

Abstract
Background:
Alzheimer’s disease (AD) is a heterogeneous disorder.
Objective:
To investigate whether cognitive AD subtypes are associated with different rates of disease progression.
Methods:
We included 1,066 probable AD patients from the Amsterdam Dementia Cohort (n = 290), Alzheimer’s Disease Neuroimaging Initiative (n = 268), Dementia Competence Network (n = 226), and University of California, San Francisco (n = 282) with available follow-up data. Patients were previously clustered into two subtypes based on their neuropsychological test results: one with most prominent memory impairment (n = 663) and one with most prominent non-memory impairment (n = 403). We examined associations between cognitive subtype and disease progression, as measured with repeated Mini-Mental State Examination (MMSE) and Clinical Dementia Rating scale sum of boxes (CDR sob), using linear mixed models. Furthermore, we investigated mortality risk associated with subtypes using Cox proportional hazard analyses.
Results:
Patients were 71 ± 9 years old; 541 (51%) were female. At baseline, pooled non-memory patients had worse MMSE scores (23.1 ± 0.1) and slightly worse CDR sob (4.4 ± 0.1) than memory patients (MMSE 24.0 ± 0.1; p <0.001; CDR sob 4.1 ± 0.1; p <0.001). During follow-up, pooled non-memory patients showed steeper annual decline in MMSE (−2.8 ± 0.1) and steeper annual increase in CDR sob (1.8 ± 0.1) than memory patients (MMSE −1.9 ± 0.1; pinteraction<0.001; CDR sob 1.3 ± 0.1; pinteraction<0.001). Furthermore, the non-memory subtype was associated with an increased risk of mortality compared with the memory subtype at trend level (HR = 1.36, CI = 1.00–1.85, p = 0.05).
Conclusions:
AD patients with most prominently non-memory impairment show faster disease progression and higher risk of mortality than patients with most prominently memory impairment.
Keywords: Alzheimer’s disease, clustering, cognition, dementia, disease progression, mortality, phenotypes, subtypes
INTRODUCTION
Alzheimer’s disease (AD) is characterized by progressive cognitive decline that most typically manifests through episodic memory impairment. As the disease progresses, other cognitive domains (i.e., executive functioning, language, visuospatial functioning, praxis, attention, and/or behavior) become affected as well [1]. Some AD patients, however, show relative sparing of memory functioning in early disease stages, with more prominent impairment in other cognitive domains, such as language, visuospatial, or executive functioning [2–4]. Atypical presentations have previously been associated with demographic, genetic, and neuroimaging/biomarker characteristics that differ from the typical memory-impaired subtype in terms of a younger age at disease onset, with patients more often having an apolipoprotein E (APOE) ε4 negative genotype, and show variability in the anatomical distribution of cortical atrophy, hypometabolism, tau deposition, and pathological findings [5–10].
AD is not only heterogeneous in terms of clinical presentation at diagnosis, but also in rate of disease progression. Previous studies suggest that patient characteristics, such as cognitive subtype [11], age of disease onset [12–15], APOE ε4 genotype [15, 16], and distribution of neurodegeneration [17] are associated with faster disease progression. However, most previous studies investigated this question comparing patients classified a priori based on their clinical or biological characteristics, which do not necessarily consider relationships between such characteristics. We previously identified two distinct subtypes in four large AD cohorts (total n = 1,982) based on their patterns of neuropsychological test scores using dual-clustering approach nonnegative matrix factorization (NMF) [18, 19]. One subtype showed most prominently memory impairment (n = 1,195, 60%), and the other subtype showed most prominently impairment on non-memory tests (n = 787, 40%). Compared to the memory subtype, the non-memory subtype had a younger age at onset, was more often APOE ε4 negative, and had more atrophy of the posterior cortex with relative sparing of the medial temporal lobe. In addition, non-memory patients reported shorter duration of complaints, while scores on the Mini-Mental State Examination (MMSE) were already lower. Based on these previous results, it could be hypothesized that disease progression is faster in non-memory patients. In the present study, we investigated this hypothesis by testing whether the non-memory AD subtype would show faster cognitive decline over time and higher risk of mortality than the more typical memory subtype.
METHODS
Patients
For the present longitudinal study, we selected n = 1,066 AD patients with available follow-up MMSE measurements (≥ one year follow-up duration) from a multi-center sample of 1,982 patients previously included in the cross-sectional clustering study [18]. For this previous clustering study patients were selected based on clinical diagnosis probable AD [20], and MMSE score > 16/30 [21], from four large cohorts; the Amsterdam Dementia cohort (ADC), Alzheimer’s Disease Neuroimaging Initiative (ADNI), Dementia Competence Network (DCN), and University of California, San Francisco (UCSF). From the 1,066 patients selected for present longitudinal analyses, 663 (62%) of patients were previously clustered as memory subtype, ranging from 52% (ADNI) to 70% (ADC and DCN), and 403 (38%) of patients were clustered as non-memory subtype, ranging from 30% (ADC and DCN) to 48% (ADNI). From the originally identified clusters, 916 patients were excluded for main analyses of the present longitudinal study due to missing follow-up MMSE measures (n = 532 memory patients, n =384 non-memory patients; baseline characteristics of excluded patients are given in the Supplementary Material).
