stevens, 2001), but we use the term sampling prior throughout this article. Let 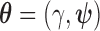 be the collection of parameters, where
be the collection of parameters, where  is the treatment effect parameter and
is the treatment effect parameter and 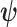 a vector of nuisance parameters. We focus on the case where the historical data informs
a vector of nuisance parameters. We focus on the case where the historical data informs 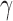 but not
but not 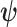 . Throughout this article, we consider the one-sided interval hypotheses
. Throughout this article, we consider the one-sided interval hypotheses 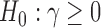 and
and 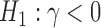 so that a negative value of
so that a negative value of 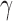 constitutes a favorable treatment effect. We write
constitutes a favorable treatment effect. We write 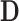 and
and 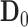 as general representations for the data from a new trial and historical trial, respectively. For this section, we write
as general representations for the data from a new trial and historical trial, respectively. For this section, we write 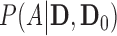 to represent the posterior probability of the event
to represent the posterior probability of the event 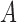 (e.g.,
(e.g., 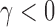 ) after observing the new trial data. Though not explicit in the notation for the posterior probability, it is important to note that the historical data influence the analysis through the chosen prior used to analyze the data. This prior is commonly referred to as the fitting (Wang and Gelfand, 2002) or analysis prior (O’Hagan and Stevens, 2001). We use the term analysis prior subsequently.
) after observing the new trial data. Though not explicit in the notation for the posterior probability, it is important to note that the historical data influence the analysis through the chosen prior used to analyze the data. This prior is commonly referred to as the fitting (Wang and Gelfand, 2002) or analysis prior (O’Hagan and Stevens, 2001). We use the term analysis prior subsequently.
2.1. Sampling priors
A sampling prior is simply a probability distribution for 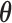 that reflects a (possibly assumed) state of knowledge about
that reflects a (possibly assumed) state of knowledge about 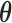 . The sampling prior, coupled with a model for the data, determine the prior-predictive distribution for future data (i.e.,
. The sampling prior, coupled with a model for the data, determine the prior-predictive distribution for future data (i.e., 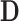 ). A sampling prior is referred to as such because it is used to sample values of
). A sampling prior is referred to as such because it is used to sample values of 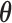 in the simulation process used to estimate operating characteristics for the trial design. We are interested in studying the type I error rate and power of the design when informative prior information on the treatment effect is to be used. In this application, one must specify sampling prior distributions for
in the simulation process used to estimate operating characteristics for the trial design. We are interested in studying the type I error rate and power of the design when informative prior information on the treatment effect is to be used. In this application, one must specify sampling prior distributions for 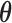 that are consistent with a true null as well as a true alternative. Let
that are consistent with a true null as well as a true alternative. Let 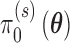 denote the null sampling prior and
denote the null sampling prior and 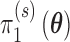 denote the alternative sampling prior. The null sampling prior will give zero weight to values of
denote the alternative sampling prior. The null sampling prior will give zero weight to values of 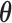 having a negative
having a negative 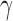 component and the alternative sampling prior will give zero weight to values of
component and the alternative sampling prior will give zero weight to values of 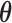 having a non-negative
having a non-negative 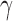 component.
component.
2.2. Defining the Bayesian type I error rate and power
For a fixed value of 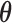 , define the null hypothesis rejection rate
, define the null hypothesis rejection rate 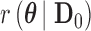 as
as 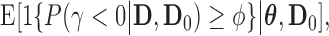 where
where 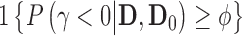 is an indicator that one accepts
is an indicator that one accepts 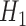 based on the posterior probability
based on the posterior probability 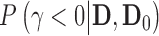 and prespecified critical value
and prespecified critical value 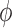 . For chosen null and alternative sampling priors, define the Bayesian type I error rate as
. For chosen null and alternative sampling priors, define the Bayesian type I error rate as 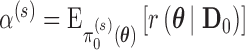 and Bayesian power as
and Bayesian power as 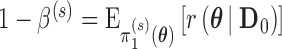 . Thus, the Bayesian type I error rate and power are weighted averages of
. Thus, the Bayesian type I error rate and power are weighted averages of 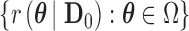 with weights determined by
with weights determined by 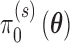 and
and 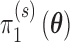 , respectively. We note that Chen and others (2011) define the Bayesian type I error rate and power as expectations of
, respectively. We note that Chen and others (2011) define the Bayesian type I error rate and power as expectations of 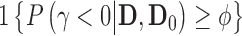 with respect to the null and alternative prior-predictive distribution for the data,
with respect to the null and alternative prior-predictive distribution for the data, 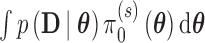 and
and 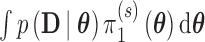 , respectively. The two definitions are equivalent since
, respectively. The two definitions are equivalent since
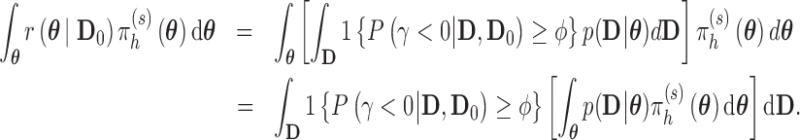 |
We have only changed the order of integration to highlight the fact that the Bayesian type I error rate and power are weighted averages of the quantities based on fixed values of  . Our recipe for simulation-based estimation of the Bayesian type I error rate and power follows closely with the development in Chen and others (2011).
. Our recipe for simulation-based estimation of the Bayesian type I error rate and power follows closely with the development in Chen and others (2011).
2.3. Default null and alternative sampling priors
The Bayesian type I error rate and power only become well-defined upon choosing a set of null and alternative sampling priors. From the Bayesian perspective, it is natural that the sampling priors reflect one’s belief about  gleaned from the historical data. Logical choices for the null and alternative sampling priors are
gleaned from the historical data. Logical choices for the null and alternative sampling priors are 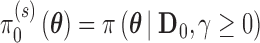 (conditional on
(conditional on 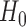 ) and
) and 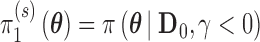 (conditional on
(conditional on 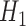 ), respectively. We refer to this pair of sampling priors as the default sampling priors.
), respectively. We refer to this pair of sampling priors as the default sampling priors.
