

Abstract
Artificial minielastin constructs have been designed which replicate the structure and function of natural elastins in a simpler context, allowing the NMR observation of structure and dynamics of elastin-like proteins with complete residue-specific resolution. We find that the alanine-rich crosslinking domains of elastin have partially helical structure, but only when capped by proline-rich hydrophobic domains. We also find that the hydrophobic domains, composed of prominent 6-residue repeats VPGVGG and APGVGV found in natural elastins appear random coil by both NMR chemical shift analysis and circular dichroism. However, these elastin hydrophobic domains exhibit structural bias for a dynamically disordered conformation that is neither helical nor beta sheet with a degree of non-random structural bias which is dependent on residue type and position in the sequence. Another non-random-coil aspect of hydrophobic domain structure lies in the fact that, in contrast to other intrinsically disordered proteins, these hydrophobic domains retain a relatively condensed conformation whether attached to crosslinking domains or not. Importantly, these domains and the proteins containing them constrict with increasing temperature by up to 30% in volume without becoming more ordered. This property is often observed in non-biological polymers, and suggests that temperature-driven constriction is a new type of protein structural change that is linked to elastin’s biological functions of coacervation-driven assembly and elastic recoil.
Graphical Abstract
INTRODUCTION
Elastin is nature’s most abundant elastomeric material. It is found in skin, the lung, veins, and in arteries where its key functional properties are flexibility and reversible recoil. Elastin evolved in early vertebrates where it provided energy storage for the beating heart in a closed circulatory system.1 The mature elastic matrix is formed when tropoelastin, nature’s most hydrophobic protein, is exported to the extracellular matrix and undergoes an inverse thermal oligomerization transition known as coacervation.2,3 Subsequently, desmosine and isodesmosine cross-links are formed following enzymatic oxidation of lysyl ε-amino groups.4
Natural elastins are organized as tandem repeats of alternating hydrophobic (H) and cross-linking (X) modules: H-X-H’-X’-…….H“.5 Individual modules are coded by separate exons and contain 9–49 amino acid residues.6 Hydrophobic modules are likely responsible for extensibility and recoil. They frequently contain quasi-repeats of 3–7 residues that are rich in hydrophobic amino acids and proline.1 Cross-link modules are rich in alanine and contain the lysyl residues that are post translationally modified to desmosine or isodesmosine that couple four lysyl side chains in a substituted pyridyl ring structure. A common motif found in cross-link modules is two lysines separated by two or three alanine residues and preceded by several more alanines, AnKA2/3K.7,8 An important feature of the elastin matrix is that it is only elastic if hydrated. Dry fibers are brittle and inelastic and swelling in water approximately doubles the mass.8,9 While it is generally accepted that elastin is an entropic elastomer, the molecular basis for entropy increase upon recoil could arise from a decrease in solvent exposure of hydrophobic residues at the protein:water interface, i.e. from hydrophobic forces10,11 and/or from an increase in the configurational entropy of the protein as in rubbers.12
We have previously applied solid-state NMR to natural, hydrated fibrils.13 The degree to which anisotropic shielding was dynamically averaged, i.e., close to that in isotropic fluids, was unexpected. Combined with the observation that 13C chemical shifts were close to random coil values, we concluded that a predominant feature of the natural protein is dynamical disorder and an absence of stable secondary structure.13 The conclusion that elastin and elastin-like proteins are intrinsically disordered has been reviewed14,15 and further supported by the observation of vanishing quadrupole coupling from 2H labeled glycyl residues in natural elastin at 37 °C.16 However, a different conclusion based on isotropic 15N chemical shifts observed in cross-linked recombinant elastin has also been reported.17
The analysis of chemical shifts in elastin13,17 has been based on residue type. A better analysis requires complete resonance assignments.18 Given the reduced spectral dispersion of a disordered protein, achieving this in a ~650 residue protein is challenging. Here, we describe an atomic resolution picture of structure and dynamics based on NMR studies with sequence specific assignments. These constructs retain the key features of natural elastins described above, they coacervate and their chemical shifts closely mirror those found in bovine elastin fibers.13 Cross-linked, these proteins will allow us to study the role of polymer dynamics in elastin recoil at atomic resolution. This approach complements results from CD spectroscopy and light scattering methods. In a related paper, we used 2Q filtered 2H NMR techniques to examine the role of the solvent in recoil.19
Proteins prepared by bacterial expression and labeled with 15N or 15N and 13C were studied using multidimensional NMR. Coacervation temperatures were determined and confirm that these constructs undergo the expected phase transition. Well-resolved solution spectra were obtained and essentially complete sequence specific assignments have been made using standard 2D and 3D NMR methods. Secondary chemical shifts and sequential NOEs provide insight into the conformational space explored by these dynamically disordered proteins at specific residues in the sequence. Comparison of NMR data from isolated modules with constructs containing hydrophobic and cross-link modules shows the effect on cross-link modules of flanking hydrophobic modules and vice versa. Hydrodynamic radii, determined by gradient NMR methods, show that these proteins are more compact than typical intrinsically disordered proteins.