In addition, n = 806 patients had available follow-up Clinical Dementia Rating scale – sum of boxes (CDR sob) [22] available with a minimum follow-up duration of one year (i.e., ADNI [n = 269], DCN [n = 366], and UCSF [n = 171]). In this selection, the memory subtype included 479 (59%) of patients, ranging from 51% (ADNI) to 66% (DCN). The non-memory subtype included 327 (41%) of patients, ranging from 34% (DCN) to 49% (ADNI). For both follow-up MMSE and CDR sob analyses we included measurements with a maximum of three years to limit the influence of survivor bias on the results. Finally, for one cohort (ADC) we were able to obtain information on survival (deceased: yes or no; date of death if deceased) from the Dutch Municipal Population Register (n = 492, 99% of original clusters).
Cohort descriptions
ADC
The ADC includes clinical data of patients visiting the outpatient memory clinic of the Amsterdam University Medical Centers (UMC) Alzheimer Center for diagnostic purposes. The first visit of patients included in the present study took place between 2008 and 2013, and patients were diagnosed with probable AD based on a standardized dementia screening [23]. The local ethical committee approved the study and all patients gave informed consent for their clinical data to be used for research purposes.
ADNI
The ADNI database is a research cohort launched in 2003, including data of patients from over 50 sites across the U.S. and Canada (http://www.adni-info.org). Patients selected in the present study enrolled in ADNI-1 or ADNI-2 between 2005 and 2013. All patients gave written informed consent.
DCN
The DCN is a collaboration of fourteen specialized German memory clinics from university hospitals (http://www.kompetenznetz-demenzen.de) [24]. Patients included in the present study first visited one of the memory clinics between 2003 and 2007, and all patients, or their legal guardians, provided written informed consent for their clinical data to be used for research purposes. The Institutional Review Board of all participating centers approved the DCN study protocol.
UCSF
The UCSF research cohort includes patients either seen in the outpatient memory clinic, or for a research assessment in the UCSF Alzheimer’s Disease Research Center [25]. First visits of patients included in the present study took place between 1998 and 2013. All patients and informants provided written informed consent for their clinical data to be used for research purposes. Surrogate consent was accepted when patients lacked capacity to provide consent themselves. The local medical ethical committee approved the study.
Statistical analysis
We compared baseline characteristics of cognitive subtypes (memory versus non-memory) pooled across the four cohorts, and for each cohort separately, using χ2, Kruskal Wallis, or t-tests where appropriate with SPSS version 20 for Mac (SPSS Inc., USA). To validate identification of cognitive subtypes in the subsets used for present study (i.e., based on availability of MMSE [main subset] or CDR sob [additional subset] measures), we repeated cluster analysis as described previously [18].
Rates of disease progression were compared between subtypes using linear mixed models (LMM) with random intercepts and slopes in RStudio for Mac version 3.2.2 (http://www.rstudio.com) with the package lme4 version 1.1–10. The outcome measure was either MMSE or CDR sob. Predictors were cognitive subtype (0 = memory, 1 = non-memory), time, and the interaction between subtype and time. We assumed an unstructured covariance matrix. For pooled analyses, we also adjusted for center as a clustering variable in the model. Analyses were adjusted for age (mean-centered), sex, and APOE ε4 genotype in a second model. Data of linear mixed models are presented as beta ± SE with p-values. Mortality rates were compared between cognitive subtypes with Cox proportional hazard analyses in SPSS, and repeated adjusting for age, sex, and APOE ε4 genotype in a second model, and in a third model additionally adjusting for baseline MMSE. We calculated hazard ratios (HRs) and 95% confidence intervals (CIs) for the non-memory subtype with the memory subtype as reference. For all analyses the significance level was set at p <0.05 for main effects and p ≤ 0.10 for interactions.
RESULTS
Table 1 shows baseline characteristics of AD subtypes (main subset with follow-up MMSE scores available). The median number of MMSE scores available was 3 (interquartile range 3–4). The median follow-up duration was 2.0 years (interquartile range 1.3–3.0 years). Compared with the memory subtype, the non-memory subtype was younger (ADC and UCSF cohorts), less often APOE ε4 positive (pooled sample, ADC, DCN, and UCSF cohorts), and had lower baseline MMSE scores (pooled sample, ADC, ADNI, and UCSF cohorts).
Table 1.
