

Abstract
TARP syndrome (talipes equinovarus, atrial septal defect, Robin sequence, and persistence of the left superior vena cava) is a rare X‐linked syndrome often resulting in pre‐ or post‐natal lethality in affected males. In 2010, RBM10 was identified as the disease‐causing gene, and we describe the first adult patient with TARP syndrome at age 28 years, hereby expanding the phenotypic spectrum. Our patient had Robin sequence, atrial septal defect, intellectual disability, scoliosis, and other findings previously associated with TARP syndrome. In addition, he had a prominent nose and nasal bridge, esotropia, displacement of lacrimal points in the cranial direction, small teeth, and chin dimple, which are the findings that have not previously been associated with TARP syndrome. Our patient was found to carry a hemizygous c.273_283delinsA RBM10 mutation in exon 4, an exon skipped in three of five protein‐coding transcripts, suggesting a possible explanation for our patient surviving to adulthood. Direct sequencing of maternal DNA indicated possible mosaicism, which was confirmed by massive parallel sequencing. One of two sisters were heterozygous for the mutation. Therefore, we recommend sisters of patients with TARP syndrome be carrier tested before family planning regardless of carrier testing results of the mother. Based on our patient and previously reported patients, we suggest TARP syndrome be considered as a possible diagnosis in males with severe or profound intellectual disability combined with septal heart defect, and Robin sequence, micrognathia, or cleft palate.
Keywords: clubfoot; heart septal defects, congenital; intellectual disability; mental retardation, X‐linked; Pierre Robin syndrome; RBM10; RNA‐binding proteins; scoliosis; TARP syndrome
1. INTRODUCTION
TARP syndrome (OMIM 311900) is a rare X‐linked syndrome first described in 1970 as Robin's syndrome (Gorlin, Cervenka, Anderson, Sauk, & Bevis, 1970). Talipes equinovarus, atrial septal defect, Robin sequence, and persistence of the left superior vena cava are the main features of this syndrome, and thus the syndrome became known as TARP syndrome in 2003 (Kurpinski, Magyari, Gorlin, Ng, & Biesecker, 2003). In 2010, RBM10 was identified as the causative gene (Johnston et al., 2010). Seven TARP syndrome families and two patients with a phenotype overlapping TARP syndrome have previously been described (Gorlin et al., 1970; Gripp et al., 2011; Johnston et al., 2010; Johnston et al., 2014; Powis et al., 2017; Wang et al., 2013). In the first reported families, all patients displayed a phenotype including at least three of the four findings in the TARP acronym (Supporting Information Table S1; Gorlin et al., 1970; Johnston et al., 2010; Gripp et al., 2011). Some later reported patients displayed two or fewer of these findings (Johnston et al., 2014; Wang et al., 2013). Additional clinical findings have been reported in patients with hemizygous loss‐of‐function mutations in RBM10 (Supporting Information Table S1). TARP syndrome often results in pre‐ or post‐natal lethality. In 2011, the oldest patient with TARP syndrome was 3 years and 7 months when reported (Gripp et al., 2011), whereas in 2013, two patients were reported to be 14 years with a phenotype overlapping TARP syndrome because of a large in‐frame deletion in the RBM10 gene (Wang et al., 2013). Until now, no adult patients with TARP syndrome have been reported. Here, we report an adult patient with TARP syndrome, thus expanding the phenotype in TARP syndrome. We also present recommendations regarding carrier testing of sisters of patients with TARP syndrome and suggest a possible cause for the milder phenotype in our patient.
2. MATERIALS AND METHODS
2.1. Clinical report
We report a male patient born in 1989, to non‐consanguineous Caucasian parents following a normal pregnancy of 42 gestational weeks. His birth weight was 2.700 kg (less than second centile for gestational age). He was diagnosed with Robin sequence shortly after birth and was hospitalized for the first 3 weeks of life because of hypotonia and feeding and breathing problems. He was diagnosed with severe to profound intellectual disability in early childhood and had not developed language at age 28 years. An atrial septal defect was surgically repaired at age 18 years. Surgery for severe thoracolumbar scoliosis was performed at age 21 years. He was referred for genetic counseling because of family planning by his two sisters. They sought genetic counseling, when he was 26 years to evaluate the recurrence risk in the family. Array‐CGH and genetic testing for fragile X syndrome were performed with normal results. Clinical examination of the patient revealed sloping forehead, prominent nasal bridge and nose, high myopia, esotropia, displacement of lacrimal points in the cranial direction, downslanted palpebral fissures, thick eyebrows, prominent supraorbital ridge, unilateral hearing loss of 75 dB, high and narrow palate, alveolar ridge overgrowth, chin dimple, and incomplete cutaneous syndactyly of the second and third toes. The patient did not have talipes equinovarus. In several echocardiographies performed before referral, persistent left superior vena cava was not described. The patient communicated by sounds of joy or disapproval. His gait was broad‐based, and he could walk unaided for short distances. No family members were reported with a similar phenotype. A search in medical databases for syndromes to explain our patient's symptoms resulted in a long list of possible but unlikely diagnoses including TARP syndrome, which was an unlikely cause because of the patient's age and scoliosis.
