

Abstract
We provide immense insulin absorption from the gastrointestinal tract, combining apical sodium-dependent bile acid transporter-mediated intestinal uptake and the lymphatic transport pathway. This strategy has proven to employ chondroitin sulfate-g-taurocholic acid coated, insulin-loaded partially uncapped liposome (IPUL-CST) for type 1 diabetes mellitus (T1DM) treatment. The loading efficiency of insulin in IPUL-CST increased significantly from 33% to 75% via the partially uncapped liposome preparation method. Moreover, the IPUL-CST revealed an improved insulin protection efficacy in GIT simulated pH and digestive enzyme conditions. The high dose of IPUL-CST in the small intestine was detected 4 hours post-oral administration using ex vivo optical imaging and fluorescence intensity. The IPUL-CST exhibited significantly enhanced intestinal absorption (oral bioavailability; 34%, Tmax; 9 hour) and reduced blood glucose levels for 16 hours in T1DM. The results demonstrated that the new investigated IPUL-CST is a promising carrier for oral insulin delivery.
Keywords: oral insulin delivery, taurocholic acid, massive intestinal absorption, chondroitin sulfate coated liposome, long acting insulin
Table of Contents Use Only
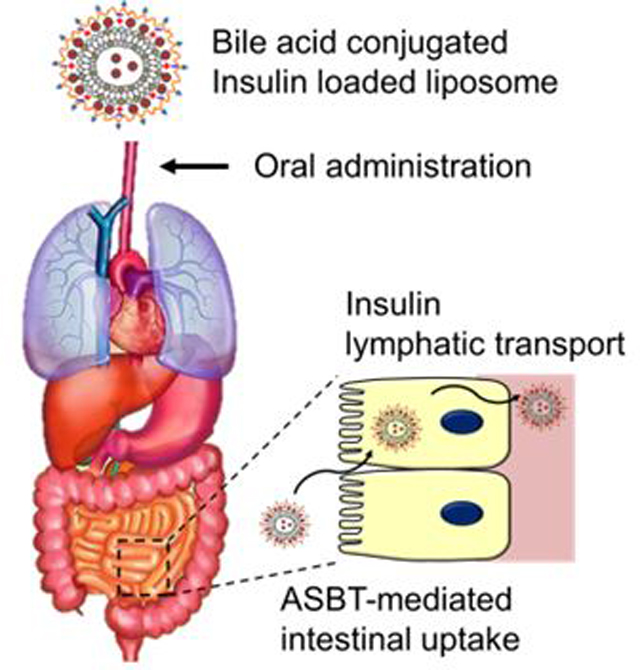
INTRODUCTION
Since being discovered in 1922, porcine insulin, human recombinant insulin, modified/engineered insulin and their pharmaceutical formulations have saved the lives of type 1 diabetes mellitus (T1DM) patients by subcutaneous (SC) injections multiple times a day.1–3 Insulin is also being used to treat advanced T2DM patients.4–5 The insulin oral delivery was first reported in 1923 and has been regarded as the most demanding technology for over 90 years.6–7 It is assumed to benefit patients with compliance,8 hepatic insulinization via portal entry (first pass effect),9 reducing peripheral hyperinsulinemia,10–11 avoiding possible hypoglycemic episodes,12 and minimizing weight gain.13 However, insulin oral technology has continued to repeat between ambition and failures,14 yielding no practical products available by far for clinical applications, despite a dozen of on-going clinical trials.
The quest for the oral delivery of insulin has been a continuous history of ambition and failure14 and, more recently, a chimera.15 The need for a flexible oral delivery platform is ever more compelling as new biologic APIs occupy major pharmaceutical markets, exclusively as parenteral infections (>USD 200 billion).16 Most oral biologic delivery strategies have employed oral enhancer technology to facilitate either transcellular and paracellular pathways, or protective digestive enzyme inhibitors.17–18 Combinations of these two strategies, plus transient disturbances of the intestinal tight junctions and gut mucoadhesion approaches have all been extensively investigated.19–20 Applied to delivering relatively small size polypeptides, such as insulin and exendin-4, these methods typically yield oral bioavailability (oBA) up to 16% in rodent models.21–22 These promising preclinical results have encouraged the development of many formulations; however, when tested in clinical trials, oral delivery technology has scarcely shown oBA higher than 1%.
The oral insulin delivery technology has been heavily relying on permeation/absorption enhancers, temporal tight junction perturbation, digestive enzyme inhibitors, mucoadhesion, and enteric coating.23–24 Nanoformulations of insulin for oral delivery are also rich in literature.21, 25–28 All articles reported relative insulin oral bioavailability (oBA) related to SC injection of up to 16% in rodent models.29–30 Similar results were observed with other small peptides/proteins not larger than insulin.31 The technology has been translated and encouraged by animal results; however, the clinical oral bioavailability hardly exceeds 1%. This limited oBA continues for decades without further improvement.
Biologics ranging from small peptides to full size antibodies become dominant in the current drug market in terms of sales volume and 9.6% of new drugs approved for marketing by the FDA is biologics.32 All are formulated as injectable, such as pre-filled syringes. No proteins larger than insulin have been challenged for oral delivery.
The serious discrepancy between preclinical and clinical oBA and the limited size of proteins urgently demand novel mechanisms for oral delivery. Should the absorption of nanoparticles, which carry an active pharmaceutical ingredient (API) and suffers from poor oBA, occurs, it would be free from most hurdles/barriers encountered in the oral delivery route and would also be free from size limitation. Receptor mediated endocytosis of nanoparticles, such as those conjugated with vitamin B12,33 and the active uptake of nanoparticles by the M cell in Peyer’s Patch can possibly be such alternatives,34 but with extremely limited uptake capacity for achieving therapeutic levels in clinical settings.
The gastrointestinal epithelial cells (enterocytes) located in the distal ileum is one of the major players, along with hepatocytes, for the enterohepatic recirculation of bile acids (BA) secreted as bile salts from the liver to emulsify the oily nutrients to help with digestion.35 The recycling capacity is around 12–18 grams per day with a recycling efficiency higher than 90%.36 Multiple transporters in ileocytes (enterocytes in the distal ileum) and hepatocytes collaboratively work for the recycling.35, 37 A recent study revealed that low molecular heparin modified with deoxycholic acid (DOCA), one of richest bile acids in rodents, enters ileocytes using an apical sodium-dependent bile acid transporter (ASBT).38 The observed entry mechanism of the modified heparin was clearly distinguished from the pumping mechanism of small-molecule bile acids of which details were only recently elucidated.