Baseline characteristics of cognitive subtypes in the main sample with follow-up MMSE scores available
| Pooled sample n = 1,066 | ADC n = 290 | ADNI n = 268 | DCN n = 226 | UCSF n = 282 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| mem n = 663 | non-mem | mem n = 203 | non-mem | mem n= 140 | non-mem | mem n= 159 | non-mem | mem n= 161 | non-mem | |
| 62% | n = 403 38% | 70% | n = 87 30% | 52% | n=128 48% | 70% | n = 67 30% | 57% | n=121 43% | |
| Age at diagnosis (y) | 70.8 ±8.8 | 70.1 ±9.9 | 66.6 ±7.5 | 63.4 ±7.9* | 75.8 ±7.2 | 74.9 ±8.0 | 70.7 ±8.0 | 71.0 ± 8.7 | 72.1 ±9.7 | 69.4 ±10.9‡ |
| Female | 332 (50%) | 209 (52%) | 98 (48%) | 43 (49%) | 59 (42%) | 59 (46%) | 89 (56%) | 34 (51%) | 86 (53%) | 73 (60%) |
| Education | 0.05 ± 0.96 | −0.07 ± 1.06 | 0.02 ±0.97 | −0.04 ±1.08 | 0.10 ±0.93 | −0.06 ±1.05 | −0.01 ± 0.97 | −0.01 ±1.09 | 0.05 ± 0.99 | −0.17 ± 1.04 |
| Duration of complaints (y) | 2 (0–16) | 2 (0–13) | 3 (0–11) | 2 (0–10) | - | - | 2 (0–16) | 2 (0–13) | - | - |
| APOE ε4 positives | 345 (71%) | 159 (58%)* | 134 (66%) | 46 (53%)‡ | 62 (45%) | 49 (38%) | 103 (75%) | 31 (55%)† | 46 (66%) | 33 (49%)‡ |
| MMSE at baseline | 23.9 ±2.8 | 23.0 ±3.1* | 23.1 ±2.9 | 21.9 ±3.2† | 23.6 ±1.9 | 23.0 ±2.0‡ | 23.8 ±2.5 | 23.9 ±2.8 | 25.1 ±3.0 | 23.2 ±4.0* |
| Follow-up time (y) | 2.1 (1.4–3.0) | 2.0 (1.2–2.6)† | 2.2 (1.2–3.2) | 2.1 (1.3–3.0) | 2.0 (1.0–2.0) | 2.0 (1.0–2.0) | 2.1 (1.9–2.8) | 2.1 (2.0–2.6) | 2.6 (1.7–4.0) | 2.5 (1.4–3.7) |
| Number of visits | 3 (3–4) | 3 (3–4) | 3 (2–4) | 3 (2–4) | 4 (3–4) | 4 (3–4) | 3 (3–4) | 3 (3–3) | 3 (2–4) | 3 (2–4) |
Data are presented in %, number (%), mean ± standard deviation, or median (interquartile range), p-values are based on i-tests, χ2, or Kruskal Wallis analyses when appropriate. Normalized values are given for education. Differences between memory and non-memory subtypes are indicated as follows:
p ≤ 0.001,
p ≤ 0.01,
p ≤ 0.05. Interpretation: Compared with the memory subtype, the non-memory subtype was younger (ADC and UCSF), less often APOE ε4 positive (pooled sample, ADC, DCN, and UCSF), had lower baseline MMSE scores (pooled sample, ADC, ADNI, and UCSF), and was characterized by a shorter follow-up duration (pooled sample).
Linear mixed models showed that pooled non-memory patients performed worse on MMSE at baseline than pooled memory patients (uncorrected model 1: MMSE 24.0 ± 0.1 versus 23.1 ± 0.1, p <0.001; Table 2). In addition, pooled non-memory patients showed faster yearly decline than memory patients (uncorrected model 1: −2.8 ± 0.1 versus −1.9 ± 0.1,pinteraction < 0.001; Table 2, Fig. 1). This effect was observed in all four cohorts when analyzed separately, with significant faster yearly decline in non-memory patients in ADNI and UCSF (both uncorrected pinteraction < 0.001; Table 2, Supplementary Figures). When analyses were corrected for age, sex, and APOE (model 2), differences at baseline in the pooled sample remained significant (p < 0.001), and faster yearly decline in the non-memory subtypes remained significant in the pooled sample, ADNI, and UCSF (all pinteraction < 0.001).
Table 2.
Estimated baseline and annual change effects of cognitive subtype on MMSE scores and CDR sob scores in the pooled sample and the separate cohorts
| Pooled sample |
ADC |
ADNI |
DCN |
UCSF |
||||||
| mem | non-mem | mem | non-mem | mem | non-mem | mem | non-mem | mem | non-mem | |
| n subjects with follow-up MMSE | 663 | 403 | 203 | 87 | 139 | 129 | 159 | 67 | 161 | 121 |
| n subjects with follow-up CDR sob Model 1 – uncorrected | 479 | 327 | n/a | n/a | 136 | 133 | 241 | 125 | 102 | 69 |
| Baseline MMSE score | 24.0 ±0.1 | 23.1 ±0.1* | 23.3 ±0.2 | 22.1 ±0.3* | 23.8 ±0.2 | 22.9 ±0.2 | 23.8 ±0.2 | 24.2 ±0.3* | 25.2 ±0.3 | 23.4 ±0.3* |
| Baseline CDR sob score | 4.1 ± 0.1 | 4.4 ±0.1* | n/a | n/a | 3.9 ±0.1 | 4.6 ±0.1* | 4.5 ±0.1 | 4.6 ±0.1* | 3.4 ±0.2 | 3.5 ±0.3* |