Figure 1 presents a histogram of the marginal posterior distribution for 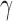 based on the historical data from the case study presented in Section 5 along with the corresponding DN and DA marginal sampling priors for
based on the historical data from the case study presented in Section 5 along with the corresponding DN and DA marginal sampling priors for 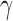 . Though we have only plotted the marginal sampling priors for
. Though we have only plotted the marginal sampling priors for 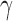 , conditioning on the null or alternative hypothesis obviously induces changes in the entire joint distribution for
, conditioning on the null or alternative hypothesis obviously induces changes in the entire joint distribution for 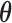 . Since the historical data are more consistent with the alternative hypothesis than the null, conditioning on the null hypothesis induces a sampling prior distribution for
. Since the historical data are more consistent with the alternative hypothesis than the null, conditioning on the null hypothesis induces a sampling prior distribution for 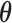 that is necessarily less consistent with the historical data, but still plausible given the historical data under the assumption that the null hypothesis is true. In other words, the DN sampling prior uses the historical data to provide a frame of reference regarding which null values of
that is necessarily less consistent with the historical data, but still plausible given the historical data under the assumption that the null hypothesis is true. In other words, the DN sampling prior uses the historical data to provide a frame of reference regarding which null values of  are realistic. This is reasonable to the extent that one believes that the historical data are pertinent to the new trial. Having a realistic sampling prior distribution for all of
are realistic. This is reasonable to the extent that one believes that the historical data are pertinent to the new trial. Having a realistic sampling prior distribution for all of  is important for design simulations even if one only borrows information through the treatment effect parameter as we have proposed. This is because the nuisance parameters (e.g., the baseline hazard parameters) may influence important secondary characteristics of the design, such as the time required to accrue the desired number of events for an event driven trial with time-to-event endpoint.
is important for design simulations even if one only borrows information through the treatment effect parameter as we have proposed. This is because the nuisance parameters (e.g., the baseline hazard parameters) may influence important secondary characteristics of the design, such as the time required to accrue the desired number of events for an event driven trial with time-to-event endpoint.
Fig. 1.
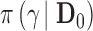 and corresponding default marginal sampling priors for
and corresponding default marginal sampling priors for 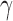 .
.
From inspecting Figure 1, one can see that the null sampling prior gives the most weight to 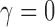 but not all the weight is given to that value. By allowing a weighted-average type I error rate to be controlled at some prespecified significance level (e.g., 0.025), as opposed to enforcing strict type I error control at the same level for
but not all the weight is given to that value. By allowing a weighted-average type I error rate to be controlled at some prespecified significance level (e.g., 0.025), as opposed to enforcing strict type I error control at the same level for 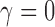 (i.e., frequentist type I error control), one permits some degree of information borrowing from the historical trial. Of course, the point-wise type I error rate will necessarily exceed the weighted-average value for some values of
(i.e., frequentist type I error control), one permits some degree of information borrowing from the historical trial. Of course, the point-wise type I error rate will necessarily exceed the weighted-average value for some values of 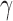 .
.
While the default sampling priors are natural, it may be desirable to modify them for a number of reasons. For example, stakeholders may feel that the tails of one or both of the default priors are unrealistic. In Appendix B of the supplementary material available at Biostatistics online, we describe using tail-truncation to modify the default sampling priors to create truncated null (TN) and alternative (TA) sampling priors. This process entails defining a constant 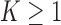 and restricting the default priors by requiring
and restricting the default priors by requiring 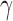 to be at least
to be at least 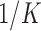 times as likely as the modal value for each of the default priors. In the simulations presented in Section 4 and the application presented in Section 5, we investigate using
times as likely as the modal value for each of the default priors. In the simulations presented in Section 4 and the application presented in Section 5, we investigate using 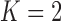 as a compromise between no borrowing and borrowing based on the default priors.
as a compromise between no borrowing and borrowing based on the default priors.
3. Constructing a size 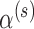 Bayesian hypothesis test
Bayesian hypothesis test
Thus far, we have defined a natural approach for using historical data to assign weights to the null and alternative parameter spaces that are used in the formulation of the Bayesian type I error rate and power. In this section, we discuss our approach for constructing a hypothesis test that provides Bayesian power no less than 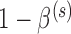 while controlling the Bayesian type I error rate at level
while controlling the Bayesian type I error rate at level 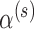 . We achieve this by constructing an informative power prior from the historical trial data where the discounting parameter is calibrated through simulation to ensure the desired properties.
. We achieve this by constructing an informative power prior from the historical trial data where the discounting parameter is calibrated through simulation to ensure the desired properties.
The class of power priors proposed by Ibrahim and Chen (2000) provide a natural means for controlling the informativeness of the prior. We consider the partial-borrowing power prior of Ibrahim and others (2012), which only borrows information through the treatment effect. The partial-borrowing power prior has the following form:
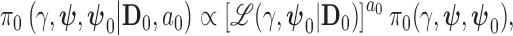 |
(3.1) |
where 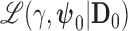 is the likelihood for the historical trial data including a shared treatment effect,
is the likelihood for the historical trial data including a shared treatment effect, 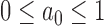 is a fixed scalar parameter, and
is a fixed scalar parameter, and 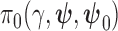 is a non-informative initial prior for all parameters. When
is a non-informative initial prior for all parameters. When 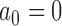 , the historical data are discarded and the power prior reduces to the initial prior. In contrast, when
, the historical data are discarded and the power prior reduces to the initial prior. In contrast, when 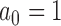 , the power prior corresponds to the posterior distribution from an analysis of the historical data using the initial prior. For intermediate values of
, the power prior corresponds to the posterior distribution from an analysis of the historical data using the initial prior. For intermediate values of 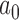 , the information in the historical data is diminished to some degree leading to a prior that is more informative than the initial prior but less informative than using the historical trial posterior as the prior for the new trial. When information is borrowed through
, the information in the historical data is diminished to some degree leading to a prior that is more informative than the initial prior but less informative than using the historical trial posterior as the prior for the new trial. When information is borrowed through 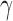 , there is little power gain for hypothesis tests about
, there is little power gain for hypothesis tests about  by also borrowing information through nuisance parameters (i.e., by assuming
by also borrowing information through nuisance parameters (i.e., by assuming 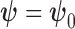 ).
).