While the specific entry and transport mechanisms are largely unknown and widely open for investigation, it was reported that a nanoparticle self-assembled from heparin-taurocholic acid (TCA)-docetaxel conjugate was orally absorbed to present an anticancer effect.39 This prompted us to test with nanoparticles carrying insulin as our first model small-size protein in chemically induced T1DM rats. This trial system was selected as a widely known animal model and is a standard assessment of the biological activity of delivered insulin.
This is the first report of insulin oral delivery and lymphatic transportation using a carrier nanoparticle (IPUL-CST), a partially uncapped cationic liposome, where insulin was loaded via diffusion and electrostatic interaction. The insulin loaded liposome was then coated with a chondroitin sulfate-TCA conjugate (CST). The apparent bioavailability after orally feeding a carrier solution reached approximately 34% with a relatively flat pharmacokinetic profile for ~24 hours in plasma.
EXPERIMENTAL SECTION
Synthesis of Chondroitin Sulfate-taurocholic Acid Conjugate.
The procedure for the synthesis of chondroitin sulfate-g-taurocholic acid (CST) is presented in Fig. S1. To synthesis of TCA-carbonate, taurocholic acid sodium salt hydrate (TCA, 925.0 μmol), 4-Dimethylaminopyridine (DMAP, 1.5 mmol), and 4-nitrophenyl chloroformate (4-NPC, 1.4 mmol) were dissolved in 10 mL of dimethylformamide (DMF). The reaction mixture was stirred for 6 h at room temperature (RT). The reacted solution was filtered and precipitated in ethyl acetate (EA). The collected pellet was dried in vacuo for 24 hours. The dried powder was dissolved in 10 mL of distilled water (DW) and then it was filtrated and lyophilized.40 To synthesis of amine-modified TCA, the TCA-carbonate (925.0 μmol), 4-methylmorpholine (4-MMP, 1.85 mmol), and ethylenediamine (EDA, 92.5 mmol) were dissolved in 5 mL of DMF. The reaction mixture was stirred for 16 hours at RT. The reacted solution was rotary evaporation to remove unreacted 4-MMP and EDA at 100 °C, and then it was precipitated by acetonitrile (ACN) with vigorous stirring. The collected pellet was dissolved in DMF, and then it was re-precipitated by EA with vigorous stirring. The collected pellet was dried in vacuo for 24 hours. To synthesis of CST, Chondroitin sulfate (CS, 10–40 kDa, injectable grade, Yantai Dongcheng Biochemicals Co.,Ltd., Shandong, China, 25 μmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC, 2.25 mmol) and N-hydroxysuccinimide (NHS, 2.25 mmol) were dissolved in 8 mL of 0.1M pH 6.0 MES buffer. The reaction mixture was stirred for 24 hours at RT. The reacted solution was filtered and three times precipitated by 60% cold ethanol, and then it was dried in vacuo for 24 hours. The synthesized CST was characterized by Mercury 400 1H NMR spectrophotometers.
Preparation of CST Coated Insulin Loaded Partially Uncapped Liposome (IPUL-CST).
As in our previous report,41 the procedure for the preparation of insulin loaded partially uncapped liposome (IPUL) is presented in Table 1. Dimethyl dioctadecyl ammonium bromide (DDAB), deoxycholic acid (DOCA), and 10 nm size of superparamagnetic iron oxide nanoparticles (SPION, Sigma-Aldrich, St. Louis, MO, USA) dissolved in 10 mL of chloroform. To make a thin file on the flask, the solvent was removed by rotary evaporation at 80 °C. The film was rehydrated in 10 mL of DW using a VWR scientific Aquasonic 75T Ultrasonic cleaner for 5 min at 65 °C. The obtained liposomes were slowly mixed using a magnetic impeller with a tailor-made magnet formed with two quarter-circles (30 mm in radius and 2 mm thick). The liposomes that stuck to the magnet were again stirred for 10 sec using a magnetic impeller at 1,500 rpm with the tailor-made magnet. After the liposome solution was magnetically stirred, ring-shaped, neodymium rare-earth magnets (10 mm in radius and 10 mm thick) were immediately attached to the bottom of the flask to remove the free SPION that had leaked from the liposomes. 10 mg of human recombinant insulin (SAFC Biosciences Inc., Lenexa, KS, USA) was dissolved in pH 3.0 DW, and then it added to liposome solution, slowly stirred at 37 °C for 2 hours, which particularly enabled facile protein encapsulation through the open pore of liposome. The insulin-loaded liposome was then purified via dialysis (Spectra/Por® MWCO 50 kDa membrane) against fresh DW for 24 hours to remove unloaded insulin. The insulin loading efficiencies was estimated using a reversed-phase high-performance liquid chromatographic (HPLC) method.42 A reversed-phase Zorbax C18 column (Agilent, 4.6 mmID, 150 mm) and gradient elution with a mobile phase composed of ACN and 0.1% trifluoroacetic acid (TFA) at a flow rate of 1 mL/min was used. Insulin identification was measured at UV 214 nm. The mobile phase changed from 70:30 (ACN:TFA, v/v) to 60:40 (v/v) in 5 min followed by isocratic elution at 60:40 (v/v) for a further five minutes. The CS or CST coated liposome (IPUL-CS and IPUL-CST) was prepared by mixing cationic charged liposome with anionic charged CS or CST complex by electrostatic interaction. In brief, 10 mL of IPUL was added to 0.2 mL of CS or CST (100 mg/mL) solution, and then stirred for 2 min. The size and zeta-potential of the each liposome was measured using a dynamic light scattering (Zetasizer 3000, Malvern Instruments, USA). Prior to the measurement, the liposome solution was stabilized at RT for 2 hours. The results were obtained with the average of three measurements. The size and shape of IPUL-CST in water was measured by TEM using a JEOL JEM-1400 Plus operated at 120 kV.
Table 1.
Pharmacokinetic of insulin in T1DM rats after SC administration of the free insulin, oral administration of IPUL-CS and IPUL-CST. Cmax: maximum plasma concentration; Tmax: time at which Cmax is attained; AUC: area under the plasma concentration-time curve.
| Formulation | Admin route |
Admin Vol (μL) |
Dose (IU/kg) |
Tmax (h) | Cmax (μIU/mL) |
AUC(0–10h)
(μIU·h/mL) |
oBA (%) |
|---|---|---|---|---|---|---|---|
| Free insulin | SC | 200 | 5 | - | 102 ± 6 | 274 ± 21 | 100 |
| IPUL-CS | Oral | 500 | 20 | 12 | 3 ± 2 | 43 ± 17 | 4 ± 2 |
| IPUL-CST | Oral | 500 | 20 | 9 | 45 ± 6 | 376 ± 43 | 34 ± 4 |
Each value represents the mean ± SEM
Synthesis of Chlorin e6-labeled Insulin (Ce6-Insulin).