| Annual change MMSE score | −1.9 ±0.1 | −2.8 ±0.1* | −2.0 ± 0.2 | −2.3 ± 0.3 | −1.5 ±0.2 | −3.1 ±0.2* | −2.4 ± 0.2 | −3.0 ±0.3 | −1.5 ±0.2 | −2.7 ± 0.2* |
| Annual change CDR sob score Model 2 – corrected for age, sex, and APOE ε4 genotype | 1.3 ±0.1 | 1.8 ± 0.1* | n/a | n/a | 1. 5±0.1 | 2.2 ±0.1* | 1.3 ±0.1 | 1.6 ±0.2 | 1.1 ± 0.1 | 1.5 ±0.2‡ |
| Baseline MMSE score | 24.2 ±0.2 | 23.4 ±0.2* | 23.6 ±0.4 | 22.4 ± 0.4* | 24.0 ±0.4 | 22.9 ± 0.4* | 24.3 ±0.4 | 25.0 ±0.5* | 25.8 ±0.6 | 23.4 ±0.6* |
| Baseline CDR sob score | 4.0 ±0.2 | 4.0 ±0.2* | n/a | n/a | 3.8 ±0.3 | 4.3 ±0.3* | 4.6 ±0.2 | 4.7 ± 0.2* | 2.6 ±0.4 | 2.6 ±0.4* |
| Annual change MMSE score | −2.0 ±0.1 | −2.8 ± 0.2* | −2.0 ± 0.2 | −2.4 ± 0.3 | −1.6 ±0.3 | −3.0 ±0.3* | −2.4 ± 0.2 | −2.9 ± 0.3 | −1.4 ±0.3 | −2.9 ± 0.3* |
| Annual change CDR sob score | 1.3 ±0.1 | 1.8 ± 0.1* | n/a | n/a | 1.5 ±0.2 | 2.1 ± 0.2† | 1.3 ±0.1 | 1.6 ±0.2 | 1.0 ±0.2 | 1.7 ±0.2† |
Data are presented in n, mean ± standard deviation (baseline scores) and uncorrected beta ± standard error (annual change). Differences between memory and non-memory subtypes are based on Kruskal Wallis analyses (baseline scores) or linear mixed models (estimated change over time). Both the uncorrected model and the model corrected for age, sex and APOE ε4 genotype are presented. Differences between memory and non-memory subtypes are indicated as follows:
p ≤ 0.001,
p ≤ 0.01,
p ≤ 0.05. Interpretation: The non-memory subtype was characterized by lower (worse) MMSE scores at baseline (all cohorts) and faster yearly decline on MMSE scores (pooled sample, ADNI, and UCSF) than the memory subtype, implying faster disease progression in the non-memory subtype. Furthermore, the non-memory subtype was characterized by higher (worse) CDR sob scores at baseline (pooled sample and UCSF) and greater yearly increase (pooled sample, ADNI, an UCSF) than the memory subtype, implying faster disease progression in the non-memory subtype.
Fig. 1.

Estimated changes over time inADphenotypes based on repeatedMMSE(left) andCDRsob (right) measures. The blue lines represent the memory phenotype; the red lines represent the non-memory phenotype. Linear mixed models showed that non-memory patients were characterized by faster yearly decline on MMSE than memory patients (2.81 ± 0.16 versus 1.92 ± 0.14, pinteraction < 0.001). In addition, non-memory patients showed faster yearly increase in CDR sob (1.79 ± 0.13 versus 1.32 ± 0.12, pinteraction < 0.001).
Similarly, when analyzing disease progression using linear mixed models with CDR sob as outcome measure, pooled non-memory patients had worse baseline CDR sob scores than those with a memory subtype (4.4 ± 0.1 versus 4.1 ± 0.1, p <0.001), and showed a faster yearly increase in CDR sob (uncorrected model 1:1.8 ± 0.1 versus 1.3 ± 0.1, pinteraction <0.001; Table 2, Fig. 1). This effect was observed in all three cohorts when analyzed separately, with significantly steeper yearly increase in non-memory patients in ADNI and UCSF (resp. pinteraction < 0.001 and pinteraction < 0.05; Table 2, Supplementary Figures). When analyses were adjusted for age, sex, and APOE (model 2), differences at baseline in the pooled sample remained significant for the pooled sample (all p < 0.001), and steeper yearly decline in the non-memory subtype remained significant in the pooled sample, ADNI, and UCSF (resp. pinteraction < 0.001, pinteraction < 0.01, and pinteraction < 0.01).
Finally, we analyzed risk of mortality in relation to cognitive subtypes. Cox proportional hazard models showed that compared with the memory subtype, patients with a non-memory subtype had an increased risk of mortality (uncorrected model 1: HR 1.36, CI= 1.00–1.85, p = 0.05, Kaplan-Meier curve shown in Fig. 2; corrected model 2 [age, sex, APOE genotype]: HR 1.66, CI 1.18–2.32, p <0.01; corrected model 3 [age, sex, APOE genotype, and baseline MMSE]: HR 1.58, CI 1.12–2.21, p<0.01).
Fig. 2.