2.2. Editorial policies and ethical considerations
Informed consent was obtained from our patient, his two sisters, and his mother.
2.3. Molecular evaluation
DNA was isolated by standard procedures from blood leukocytes. Nucleotides were numbered according to the Human Genome Variation Society guidelines using the NM_005676.4 reference sequence (den Dunnen et al., 2016). Diagnostic exome sequencing with analysis of 749 genes associated with intellectual disability (gene panel version DG‐2.5) was performed at Genome Diagnostics Nijmegen (Nijmegen, The Netherlands) on DNA from our patient.
2.4. Verification and carrier testing
Mutation genotyping of our patient and his family was performed by direct sequencing analysis of exon 4 of the RBM10 gene using forward primer 5′‐GTCTGGTGTCACTCATCTCATTCTG‐3′ and reverse primer 5′‐GCACGTACGTCATCCTCAGT‐3′. The PCR amplification was performed using OneTaq Hot Start DNA polymerase with GC buffer (New England Biolabs, Ipswich, MA) according to the manufacturer's instructions.
For targeted massive parallel sequencing, amplicon libraries were prepared using the Ion AmpliSeq library kit with target amplification of the RBM10 gene included in the Ion Ampliseq™ cardiovascular research panel (Thermo Fisher Scientific Inc., Waltham, MA). Template preparation was performed on the Ion Chef and sequencing on the Ion Proton (Thermo Fisher Scientific Inc.). Manual inspection and visualization of BAM files was achieved using the Genome Browse function of VarSeq, Version 1.4.4 (Golden Helix Inc., Bozeman, MT).
3. RESULTS
The diagnostic exome sequencing revealed the hemizygous c.273_283delinsA mutation in exon 4 of the RBM10 gene in the patient, a mutation not previously reported. The subsequent direct sequencing confirmed the c.273_283delinsA mutation in a hemizygous state in the patient and revealed the same mutation in a heterozygous state in one of the two sisters. The other sister and the mother were tested negative for the mutation by direct sequencing analysis (Figure 1). However, the mother's electropherogram suggested possible mosaicism. Maternal mosaicism was confirmed by targeted massive parallel sequencing where 4 of 16 reads were found to be mutated (Supporting Information Figure S1). The degree of mosaicism could not be determined because of low‐sequencing depth.
Figure 1.
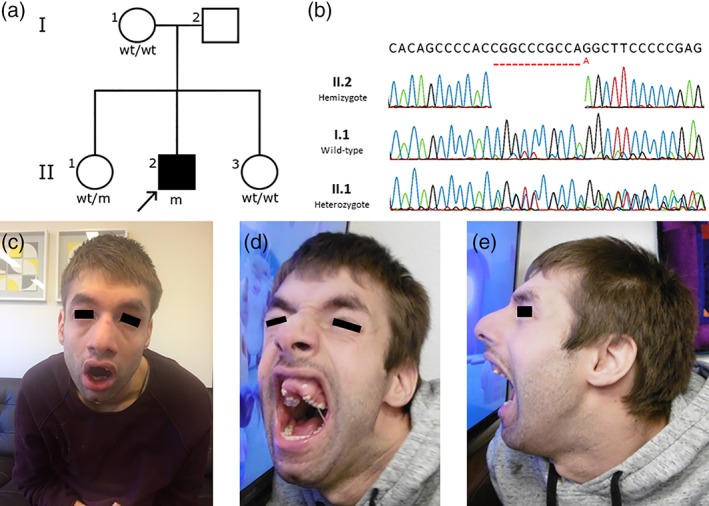
(a) Pedigree of the family wt, wild‐type; m: c.273_283delinsA mutation in the RBM10 gene. (b) Direct sequencing electropherograms of part of exon 4 in the RBM10 gene in the index patient (II.2), the mother (I.1), and a sister (II.1). The electropherogram of I.1 suggests mosaicism. (c–e) Photographs of the index patient. Features include wide mouth, sloping forehead, prominent nasal bridge, prominent nose, thick eyebrows, prominent supraorbital ridge, high and narrow palate, alveolar ridge overgrowth, chin dimple, and micrognathia. Upper front teeth are missing because of trauma. The black bars covering the eyes are by request of the legal guardian of the patient [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
We report an adult patient with TARP syndrome. Previously reported patients with TARP syndrome demonstrate variability of the phenotype (Supporting Information Table S1). The first reported families of TARP syndrome described the syndrome as a pre‐ or post‐natal lethal condition (Gorlin et al., 1970; Johnston et al., 2010). However, later reported families (Gripp et al., 2011; Johnston et al., 2014) have included patients surviving into early childhood. Two patients were reported at age 14 years, with a phenotype‐overlapping TARP syndrome and a large in‐frame RBM10 deletion (Wang et al., 2013). Our patient has two of the four findings corresponding to the TARP acronym and other findings previously associated with TARP syndrome (Supporting Information Table S1; Gorlin et al., 1970; Johnston et al., 2010; Gripp et al., 2011; Wang et al., 2013; Johnston et al., 2014; Powis et al., 2017). He also presented with alveolar ridge overgrowth, prominent nasal bridge and nose, chin dimple, esotropia, and displacement of lacrimal points in the cranial direction, which are likely part of the phenotypical spectrum of TARP syndrome in adults.
