

Abstract
LIM kinases modulate multiple aspects of cancer development, including cell proliferation and survival. As the mechanisms of LIMK‐associated tumorigenesis are still unclear, we analyzed the tumorigenic functions of LIM kinase 2 (LIMK2) in human bladder cancer (BC) and explored whether the newly identified LIMK2 3´‐UTR SNP rs2073859 (G‐to‐A allele) is correlated with clinical features. Expression levels of LIMK2 in 38 human BC tissues and eight cell lines were examined using quantitative real‐time PCR and immunohistochemistry. LIMK2 was overexpressed in most BC tissues (27/38, 71%) and BC‐derived cell lines (6/8), and was more frequently overexpessed in high‐grade than low‐grade BC (80% vs. 47%). The effects of LIMK2 on BC cell proliferation, survival and migration, were studied by overexpression and RNA interference approaches in vitro and in vivo. LIMK2 overexpression promoted proliferation, migration and invasion of BC cells, while LIMK2 depletion inhibited cell invasion and viability and induced growth arrest in vitro and in vivo. PCR‐Restriction Fragment Length Polymorphism (RFLP) was used to genotype LIMK2 SNP rs2073859 and multivariate logistic regression applied to assess the relationship between allele frequency and clinical features in 139 BC patients. Functional analyses localized SNP rs2073859 within the microRNA‐135a seed‐binding region and revealed significantly lower LIMK2 G allele expression. The frequency of A genotypes (AG + AA) was higher in the BC group than normal controls and correlated with risks of high‐grade and high‐stage BC. In conclusion, LIMK2 may function as an oncogene in human BC, while allele‐specific regulation by microRNA‐135a may influence disease risk.
Keywords: LIMK2, microRNA‐135a, SNP, bladder cancer
Short abstract
What's new?
Genetic variations in microRNA (miRNA)‐binding regions are suspected to influence tumorigenesis. In this study, in tissues from bladder cancer (BC) patients, the single nucleotide polymorphism rs2073859 (G‐to‐A allele) in the 3’‐UTR miRNA‐135a‐binding site of the oncogene LIM kinase 2 (LIMK2) was associated with gene overexpression and higher clinical grade. A high frequency of rs2073859 A genotypes was detected in BC tissues. In BC cells and mice, LIMK2 overexpression promoted cell proliferation, migration, and invasion and accelerated BC tumor growth. The findings suggest that LIMK2 normally is downregulated by miRNA‐135a and becomes tumorigenic following miRNA‐135a disruption by rs2073859 variant A.
Introduction
Bladder cancer (BC) is the most common urological tumor in China, however, the mechanisms of bladder cancer tumorigenesis are not well understood. Tumor microenvironment, oncogenes and tumor suppressors have also been reported to play important roles in BC.1
LIM kinase 2 (LIMK2) belongs to the LIM kinase (LIMK) family of serine/threonine kinases.2 The LIMKs are key regulators of actin dynamics through phosphorylation and inactivation of the actin depolymerizing factor cofilin.3, 4, 5 Given their cytoskeleton‐associated functions and indications of elevated expression in various cancer types, research has largely focused on their roles in tumorigenesis. Indeed, recent studies have shown that the LIMks modulate activities that contribute to cancer development, including cell proliferation and survival.6, 7, 8, 9 However, the underlying molecular mechanisms remain unknown.
Single nucleotide polymorphisms (SNPs) located within microRNA‐binding sites of oncogenes and tumor suppressors have been linked to the tumorigenesis process. MicroRNA‐binding SNPs could alter the thermodynamic interaction between the microRNA and the mRNA sequence, strengthening or weakening binding and thereby modulating output of protein expression,10 and are thus associated with susceptibility to cancer development. In a previous study, our group suggested that SNP (1805C/T) in the miR‐181a binding site of the Mel‐18 gene is related to certain clinical features of prostate cancer.11
In our study, we report that LIMK2 may function as an oncogene in human BC. Moreover, we show that the miR‐135a‐binding SNP rs2073859 (G‐to‐A allele) located within the 3’UTR of the LIMK2 gene is associated with LIMK2 overexpression and higher clinical grade. Thus, this SNP may be a promising prognostic factor for BC.
Materials and Methods
Patients and tumor specimens
A total of 139 patients with BC and 101 normal patients (non‐bladder cancer patients) treated at Guangzhou General Hospital of Guangzhou Military Command (China) between April 2001 and April 2015 were enrolled in our study (Table 1). Among these, a total of 38 fresh bladder tumor samples and their normal adjacent tissues were obtained from biopsy or surgical resection. Samples were immediately frozen in liquid nitrogen and stored at −80°C for subsequent quantitative real‐time PCR (qRT‐PCR) and immunohistochemistry. All samples were diagnosed histologically with specimens and analyzed in accordance with institutional guidelines on the use of human tissue. Samples included 23 non‐muscle‐invasive bladder tumors, 15 muscle‐invasive bladder tumors and 38 normal adjacent tissues. The grading system classified 28 tumors as low grade and 10 as high grade. Tumor stages and grades were assigned according to the TNM classification system and WHO criteria.12 All aspects of the present study were approved by the institutional ethics committee of Guangzhou General Hospital of Guangzhou Military Command, China.
Table 1.
Genotype frequencies of the LIMK2 polymorphism in controls and bladder cancer groups
| LIMK SNP | Controls | Bladder cancer | AOR1(95%CI2) | p |
|---|---|---|---|---|
| N3(%) | N3(%) | |||
| GG | 40(39.6) | 36(25.9) | 1.0(reference) | |
| AG | 42(41.6) | 62(44.6) | 1.54(0.96–4.79) | 0.112 |
| AA | 19(18.8) | 41(29.5) | 3.09(1.30–7.35) | 0.018 |
| AA+AG(against GG) | 61(60.4) | 103(74.1) | 2.90(1.02–8.25) | 0.029 |
| GG + AG(against AA) | 82(81.2) | 98(70.5) | 0.94(0.69–1.83) | 0.064 |
adjusted odds ratio for age and gender.
95% confidence interval.
Numbers of people.