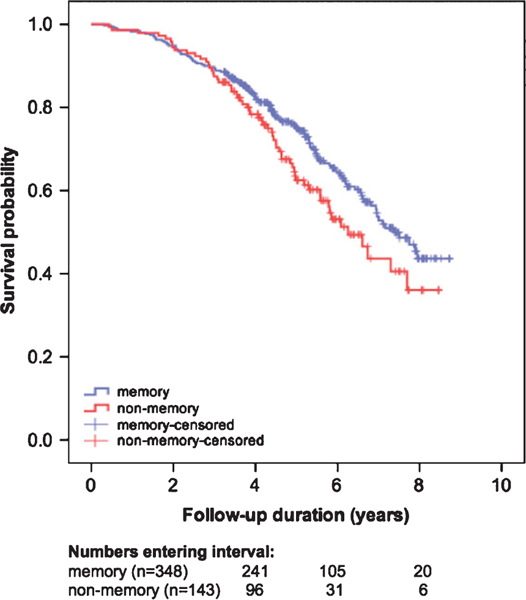
Kaplan-Meier curves visualizing mortality in AD phenotypes. Numbers of entering the intervals 4, 6, and 8 years are depicted below the figure. Cox proportional hazard models showed that compared with the memory subtype, patients with a non-memory subtype had an increased risk of mortality (uncorrected model 1: HR 1.36, CI = 1.00–1.85, p = 0.05; corrected model 2 [age, sex, APOE genotype]: HR 1.66, CI 1.18–2.32, p <0.01; corrected model 3 [age, sex, APOE genotype, and baseline MMSE]: HR 1.58, CI 1.12–2.21, p <0.01).
DISCUSSION
Our main finding is that in four large and independent cohorts, AD patients with a non-memory subtype consistently showed faster disease progression than those with a memory subtype in terms of rate of decline based on MMSE, CDR sob, and risk of mortality. This suggests that patients with a non-memory subtype may have a more aggressive form of the disease.
Several studies have previously investigated whether more rapid disease progression in AD is associated with specific patient characteristics (e.g., the role of age at onset or APOE ε4 genotype on disease progression). Some studies have found faster disease progression in AD patients with younger age at onset [12–14, 16], APOE ε4 negative genotype, relatively spared hippocampi [26], lower baseline MMSE scores, or a combination of these features [15–17]. However, characteristics of a single variable may not fully reflect biological processes that underlie disease heterogeneity. An alternative approach is to determine subtypes based on neuropsychological profile. A previous study for example employing such an approach reported 2.2 times faster decline on MMSE in dysexecutive AD (n = 165)—defined as having executive performance >1.5 SD worse than memory performance—compared with amnestic AD (n = 157) [27]. We analyzed disease progression in AD subtypes that were previously identified using an unbiased, data-driven method. Two subtypes were robustly identified across four large cohorts that showed were associated with distinct cognitive profiles, demographics, and neurobiological characteristics. We now further extend these findings and show that these identified subtypes differ in disease progression rates, suggesting that heterogeneity in disease progression is associated with subtype specific underlying disease mechanisms.
The non-memory subtype showed the fastest disease progression, and consistently so across four cohorts that differed in patient population (e.g., geographically, age, disease severity) and setting (memory clinic versus research cohort). Analyses for each cohort separately showed faster disease progression in the non-memory subtypes, significant in ADNI and UCSF (pinteraction < 0.05 for both MMSE and CDR sob, data shown in the Supplementary Material). Faster disease progression in the non-memory subtype supports the hypothesis that AD is a heterogeneous disease in terms of in disease progression, and that the subtype prone for faster disease progression is associated with younger age, APOE ε4 negative genotype, relative sparing of the hippocampus, and more severe posterior atrophy. Given that amyloid-β plaques show a plateau effect at very early, preclinical stages of AD, a potential explanation in the underlying disease mechanism that is involved in faster disease progression in the non-memory AD subtype might be found in the tau cascade, since the amount of neurofibrillary tangles rather than amyloid plaques have robustly been associated with disease duration [28]. According to the staging of Braak and Braak, the nidus of neurofibrillary tau tangles are typically posited in the entorhinal cortex, which then spreads via the hippocampus to the association cortex and finally to other cortical areas [29]. However, this typical pathological pattern of tau spreading is not always the case, since a previous autopsy study demonstrated that AD subjects exist that show relative sparing of neurofibrillary tangle burden in the hippocampus, and more prominent tau deposition in cortical areas [10]. In line with our non-memory subtype, these hippocampus-spared AD patients were younger at death, showed rapid disease progression, and showed more often focal cortical clinical syndromes. Another potential explanation of heterogeneity in disease progression could be found in the diversity of genes associated with increased risk for developing sporadic AD. This potential explanation is strengthened by the finding that non-memory AD patients less often carried an APOE ε4 allele—the most important risk gene in non-familial AD—and that other susceptible loci are associated with different pathways underlying AD (such as hippocampal synaptic function, cytoskeletal function and axonal transport, regulation of gene expression, and post-translational modification of proteins) [30]. Possibly, memory AD patients carry more risk genes involved in hippocampal synaptic functioning. Patients with a non-memory AD subtype might carry another combination of risk genes, associated with selective vulnerability of (specific) networks and cortical areas, and more aggressive disease mechanisms. Future research should further examine the neurobiological/neuropathological correlates of the subtypes that we identified in this study. Furthermore, it would be interesting to further explore heterogeneity within the non-memory subtype, since previous studies demonstrated presence of more extreme AD subtypes with most prominently impaired non-memory domains [2–4], with distinct neurobiological correlates, e.g., distribution of neurodegeneration.