In addition to specifying an analysis prior, one must also construct a decision rule that emits a size  hypothesis test with respect to the Bayesian type I error rate. There are many ways to do this. We suggest setting the posterior probability critical value
hypothesis test with respect to the Bayesian type I error rate. There are many ways to do this. We suggest setting the posterior probability critical value 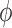 to
to 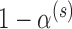 and then maximizing the borrowing parameter
and then maximizing the borrowing parameter 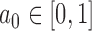 subject to the Bayesian type I error rate restriction. This approach is motivated by the fact that the posterior probability
subject to the Bayesian type I error rate restriction. This approach is motivated by the fact that the posterior probability 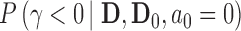 (or simply
(or simply 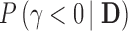 ) is asymptotically equivalent to a frequentist p-value when
) is asymptotically equivalent to a frequentist p-value when 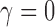 . In other words,
. In other words, 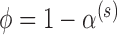 is the asymptotically correct choice to achieve frequentist type I error control at level
is the asymptotically correct choice to achieve frequentist type I error control at level 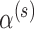 when there is no borrowing. Thus, the mathematical exercise of maximizing
when there is no borrowing. Thus, the mathematical exercise of maximizing 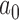 can be conceptualized as starting with a hypothesis test that asymptotically controls the classical frequentist type I error rate at level
can be conceptualized as starting with a hypothesis test that asymptotically controls the classical frequentist type I error rate at level 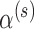 (i.e., based on
(i.e., based on 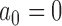 ) and then borrowing increasing amounts of information from the historical data until the test is size
) and then borrowing increasing amounts of information from the historical data until the test is size 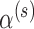 with respect to the Bayesian type I error rate (or until
with respect to the Bayesian type I error rate (or until 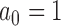 ). In Appendix C of the supplementary material available at Biostatistics online, we provide a recipe for conducting simulation studies to identify the minimum sample size required for the new trial and corresponding maximum value of
). In Appendix C of the supplementary material available at Biostatistics online, we provide a recipe for conducting simulation studies to identify the minimum sample size required for the new trial and corresponding maximum value of 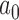 , subject to Bayesian type I error and power requirements. In the same appendix, we describe how
, subject to Bayesian type I error and power requirements. In the same appendix, we describe how 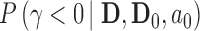 can be approximated without MCMC to make large-scale design simulations less computationally demanding.
can be approximated without MCMC to make large-scale design simulations less computationally demanding.
4. Simulation study
In this section, we present results from a simulation study designed to illustrate properties of designs based on the Bayesian type I error rate and power and to demonstrate that designs based on the power prior with fixed 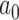 are as powerful as designs based on more complicated meta-analytic priors under the constraint of Bayesian type I error control when borrowing information on a treatment effect. For ease of exposition, we consider a simple normal model for the new and historical trial data. The model contains a single mean parameter
are as powerful as designs based on more complicated meta-analytic priors under the constraint of Bayesian type I error control when borrowing information on a treatment effect. For ease of exposition, we consider a simple normal model for the new and historical trial data. The model contains a single mean parameter 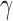 and standard deviation (SD) parameter
and standard deviation (SD) parameter 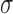 which we assume to be known and, without loss of generality, equal to 1. Our goal is to test the hypothesis
which we assume to be known and, without loss of generality, equal to 1. Our goal is to test the hypothesis 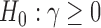 and
and 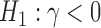 . In Appendix D of the supplementary material available at Biostatistics online, we explain how simulation studies using the simple normal model might inform our intuition for a wide class of realistic data models used in practice.
. In Appendix D of the supplementary material available at Biostatistics online, we explain how simulation studies using the simple normal model might inform our intuition for a wide class of realistic data models used in practice.
We first generated five hypothetical historical data sets with sample size 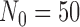 ranging from strongly favoring treatment efficacy to mildly favoring treatment efficacy. In particular, we generated the historical data sets such that
ranging from strongly favoring treatment efficacy to mildly favoring treatment efficacy. In particular, we generated the historical data sets such that 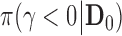 = 0.990, 0.975, 0.95, 0.90, and 0.85. Figure S1 of supplementary material available at Biostatistics online presents the historical posterior distributions for
= 0.990, 0.975, 0.95, 0.90, and 0.85. Figure S1 of supplementary material available at Biostatistics online presents the historical posterior distributions for 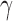 for three of these simulated data sets. The regions associated with TN and TA sampling priors (based on taking
for three of these simulated data sets. The regions associated with TN and TA sampling priors (based on taking 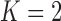 ) are shaded on the figure as well.
) are shaded on the figure as well.
For each historical data set, we identified the sample size required for a future trial, denoted by 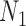 , and corresponding value of
, and corresponding value of 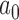 such that the design controls the Bayesian type I error rate at level
such that the design controls the Bayesian type I error rate at level 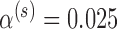 while assuring Bayesian power
while assuring Bayesian power 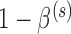 is either 0.80 (for the three most informative cases) or 0.70 (for the two least informative cases). The Bayesian type I error rate and power were defined using TN and TA sampling priors. We also computed the Bayesian type I error rate associated with a point-mass null sampling prior that places all mass at
is either 0.80 (for the three most informative cases) or 0.70 (for the two least informative cases). The Bayesian type I error rate and power were defined using TN and TA sampling priors. We also computed the Bayesian type I error rate associated with a point-mass null sampling prior that places all mass at 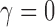 and the Bayesian power associated with a point-mass alternative (PA) sampling prior centered at
and the Bayesian power associated with a point-mass alternative (PA) sampling prior centered at 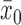 , the historical trial sample mean.
, the historical trial sample mean.
Having identified the optimal choice of 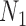 and
and 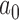 for each historical data set using the proposed design method, we then evaluated whether the design could be improved by using an analysis prior that allows the new trial data to influence the amount of borrowing (i.e., a meta-analytic prior). We considered the NPP (Duan and others, 2006) which models
for each historical data set using the proposed design method, we then evaluated whether the design could be improved by using an analysis prior that allows the new trial data to influence the amount of borrowing (i.e., a meta-analytic prior). We considered the NPP (Duan and others, 2006) which models 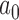 as a random variable and gives it a prior, a two-part mixture prior similar to the robust MAP prior (Schmidli and others, 2014), and a power prior where
as a random variable and gives it a prior, a two-part mixture prior similar to the robust MAP prior (Schmidli and others, 2014), and a power prior where 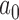 is calculated at the time of the analysis based on a statistic that measures prior-data conflict. We refer to the last type of prior as a supervised-borrowing power prior. We also considered an approach that fixes
is calculated at the time of the analysis based on a statistic that measures prior-data conflict. We refer to the last type of prior as a supervised-borrowing power prior. We also considered an approach that fixes 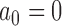 and modifies the posterior probability critical value (i.e.,
and modifies the posterior probability critical value (i.e., 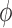 ) to obtain a size
) to obtain a size 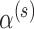 Bayesian hypothesis test. This approach makes no use of the historical data in the analysis prior. For all methods based on a power prior, we used the initial prior
Bayesian hypothesis test. This approach makes no use of the historical data in the analysis prior. For all methods based on a power prior, we used the initial prior 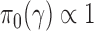 for which it is easy to see that the posterior distribution is proper once at least one data point is observed.
for which it is easy to see that the posterior distribution is proper once at least one data point is observed.