Cell lines and culture
Eight BC‐derived cell lines were also examined. The BC lines TCCSUP, T24 and 5,637 and the normal bladder cell line SV‐HUC‐1 were obtained from American Tissue Type Culture Collection (ATCC, Manassas, VA). The BC cell line EJ was obtained from Japanese Cancer Research Resources Bank (JCRB, Tokyo, Japan) and the BC line BIU‐87 from China Center for Type Culture Collection (CCTCC, Wuhan, China). The BC lines J82, UM‐UC‐3, and RT4 as well as HEK‐293 T cells were kindly provided by Dr. He W (Department of Urology, Sun Yat‐sen Memorial Hospital, Guangzhou, China). All lines were cultured in a humidified air atmosphere containing 5% CO2 at 37°C, and all culture media were supplemented with 10% fetal bovine serum (Hyclone, Logan, UT). The T24 line was cultured in McCoy's 5a medium (modified), 5,637 cells in RPMI 1640 medium, J82, UM‐UC‐3, and TCCSUP cells in Eagle's minimum essential medium (EMEM, Hyclone), and BIU‐87, RT4, EJ, SV‐HUC‐1, and HEK‐293 T cells in Dulbecco's modified Eagle's medium (DMEM, Hyclone).
RNA Extraction and quantitative real‐time PCR
Total RNA was extracted from bladder tissue samples and cell lines using the RNA Universal Tissue Kit (Qiagen, Valencia CA) following the manufacturer's instructions. Primers used for quantitative RT‐PCR were as follows: (forward and reverse) LIMK2 5′‐GGGTGAAGATGTCTGGAG‐3′ and 5′‐TCGTTGACAGTCCTGTACC‐3′, GAPDH 5′‐GGGAAACTGTGGCGTGAT‐3′ and 5′‐GAGTGGGTGTCGCTGTTGA‐3′. GAPDH was used as the internal control and all LIMK2 expression values were normalized relative to GAPDH transcript levels.
Immunohistochemistry
Immunohistochemistry was conducted on 5‐mm formalin‐fixed, paraffin‐embedded tissue sections from BC tumor samples (n = 29, Guangzhou General Hospital of Guangzhou Military Command, China) using antibodies against LIMK2 (1:400, Abcam, Cambridge, MA). Immunostaining was performed using the ChemMate™ DAKO EnVision™ Detection Kit (DakoCytomation, Glostrup, Denmark) as described previously.13, 14 Subsequently, sections were counterstained with hematoxylin (Zymed Laboratories, South San Francisco, CA) and mounted in nonaqueous mounting medium. The primary antibody was omitted for the negative controls.
Stable cell lines
Full‐length LIMK2 cDNA was cloned into the pLVX‐mCMV‐ZsGreen‐puro lentiviral vector. A pLVX‐shRNA2 lentiviral vector expressing LIMK2‐shRNA (The primers of LIMK2 shRNA: Forward,5’‐CCGGGCTATTCACAGCAGATCTTCTCGAGAAGATCTGCTGTGAATAGCTTTTTG‐3′, Reverse 5′‐AATTCAAAAAGCTATTCACAGCAGATCTTCTCGAGAAGATCTGCTGTGAATAGC‐3′) and a non‐target shRNA control vector (scramble) were obtained from Sigma (St. Louis, MO). Lentiviruses were produced according to the manufacturers’ manual. UM‐UC‐3 cells stably expressing LIMK2 or LIMK2‐shRNA were obtained by infection with pLVX‐mCMV‐ZsGreen‐puro containing LIMK2 DNA or pLVX‐shRNA2‐LIMK2 and selected in 6 μg/ml puromycin for 2 weeks. The pLVX‐mCMV‐ZsGreen‐puro vector and the nontarget shRNA control vector were used to generate the control cell lines after the same protocol.
Cell proliferation assay
The MTT (3, 4, 5‐dimethylthiazol‐2, 5 biphenyl tetrazolium bromide; Invitrogen, Carlsbad, CA) and EdU assays were used to evaluate cell proliferation as described previously.15
Wound healing assay
UM‐UC‐3 cells stably overexpressing LIMK2 or LIMK2‐shRNA were seeded in 30‐mm dishes at 1 × 105 cells/dish in 2 ml EMEM. At confluence, cell monolayers were scratched with a 200 μl pipette tip, and culture continued in the presence of 3% FBS. The scratched monolayer cultures were photographed using an inverted microscope at 0, 10 and 20 hr. Cells migrating into the wound surface and the average distance of migration were determined at designated time points (0 hr and 10 hr).
Invasion assay
Transwell chambers containing filters coated with an extracellular matrix on the upper surface (BD‐Biocoat Matrigel 24‐well invasion chambers, BD Biosciences) were used to examine BC invasive capacity according to the manufacturer's protocol. Briefly, UM‐UC‐3 cells stably expressing LIMK2, LIMK2‐shRNA, empty vector or scrambled shRNA (1 × 105) were plated on the upper chamber membrane in serum‐free medium and incubated at 37°C under a 5% CO2 atmosphere for 48 hr. Cells that had penetrated to the bottom side of the membrane were then fixed in 4% paraformaldehyde (PFA), stained using crystal violet and counted. Each reported value represents the mean of three independent experiments with triplicate determinations. The invasion index was measured as relative migration of cells across the Matrigel‐coated membrane.
Anchorage‐independent growth
UM‐UC‐3 cells stably expressing LIMK2 or LIMK2‐shRNA, empty vector or scrambled shRNA were suspended in 2 ml top agar medium (EMEM supplied with 0.3% agar) (Sigma, St Louis, MO), and layered over 1.5 ml bottom agar medium (EMEM supplied with 1.0% agar) (Sigma, St Louis, MO) in 35‐mm dishes. After 10 days, the cells were photographed under inverted microscope and the number of colonies was counted. All results are from three independent experiments with internal triplicate repeats.
Three‐dimension spheroid invasion assay
Growth factor‐reduced Matrigel (BD Biosciences) was thawed on ice and diluted to 4 mg/ml with ice‐cold serum‐free EMEM. After vortexing, 50‐μL aliquots were added to each well of 24‐well plates and incubated at 37°C for 1 hr to allow gelling. UM‐UC‐3 cells stably expressing LIMK2, LIMK2‐shRNA, empty vector or scrambled shRNA (1 × 104) were then seeded into each well. Invasion was monitored by taking pictures under a light microscope (OLYMPUS, CKX41, U‐CTR30–2) immediately after implantation and after 8 days. Depicted spheroids are representative of three independent experiments as described.16
Cell cycles and cell apoptosis analysis
UM‐UC‐3 cells stably expressing LIMK2,LIMK2‐shRNA, empty vector or scrambled shRNA were collected and fixed in 70% ethanol at 4°C overnight, followed by staining with propidium iodide (PI) (Sigma) under darkness at 4°C for 30 min. The cell cycle was analyzed as described previously.15 For detection of apoptotic cells, samples were fixed with 4% paraformaldehyde for 1 hr at room temperature. After washing with PBS, the fixed specimens were permeabilized in 0.1% sodium citrate containing 0.1% Triton X‐100 for 2 min on ice and incubated with a mixture of TdT solution and fluorescein‐dUTP solution in a humidified chamber for 1 hr at 37°C. The number of TUNEL‐positive cell nuclei was quantified.