Our hypothesis coincides with findings of a previous study demonstrating that within late-onset AD patients the cognitive spectrum from memory to executive functioning as a continuous subtype is associated with a specific pattern of heritability [31]. Further research is needed to investigate heterogeneity in genetic risk profiles, corresponding protein characteristics, and disease mechanisms that underlie clinical AD subtypes.
Among the limitations of our study is that patients with faster disease progression have a higher probability to be lost to follow-up (i.e., survivor bias), because they have more severe complaints (patients’ argument), show floor effects on neuropsychological tests (doctor’s argument), or because they were admitted in a nursing home, or deceased. Although we limited the influence of survivor bias on our results by including only measurements of the first three years follow-up, we cannot exclude the possibility that survivor bias may have caused an underestimation of our found disease progression, more prominently in the non-memory subtype that was associated with shorter follow-up duration. Another possible limitation could be that we were only able to analyze disease progression in subsets of original AD subtypes that had available follow-up measurements (i.e., selection bias). To estimate the influence of selection bias on our results, we compared included patients with excluded patients in terms of baseline characteristics (see Supplementary Material). Briefly, excluded patients differed from included patients, most importantly in terms of worse MMSE scores at baseline. Since lower MMSE scores at diagnosis are associated with faster disease progression [17], we think that this selection bias potentially has led to an underestimation of our disease progression results. In addition to lower MMSE scores at baseline, excluded patients also reported shorter disease duration, which is suggestive of faster disease progression, further supporting the notion that selection bias may have resulted in an underestimation of the observed effect on disease progression. We repeated NMF clustering on the subsets selected based on availability of follow-up MMSE or CDR sob measures to study the stability of the cluster results and found that most individuals were labelled quite similarly as the original clustering solution (Supplementary Table 2). We further investigated characteristics of patients that changed cluster assignment in the DCN, since agreement of membership for the memory subtype was least consistent in this cohort when NMF was repeated in the subset with follow-up MMSE measures available (69% of patients were consistently assigned membership to the memory subtype; 31% of former memory patients were now assigned to the non-memory subtype in subset analysis). Further scrutiny of these results pointed out that the clustering of test scores was largely similar as before, and that inconsistent patients were characterized by intermediate ‘H’ values (see Supplementary Figure 2A, B), which indicate how well a patient fits to one of two subtypes (the lower the better fitting to the non-memory subtype; the higher the better fitting to the memory subtype). The H values of inconsistent patients were around 0.5 indicating that these matches either component, and they significantly differed from patients that were consistently assigned to the non-memory phenotype (p <0.001 using Kruskal-Wallis tests). For practical reasons, we dichotomized individuals as belonging to one or the other subtype; however, future research should further investigate subtyping based on continuous values, which take into account more information. Another potential limitation is that not all patients with follow-up MMSE measures did have follow-up CDR sob measures available as well, resulting in two subsets in which either follow-up MMSE or CDR sob could be assessed. Also, a potential limitation is that we have measured disease progression with changes in MMSE and CDR sob scores. Both these instruments are not designed to measure disease progression, therefore lacking sensitivity to capture changes over time. Although these assessments do complement each other well and are widely used, it cannot be excluded that our study might have provided stronger conclusions when an appropriate scale measuring disease progression in the broad clinical spectrum of AD would have been available in our included sample. Furthermore, we used a data-driven clustering approach, which does not take into account clinical interpretations of the data, whereas other approaches such as, e.g., confirmatory factor analysis are able to take those into account. What the best approach is to identify subtypes remains uncertain; for example, a clinical interpretation may not accurately reflect pathological changes, and so an advantage of using data-driven approaches for clustering is that these are unbiased. Our study on the other hand demonstrated that NMF has found to be reproducible in four AD cohorts, that differed in terms of patient population (e.g., age, disease severity, geographic location) and composition and extensiveness of neuropsychological test battery.
Strengths of our study include the notion that we could compare disease progression between data-driven based cognitive subtypes of AD, and that we were able to show robustness of the results across four large and independent cohorts. Because these data-driven subtypes were consistently associated with distinct rate of disease progression, we considered our results to be robust. Furthermore, results obtained based on follow-up MMSE scores were not only validated by repeating analyses in multiple cohorts, but also by repeating analyses based on follow-up CDR sob scores (in three cohorts) and by performing survival analyses (one cohort). Our results have important implications for daily clinical practice since they provide physicians with further insight in expected disease progression of patients based on their cognitive profile at diagnosis. Furthermore, we show that clinical trials exploring long-term effects of an intervention on disease progression might benefit of taking these AD subtypes into account.
In conclusion, we found in four large cohorts that the non-memory AD subtype—previously identified using a data-driven approach and associated with distinct demographical and neurobiological characteristics—is characterized by faster disease progression compared with the memory AD subtype.
Supplementary Material
ACKNOWLEDGEMENTS
Research of the Alzheimer center Amsterdam is part of the neurodegeneration research program of Amsterdam Neuroscience. The Alzheimer Center Amsterdam is supported by Stichting Alzheimer Nederland and Stichting VUmc fonds. The clinical database structure of the Amsterdam Dementia Cohort (ADC) was developed with funding from Stichting Dioraphte.
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12–2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujire-bio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (http://www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. The Dementia Competence Network (DCN) was supported by a grant from the German Federal Ministry of Education and Research (BMBF) (01GI0420).