4.1. Specification of meta-analytic priors
The NPP has the following form 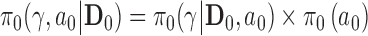 with
with 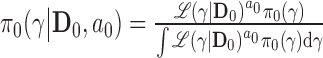 , where
, where 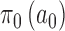 and
and 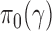 are initial priors. We optimized the design based on the NPP by identifying an ideal choice for
are initial priors. We optimized the design based on the NPP by identifying an ideal choice for 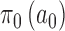 from among the family of beta distributions indexed by mean parameter
from among the family of beta distributions indexed by mean parameter 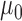 and dispersion parameter
and dispersion parameter 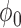 . The optimal settings for both
. The optimal settings for both 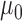 and
and 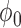 were determined using a grid search over the discrete parameter space defined by
were determined using a grid search over the discrete parameter space defined by 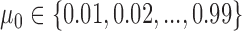 and
and 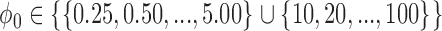 . Note that as
. Note that as 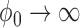 , the NPP becomes equivalent to a power prior with
, the NPP becomes equivalent to a power prior with 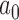 fixed and equal to
fixed and equal to 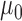 .
.
The two-part mixture prior we considered is closely related to the robust MAP prior proposed by Schmidli and others (2014) but was tailored to deal with the problem of borrowing information on a treatment effect rather than historical controls. In our context, a robust MAP prior could be defined as 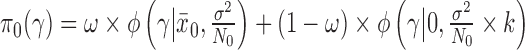 , where
, where 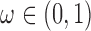 is a mixing weight,
is a mixing weight, 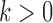 is a variance inflation factor that controls the level of informativeness of the robust component of the prior, and
is a variance inflation factor that controls the level of informativeness of the robust component of the prior, and 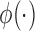 is a normal density. To identify an optimal value of
is a normal density. To identify an optimal value of 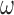 and
and 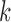 , we performed a grid search over
, we performed a grid search over 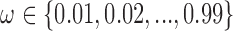 and
and 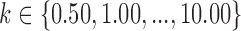 .
.
For the supervised borrowing power prior, we set 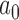 to
to 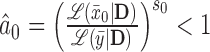 , where
, where 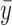 is the sample mean from the new trial and
is the sample mean from the new trial and 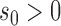 is a calibration parameter. The chosen statistic has several nice properties. Specifically, when
is a calibration parameter. The chosen statistic has several nice properties. Specifically, when 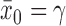 , then as
, then as  it is easy to see that
it is easy to see that 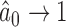 . In addition, for fixed
. In addition, for fixed 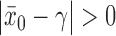 , as
, as 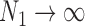 it is easy to see that
it is easy to see that 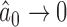 . For
. For 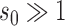 , the historical data will be discounted greatly for even modest absolute differences
, the historical data will be discounted greatly for even modest absolute differences 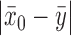 . For
. For 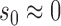 , virtually all the information will be borrowed regardless of the magnitude of
, virtually all the information will be borrowed regardless of the magnitude of 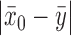 . To identify an optimal value of
. To identify an optimal value of 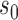 , we performed a grid search over
, we performed a grid search over 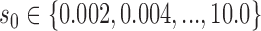 .
.
4.2. Simulation results
The estimated Bayesian type I error rate and power based on TN and TA sampling priors for the optimal power prior with fixed 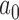 (FPP), optimal NPP, optimal supervised borrowing power prior (SPP), optimal robust MAP prior (MAP), and for the approach that takes
(FPP), optimal NPP, optimal supervised borrowing power prior (SPP), optimal robust MAP prior (MAP), and for the approach that takes 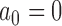 and modifies the posterior probability critical value (MCV) are provided in Table 1. The corresponding estimates based on point-mass null and alternative sampling priors are provided in Table S1 of supplementary material available at Biostatistics online. In each case, the type I error rate and power were estimated using
and modifies the posterior probability critical value (MCV) are provided in Table 1. The corresponding estimates based on point-mass null and alternative sampling priors are provided in Table S1 of supplementary material available at Biostatistics online. In each case, the type I error rate and power were estimated using 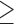 200 000 simulation studies, resulting in highly precise estimates of these operating characteristics.
200 000 simulation studies, resulting in highly precise estimates of these operating characteristics.
Table 1.
Bayesian type I error rate and power using truncated sampling priors (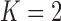 )
)
| Type I error rate | Power | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
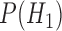
|
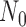
|
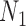
|
FPP | MAP | SPP | NPP | MCV | FPP | MAP | SPP | NPP | MCV |
| 0.990 | 50 | 77 | 0.025 | 0.025 | 0.025 | 0.024 | 0.025 | 0.798 | 0.797 | 0.798 | 0.797 | 0.798 |
| 0.975 | 50 | 115 | 0.025 | 0.025 | 0.025 | 0.024 | 0.025 | 0.805 | 0.804 | 0.805 | 0.804 | 0.805 |
| 0.950 | 50 | 168 | 0.025 | 0.025 | 0.025 | 0.025 | 0.025 | 0.800 | 0.799 | 0.799 | 0.797 | 0.800 |
| 0.900 | 50 | 192 | 0.025 | 0.025 | 0.025 | 0.025 | 0.025 | 0.702 | 0.701 | 0.702 | 0.704 | 0.702 |
| 0.850 | 50 | 320 | 0.016 | 0.015 | 0.015 | 0.015 | 0.025 | 0.701 | 0.700 | 0.700 | 0.700 | 0.738 |
The fact that Bayesian type I error rates agree for all methods in Table 1 is by construction. The only exception is for the scenario where 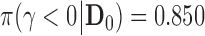 . In that case, all the methods that make use of the historical data in the analysis prior (i.e., FPP, MAP, SPP, and NPP) result in borrowing essentially all the information in the historical data without attaining a size
. In that case, all the methods that make use of the historical data in the analysis prior (i.e., FPP, MAP, SPP, and NPP) result in borrowing essentially all the information in the historical data without attaining a size 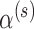 test (Bayesian type I error rate
test (Bayesian type I error rate  1.5%). In contrast, the MCV approach can always produce a size
1.5%). In contrast, the MCV approach can always produce a size 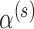 test since that approach is not restricted by the amount of information in the historical data. Of course, such a restriction is quite reasonable, and so we would not advocate for the MCV approach in general. Our reason for including it in the simulation is to convey that one can produce a decision rule that controls the Bayesian type I error rate at level
test since that approach is not restricted by the amount of information in the historical data. Of course, such a restriction is quite reasonable, and so we would not advocate for the MCV approach in general. Our reason for including it in the simulation is to convey that one can produce a decision rule that controls the Bayesian type I error rate at level 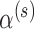 with or without using the historical data in the analysis prior and that those decision rules are equivalent in terms of power. This is an important point as it highlights the fact that even though our approach uses the historical data to define the sampling priors and in the analysis prior, there is effectively only a single use of the historical data: to define the weights associated with the Bayesian type I error rate and power. We chose to incorporate the historical data into a power prior because, by doing so, we are able to quantify the fraction of the prior information that is incorporated in the analysis.
with or without using the historical data in the analysis prior and that those decision rules are equivalent in terms of power. This is an important point as it highlights the fact that even though our approach uses the historical data to define the sampling priors and in the analysis prior, there is effectively only a single use of the historical data: to define the weights associated with the Bayesian type I error rate and power. We chose to incorporate the historical data into a power prior because, by doing so, we are able to quantify the fraction of the prior information that is incorporated in the analysis.