Tumorigenesis in nude mice
Stably transfected UM‐UC‐3 cells of each line (2 × 106 in 200 μl normal saline) were injected subcutaneously into 4‐week‐old female BALB/c nude mice obtained from the Experimental Center of Southern Medical University. Each cell line was injected into five animals. Tumor dimensions were measured weekly using a Vernier caliper and volumes calculated according to the formula V = πLW 2 /6. All mice were sacrificed 30 days after cell injection and the final tumor dimensions and weights recorded.
Western blot
Cells were lysed in RIPA buffer (50 mMTris‐Cl [pH 7.4], 150 mM NaCl,1% TritonX‐100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 1 mM EDTA) supplemented with protease and phosphatase inhibitor cocktail (Sigma). Lysate protein concentration was measured using a BCA Protein Assay Kit (KEYGEN, China). Proteins were separated on 12% SDS‐polyacrylamide gels, and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with 5% skim milk for 1 hr at room temperature, and probed with different primary antibodies overnight at 4°C. Target proteins and antibody titers were as follows: LIMK2 (1:300, Santa Cruz Biotechnology, Dallas TX, sc‐8,389), p27 (1:1000, Cell Signaling, Danvers MA, 2552), cyclin D1 (1:300, Santa Cruz, sc‐8,396), Ki67 (1:1000, Cell Signaling, D2H10), cofilin (1:1000, Abcam), p‐cofilin (1:500, Abcam), and β‐actin (1:500, Santa Cruz, sc‐4,778). After primary antibody incubation, blots were washed three times with Tris‐buffered saline with 1% Triton X (TBST), incubated with appropriate secondary antibodies for 1 hr at room temperature, and washed again three times in TBST. Immunoblotting was visualized using Electro‐Chemi‐Luminescence (ECL) reagent.
Bioinformatics
The NCBI SNP database was used to find SNPs located within the 3´‐UTR of the LIMK2 gene. MicroRNA targets were predicted using the online tools www.microrna.org/ and targetscan.org/. The online mRNA secondary structure prediction tool RNAhybrid (bibiserv2.cebitec.uni‐bielefeld.de/rnahybrid) was used to predict the mean free energy (MFE) change associated with miRNA binding.
3´‐UTR luciferase assay
Functional interactions between miRNAs and 3´‐UTRs were tested in vitro using reporter constructs containing 3´‐UTR fragments bearing either the G and A allele of rs2073859 cloned downstream of Renilla Luciferase. These constructs were created through genomic DNA amplification followed by cloning into the psiCHECK‐2 vector (Promega, Madison, WI) modified using Gateway technology (Invitrogen). The LIMK2 reporter was generated based on Refseq NM_005569.3 and encompassed the first 251 bp of the 3´‐UTR, which is centered aroundrs2073859 at position 126 bp. The presence of rs2073859‐G was verified and mutagenized to generate rs2073859‐A. The predicted site for miR‐135a binding was mutated from AACAAGCC to GCCAGAAC to abolish miRNA binding as a control. For the luciferase reporter assay, HEK‐293 T cells (2 × 104/well) were seeded into 24‐well plates. Cells were cotransfected for 24 hr with 0.5 mg reporter (psiCHECK‐2‐3´‐UTR‐G, psiCHECK‐2‐3´‐UTR‐A or psiCHECK‐2) and miR‐135a or scrambled miRNA (20 μM) using Lipofectamine 2000 (Invitrogen). Assays were performed 48 hr after transfection start using the Dual‐Luciferase Reporter Assay System (Promega). Luciferase activity was detected using the GloMax‐Multi Detection System (Promega).
LIMK2 genotyping analysis
Total DNA was extracted from the tumor samples and cell lines using QIAamp reagent (Qiagen, Germantown, MD) according to the manufacturer's protocol. LIMK2 genotyping was conducted using a PCR‐RFLP method as described reviously17 (Supporting Information Fig. S1). The PCR primer sequences were 5´‐TATTGGATTTACAGACAGTAACTA‐3′ and 5´‐TTCCTGTGGATTGGCGGAATG‐3′.
Measurement of LIMK2 rs2073859 G and A allele expression levels
The cDNAs from three BC‐derived cell lines (TCCSUP, UM‐UC3 and EJ) and 10 tissue samples (10 cases) heterozygous for the rs2073859 G/A SNP were subjected to PCR using primers from the TaqMan1 SNP Genotyping Assay kit (ABI Applied Biosystems, Foster City, CA). The real‐time intensity of fluorescence (from VIC for 1805G and FAM for 1805A) was measured using the TaqMan1 Gene Expression Master Mix (ABI Applied Biosystems) as described.11 Genomic DNA was also extracted from cell lines and cancer tissues as an internal control.
LIMK2 mRNA half‐life
qPCR was also used to measure the half‐life of LIMK2 mRNA. BIU‐87(GG) and 5,637(AA) BC cells were plated onto 10‐cm dishes at 1 × 106/dish 1 day before treatment with the transcription inhibitor actinomycin D (5 mg/ml). Cells were lysed using TRIzol after 0, 4, 8, 12, 24 and 48 hr in actinomycin D. Total RNA was extracted and the LIMK mRNA level was quantified by qPCR using the Taqman assay as described for measures of LIMK2 alleles.
Statistical analysis
All data were entered into an access database and analyzed using statistical product and service solutions (SPSS) (version 18.0 J). Differences between groups were analyzed using Student's t test or one‐way analysis of variance. The adjusted odds ratios (aORs) and 95% confidence intervals (CIs) for the relationships between LIMK2 3´‐UTR genotype and clinical/histological features were determined by multivariate logistic regression. All statistical tests were two‐sided. Differences were considered statistically significant at p < 0.05.