Additional funding related to randomized clinical trials came from Janssen-Cilag and Merz Pharmaceuticals. The latter funds were exclusively used for personnel, pharmaceuticals, blistering and shipment of medication, monitoring and as capitation fees for recruiting centers.
Footnotes
Authors’ disclosures available online (http://www.j-alz.com/manuscript-disclosures/17-1088r1).
SUPPLEMENTARY MATERIAL
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-171088.
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
REFERENCES
- [1].McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH (2011) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 3, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, Manes F, Dronkers NF, Vandenberghe R, Rascovsky K, Patterson K, Miller BL, Knopman DS, Hodges JR, Mesulam MM, Grossman M (2011) Classification of primary progressive aphasia and its variants. Neurology 11, 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Benson DF, Davis RJ, Snyder BD (1988) Posterior cortical atrophy. Arch Neurol 45, 789–793. [DOI] [PubMed] [Google Scholar]
- [4].Ossenkoppele R, Pijnenburg YAL, Perry DC, Cohn-Sheehy BI, Scheltens NME, Vogel JW, Kramer JH, van der Vlies AE, La Joie R, Rosen HJ, van der Flier WM, Grinberg LT, Rozemuller AJ, Huang EJ, van Berckel BNM, Miller BL, Barkhof F, Jagust WJ, Scheltens P, Seeley WW, Rabi-novici GD (2015) The behavioural/dysexecutive variant of Alzheimer’s disease: Clinical, neuroimaging and pathological features. Brain 138, 2732–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Van Der Flier WM, Schoonenboom SNM, Pijnenburg YAL, Fox NC, Scheltens P (2006) The effect of APOE genotype on clinical phenotype in Alzheimer disease. Neurology 67, 526–527. [DOI] [PubMed] [Google Scholar]
- [6].van der Vlies AE, Pijnenburg YAL, Koene T, Klein M, Kok A, Scheltens P, van der Flier WM (2007) Cognitive impairment in Alzheimer’s disease is modified by APOE genotype. Dement Geriatr Cogn Disord 24, 98–103. [DOI] [PubMed] [Google Scholar]
- [7].Smits LL, Tijms BM, Benedictus MR, Koedam ELGE, Koene T, Reuling IEW, Barkhof F, Scheltens P, Pijnen-burg YAL, Wattjes MP, van der Flier WM (2014) Regional atrophy is associated with impairment in distinct cognitive domains in Alzheimer’s disease. Alzheimers Dement 10, S299–305. [DOI] [PubMed] [Google Scholar]
- [8].Ossenkoppele R, Cohn-Sheehy BI, La Joie R, Vogel JW, Möller C, Lehmann M, van Berckel BNM, Seeley WW, Pij-nenburg YA, Gorno-Tempini ML, Kramer JH, Barkhof F, Rosen HJ, van der Flier WM, Jagust WJ, Miller BL, Scheltens P, Rabinovici GD (2015) Atrophy patterns in early clinical stages across distinct phenotypes of Alzheimer’s disease. Hum Brain Mapp 36, 4421–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ossenkoppele R, Mattsson N, Teunissen CE, Barkhof F, Pijnenburg Y, Scheltens P, van derFlier WM, Rabinovici GD (2015) Cerebrospinal fluid biomarkers and cerebral atrophy in distinct clinical variants of probable Alzheimer’s disease. Neurobiol Aging 36, 2340–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW (2011) Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: A retrospective study. Lancet Neurol 10, 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mez J, Cosentino S, Brickman AM, Huey ED, Mayeux R (2013) Different demographic, genetic, and longitudinal traits in language versus memory Alzheimer’s subgroups. J Alzheimers Dis 37, 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jacobs D, Sano M, Marder K, Bell K, Bylsma F, Lafleche G, Albert M, Brandt J, Stern Y (1994) Age at onset of Alzheimer’s disease: Relation to pattern of cognitive dysfunction and rate of decline. Neurology 44, 1215–1220. [DOI] [PubMed] [Google Scholar]
- [13].Holland D, Desikan RS, Dale AM, McEvoy LK, Alzheimer’s Disease Neuroimaging Initiative (2012) Rates of decline in Alzheimer disease decrease with age. PLoS One 7, e42325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Panegyres PK, Chen H-Y (2013) Differences between early and late onset Alzheimer’s disease. Am J Neurodegener Dis 2, 300–306. [PMC free article] [PubMed] [Google Scholar]
- [15].Chang Y-L, Fennema-Notestine C, Holland D, McEvoy LK, Stricker NH, Salmon DP, Dale AM, Bondi MW, Alzheimer’s Disease Neuroimaging Initiative (2014) APOE interacts with age to modify rate of decline in cognitive and brain changes in Alzheimer’s disease. Alzheimers Dement 10, 336–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].van der Vlies AE, Koedam ELGE, Pijnenburg YAL, Twisk JWR, Scheltens P, Van Der Flier WM (2009) Most rapid cognitive decline in APOE epsilon4 negative Alzheimer’s disease with early onset. Psychol Med 39, 1907–1911. [DOI] [PubMed] [Google Scholar]
- [17].Sluimer JD, Vrenken H, Blankenstein MA, Fox NC, Scheltens P, Barkhof F, Van Der Flier WM (2008) Whole-brain atrophy rate in Alzheimer disease: Identifying fast progressors. Neurology 70, 1836–1841. [DOI] [PubMed] [Google Scholar]