Chlorin e6 (Ce6, 0.1 mM), EDC (5 mM), and NHS (5 mM) were dissolved in 10 mL of DW. After 30 min, excess insulin (10 mM) was added to reaction mixture, stirred at RT for 3 days. The resulting solution was then purified via dialysis (Spectra/Por MWCO 50 kDa membrane) against fresh DW for 2 days to remove unconjugated chemicals. The obtained solution was lyophilized.
Insulin Release Study.
In vitro insulin release was measured by a dialysis method. In brief, the IPUL, IPUL-CS, and IPUL-CST were dispersed in release medium. The suspension was put into a dialysis membrane (Spectra/Por® MWCO 50 kDa). The dialysis membrane bag was sealed and subsequently immersed in a vial containing fresh release medium. The release of insulin from the liposomes was induced under mechanical shaking (100 rev./min) at 37 °C. The outer phase of the dialysis membrane bag was extracted and replaced with fresh buffer solution at predetermined time intervals (0–24 hours). The insulin concentration of the extracted solution was calculated by HPLC method (n=3).
Insulin Protection Efficacy in GIT Simulated Conditions.
To evaluate the insulin protects efficacy of each liposomes, 0.5 mL of liposome solution was diluted in 50 mL of digestive media such as simulated gastric fluid (pH 1.2, containing 1% pepsin), intestinal fluid (pH 6.8, containing 1% trypsin), or α-chymotrypsin solution (100 μg/mL, in phosphate buffer, pH 7.8). The temperature was maintained at 37 ± 1.0°C and stirred at 100 rpm. At each hour, 200 μL of the solution was obtained and measured the insulin concentration by HPLC method (n=3).
ASBT-mediated Cellular Uptake Efficacy.
ASBT expressed Caco-2 cells (human epithelial colorectal adenocarcinoma) were maintained in Dulbeco’s Modified Eagle’s Medium (DMEM) containing 1% penicillin–streptomycin and 10% fetal bovine serum (FBS) in a humidified standard incubator at 37°C with a 5% CO2 atmosphere. Caco-2 cells were seed in 6-well plates with cover glass at a density of 1×105 cell per well, incubated for 24 hours. The free Ce6-insulin, Ce6-insulin loaded IPUL, IPUL-CS, and IPUL-CST (Ce6 concentration; 10 μg/mL) were treated to each well, incubated for 4 hours. Each well was washed with PBS three times. After washing, cells were fixed with 4% paraformaldehyde immediately and nuclei were stained with hoechst (blue). The localization of insulin was observed using a confocal laser scanning microscope (n=3). To flow cytometry analysis, Caco-2 cells were seed in 6-well plates at a density of 1×105 cell per well, incubated for 24 hours. The Ce6-insulin loaded IPUL, IPUL-CS, and IPUL-CST (Ce6 concentration; 10 μg/mL) were treated to each well, incubated for 4 hours. After 4 hours, cells were lifted with DPBS containing 2% FBS (FACS buffer) and washed three times with FACS buffer. 5×104 cells per sample were analyzed by FACSCalibur™ Flow Cytometer (Becton Dickinson, USA) and subsequent data analysis was performed using FlowJo software.
Biodistribution Analysis.
All animal experiments were approved by Institutional Animal Care and Use Committee (IACUC) of University of Utah and the Catholic University of Korea. Ce6-insulin (SC administration dose; 5 IU/kg per mice), Ce6-insulin loaded IPUL-CS, and IPUL-CST (oral administration dose; 5 IU/kg per mice) were injected into 6–8 weeks BALB/c nude mice. At 4 hours post-administration, each organ was harvested. The images were obtained using a 12-bit CCD camera (Image Station 4000 MM; Kodak, New Haven, CT). To analysis of insulin contents in each organ, after 4 hours post-administration, each organ harvested and suspended in 70% ethanol with 0.3 N HCl and then homogenized to extract Ce6-insulin. Following centrifugation, Ce6-insulin fluorescence (excitation at 410 nm, emission at 670 nm) in the supernatant was measured using a fluorescence plate reader (n=5).
In vivo Pharmacokinetic Study.
To establish the T1DM model, male SD rats (200–225 g) were rendered diabetic by intraperitoneal injection of streptozotocin (STZ, dissolved in 10 mM citrate buffer at pH 4.5) at a dose of 75 mg/kg body weight.43 Rats were considered diabetic when their fasting blood glucose level was higher than 350 mg/dL. To evaluate the blood insulin level and oral bioavailability of IPUL-CS and IPUL-CST, 200–225 g of male Sprague-Dawley (SD) rats were randomly divided into three groups (n=5). They were fasted for 8 h before and 4 hours after administration with free access to water. The following formulations were administered to T1DM rats individually: IPUL-CS (oral gavage, 20.0 IU/kg), IPUL-CST (oral gavage, 20.0 IU/kg), and free insulin (SC injection, 5.0 IU/kg). Blood samples (200 μL) were collected from the jugular vein, and then centrifuged (3,200 rpm, 4 °C for 10 min) and subsequently quantified using insulin ELISA kit (Crystal Chem, Downers Grove, IL, USA). The relative oral bioavailability (oBA) value can be expressed by eqn (1).44
| (1) |
In vivo IPUL-CST Absorption Pathway.
To evaluate the absoprtion pathway, the IPUL-CST (20 IU/kg) was administered oral gavage (o.g.) into lymph fistula rat model (n=3).45 Lymph samples (200 μL) were collected for 6 hours and then centrifuged (3,200 rpm, 4 °C for 10 min) and subsequently quantified by insulin ELISA kit. The size and shape of IPUL-CST in plasma and lymph were measured by TEM using a JEOL JEM-1400 Plus operated at 120 kV.
In vivo Blood Glucose Levels.