Results
LIMK2 overexpression in human bladder cancer tissues and cell lines
Expression of LIMK2 mRNA was elevated in 27of 38 BC tissue samples (71%) compared to normal adjacent tissue (NAT) (Fig. 1 a) as measured by qRT‐PCR. Similarly, LIMK2 mRNA expression was higher in 6 of 8 BC cell lines (BIU‐87, TCCSUP, J82, UM‐UC‐3, T24, EJ, 5637 and RT4) than in the normal bladder cell line SV‐HUC‐1 (Fig. 1 b). Immunohistochemistry using a LIMK2‐specific antibody revealed that the expression of LIMK2 protein was elevated in a significantly larger proportion of high‐grade BC samples (8/10) than low‐grade samples (9/19) (p = 0.009, Fig. 1 c).
Figure 1.
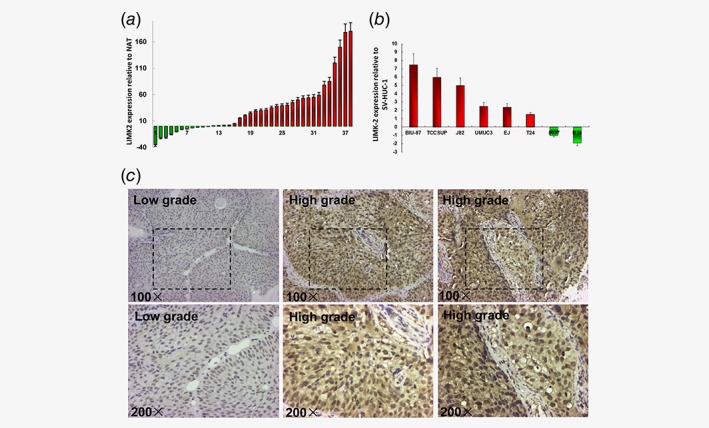
LIMK2 expression in human bladder cell lines, tumors or normal adjacent tissues. (a) Expression of LIMK2 in 38 bladder cancer tissue samples relative to normal adjacent tissues (NAT). Columns above the X‐axis indicate higher expression of LIMK2 while those below the X‐axis indicate lower expression of LIMK2 relative to NAT. (b) LIMK2 mRNA expression levels in bladder cancer cell lines relative to normal urothelial cells (SV‐HUC‐1). Columns above the X‐axis indicate overexpression of LIMK2 while those below the X‐axis indicate under expression of LIMK2 relative to normal cells. (c) Representative LIMK2 immunohistochemical staining of bladder cancer tissues. Images are × 100 and × 200. Staining: brown, LIMK2.
LIMK2 promotes cell motility, invasion and anchorage‐independent growth
As shown in Figures 2 a and 2b, LIMK2 overexpression significantly enhanced UM‐UC‐3 cell migration and invasion as revealed by cell wound healing and transwell migration assays. Conversely, LIMK2‐shRNA inhibited migration and invasion compared to a scrambled control shRNA. Further, LIMK2 had marked effects on cell morphology and colony forming capacity. In the three‐dimensional (3D) spheroid invasion assay, control UM‐UC‐3 cells infected with empty vector were immotile and had a smooth spheroid structure after culture in Matrigel for 8 days (Fig. 2 c). UM‐UC‐3 cells overexpressing LIMK2 generated more numerous and larger colonies than control cells and cells stably expressing LIMK2‐targeted shRNA in the anchorage‐independent growth assay (Fig. 2 d). Conversely, LIMK2‐infected cells exhibited active invasive behaviors characterized by outward projections from individual cells. Taken together, these results suggest that overexpression of LIMK2 promotes the metastatic and invasive capacities of UM‐UC‐3 cells.
Figure 2.
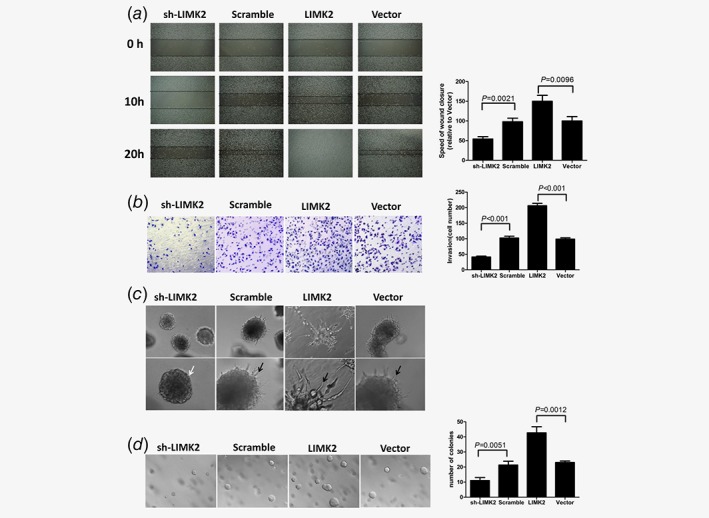
Effect of LIMK2 expression level on UM‐UC‐3 bladder cancer cell metastasis potential. Cells were stably transfected with LIMK2‐targeted shRNA (sh‐LIMK2), scrambled control siRNA (Scramble), LIMK2 overexpression vector (LIMK2) or empty vector (Vector). (a) Wound healing assay. UM‐UC‐3 cells invasion was analyzed by video microscopy. Left panel: representative pictures of wound closure. Right panel: Speed of wound closure relative to empty vector (mean ± S.D). (b) Invasion assay. The invasive properties were analyzed in a Boyden chamber coated with Matrigel. (Left) Cells that adhered to the lower surface of the filter are shown. (Right) Results presented as mean cell number ± S.D. (c) Representative micrographs of cultured cells after 8 days of culture in 3D spheroid invasion assays. (d). Anchorage‐independent growth assay. Colonies >0.1 mm in diameter were counted under a microscopic field (Left). Results presented as mean ± S.D. (Right).
LIMK2 promotes proliferation and cell cycle progression
Cells overexpressing LIMK2 also showed significantly accelerated proliferation compared to cells expressing empty vector as revealed by MTT and EdU assays. Conversely, LIMK2‐shRNA inhibited cell proliferation (Figs. 3 a and b). Flow cytometry revealed that LIMK2 overexpression induced a G1 to S shift in the cell cycle (Fig. 3 c). These results indicate that overexpression of LIMK2 promotes the proliferation and cell cycle progression of BC cells in vitro.
Figure 3.