The characteristics of the priors resulting in optimally powered designs are provided in Table 2. For both the MAP and NPP priors, there are multiple hyperparameters that can be manipulated and so, not surprisingly, there were numerous choices for the hyperparameters that led to designs with nearly identical operating characteristics (none better than designs based on the FPP). For inclusion in Table 2, we selected from designs with 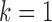 for the MAP prior and
for the MAP prior and 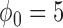 for the NPP priors. For all classes of priors, the prior characteristics that lead to a controlled type I error rate depend on the level of informativeness of the historical data and the sample size in the new study. Thus, there is no silver bullet prior that provides precise performance with respect to Bayesian type I error control while providing consistently high Bayesian power. If one cares about controlling type I error rates in a precise way, any prior will have to be calibrated to ensure good properties and the authors would argue that this fact makes more complicated meta-analytic priors significantly less appealing as general use tools in this setting.
for the NPP priors. For all classes of priors, the prior characteristics that lead to a controlled type I error rate depend on the level of informativeness of the historical data and the sample size in the new study. Thus, there is no silver bullet prior that provides precise performance with respect to Bayesian type I error control while providing consistently high Bayesian power. If one cares about controlling type I error rates in a precise way, any prior will have to be calibrated to ensure good properties and the authors would argue that this fact makes more complicated meta-analytic priors significantly less appealing as general use tools in this setting.
Table 2.
Characteristics of analysis priors for designs based on truncated sampling priors
| FPP | MAP | SPP | NPP | MCV | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Prior | Med. Post. 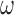
|
Med. 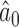
|
Med. 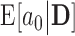
|
|||||||||

|
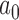
|
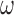
|
Null | Alt |
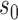
|
Null | Alt |
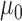
|
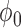
|
Null | Alt |
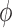
|
| 0.990 | 0.12 | 0.60 | 0.20 | 0.89 | 3.76 | 0.00 | 0.24 | 0.06 | 5.00 | 0.10 | 0.14 | 0.965 |
| 0.975 | 0.23 | 0.64 | 0.28 | 0.87 | 2.02 | 0.00 | 0.40 | 0.19 | 5.00 | 0.21 | 0.25 | 0.961 |
| 0.950 | 0.44 | 0.74 | 0.45 | 0.89 | 0.96 | 0.00 | 0.58 | 0.41 | 5.00 | 0.41 | 0.45 | 0.955 |
| 0.900 | 0.82 | 0.91 | 0.81 | 0.95 | 0.52 | 0.11 | 0.72 | 0.82 | 5.00 | 0.82 | 0.83 | 0.948 |
| 0.850 | 1.00 | 0.99 | 0.98 | 0.99 | 0.02 | 0.90 | 0.98 | 0.99 | 5.00 | 0.99 | 0.99 | 0.933 |
Med., Median; Post., Posterior.
5. Application: design of a follow-up trial in high-risk melanoma
The E1684 trial was a randomized controlled trial conducted to assess the utility of interferon alfa-2b (INF) as an adjuvant therapy following surgery for deep primary or regionally metastatic melanoma. A detailed analysis of the trial was given by Kirkwood and others (1996). The design and primary analysis were stratified by disease stage according to four groups (i) deep primary melanomas of Breslow depth more than 4 mm, (ii) primary melanomas of any tumor stage in the presence of N1 regional lymph node metastasis detected at elective lymph node dissection with clinically inapparent regional lymph node metastasis, (iii) clinically apparent N1 regional lymph node involvement synchronous with primary melanoma of T1–4, and (iv) regional lymph node recurrence at any interval after appropriate surgery for primary melanoma of any depth. Subjects treated with INF demonstrated a statistically significant prolongation of relapse-free survival compared to those receiving the standard of care (SOC) based on a stratified log-rank test (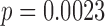 , one-sided). To demonstrate our methodology, we restrict our attention to subjects from the fourth stratum, treating this group as a hypothetical historical trial that demonstrated inconclusive efficacy.
, one-sided). To demonstrate our methodology, we restrict our attention to subjects from the fourth stratum, treating this group as a hypothetical historical trial that demonstrated inconclusive efficacy.
Table S2 of supplementary material available at Biostatistics online presents summary relapse-free survival data by treatment group and number of positive nodes at lymphadenectomy. The number of positive lymph nodes was used as a stratification variable in our design model due to its prognostic value. We analyzed the historical trial data using a stratified Cox model (Cox, 1972) with a piecewise constant baseline hazard. Use of the stratified Cox model is ubiquitous in the analysis of time-to-event data and modeling the baseline hazard with a piecewise constant function is a common approach for Bayesian analyzes (Ibrahim and others, 2001). We assume a model with a common treatment effect and stratum-specific baseline hazard for this exercise. To identify the best model for the baseline hazard, we considered all possible models with 10 or fewer components (per stratum) defined using the deciles from the observed event-time distribution as the set of possible change points. Each model was fit with MCMC using a non-informative normal prior for the treatment effect (mean zero and variance 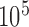 ) and an independent non-informative gamma prior for each baseline hazard parameter (shape and inverse scale parameters equal to
) and an independent non-informative gamma prior for each baseline hazard parameter (shape and inverse scale parameters equal to 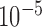 ). The best model was selected using the deviance information criterion (DIC) (Spiegelhalter and others, 2002).
). The best model was selected using the deviance information criterion (DIC) (Spiegelhalter and others, 2002).
Table S3 of supplementary material available at Biostatistics online presents the posterior mean, the posterior SD, and 95% highest posterior density (HPD) interval for the treatment effect parameter (log hazard ratio for treatment versus control, denoted by 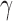 ) and for the baseline hazard parameters (i.e.,
) and for the baseline hazard parameters (i.e., 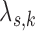 with
with 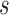 indexing stratum and
indexing stratum and 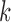 indexing baseline hazard component) using the best model based on 100 000 MCMC samples. The right endpoints for the time axis partition for each stratum (denote as
indexing baseline hazard component) using the best model based on 100 000 MCMC samples. The right endpoints for the time axis partition for each stratum (denote as 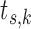 ) are given in the rightmost column of Table S3 of supplementary material available at Biostatistics online. It is clear from the HPD interval for the treatment effect that the historical trial data suggest the treatment is efficacious but that the evidence is not overwhelming by traditional criteria. Thus, it is reasonable to assume that if these data were collected in a clinical trial, an additional trial might be conducted, the design and analysis of which would be informed by these data. Since the comparator group is a potentially evolving standard of care, there is the potential for the relative efficacy of the investigational therapy to change as the SOC evolves, thus motivating the need to balance the informativeness of the prior with the desire to limit the probability of a type I error.