No loss‐of‐function mutations in RBM10 are registered in the Exome Aggregation Consortium (ExAC) database or the Genome Aggregation Database (gnomAD) (Lek et al., 2016). All previously reported hemizygous loss‐of‐function RBM10 mutations have been associated with TARP syndrome (Gripp et al., 2011; Johnston et al., 2010, 2014; Powis et al., 2017; Wang et al., 2013). The novel c.273_283delinsA RBM10 mutation creates a frameshift at codon 91 which produces a premature stop codon 40 positions downstream (p.(Gly92Alafs*39)). This is likely to shorten RBM10 mRNA that may undergo nonsense‐mediated decay and thus result in no or little protein production. The c.273_283delinsA mutation, however, is the first reported loss‐of‐function mutation in exon 4, which is skipped in three of five NCBI‐curated protein‐coding RBM10 transcripts (Figure 2) (O'Leary et al., 2016). The three transcripts without exon 4 are likely unaffected by the mutation. Therefore, we suggest alternative splicing as a possible cause for the milder phenotype in our patient, resulting in his survival into adulthood. Further studies on the expression of the different RBM10 transcripts in different tissues are needed to investigate this further.
Figure 2.
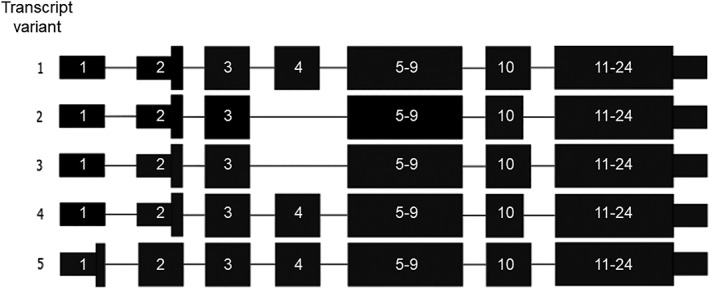
Protein‐coding transcripts of RBM10 curated by NCBI staff. The c.273_283delinsA mutation (this case report) is the first loss‐of‐function mutation identified in exon 4. The four other transcript variants differ from transcript variant 1 (NM_005676.4) in three ways. The starting codon is in exon 1 in transcript variant 5 (NM_001204468.1). Exon 4 is skipped in transcript variants 2 (NM_152856.2) and 3 (NM_001204466.1). Exon 10 is shortened by three bases in transcript variants 2 and 4 (NM_001204467.1)
The RBM10 gene is included in some massive parallel sequencing gene panels for intellectual disability, congenital heart malformations, and malformation syndromes with cleft palate. The inclusion of the RBM10 gene in the diagnostic gene panel chosen was essential for reaching the right diagnosis in our patient, as our initial search in medical databases, based on the patient's clinical findings, gave a long list of possible diagnoses but did not guide us to a likely diagnosis. TARP syndrome was initially believed to be an unlikely diagnosis owing to our patient's age and scoliosis. Inclusion of RBM10 in additional gene panels may result in identification of more adult patients with TARP syndrome, possibly expanding the phenotype further in the near future. Milder mutations, such as missense mutations, may theoretically cause a milder phenotype. Based on our patient and previously reported patients, we suggest TARP syndrome be considered in men with severe or profound intellectual disability, atrial or ventricular septal defect, and Robin sequence, micrognathia, or cleft palate.
5. CONCLUSION
We report the first adult male with TARP syndrome and suggest alternative splicing as a possible cause for the milder phenotype in our patient, resulting in his survival into adulthood. Based on our patient and previously reported patients, TARP syndrome should be considered as a possible diagnosis in males with severe or profound intellectual disability combined with atrial or ventricular septal defect and Robin sequence, micrognathia, or cleft palate. Because of the risk of low‐grade mosaicism in female carriers, preconception carrier testing should be offered to sisters of patients with TARP syndrome, as a part of family planning, regardless of whether the mutation has been identified in the mother despite adequate carrier testing.