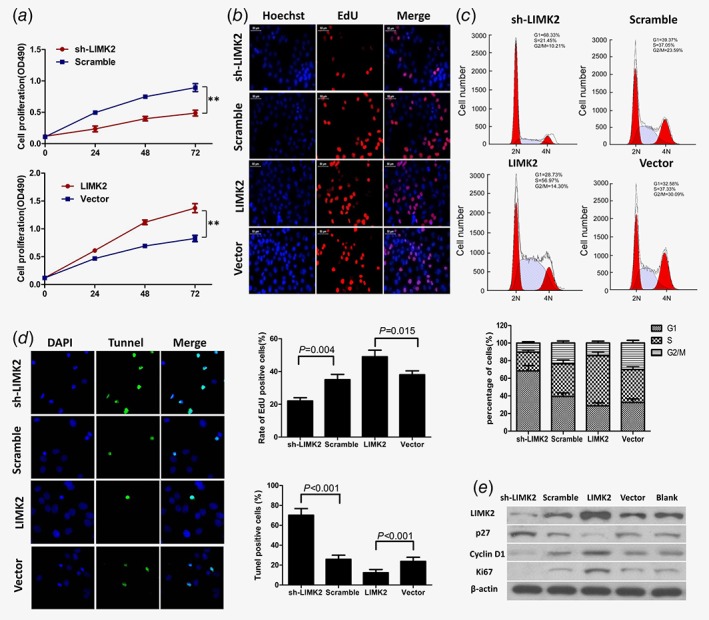
LIMK2 overexpression in UM‐UC‐3 cells promotes proliferation and cell cycle progression, inhibits apoptosis, and affects expression of multiple cell cycle regulators. Cells were stably transfected with LIMK2‐targeted shRNA (sh‐LIMK2), scrambled control siRNA (Scramble), LIMK2 overexpression vector (LIMK2), or empty vector (Vector). (a and b) Cell proliferation measured by MTT and EdU assays. (c) Cell cycle distribution of UM‐UC‐3 cells measured by flow cytometry. (d) Cell apoptosis measured by TUNEL assays. (e) Western blotting of LIMK2, p27, cyclinD1 and ki67. β‐actin was used as a loading control.
LIMK2 inhibits apoptosis and affects expression of cell cycle regulators
The proportion of apoptotic UM‐UC‐3 cells was significantly increased by LIMK2‐shRNA and reduced by LIMK2 overexpression according to the TUNEL assay (Fig. 3 d). We further investigated the effects of LIMK2 on the expression of genes that regulate the cell cycle and proliferation (Fig. 3 e). The cell cycle inhibitor p27Kip1 was downregulated and Cyclin D1 upregulated in LIMK2‐overexpressing UM‐UC‐3 cells compared to empty vector‐expressing (control) UM‐UC‐3 cells. Conversely, Cyclin D1 was downregulated and p27Kip1 upregulated in shRNA‐mediated LIMK2 knockdown cells (Fig. 3 e). Consistent with altered expression of cell cycle regulators, expression of the proliferation marker Ki67 was upregulated in LIMK2‐overexpressing cells and downregulated in shRNA‐mediated LIMK2 knockdown cells (Fig. 3 e). The Rho/ROCK/LIMK/cofilin pathway is one of the major signaling pathways involved in tumor metastasis through regulation of the actin cytoskeleton. Thus, we examined the relationship between LIMK2 expression and cofilin activity (p‐Cofilin and cofilin). Western blotting showed that cofilin expression not vary significantly with LIMK2 expression, while as expected LIMK2 overexpression increased p‐cofilin in BC cells (Supporting Information Fig. S2).
LIMK2 accelerates tumor growth in vivo
In the subcutaneous nude mouse model, tumors were palpable 6 days after injection of UM‐UC‐3 cells stably overexpressing LIMK2, empty vector or scrambled siRNA, but were not palpable until 9 days after injection of cells stably expressing LIMK2‐shRNA. All mice developed tumors after 30 days (Fig. 4 a). LIMK2 protein overexpression and shRNA‐mediated knockdown in subcutaneous xenografts was confirmed by Western blotting (Fig. 4 b). Tumors derived from UM‐UC‐3 cells overexpressing LIMK2 exhibited greater volume and higher weight than tumors derived from UM‐UC‐3 cells transfected with empty vector. Conversely, there was a dramatic decrease in tumor volume and weight in the LIMK2‐shRNA group compared to scrambled vector (Figs. 4 c and 4d). Thus, LIMK2 overexpression may accelerate and knockdown may suppress BC tumorigenicity in vivo.
Figure 4.
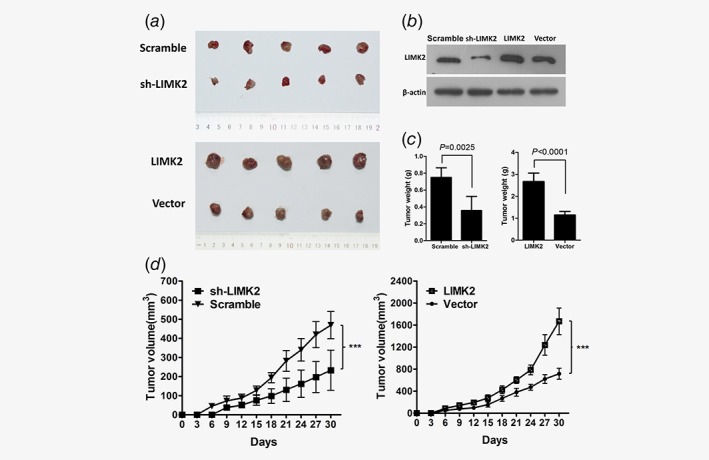
Effects of LIMK2 expression level on the growth of UM‐UC‐3‐derived xenograft tumors in nude mice. UM‐UC‐3 cells were transfected with shRNA LIMK2, scrambled shRNA, LIMK2 overexpression vector, or empty vector. (a) Images of tumor xenografts in nude mice. (b) Expression of LIMK2 in tumor xenografts of nude mice. (c and d). Effects of LIMK2 expression level on tumor weight (c) and (d) size expressed as mean ± SD.
SNP rs2073859 G/A is located within the miR‐135a binding site of the LIMK2 3´‐UTR
It has been reported that the 3´‐UTR of the LIMK2 gene contains potential binding sites for miR‐135a (http://www.targetscan.org andhttp://www.microrna.org) (Fig. 5 a). RNA folding and hybridization models predicted that the rs2073859 G‐to‐A allele substitution leads to a minimum free energy (MFE) change from −15.2 to −10.3 kcal/mol, indicating that the G allele has a higher binding affinity for miR‐135a than the A allele (Fig. 5 b). Indeed, significantly greater binding of miR‐135a to the G allele (and ensuing instability) was demonstrated by lower luciferase activity in the presence of the G allele compared to the A allele (Fig. 5 c).