To clearfy the T1DM therapeutic effect of IPUL-CST, non-diabetic control rats (NC), diabetic control (DC), insulin unloaded IPUL-CST, a mixture of insulin unloaded IPUL-CST and free insulin, and SC administration of free insulin (5 IU/kg) were used as controls (n=5). Prior to all drug administration, they were water fated 8 h before and remained water fasted after 12 hours post-administrations. Blood samples were collected from the tail vein of rats pre-and post-drug administration. The blood glucose level was immediately measured using a blood glucose meter (Contour next EZ, Bayer).
Statistical Analysis.
The results from the in vitro and ex vivo studies are expressed as the mean ± standard deviation (SD). The results from the in vivo studies are expressed as the mean ± standard error of the mean (SEM). Differences between the values were assessed using Student’s t-test.
RESULTS AND DISCUSSION
Synthesis of CST.
In order to use liposome shielding and intestinal absorption enhancing materials, we synthesized chondroitin sulfate-g-taurocholic acid (CST) using a previous report.39 The procedure for the synthesis of CST is presented in Figure S1. The hydroxyl group of TCA was modified to primary amine. The amine-modified TCA was coupled with carboxylic groups of CS through a conventional carbodiimide coupling reaction. Successful synthesis of CST was confirmed by a -CH3 peak of TCA appearing at 0.5–1 ppm using 1H-NMR (Figure S2). The degree of TCA conjugation on CS could be estimated by comparing the intensities of the -CH3 peak at 1.8 ppm from CS. The degree of TCA was determined as 16 units per 100 repeat units of CS.
Preparation and Characterization of IPUL.
We prepared the four compositions and characterizations of liposome as Table S1. These liposomes were prepared according to our previous report.41 Figure 1a shows the schematic representation of IPUL-CSG. The conventional liposome (CL) was prepared without SPION, following the conventional thin film rehydration method,46 using the same volume of DDAB and DOCA. The IPUL showed a 1.7-fold higher loading efficiency (58%) than CL (33%) due to the partially uncapped method. The partially uncapped method exhibited the SPION dispersed in the liposome, which can make magnetic shear stress consequently squeeze the liposome surface and tear it, to form open lipid bilayer holes.41 This allows insulin to be encapsulated not only in the outside of liposomes but also in the internal space. Thus, the IPUL-CS and IPUL-CST exhibited a maximum 77% loading efficiency. This indicates that the loaded insulin is located at both the internal and external stage of the IPUL. The average size of IPUL-CS (176 nm size) and IPUL-CST (194 nm size) increased more than CL (149 nm size) and IPUL (162 nm size) due to not having any surface coating and insulin loading. The size and shape of IPUL, IPUL-CS, and IPUL-CST in distilled water was measured by transmission electron microscopy (TEM) (Figure 1b and Figure S3). The zeta-potential of IPUL-CS and IPUL-CST changed from cationic to anionic as the insulin-loaded cationic liposome form complexed with anionic CS or CST. These results indicate that IPUL-CST has a 2.2-fold higher insulin loading efficiency than CL and also has a suitable nanoparticle size for oral insulin delivery.
Figure 1.
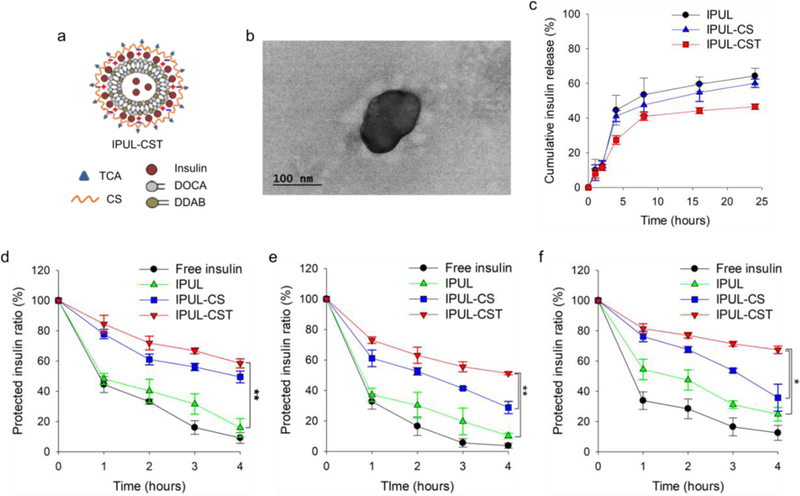
a) Schematic representation of IPUL-CST. b) TEM images of IPUL-CST in distilled water. c) Drug release profile of IPUL, IPUL-CS, and IPUL-CST, at pH 1.2 (for 2 hours incubation) and pH 7.4 (after 2 h incubation at pH 1.2). Protection of insulin at d) pepsin (pH 1.2), e) trypsin (pH 6.8), and f) α-chymotrypsin (pH 7.8) (n=3). *P <0.01, **P <0.001.
Insulin Protection from GIT Simulated Conditions.
We measured the insulin release profile under GIT simulated conditions. The IPUL, IPUL-CS, and IPUL-CST showed 64%, 60%, 47% of cumulative insulin release for 24 hours, respectively (Figure 1c). Only a small amount of insulin was released from liposome, 10–14% at a pH of 1.2. This result indicates that these liposomes are generally stable and do not lose a significant amount of insulin when exposed to an environment of low pH, such as in the stomach. To prove the insulin protection capacity of liposomes from enzymatic digestion, insulin remaining ratio was evaluated in various pH medium containing pepsin, trypsin, and α-chymotrypsin (Figure 1d-f). The free insulin was used as a negative control. The IPUL showed rapid insulin degradation within 4 hours at three enzyme conditions. However, the insulin degradation of IPUL-CS and IPUL-CST decreased less than IPUL. Notably, the IPUL-CST exhibited a maximum 5-fold higher insulin protection efficacy than IPUL (P value > 0.01). These results suggest that CST coated liposome have improved insulin protection ability compared to non-coated liposome in various enzymatic environments. This insulin protection ability plays an important role in the increase of insulin absorption in the GIT because it increases the opportunity to reach the small intestine.
ASBT-mediated Epithelial Cellular Uptake.
To evaluate the ASBT-mediated epithelial cellular uptake efficacy of liposome using florescence probes, we prepared chlorin e6 labeled-insulin (Ce6-insulin) loaded liposomes. The free Ce6-insulin, Ce6-insulin loaded IPUL, IPUL-CS, and IPUL-CST were treated and observed in ASBT expressed Caco-2 cells (Figure 2a). The IPUL-CST exhibited the highest cellular uptake efficacy compared to IPUL and IPUL-CS (Figure 2b). This evidence indicates that the TCA can lead to nanoparticle oral absorption enhancement.