- [18].Scheltens NME, Tijms BM, Koene T, Barkhof F, Teu-nissen CE, Wolfsgruber S, Wagner M, Kornhuber J, Peters O, Cohn-Sheehy BI, Rabinovici GD, Miller BL, Kramer JH, Scheltens P, van der Flier WM, Alzheimer’s Disease Neuroimaging Initiative, German Dementia Competence Network, University of California San Francisco Memory and Aging Center, Amsterdam Dementia Cohort (2017) Cognitive subtypes of probable Alzheimer’s disease robustly identified in four cohorts. Alzheimers Dement 13, 1226–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gaujoux R, Seoighe C (2010) A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services TaskForce on Alzheimer’s Disease. Neurology 34,939–944. [DOI] [PubMed] [Google Scholar]
- [21].Folstein MF, Folstein SE, McHugh PR (1975) “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12, 189–198. [DOI] [PubMed] [Google Scholar]
- [22].Morris JC (1993) The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology 43, 2412–2414. [DOI] [PubMed] [Google Scholar]
- [23].van der Flier WM, Pijnenburg YAL, Prins N, Lemstra AW, Bouwman FH, Teunissen CE, van Berckel BNM, Stam CJ, Barkhof F, Visser PJ, van Egmond E, Scheltens P (2014) Optimizing patient care and research: The Amsterdam Dementia Cohort. J Alzheimers Dis 41, 313–327. [DOI] [PubMed] [Google Scholar]
- [24].Kornhuber J, Schmidtke K, Frolich L, Perneczky R, Wolf S, Hampel H, Jessen F, Heuser I, Peters O, Weih M, Jahn H, Luckhaus C, Hüll M, Gertz H-J, Schröder J, Pantel J, Rienhoff O, Seuchter SA, Rüther E, Henn F, Maier W, Wilt-fang J (2009) Early and differential diagnosis of dementia and mild cognitive impairment: Design and cohort baseline characteristics of the German Dementia Competence Network. Dement Geriatr Cogn Disord 27, 404–417. [DOI] [PubMed] [Google Scholar]
- [25].Kramer JH, Jurik J, Sha SJ, Rankin KP, Rosen HJ, Johnson JK, Miller BL (2003) Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol 16, 211–218. [DOI] [PubMed] [Google Scholar]
- [26].Byun MS, Kim SE, Park J, Yi D, Choe YM, Sohn BK, Choi HJ, Baek H, Han JY, Woo JI, Lee DY, Alzheimer’s Disease Neuroimaging Initiative (2015) Heterogeneity of regional brain atrophy patterns associated with distinct progression rates in Alzheimer’s disease. PLoS One 10, e0142756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mez J, Cosentino S, Brickman AM, Huey ED, Manly JJ, Mayeux R (2013) Faster cognitive and functional decline in dysexecutive versus amnestic Alzheimer’s subgroups: A longitudinal analysis of the National Alzheimer’s Coordinating Center (NACC) database. PLoS One 8, e65246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639. [DOI] [PubMed] [Google Scholar]
- [29].Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. [DOI] [PubMed] [Google Scholar]
- [30].Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin CF, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau MT, Choi SH, Reitz C, Pasquier F, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Letenneur L, Moron FJ, Rubin-sztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fiévet N, Huentelman MW, Gill M, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuiness B, Larson EB, Green R, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossú P, Spalletta G, Proitsi P, Collinge J, Sorbi S, Sanchez-Garcia F, Fox NC, Hardy J, Deniz Naranjo MC, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Matthews F, European Alzheimer’s Disease Initiative (EADI), Genetic and Environmental Risk in Alzheimer’s Disease, Alzheimer’s Disease Genetic Consortium, Cohorts for Heart and Aging Research in Genomic Epidemiology, Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Lannefelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alvarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson KL, Gu W, Razquin C, Pastor P, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O’Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley TH, Bennett DA, Harris TB, Fratiglioni L, Holmes C, de Bruijn RF, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie K, Lunetta KL, Kauwe JS, Boerwinkle E, Riemenschneider M, Boada M, Hiltuenen M, Martin ER, Schmidt R, Rujescu D, Wang LS, Dartigues JF, Mayeux R, Tzourio C, Hofman A, Nöthen MM, Graff C, Psaty BM, Jones L, Haines JL, Holmans PA, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, van Duijn CM, Van Broeckhoven C, Moskvina V, Seshadri S, Williams J, Schellenberg GD, Amouyel P (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mez J, Mukherjee S, Thornton T, Fardo DW, Trittschuh E, Sutti S, Sherva R, Kauwe JS, Naj AC, Beecham GW, Gross A, Saykin AJ, Green RC, Crane PK, Executive Prominent Alzheimer’s Disease: Genetics and Risk Factors (EPAD:GRF), Alzheimer’s Disease Neuroimaging Initiative (ADNI1), Alzheimer’s Disease Genetics Consortium (ADGC) (2016) The executive prominent/memory prominent spectrum in Alzheimer’s disease is highly heritable. Neurobiol Aging 41, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.