Figure 5.
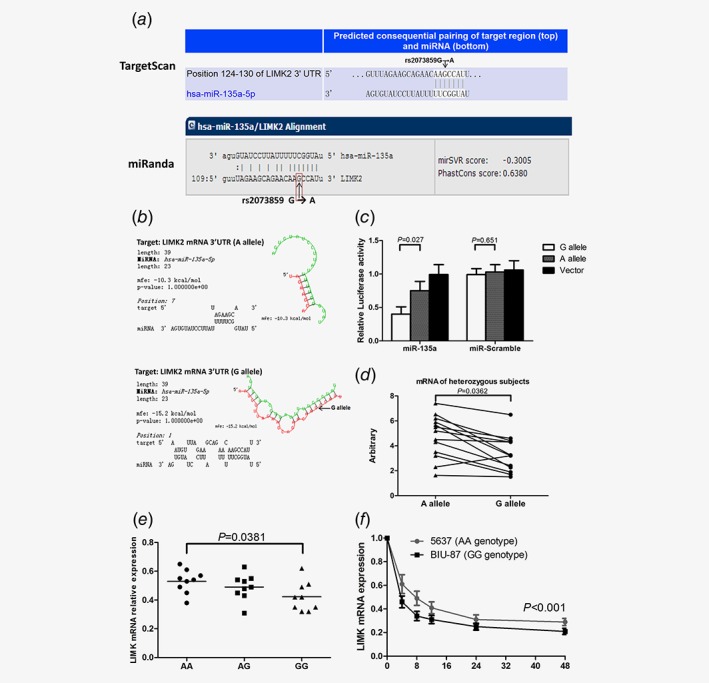
The LIMK2 3´‐UTR SNP rs2073859 (A/G) influences miR‐135a binding and differentially influences LIMK2 expression. (a) The SNP rs2073859 is predicted to lie within the miR‐135a binding site. (b) The minimum free energy (MFE) change predicts stronger binding of miRNA‐135a to the G allele.(c) 3´‐UTR luciferase assay for miR‐135a binding to the G and A alleles of rs2073859.(d) Expression levels of the LIMK2 mRNA harboring rs2073859G or rs2073859A in heterozygous samples (TCCSUP, UM‐UC3 and EJ cells plus 10 tissue samples). (e) Correlation analysis of SNP variants and relative LIMK2 mRNA expression. (f) Half‐life of LIMK2 mRNA in BIU‐87 (GG genotype) and 5,637 (AA genotype) cells.
SNP rs2073859 G/A affects LIMK2 expression
We examined mRNA levels of the LIMK2 G and A alleles in 10 heterozygous BC tissues and 3 heterozygous cell lines using the Taqman assay. Expression of LIMK2 A allele mRNA was significantly higher than G allele mRNA in both cancer tissues and tumor‐derived cell lines (Fig. 5 d), suggesting a greater negative regulation of the LIMK2 G allele translation by stronger miR‐135a binding. Relative LIMK2 mRNA expression was lower in subjects with the GG genotype than the AA genotype (Fig. 5 e), which was consistent with the Western blotting analysis result in 8 BC cell lines (BIU‐87, J82, TCCSUP, UM‐UC‐3, EJ, T24 and 5,637, RT4) and the normal bladder cell line SV‐HUC‐1(Supporting Information Fig. S3). We also compared LIMK2 mRNA half‐life between the homozygous 5,637(AA) and BIU‐87(GG) BC cell lines after treatment with actinomycin D, and found a 2.97‐fold longer half‐life for the AA genotype (11 hr vs. about 3.7 hr for the GG genotype) (Fig. 5 f), consistent with greater miR‐135a‐induced G allele transcript instability due to stronger 3´‐UTR binding.18
Association between the rs2073859 G/A genotype frequency and risk of bladder cancer
The SNP genotype distribution was in agreement with Hardy–Weinberg equilibrium (p > 0.05). The combined frequency of A genotypes (AG + AA) was higher in BC tissues than normal controls (Table 1). Further, the A genotype (AG + AA) may increase the risk of high‐grade cancer (Table 2) and high‐stage cancer (T2–T4, muscle invasive type, Table 3) compared to the GG genotype.
Table 2.
Genotype frequencies of the LIMK2 polymorphism in bladder cancer subgroups (low grade and high grade groups)
| LIMK SNP | Low grade | High grade | AOR1(95%CI2) | p |
|---|---|---|---|---|
| N3(%) | N3(%) | |||
| GG | 23(35.4) | 13(17.6) | 1.0(reference) | |
| AG | 28(43.1) | 34(45.9) | 1.54(0.89–2.68) | 0.074 |
| AA | 14(21.5) | 27(36.5) | 3.09(1.30–7.35) | 0.009 |
| AA+AG(against GG) | 42(64.6) | 61(82.4) | 2.77(1.16–5.69) | 0.017 |
| GG + AG(against AA) | 51(78.5) | 47(63.5) | 0.88(0.86–2.79) | 0.054 |
adjusted odds ratio for age and gender.
95% confidence interval.
Numbers of people.
Table 3.
Genotype frequencies of the LIMK2 polymorphism in bladder cancer subgroups (Non‐muscle invasive and Muscle‐invasive groups)
| LIMK SNP | Low stage | High stage | AOR1(95%CI2) | p |
|---|---|---|---|---|
| N3(%) | N3(%) | |||
| GG | 26(33.3) | 10(16.4) | 1.0(reference) | |
| AG | 31(39.7) | 31(50.8) | 2.35(1.97–5.60) | 0.024 |
| AA | 21(26.9) | 20(32.8) | 2.70(0.91–8.02) | 0.054 |
| AA+AG(against GG) | 52(66.7) | 51(83.6) | 2.47(1.49–5.67) | 0.038 |
| GG + AG(against AA) | 57(73.1) | 41(67.2) | 0.92(0.63–2.29) | 0.452 |
adjusted odds ratio for age and gender.
95% confidence interval.
Numbers of people.