Supporting information
Table S1 Features associated with TARP syndrome1
Figure S1 Result of targeted Massive Parallel Sequencing of nucleotides 263 to 293 in the RBM10 gene's exon 4 (NM_005676.4) in the mother of the index patient (I.1 in Figure 1B). Please note that 4 out of 16 reads contain the c.273_283delinsA mutation confirming that the mother is mosaic for this mutation. Due to the low number of reads, the degree of mosaicism is not possible to estimate. Red, yellow and blue squares identify a change at the specific position from the reference sequence (turquoise squares) to an A, C and T, respectively. The orange symbols identify insertions. Purple squares identify deletions of the specific bases
ACKNOWLEDGMENTS
The authors would like to thank the family members for their participation and staff members at the Centre for Deafblindness and Hearing Loss, Aalborg, Denmark, for their cooperation. The authors would also like to thank the Exome Aggregation Consortium (ExAC) and the Genome Aggregation Database (gnomAD) and the groups that provided exome and genome variant data to these resources. A full list of contributing groups can be found at http://exac.broadinstitute.org/about and http://gnomad.broadinstitute.org/about, respectively.
Højland AT, Lolas I, Okkels H, et al. First reported adult patient with TARP syndrome: A case report. Am J Med Genet Part A. 2018;176A:2915–2918. 10.1002/ajmg.a.40638
REFERENCES
- den Dunnen, J. T. , Dalgleish, R. , Maglott, D. R. , Hart, R. K. , Greenblatt, M. S. , McGowan‐Jordan, J. , … Taschner, P. E. M. (2016). HGVS recommendations for the description of sequence variants: 2016 update. Human Mutation, 37, 564–569. [DOI] [PubMed] [Google Scholar]
- Gorlin, R. J. , Cervenka, J. , Anderson, R. C. , Sauk, J. J. , & Bevis, W. D. (1970). Robin's syndrome. A probably X‐linked recessive subvariety exhibiting persistence of left superior vena cava and atrial septal defect. American Journal of Diseases of Children, 119, 176–178. [PubMed] [Google Scholar]
- Gripp, K. W. , Hopkins, E. , Johnston, J. J. , Krause, C. , Dobyns, W. B. , & Biesecker, L. G. (2011). Long‐term survival in TARP syndrome and confirmation of RBM10 as the disease‐causing gene. American Journal of Medical Genetics. Part A, 155, 2516–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J. J. , Sapp, J. C. , Curry, C. , Horton, M. , Leon, E. , Cusmano‐Ozog, K. , … Biesecker, L. G. (2014). Expansion of the TARP syndrome phenotype associated with de novo mutations and mosaicism. American Journal of Medical Genetics. Part A, 164, 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J. J. , Teer, J. K. , Cherukuri, P. F. , Hansen, N. F. , Loftus, S. K. , Chong, K. , … Biesecker, L. G. (2010). Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. American Journal of Human Genetics, 86, 743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurpinski, K. T. , Magyari, P. A. , Gorlin, R. J. , Ng, D. , & Biesecker, L. G. (2003). Designation of the TARP syndrome and linkage to Xp11.23‐q13.3 without samples from affected patients. American Journal of Medical Genetics. Part A, 120, 1–4. [DOI] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary, N. A. , Wright, M. W. , Brister, J. R. , Ciufo, S. , Haddad, D. , McVeigh, R. , … Pruitt, K. D. (2016). Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Research, 44, D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powis, Z. , Hart, A. , Cherny, S. , Petrik, I. , Palmaer, E. , Tang, S. , & Jones, C. (2017). Clinical diagnostic exome evaluation for an infant with a lethal disorder: Genetic diagnosis of TARP syndrome and expansion of the phenotype in a patient with a newly reported RBM10 alteration. BMC Medical Genetics, 18, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Gogol‐Doring, A. , Hu, H. , Frohler, S. , Ma, Y. , Jens, M. , … Chen, W. (2013). Integrative analysis revealed the molecular mechanism underlying RBM10‐mediated splicing regulation. EMBO Molecular Medicine, 5, 1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Features associated with TARP syndrome1
Figure S1 Result of targeted Massive Parallel Sequencing of nucleotides 263 to 293 in the RBM10 gene's exon 4 (NM_005676.4) in the mother of the index patient (I.1 in Figure 1B). Please note that 4 out of 16 reads contain the c.273_283delinsA mutation confirming that the mother is mosaic for this mutation. Due to the low number of reads, the degree of mosaicism is not possible to estimate. Red, yellow and blue squares identify a change at the specific position from the reference sequence (turquoise squares) to an A, C and T, respectively. The orange symbols identify insertions. Purple squares identify deletions of the specific bases