Figure 2.
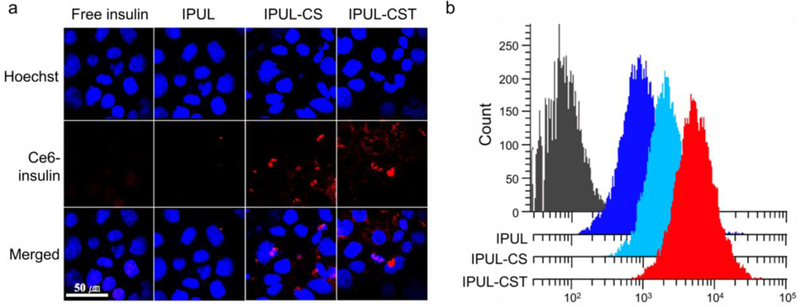
a) Confocal microscopy images of Caco-2 cells after 4 h incubation with Ce6-labeled insulin (Ce6-insulin) loaded IPUL, IPUL-CS, and IPUL-CST (10 μg/mL of Ce6-insulin concentration). b) Cellular uptake efficacy of Ce6-insulin loaded IPUL, IPUL-CS, and IPUL-CST using flow cytometry.
Ex vivo Biodistribution.
To visualize and quantify IPUL-CST in each organ, we synthesized the pH-insensitive fluorescent dye of chlorin e6-insulin (Ce6-insulin) conjugate. The Ce6-insulin, Ce6-insulin loaded IPUL-CS and IPUL-CST were administered in Balb/c nude mice. These mice were sacrificed and each organ was harvested 4 hours post-administration. The IPUL-CS displayed the highest accumulation in the duodenum and jejunum, while IPUL-CST displayed high accumulation in not only the duodenum and jejunum, but also the ileum due to ASBT being the most highly expressed in the ileum (Figure 3a).47 To clearly quantify insulin in each organ, we analyzed for insulin contents by fluorescent spectrophotometry, and the data is presented as the injected dose accumulated per mass unit of organ (Figure 3b). A large amount of IPUL-CST was measured in the duodenum, jejunum, and ileum, which is consistent with Figure 3a (P value < 0.01). These results suggest that the IPUL-CST can have a TCA-mediated interaction with the ASBT of the ileum in the in vivo environment.
Figure 3.
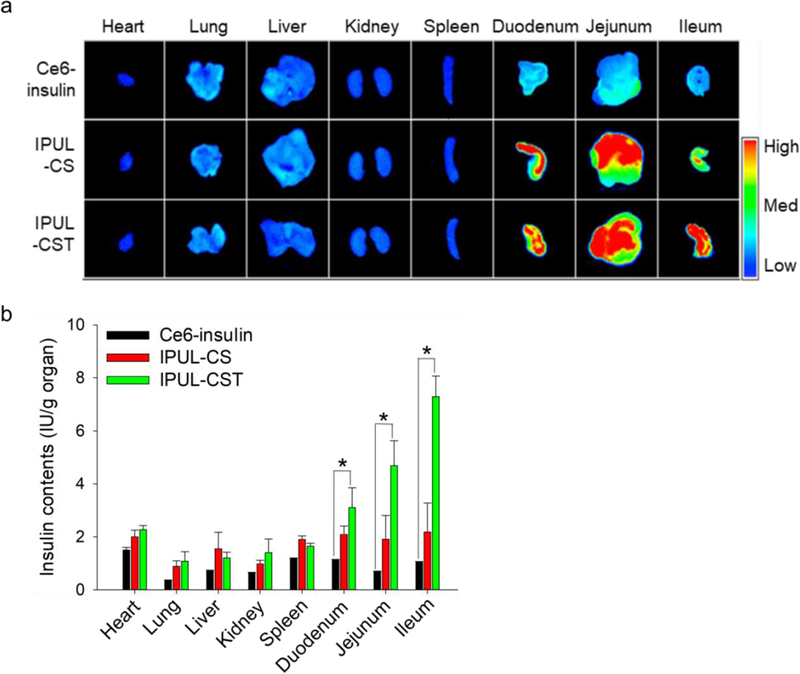
a) Near-infrared fluorescence images and b) fluorescence signal of isolated main organs from 5 IU/kg of each sample administered Balb/c nude mice (n=5). Ce6-insulin was administered by SC injection. IPUL-CS and IPUL-CSG were administered o.g. (n=5). *P <0.01.
In vivo Pharmacokinetic study and Absorption Pathway.
To evaluate the insulin absorption efficacy of IPUL-CST in GIT, we measured the time-dependent plasma insulin levels and the related pharmacokinetic parameters (Table 1). The plasma insulin levels of the SC injected insulin solution increased dramatically and rapidly decreased within 6 hours post-administration, while the IPUL-CST exhibited a sustained increase for 9 hours and slowly decreased 24 hours post-administration (Figure 4a and Figure S4a). The IPUL-CS was used as a negative control group. The group orally administered with IPUL-CST exhibited an 8-fold higher AUC than the group orally administered with IPUL-CS. The IPUL-CST exhibited a significantly enhanced oral bioavailability (oBA; 34%) by overcoming numerous difficulties encountered with the intestinal absorption of insulin. The presence of IPUL-CST in the plasma was confirmed by TEM imaging (Figure 4b), indicating IPUL-CST absorbed in a GIT intact form. These results demonstrate that the IPUL-CST formulation exhibited ASBT-mediated sustained absorption and release of insulin in the GIT.
Figure 4.