Discussion
The well characterized functions of LIM kinases in actin cytoskeleton dynamics suggest potential application of LIMK modulators in anti‐metastasis therapy.19, 20, 21 Members of the LIMK family, including LIMK1 and LIMK2, are serine kinases that exert important effects on the regulation of the actin cytoskeleton through the phosphorylation of cofilin. Accumulating evidence implicats LIMks in multiple aspects of cancer development, including cell proliferation, survival and cell cycle progression.6, 7, 8, 14, 15 What arouse our attention was that knockdown of LIMK2 expression in NIH3T3 mouse fibroblasts caused an increase in cyclin D1 levels and in addition, the increased level of cyclin A was specific to LIMK2 activation by ROCK but not LIMK1 activation.22 LIMK2 has also been reported to promote metastatic behavior of fibrosarcoma cells and angiogenesis of pancreatic cancer cells,23, 24 and is a crucial regulator and effector of Aurora‐A kinase‐mediated malignancy2. Conversely, LIMK2 knockdown increased cell cycle arrest and apoptosis induced by microtubule targeting drugs.25 Downregulation of LIMK2 expression increased the sensitivity of neuroblastoma SH–EP cells to vincristine and vinblastine, while overexpression of LIMK2 increased resistance to vincristine.26 Collectively, these findings implicate LIMK2 overexpression in multiple aspects of tumorigenesis; however, Lourenco et al. reported that elevated LIMK2 expression in colon cancers was correlated with better prognosis, suggesting the possibility that LIMK2 has distinct regulatory effects in different forms of cancer.14
Despite evidence for multiple functions in tumorigenesis, no previous study had examined the relationship between LIMK2 expression and human BC behavior. We found that LIMK2 was overexpressed in the majority of human BC tissues and cell lines examined, strongly implicating LIMK2 as an oncogene in bladder carcinogenesis. Indeed, LIMK2 overexpression promoted the proliferation, cell motility, migration, invasion, colony formation and anti‐apoptotic activity of bladder UM‐UC‐3 BC cells, while LIMK2 depletion inhibited cell invasion and cell viability, and induced growth arrest. LIMK2 suppression also led to the promotion of UM‐UC‐3 cells apoptosis. Transplanted UM‐UC‐3 cells overexpressing LIMK2 produced larger tumors while LIMK2‐knockdown UM‐UC‐3 cells yielded smaller tumors than control cells in the subcutaneous nude mouse model. Overexpression of LIMK2 also dysregulated the expression levels of p27, Cyclin D1 and Ki67, indicating a putative association between LIMK2 and the PI3K/Akt signaling pathway. Moreover, LIMK2 overexpression also enhanced phosphorylation and inactivation of the actin depolymerizing factor cofilin in BC tissue. These findings are in line with previous studies demonstrating a role for LIMK2 in radiation‐induced cell cycle arrest.27, 28 For instance, Hsu et al. reported that depletion of LIMK2 promoted G2/M arrest after DNA damage.28 Further studies are required to elucidate the molecular signaling pathways linking LIMK2 elevation to tumor progression.
A genome‐wide analysis of SNPs located within the miRNA‐binding sites of various human gene 3´‐UTRs identified 12 with different allele frequencies in human cancers compared to the dbSNP database. We found that HOXB5, a transcription factor associated with stemness, was upregulated in BC and that upregulation was associated with the G variant of a miRNA‐binding SNP (1010A/G) located within the 3´‐UTR.13 Thus, miRNA‐binding SNPs within 3´‐UTRs are potential valuable prognostic markers for various cancers, including BC. SNPs affecting miR‐135a binding may be functionally significant in multiple cancer phases and types. MiR‐135a downregulates multiple parameters of cancer progression, and its expression is decreased in metastatic breast, gastric and prostate tumors. Further, MiR‐135a inhibits migration and invasion and regulates epithelial–mesenchymal transition (EMT)‐related marker genes in lung cancer cells.29, 30, 31 Functional analyses showed that LIMK2 SNP rs2073859 is within the miR‐135a seed‐binding region. The combined frequency of A genotypes (AG + AA) was higher in cancerous bladder tissues than in normal bladder tissue and correlated with the risks of high grade and high stage. MiR‐135a negatively regulates LIMK2, with rs2073859 variant A leading to loss of miR‐135a regulation, increased LIMK2 expression, and ultimately elevated disease risk (Supporting Information Fig. S4).
In conclusion, our study suggests that LIMK2 may act as an oncogene in BC and reveals a potentially important genetic risk factor, disruption of normal LIMK2 downregulation by miR‐135a. Additional population and functional studies are warranted to confirm our findings.
Supporting information
Supplementary Figure 1. Representative screening for LIMK2 genotypes. Patterns for each of the genotypes GG, AA, and AG screened by PCR‐RFLP. Arrows indicate the size of each fragment (bp) at the rs2073859 polymorphic site. M: Molecular size markers.
Supplementary Figure 2. Western blotting of cofilin and p‐cofilin in LIMK2‐overexpressing UM‐UC‐3 cells or shRNA‐mediated LIMK2 knockdown cells. GAPDH was used as a loading control.
Supplementary Figure 3. LIMK2 protein expression levels in in 8 BC cell lines (BIU‐87, J82, TCCSUP, UM‐UC‐3, EJ, T24, and 5,637, RT4) and the normal bladder cell line SV‐HUC‐1.
Supplementary Figure 4. A model for microRNA‐mediated regulation of LIMK2.
Acknowledgments
The research is funded by the National Natural Science Foundation of China under grant agreement number 81372744.
W.W., C.Y. and X.Q. are equal first authors.
W.W. and H.N are equal senior authors
Contributor Information
Wei Wang, Email: wangweiccc@hotmail.com.
Haibo Nie, Email: niehaibo98@163.com.