) are given in the rightmost column of Table S3 of supplementary material available at Biostatistics online. It is clear from the HPD interval for the treatment effect that the historical trial data suggest the treatment is efficacious but that the evidence is not overwhelming by traditional criteria. Thus, it is reasonable to assume that if these data were collected in a clinical trial, an additional trial might be conducted, the design and analysis of which would be informed by these data. Since the comparator group is a potentially evolving standard of care, there is the potential for the relative efficacy of the investigational therapy to change as the SOC evolves, thus motivating the need to balance the informativeness of the prior with the desire to limit the probability of a type I error.
In the remainder of this section, our primary goal is to demonstrate the application of our Bayesian design procedure using the E1684 data. To that end, we consider a design that: (i) controls the Bayesian type I error rate at no more than 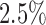 based on the DN sampling prior, (ii) controls the Bayesian type I error rate at no more than 2.5% based on the TN sampling prior using
based on the DN sampling prior, (ii) controls the Bayesian type I error rate at no more than 2.5% based on the TN sampling prior using 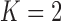 , and (iii) controls the Bayesian type I error rate at no more than 2.5% based on the limiting case of the TN sampling prior that fixes
, and (iii) controls the Bayesian type I error rate at no more than 2.5% based on the limiting case of the TN sampling prior that fixes 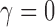 (i.e.,
(i.e., 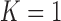 ). We refer to this limiting case as the frequentist-like null (FN) sampling prior. For power, we consider a design that: (i) provides 80% Bayesian power based on a PA sampling prior with parameters set to the posterior means in Table S3 of supplementary material available at Biostatistics online, (ii) provides 80% Bayesian power based on the TA sampling prior using
). We refer to this limiting case as the frequentist-like null (FN) sampling prior. For power, we consider a design that: (i) provides 80% Bayesian power based on a PA sampling prior with parameters set to the posterior means in Table S3 of supplementary material available at Biostatistics online, (ii) provides 80% Bayesian power based on the TA sampling prior using 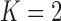 , and (iii) provides 80% Bayesian power based on the DA sampling prior.
, and (iii) provides 80% Bayesian power based on the DA sampling prior.
For all simulation studies, the generative model for the baseline hazard in the new trial used the time axis partition from Table S3 of supplementary material available at Biostatistics online. Let 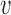 be the number of events at which the new trial will stop and
be the number of events at which the new trial will stop and 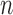 be the total number of subjects enrolled. In time-to-event trials, the number of events is the key determinant of power. For each possible
be the total number of subjects enrolled. In time-to-event trials, the number of events is the key determinant of power. For each possible 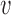 , we took
, we took 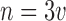 and simulated uniform accrual over a 4-year period with no censoring other than administrative censoring that occurred when the planned number of events had been reached. Subjects were allocated to strata in proportions similar to the historical trial (i.e.,
and simulated uniform accrual over a 4-year period with no censoring other than administrative censoring that occurred when the planned number of events had been reached. Subjects were allocated to strata in proportions similar to the historical trial (i.e., 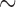 50% to stratum one) and balanced randomization was used. The time axis partition from Table S3 of supplementary material available at Biostatistics online was used in the fitted model.
50% to stratum one) and balanced randomization was used. The time axis partition from Table S3 of supplementary material available at Biostatistics online was used in the fitted model.
For a given null sampling prior, the first step in the design process is to find the maximum value of  that yields Bayesian type I error control for each value of
that yields Bayesian type I error control for each value of 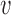 in the set under consideration. To do this, we performed 100 000 simulation studies for each
in the set under consideration. To do this, we performed 100 000 simulation studies for each 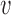 to estimate the Bayesian type I error rate over a range of
to estimate the Bayesian type I error rate over a range of 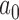 values. We then used LOESS methods to smooth these estimates and to interpolate the precise value of
values. We then used LOESS methods to smooth these estimates and to interpolate the precise value of 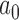 that corresponded to the desired error rate. Figure 2 presents LOESS curves of the Bayesian type I error rate as a function of
that corresponded to the desired error rate. Figure 2 presents LOESS curves of the Bayesian type I error rate as a function of 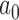 for each null sampling prior (DN, FN, TN) and for
for each null sampling prior (DN, FN, TN) and for 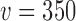 ,
, 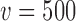 , and
, and 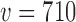 . As expected, it is clear that no information can be borrowed from the historical trial when the type I error rate is based on the FN sampling prior. The estimates obtained for
. As expected, it is clear that no information can be borrowed from the historical trial when the type I error rate is based on the FN sampling prior. The estimates obtained for 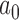 using this null sampling prior were always approximately zero for every value of
using this null sampling prior were always approximately zero for every value of  we considered (
we considered (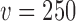 to
to 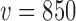 ). In contrast, we see that when either of the TN or DN sampling priors is used to define the Bayesian type I error rate, we are able to borrow much and sometimes all of the information in the historical data without surpassing the threshold. Note that a relatively large number of events are required for the future trial to allow borrowing all the information from the historical trial when using the DN sampling prior (
). In contrast, we see that when either of the TN or DN sampling priors is used to define the Bayesian type I error rate, we are able to borrow much and sometimes all of the information in the historical data without surpassing the threshold. Note that a relatively large number of events are required for the future trial to allow borrowing all the information from the historical trial when using the DN sampling prior (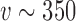 ) and TN sampling prior (
) and TN sampling prior (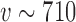 ). Thus, although our Bayesian version of type I error control is less restrictive than frequentist type I error control, there is still significant restriction on the amount of information that can be borrowed.
). Thus, although our Bayesian version of type I error control is less restrictive than frequentist type I error control, there is still significant restriction on the amount of information that can be borrowed.
Fig. 2.
LOESS curves and point estimates for Bayesian type I error rate as a function of  for
for  , and
, and 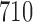 for each null sampling prior. Each point estimate is based on 100 000 simulation studies.
for each null sampling prior. Each point estimate is based on 100 000 simulation studies.