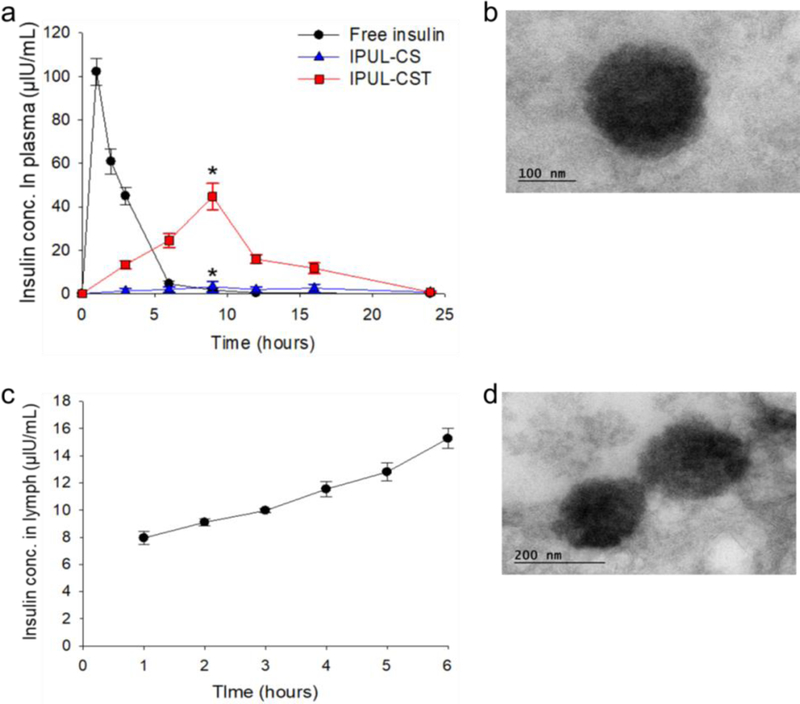
a) Plasma insulin level versus time profiles of T1DM SD rats. Free insulin (5 IU/kg) was administered by SC injection. IPUL-CS (20 IU/kg) and IPUL-CSG (20 IU/kg) were administered o.g. (n=5). *P < 0.001. b) TEM images of IPUL-CST in plasma at 6 hours post-administration o.g. with 20 IU/kg of IPUL-CST into SD rats. c) Lymph insulin concentration versus time profiles in fistula SD rat model (n=3). c) TEM images of IPUL-CST in cisterna chyli at 6 hours post-administration o.g. with 20 IU/kg of IPUL-CST into SD rats.
To investigate the cause of high oBA in IPUL-CST, the absorption pathway of IPUL-CST was clearly confirmed by using a lymph fistula rat model.45 Insulin was found to increase gradually in the lymph 6 hours post-administration (Figure 4c and Figure S4b). The presence of IPUL-CST in the lymph was also confirmed by TEM imaging (Figure 4d). The lymphatic transport of IPUL-CST is presumed to share the chylomicron transport pathway, the lipid transport pathway, according to previous study.48 These results suggest that IPUL-CST absorbed from the distal ileum is transmitted through the lymphatic pathway to systemic circulation. The pathway of API delivery through the lymphatic pathway is thought to be an increase in oBA because it can avoid the first pass effect in the liver.
T1DM Treatment.
The most important part of our strategy is T1DM treatment through oral insulin delivery in vivo. To prove this, various doses of IPUL-CST was administered o.g. in fasted T1DM rats (Figure 5a). The blood glucose level showed that it was dose-dependent on IPUL-CST (P value < 0.01). At 24 hours post-administration, the blood glucose level returned to the initial glucose level. In further study, insulin dose in oral administration was fixed with 20 IU/kg per rat. To evaluate the T1DM therapeutic efficacy, the free insulin (5 IU/kg per rat, SC injection), IPUL-CS and IPUL-CST (o.g.) were administered in fasted T1DM rats. The blood glucose level of the free insulin solution rapidly decreased, with a maximum decrease to 72% 2 hours post-administration and then returning to the base level 9 hours post-administration (Figure 5b). This short-term effect and extreme drop in blood glucose levels need repeated insulin administration and cause undesirable side effects.48 However, the IPUL-CST exhibited gradually decreased blood glucose levels (maximum decrease to 34% at 12 hours post-administration, P value < 0.001) 16 hours post-administration, which is consistent with Figure 4a, while the IPUL-CS showed no decrease in blood glucose levels, indicating poor oral absorption due to the IPUL-CS absence of a suitable oral absorption enhancing agent. This result is consistent with the above changes in blood insulin patterns and indicates that the blood glucose change is caused by absorbed insulin. To clarify the T1DM therapeutic effect of IPUL-CST, the insulin unloaded IPUL-CST, a physical mixture of insulin unloaded IPUL-CST, and free insulin was administered o.g. to fasted T1DM rats, and all of these administered groups showed no decrease in blood glucose levels (Figure S5). These results indicate that TCA can be absorbed through ASBT-mediation in the enterocyte of the distal ileum, and the IPUL-CST can be used as a suitable oral T1DM treatment agent.
Figure 5.
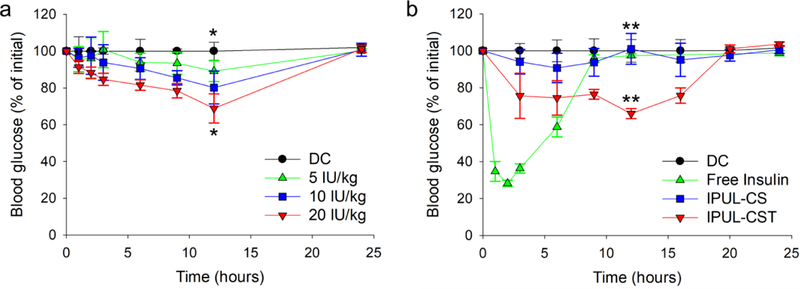
Blood glucose levels versus time profiles for T1DM rats. a) Varying dose of IPUL-CST were administered o.g. (5–20 IU/kg, n=5). b) Free insulin (5 IU/kg) was injected by SC. IPUL-CS and IPUL-CST were administered o.g. (20 IU/kg, n=5). Diabetic control (DC) is the group of PBS administered T1DM rats (n=5). *P <0.01. **P <0.001.
CONCLUSIONS
We provide a new chondroitin sulfate-g-taurocholic acid coated insulin-loaded partially uncapped liposome (IPUL-CST), which has been developed to overcome the strong degradation and poor oBA issue in using the oral administration route of insulin. The in vitro enzymatic digestion studies demonstrated the insulin protection efficacy of IPUL-CST in various enzymatic environments. The IPUL-CST exhibited significantly improved insulin oral bioavailability of up to 34% and reduced the sustained blood glucose at least 16 hours post oral administration in T1DM rats, which provides a unique approach to the non-invasive and direct delivery of nanocarriers to the central lymphatic system. Importantly, the pharmacokinetic evaluation of IPUL-CST in T1DM rats suggested that this system can be used as a suitable diabetic treatment method as well as may be employed as a potential platform for the oral delivery of various API.
Supplementary Material
ACKNOWLEDGMENTS
This work was partially supported by the National Institutes of Health (NIH DK114015–01).