References
- 1. Feber A, Clark J, Goodwin G, et al. Amplification and overexpression of E2F3 in human bladder cancer. Oncogene 2004;23:1627–30. [DOI] [PubMed] [Google Scholar]
- 2. Johnson EO, Chang KH, Ghosh S, et al. LIMK2 is a crucial regulator and effector of Aurora‐A‐kinase‐mediated malignancy. J Cell Sci 2012;125:1204–16. [DOI] [PubMed] [Google Scholar]
- 3. Maekawa M, Ishizaki T, Boku S, et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM‐kinase. Science 1999;285:895–8. [DOI] [PubMed] [Google Scholar]
- 4. Sumi T, Matsumoto K, Takai Y, et al. Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho‐ and Cdc42‐activated LIM‐kinase 2. J Cell Biol 1999;147:1519–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang N, Higuchi O, Ohashi K, et al. Cofilin phosphorylation by LIM‐kinase 1 and its role in Rac‐mediated actin reorganization. Nature 1998;393:809–12. [DOI] [PubMed] [Google Scholar]
- 6. Oku Y, Tareyanagi C, Takaya S, et al. Multimodal effects of small molecule ROCK and LIMK inhibitors on mitosis, and their implication as anti‐leukemia agents. PLoS One. 2014;9:e92402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mardilovich K, Gabrielsen M, McGarry L, et al. Elevated LIM kinase 1 in nonmetastatic prostate cancer reflects its role in facilitating androgen receptor nuclear translocation. Mol Cancer Ther 2015;14:246–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Prudent R, Vassal‐Stermann E, Nguyen CH, et al. Pharmacological inhibition of LIM kinase stabilizes microtubules and inhibits neoplastic growth. Cancer Res 2012;72:4429–39. [DOI] [PubMed] [Google Scholar]
- 9. Lagoutte E, Villeneuve C, Lafanechere L, et al. LIMK regulates tumor‐cell invasion and matrix degradation through tyrosine phosphorylation of MT1‐MMP. Sci Rep 2016;6:24925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer 2010;10:389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang W, Yuasa T, Tsuchiya N, et al. The novel tumor‐suppressor Mel‐18 in prostate cancer: its functional polymorphism, expression and clinical significance. Int J Cancer 2009;125:2836–43. [DOI] [PubMed] [Google Scholar]
- 12. Calabro F, Lorusso V, Rosati G, et al. Gemcitabine and paclitaxel every 2 weeks in patients with previously untreated urothelial carcinoma. Cancer 2009;115:2652–9. [DOI] [PubMed] [Google Scholar]
- 13. Luo J, Cai Q, Wang W, et al. A microRNA‐7 binding site polymorphism in HOXB5 leads to differential gene expression in bladder cancer. PLoS One. 2012;7:e40127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lourenco FC, Munro J, Brown J, et al. Reduced LIMK2 expression in colorectal cancer reflects its role in limiting stem cell proliferation. Gut 2014;63:480–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu Y, Wang W, Hu W, et al. MicroRNA‐205 suppresses the growth of adrenocortical carcinoma SW‐13 cells via targeting Bcl‐2. Oncol Rep 2015;34:3104–10. [DOI] [PubMed] [Google Scholar]
- 16. Song L, Li W, Zhang H, et al. Over‐expression of AEG‐1 significantly associates with tumour aggressiveness and poor prognosis in human non‐small cell lung cancer. J Pathol 2009;219:317–26. [DOI] [PubMed] [Google Scholar]
- 17. Tsuchiya N, Wang L, Suzuki H, et al. Impact of IGF‐I and CYP19 gene polymorphisms on the survival of patients with metastatic prostate cancer. J Clin Oncol 2006;24:1982–9. [DOI] [PubMed] [Google Scholar]
- 18. Nicoloso MS, Sun H, Spizzo R, et al. Single‐nucleotide polymorphisms inside microRNA target sites influence tumor susceptibility. Cancer Res 2010;70:2789–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Manetti F. Recent findings confirm LIM domain kinases as emerging target candidates for cancer therapy. Curr Cancer Drug Targets 2012;12:543–60. [DOI] [PubMed] [Google Scholar]
- 20. Manetti F. LIM kinases are attractive targets with many macromolecular partners and only a few small molecule regulators. Med Res Rev 2012;32:968–98. [DOI] [PubMed] [Google Scholar]
- 21. Rak R, Haklai R, Elad‐Tzfadia G, et al. Novel LIMK2 inhibitor blocks panc‐1 tumor growth in a mouse xenograft model. Oncoscience 2014;1:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Croft DR, Olson MF. The Rho GTPase effector ROCK regulates cyclin A, cyclin D1, and p27Kip1 levels by distinct mechanisms. Mol Cell Biol 2006;26:4612–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suyama E, Wadhwa R, Kawasaki H, et al. LIM kinase‐2 targeting as a possible anti‐metastasis therapy. J Gene Med 2004;6:357–63. [DOI] [PubMed] [Google Scholar]
- 24. Vlecken DH, Bagowski CP. LIMK1 and LIMK2 are important for metastatic behavior and tumor cell‐induced angiogenesis of pancreatic cancer cells. Zebrafish 2009;6:433–9. [DOI] [PubMed] [Google Scholar]
- 25. Gamell C, Schofield AV, Suryadinata R, et al. LIMK2 mediates resistance to chemotherapeutic drugs in neuroblastoma cells through regulation of drug‐induced cell cycle arrest. PLoS One. 2013;8:e72850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Po'uha ST, Shum MS, Goebel A, et al. LIM‐kinase 2, a regulator of actin dynamics, is involved in mitotic spindle integrity and sensitivity to microtubule‐destabilizing drugs. Oncogene 2010;29:597–607. [DOI] [PubMed] [Google Scholar]
- 27. Croft DR, Crighton D, Samuel MS, et al. p53‐mediated transcriptional regulation and activation of the actin cytoskeleton regulatory RhoC to LIMK2 signaling pathway promotes cell survival. Cell Res 2011;21:666–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hsu FF, Lin TY, Chen JY, et al. p53‐Mediated transactivation of LIMK2b links actin dynamics to cell cycle checkpoint control. Oncogene 2010;29:2864–76. [DOI] [PubMed] [Google Scholar]
- 29. Tribollet V, Barenton B, Kroiss A, et al. miR‐135a inhibits the invasion of cancer cells via suppression of ERRalpha. PLoS One 2016;11:e0156445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang C, Chen X, Chen X, et al. miR‐135a acts as a tumor suppressor in gastric cancer in part by targeting KIFC1. Onco Targets Ther 2016;9:3555–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shi H, Ji Y, Zhang D, et al. MiR‐135a inhibits migration and invasion and regulates EMT‐related marker genes by targeting KLF8 in lung cancer cells. Biochem Biophys Res Commun 2015;465:125–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Representative screening for LIMK2 genotypes. Patterns for each of the genotypes GG, AA, and AG screened by PCR‐RFLP. Arrows indicate the size of each fragment (bp) at the rs2073859 polymorphic site. M: Molecular size markers.
Supplementary Figure 2. Western blotting of cofilin and p‐cofilin in LIMK2‐overexpressing UM‐UC‐3 cells or shRNA‐mediated LIMK2 knockdown cells. GAPDH was used as a loading control.
Supplementary Figure 3. LIMK2 protein expression levels in in 8 BC cell lines (BIU‐87, J82, TCCSUP, UM‐UC‐3, EJ, T24, and 5,637, RT4) and the normal bladder cell line SV‐HUC‐1.
Supplementary Figure 4. A model for microRNA‐mediated regulation of LIMK2.