At a certain point, the worst-case Bayesian type I error rate (i.e., based on the FN sampling prior) may become too large to satisfy stakeholders even if the average type I error rate based on the DN (or TN) sampling prior is reasonable. We recommend always exploring the worst-case type I error rate since it has traditionally been the focus in clinical trials. This can be easily accomplished by calculating the Bayesian type I error rate based on the FN sampling prior using the value of 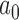 identified using the null sampling prior chosen for design. Figure S2 of supplementary material available at Biostatistics online presents the worst-case Bayesian type I error rate associated with use of both the DN and TN sampling priors. As shown in the figure, the worst-case type I error rate is approximately two to three times the average rate which was consistent with the simulation studies presented in Section 4.
identified using the null sampling prior chosen for design. Figure S2 of supplementary material available at Biostatistics online presents the worst-case Bayesian type I error rate associated with use of both the DN and TN sampling priors. As shown in the figure, the worst-case type I error rate is approximately two to three times the average rate which was consistent with the simulation studies presented in Section 4.
After identifying the maximum amount of information that can be borrowed from the historical trial for each possible value of 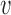 , the next step is to determine the smallest value of
, the next step is to determine the smallest value of 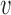 that provides adequate power under a chosen alternative sampling prior. For each alternative sampling prior considered, we performed 100 000 simulation studies using each
that provides adequate power under a chosen alternative sampling prior. For each alternative sampling prior considered, we performed 100 000 simulation studies using each 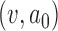 pair to estimate the Bayesian power. We then used LOESS methods to smooth the estimated Bayesian power curves and to interpolate a pair of values
pair to estimate the Bayesian power. We then used LOESS methods to smooth the estimated Bayesian power curves and to interpolate a pair of values  that provided 80% power. Figure 3 presents LOESS curves of the Bayesian power as a function of
that provided 80% power. Figure 3 presents LOESS curves of the Bayesian power as a function of 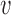 for each combination of null sampling prior (DN, TN, and FN) and alternative sampling prior (DA, TA, and PA).
for each combination of null sampling prior (DN, TN, and FN) and alternative sampling prior (DA, TA, and PA).
Fig. 3.
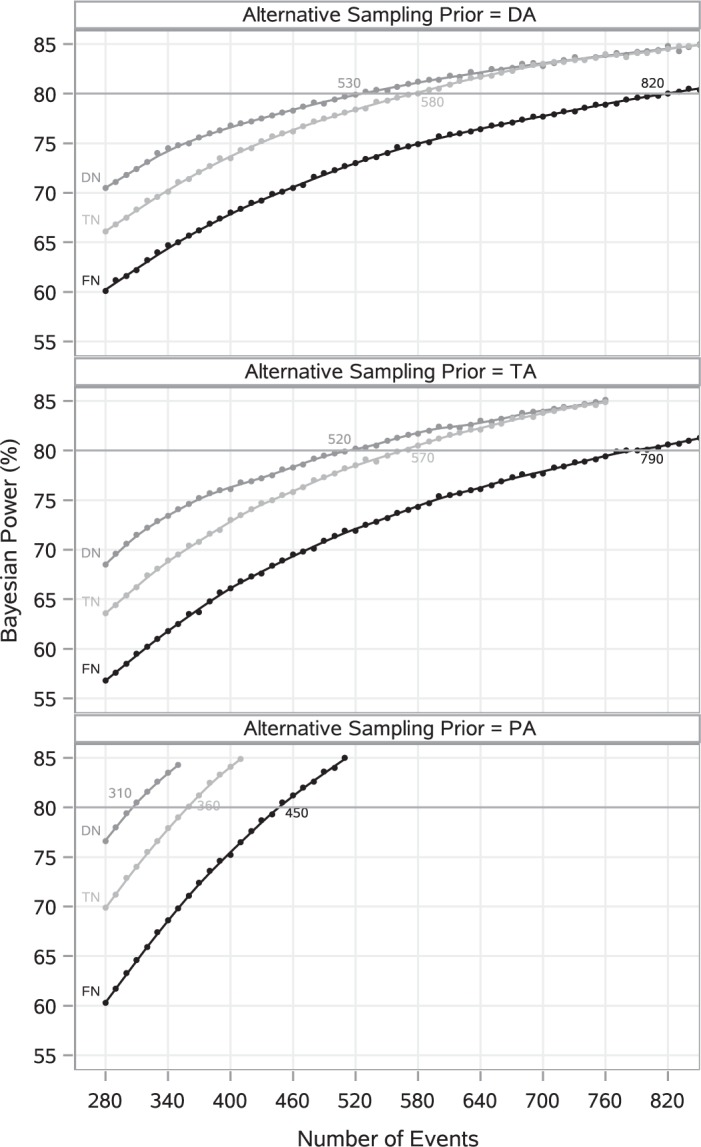
LOESS curves and point estimates for Bayesian power as a function of 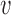 for each combination of null sampling prior and alternative sampling prior. Each point estimate is based on 100 000 simulation studies.
for each combination of null sampling prior and alternative sampling prior. Each point estimate is based on 100 000 simulation studies.
When no borrowing is permitted (i.e., the FN sampling prior is used), 820, 790, and 450 events are required to have 80% Bayesian power based on the DA, TA, and optimistic PA sampling priors, respectively. This case is instructive because it allows us to consider the implications of using Bayesian power as compared to the traditional fixed-point power approach while still adhering to traditional frequentist type I error control. When Bayesian power is based on the DA or TA sampling prior, the required number of events is much more conservative compared to the traditional approach. Regardless of the alternative sampling prior chosen for power analysis, we see that when Bayesian type I error control is based on the DN or TN sampling prior, far fewer events are needed for the new trial compared to the case where it is based on the FN sampling prior.
All of the results presented in this section were obtained using the asymptotic approximation described in Appendix C of the supplementary material available at Biostatistics online. Using exact Bayesian inference with MCMC, we performed 50 000 confirmatory simulation studies for each number of events shown in Figure 3 (along with their associated values of 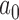 ) to verify the accuracy of the estimated Bayesian type I error rate and power. In all cases the estimated Bayesian power was within 1.0% of the targeted level and the estimated Bayesian type I error rate was within 0.25% of the targeted rate.
) to verify the accuracy of the estimated Bayesian type I error rate and power. In all cases the estimated Bayesian power was within 1.0% of the targeted level and the estimated Bayesian type I error rate was within 0.25% of the targeted rate.
6. Discussion
The results that we have presented in this article confirm that in the presence of information on a treatment effect, one must either disregard that information in the analysis of a new trial or relax the classical frequentist approach to type I error control. Some would argue that the entire notion of type I error control is non-Bayesian. However, in the authors’ opinion, any time information on a treatment effect (or any other parameter for that matter) is incorporated from outside a randomized, controlled trial, the pertinence of that information to the problem at hand will likely not be indisputable. This is because many of Pocock’s insightful criteria (Pocock, 1976) for justifiably borrowing information from historical controls (or on all subjects) will either be partially unverifiable or not met outright. If we are to therefore admit evidence of a treatment effect on grounds other than classic Bayesian exchangeability assumptions, we need statistical procedures that help balance the potential efficiency gains of doing so with unintended negative consequences that will arise when in reality, despite sound rationale, the prior information is not pertinent.