REFERENCES
- 1.Mortensen HB; Robertson KJ; Aanstoot HJ; Danne T; Holl R; Hougaard P; Atchison J; Chiarelli F; Daneman D; Dinesen B, Insulin Management and Metabolic Control of Type 1 Diabetes Mellitus in Childhood and Adolescence in 18 Countries. Diabet. Med. 1998, 15 (9), 752–759. [DOI] [PubMed] [Google Scholar]
- 2.Trial–Type DP; Group DS, Effects of Insulin in Relatives of Patients with Type 1 Diabetes Mellitus. N Engl. J. Med. 2002, 2002 (346), 1685–1691. [DOI] [PubMed] [Google Scholar]
- 3.Malmberg K, Prospective Randomised Study of Intensive Insulin Treatment on Long Term Survival After Acute Myocardial Infarction in Patients with Diabetes Mellitus. Bmj 1997, 314 (7093), 1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raskin P; Rendell M; Riddle MC; Dole JF; Freed MI; Rosenstock J, A Randomized Trial of Rosiglitazone Therapy in Patients with Inadequately Controlled Insulin-Treated Type 2 Diabetes. Diabet. Care 2001, 24 (7), 1226–1232. [DOI] [PubMed] [Google Scholar]
- 5.Turner RC; Cull CA; Frighi V; Holman RR; Group UPDS, Glycemic Control with Diet, Sulfonylurea, Metformin, or Insulin in Patients with Type 2 Diabetes Mellitus: Progressive Requirement for Multiple Therapies (UKPDS 49). Jama 1999, 281 (21), 2005–2012. [DOI] [PubMed] [Google Scholar]
- 6.Collip J, Frederick Grant Banting, Discoverer of Insulin. Sci. Mon. 1941, 52, 472–474. [Google Scholar]
- 7.Harrison G, Insulin in Alcoholic Solution by The Mouth. Br. Med. J. 1923, 2 (3286), 1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’neill V; Twelves C, Oral Cancer Treatment: Developments in Cheomotherapy and Beyond. Br. J. Cancer 2002, 87 (9), 933–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clement S; Dandona P; Still JG; Kosutic G, Oral Modified Insulin (HIM2) in Patients with Type 1 Diabetes Mellitus: Results From a Phase I/II Clinical Trial. Metabolism 2004, 53 (1), 54–58. [DOI] [PubMed] [Google Scholar]
- 10.Kisel M; Kulik L; Tsybovsky I; Vlasov A; Vorob’Yov M; Kholodova E; Zabarovskaya Z, Liposomes with Phosphatidylethanol as A Carrier for Oral Delivery of Insulin: Studies in the Rat. Int. J. Pharm. 2001, 216 (1), 105–114. [DOI] [PubMed] [Google Scholar]
- 11.Kennedy FP, Recent Developments in Insulin Delivery Techniques. Drugs 1991, 42 (2), 213–227. [DOI] [PubMed] [Google Scholar]
- 12.Sarmento B; Ribeiro A; Veiga F; Sampaio P; Neufeld R; Ferreira D, Alginate/chitosan Nanoparticles are Effective for Oral Insulin Delivery. Pharm. Res. 2007, 24 (12), 2198–2206. [DOI] [PubMed] [Google Scholar]
- 13.Rosenstock J; Davtes M; Home PD; Larsen J; Tamer SC; Schernthaner G, Insulin Detemir Added to Oral Anti-Diabetic Drugs in Type 2 Diabetes Provides Glycemic Control Comparable to Insulin Glargine with Less Weight Gain. Diabetes 2006, 55, A132. [Google Scholar]
- 14.Berger M, Oral Insulin 1922–1992: the History of Continuous Ambition and Failure. Front. Insulin Pharmacol. 1993, 144–8. [Google Scholar]
- 15.Florence AT, Oral Insulin Delivery: A Chimera? Int. J. Pharm. 2015, 1 (495), 218–219. [DOI] [PubMed] [Google Scholar]
- 16.Walsh G, Biopharmaceutical benchmarks 2014. Nat. Biotech. 2014, 32 (10), 992–1000. [DOI] [PubMed] [Google Scholar]
- 17.Thanou M; Verhoef J; Junginger H, Oral Drug Absorption Enhancement by Chitosan and Its Derivatives. Adv. Drug Deliv. Rev. 2001, 52 (2), 117–126. [DOI] [PubMed] [Google Scholar]
- 18.Bernkop-Schnürch A, The Use of Inhibitory Agents to Overcome the Enzymatic Barrier to Perorally Administered Therapeutic Peptides and Proteins. J. Control. Release 1998, 52 (1), 1–16. [DOI] [PubMed] [Google Scholar]
- 19.Salama NN; Eddington ND; Fasano A, Tight Junction Modulation and Its Relationship to Drug Delivery. Adv. Drug Deliv. Rev. 2006, 58 (1), 15–28. [DOI] [PubMed] [Google Scholar]
- 20.Ponchel G; Irache J-M, Specific and Non-Specific Bioadhesive Particulate Systems for Oral Delivery to the Gastrointestinal Tract. Adv. Drug Deliv. Rev. 1998, 34 (2), 191–219. [DOI] [PubMed] [Google Scholar]
- 21.Damgé C; Maincent P; Ubrich N, Oral Delivery of Insulin Associated to Polymeric Nanoparticles in Diabetic Rats. J. Control. Release 2007, 117 (2), 163–170. [DOI] [PubMed] [Google Scholar]
- 22.Carino GP; Mathiowitz E, Oral Insulin Delivery. Adv. Drug Deliv. Rev. 1999, 35 (2), 249–257. [DOI] [PubMed] [Google Scholar]
- 23.Cui F; He C; Yin L; Qian F; He M; Tang C; Yin C, Nanoparticles Incorporated in Bilaminated Films: A Smart Drug Delivery System for Oral Formulations. Biomacromolecules 2007, 8 (9), 2845–2850. [DOI] [PubMed] [Google Scholar]
- 24.Russell-Jones G, Use of Targeting Agents to Increase Uptake and Localization of Drugs to the Intestinal Epithelium. J. Drug Target. 2004, 12 (2), 113–123. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y; Wei W; Lv P; Wang L; Ma G, Preparation and Evaluation of Alginate–Chitosan Microspheres for Oral Delivery of Insulin. Eur. J. Pharm. Biopharm. 2011, 77 (1), 11–19. [DOI] [PubMed] [Google Scholar]
- 26.Iwanaga K; Ono S; Narioka K; Morimoto K; Kakemi M; Yamashita S; Nango M; Oku N, Oral Delivery of Insulin by Using Surface Coating Liposomes: Improvement of Stability of Insulin in GI Tract. Int. J. Pharm. 1997, 157 (1), 73–80. [Google Scholar]
- 27.Chalasani KB; Russell-Jones GJ; Jain AK; Diwan PV; Jain SK, Effective Oral Delivery of Insulin in Animal Models Using Vitamin B12-Coated Dextran Nanoparticles. J. Control. Release 2007, 122 (2), 141–150. [DOI] [PubMed] [Google Scholar]
- 28.Lowman A; Morishita M; Kajita M; Nagai T; Peppas N, Oral Delivery of Insulin Using pH‐Responsive Complexation Gels. J. Pharm. Sci. 1999, 88 (9), 933–937. [DOI] [PubMed] [Google Scholar]
- 29.Cui F; Shi K; Zhang L; Tao A; Kawashima Y, Biodegradable Nanoparticles Loaded with Insulin–Phospholipid Complex for Oral Delivery: Preparation, In vitro Characterization and In vivo Evaluation. J. Control. Release 2006, 114 (2), 242–250. [DOI] [PubMed] [Google Scholar]
- 30.Sonaje K; Lin Y-H; Juang J-H; Wey S-P; Chen C-T; Sung H-W, In vivo Evaluation of Safety and Efficacy of Self-Assembled Nanoparticles for Oral Insulin Delivery. Biomaterials 2009, 30 (12), 2329–2339. [DOI] [PubMed] [Google Scholar]
- 31.Mizuma T; Ohta K; Koyanagi A; Awazu S, Improvement of Intestinal Absorption of Leucine Enkephalin by Sugar Coupling and Peptidase Inhibitors. J. Pharm. Sci. 1996, 85 (8), 854–857. [DOI] [PubMed] [Google Scholar]
- 32.Thomas D; Burns J; Audette J; Carrol A; Dow-Hygelund C; Hay M, Clinical Development Success Rates 2006–2015. San Diego: Biomedtracker/Washington, DC: BIO/Bend: Amplion; 2016. [Google Scholar]
- 33.Petrus AK; Fairchild TJ; Doyle RP, Traveling the Vitamin B12 Pathway: Oral Delivery of Protein and Peptide Drugs. Angew. Chem. Int. Ed. 2009, 48 (6), 1022–1028. [DOI] [PubMed] [Google Scholar]
- 34.des Rieux A; Fievez V; Garinot M; Schneider Y-J; Préat V, Nanoparticles as Potential Oral Delivery Systems of Proteins and Vaccines: A Mechanistic Approach. J. Control. Release 2006, 116 (1), 1–27. [DOI] [PubMed] [Google Scholar]
- 35.Dawson PA; Lan T; Rao A, Bile Acid Transporters. J. Lipid Res. 2009, 50 (12), 2340–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grundy SM; Ahrens E; Miettinen TA, Quantitative Isolation and Gas–Liquid Chromatographic Analysis of Total Fecal Bile Acids. J. Lipid Res. 1965, 6 (3), 397–410. [PubMed] [Google Scholar]
- 37.Hofmann AF, The Continuing Importance of Bile Acids in Liver and Intestinal Disease. Arc. Intern. Med. 1999, 159 (22), 2647–2658. [DOI] [PubMed] [Google Scholar]
- 38.Al-Hilal TA; Chung SW; Alam F; Park J; Lee KE; Jeon H; Kim K; Kwon IC; Kim I-S; Kim SY, Functional Transformations of Bile Acid Transporters Induced by High-Affinity Macromolecules. Sci. Rep. 2014, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khatun Z; Nurunnabi M; Reeck GR; Cho KJ; Lee Y. k., Oral Delivery of Taurocholic Acid Linked Heparin–Docetaxel Conjugates for Cancer Therapy. J. Control. Release 2013, 170 (1), 74–82. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki AZ; Watanabe T; Kawamoto M; Nishiyama K; Yamashita H; Ishii M; Iwamura M; Furuta T, Coumarin-4-Ylmethoxycarbonyls as Phototriggers for Alcohols and Phenols. Org. Lett. 2003, 5 (25), 4867–4870. [DOI] [PubMed] [Google Scholar]
- 41.Kwag DS; Park K; Youn YS; Lee ES, Facile Synthesis of Partially Uncapped Liposomes. Colloids. Surf. B. Biointerfaces 2015, 135, 143–149. [DOI] [PubMed] [Google Scholar]
- 42.Sarmento B; Ribeiro A; Veiga F; Ferreira D, Development and Validation of A Rapid Reversed-Phase HPLC Method for the Determination of Insulin From Nanoparticulate Systems. Biomed. Chromatogr. 2006, 20 (9), 898–903. [DOI] [PubMed] [Google Scholar]
- 43.Chung H; Kim J.-s.; Um J; Kwon I; Jeong S, Self-Assembled “Nanocubicle” as A Carrier for Peroral Insulin Delivery. Diabetologia 2002, 45 (3), 448–451. [DOI] [PubMed] [Google Scholar]
- 44.Teply BA; Tong R; Jeong SY; Luther G; Sherifi I; Yim CH; Khademhosseini A; Farokhzad OC; Langer RS; Cheng J, The Use of Charge-Coupled Polymeric Microparticles and Micromagnets for Modulating the Bioavailability of Orally Delivered Macromolecules. Biomaterials 2008, 29 (9), 1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoder SM; Kindel TL; Tso P, Using the Lymph Fistula Rat Model to Study Incretin Secretion. Vitam. Horm. 2010, 84, 221–249. [DOI] [PubMed] [Google Scholar]
- 46.Kumar M; Grzelakowski M; Zilles J; Clark M; Meier W, Highly Permeable Polymeric Membranes Based on the Incorporation of the Functional Water Channel Protein Aquaporin Z. Proc. Natl. Acad. Sci. 2007, 104 (52), 20719–20724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dawson PA, Role of the intestinal bile acid transporters in bile acid and drug disposition. Handb Exp Pharmacol, 2011, 201, 169–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim KS; Suzuki K; Cho H; Youn YS; Bae YH, Oral Nanoparticles Exhibit Specific High-Efficiency Intestinal Uptake and Lymphatic Transport. ACS Nano 2018, DOI: 10.1021/acsnano.8b04315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yanai H; Adachi H; Katsuyama H; Moriyama S; Hamasaki H; Sako A, Causative Anti-Diabetic Drugs and the Underlying Clinical Factors for Hypoglycemia in Patients with Diabetes. World J. Diabetes 2015, 6 (1), 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.