In the results presented in Section 4.2, we compared designs based on the partial-borrowing power prior to those based on meta-analytic priors (e.g., a robust mixture prior) under a common type I error constraint. In those simulations, we did not allow for the possibility of adapting the designs when the meta-analytic priors were used. However, if the amount of information being borrowed at the time of the analysis is less than expected, one may expect that by adaptively increasing sample size in the new trial and postponing the analysis, one could obtain a more efficient design. Such an approach is not feasible for the partial-borrowing power prior with fixed 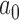 . In fact, this type strategy arguably leads to a less efficient design. We discuss this in more detail in Appendix E of the supplementary material available at Biostatistics online.
. In fact, this type strategy arguably leads to a less efficient design. We discuss this in more detail in Appendix E of the supplementary material available at Biostatistics online.
The apparent conflict between frequentist type I error control and Bayesian analysis with an informative prior for the treatment effect is undeniable. In contrast, when one borrows information only on the nuisance parameters (i.e., only on control subjects) the conflict is less obvious. One cannot simply condition on the null hypothesis as a means of constructing a null sampling prior distribution that can be used to define a meaningful Bayesian type I error rate. In future work, we will consider designs that borrow information on control subjects only, giving practical advice for defining and using the DN and DA sampling priors in that setting.
It may be desirable to borrow information from multiple historical trials through a fixed prior specified a priori. It is conceptually straightforward to adapt our methodology to this situation. For example, one could use a hierarchical model to synthesize information across historical data sets (e.g., Cox model with a random effect for study) and obtain the posterior distribution for the treatment effect through sampling. This posterior distribution could then be approximated with high precision using a normal distribution, 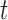 -distribution, or finite-mixture of normal distributions. This analytic form could then be used in place of the historical trial likelihoods in a power prior-like framework. Of course, this approach would likely require MCMC methods to fit the model.
-distribution, or finite-mixture of normal distributions. This analytic form could then be used in place of the historical trial likelihoods in a power prior-like framework. Of course, this approach would likely require MCMC methods to fit the model.
Supplementary Material
Acknowledgments
The authors wish to thank the editor, associate editor, and three reviewers for helpful comments and suggestions which have led to an improvement of this article. Conflict of Interest: None declared.
Funding
National Institutes of Health (NIH) (#GM 70335 and P01CA142538), in part.
References
- Brown B. W., Herson J., Atkinson E. N. and Rozell M. E. (1987). Projection from previous studies: a Bayesian and frequentist compromise. Controlled Clinical Trials 8, 29–44. [DOI] [PubMed] [Google Scholar]
- Chen M.-H., Ibrahim J. G., Lam P., Yu A. and Zhang Y. (2011). Bayesian design of noninferiority trials for medical devices using historical data. Biometrics 67, 1163–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.-H., Ibrahim J. G., Amy X. H., Liu T. and Hennessey V. (2014a). Bayesian sequential meta-analysis design in evaluating cardiovascular risk in a new antidiabetic drug development program. Statistics in Medicine 33, 1600–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.-H., Ibrahim J. G., Zeng D., Hu K. and Jia C. (2014b). Bayesian design of superiority clinical trials for recurrent events data with applications to bleeding and transfusion events in myelodysplastic syndrome. Biometrics 70, 1003–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D. R. (1972). Regression models and life tables. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 34, 187–220. [Google Scholar]
- Duan Y., Ye K. and Smith E. P. (2006). Evaluating water quality using power priors to incorporate historical information. Environmetrics 17, 95–106. [Google Scholar]
- Hobbs B. P., Carlin B. P., Mandrekar S. J. and Sargent G. J. (2011). Hierarchical commensurate and power prior models for adaptive incorporation of historical information in clinical trials. Biometrics 67, 1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim J. G. and Chen M.-H. (2000). Power prior distributions for regression models. Statistical Science 15, 46–60. [Google Scholar]
- Ibrahim J. G., Chen M.-H. and Sinha D. (2001). Bayesian Survival Analysis. New York: Springer Science & Business Media. [Google Scholar]
- Ibrahim J. G., Chen M.-H., Xia H. A. and Liu T. (2012). Bayesian meta-experimental design: evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. Biometrics 68, 578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood J. M., Strawderman M. H., Ernstoff M. S., Smith T. J., Borden E. C. and Blum R. H. (1996). Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. Journal of Clinical Oncology 14, 7–17. [DOI] [PubMed] [Google Scholar]
- O’Hagan A. and Stevens J. (2001). Bayesian assessment of sample size for clinical trials of cost-effectiveness. Medical Decision Making 21, 219–230. [DOI] [PubMed] [Google Scholar]
- Pan H., Yuan Y. and Xia J. (2016). A calibrated power prior approach to borrow information from historical data with application to biosimilar clinical trials. Journal of the Royal Statistical Society: Series C (Applied Statistics) 66, 979–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennello G. and Thompson L. (2007). Experience with reviewing Bayesian medical device trials. Journal of Biopharmaceutical Statistics 18, 81–115. [DOI] [PubMed] [Google Scholar]
- Pocock S. (1976). The combination of randomized and historical controls in clinical trials. Journal of Chronic Diseases 29, 175–188. [DOI] [PubMed] [Google Scholar]
- Rubin D. B. and Stern H. S. (1998). Sample size determination using posterior predictive distributions. Sankhyā: The Indian Journal of Statistics, Series B 60, 161–175. [Google Scholar]
- Schmidli H., Gsteiger S., Roychoudhury S., O’Hagan A., Spiegelhalter D. and Neuenschwander B. (2014). Robust meta-analytic-predictive priors in clinical trials with historical control information. Biometrics 70, 1023–1032. [DOI] [PubMed] [Google Scholar]
- Spiegelhalter D. J. and Freedman L. S. (1986). A predictive approach to selecting the size of a clinical trial, based on subjective clinical opinion. Statistics in Medicine 5, 1–13. [DOI] [PubMed] [Google Scholar]
- Spiegelhalter D. J., Best N. G., Carlin B. P. and Van Der Linde A. (2002). Bayesian measures of model complexity and fit. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 64, 583–639. [Google Scholar]
- Wang F. and Gelfand A. E. (2002). A simulation-based approach to Bayesian sample size determination for performance under a given model and for separating models. Statistical Science 17, 193–208. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.