RESULTS
Protein Design.
To make proteins that are amenable to NMR studies and to retain key structural properties of natural elastins, we have combined two approaches. First, quasi-repeats in hydrophobic modules are replaced by exact repeats. This has been widely implemented in elastin-like proteins.15,20–26 Examples are high molecular weight polymers with the formula [(VPGVG)4VPGKG]n 27,28 in which the cross-link modules found in natural elastins are replaced by single lysine substitutions at selected sites in the hydrophobic repeats. Second, to retain the tandem alternation of hydrophobic and cross-link modules, Keeley and co-workers pioneered the minielastin approach wherein the construct contains a limited number of naturally occurring modules.29,30 For example, extensive studies are reported for a construct, 202424, that contains three hydrophobic modules coded by human exons 20 or 24 that flank cross-link modules coded by exon 21 fused with exon 23.31 This minielastin, and others like it, coacervate (self-assemble) and have native-like mechanical properties when cross-linked.32,33
The four minielastins and constituent modules that we have expressed and studied are summarized in Table 1. Hydrophobic modules, 20’ and 24’, contain only the most common 6-residue repeat, VPGVGG or APGVGV, from the hydrophobic module coded by human exon 20 or 24.6 The number of repeats in each was chosen to approximate the length of the natural sequence. Average, per residue Kyte-Doolittle hydrophobicities34 of 20’ and 24’, Table 1, are also similar to sequences of 20 and 24 (1.2 and 1.3, respectively). The cross-link module, 2123, is that used by Keeley in the 202424 construct. It has two AnKA2/3K sequences; a motif found in more than half of human elastin’s cross-link modules.7 The other crosslink module used here, X’, contains a single AnKA2/3K motif. Note that 2123 is homologous to two X’ sequences connected by GVGTP. Minielastins 1-4 combine these modules in different ways. Protein 1 is closely similar to the extensively studied minielastin 20242431,35. Here, we have replaced the naturally occurring hydrophobic modules with the symmetric analogues, 20’ and 24’. Minielastins 2 and 3 are variations on 1 that increase the hydrophobicity of the construct by either increasing the lengths of the terminal hydrophobic modules (2) or decreasing the length of the cross-link modules (3). 4 is a minimalist minielastin. It has a single type of hydrophobic module, 24’, that is alternated with the short cross-link module X’. We note that a study of minielastins similar to those studied here was recently published by Sharpe and coworkers36 with some results that differ from those reported here. The minielastins differ from those studied here in two ways. The hydrophobic modules have a 5-residue repeat (VPGVG) and the terminal modules are cross-link rather than hydrophobic modules.
Table 1.
Primary sequences, number of residues, residue averaged Kyte-Doolittle hydrophobicities (hyd) and coacervation temperatures (Tc) of constituent modules and minielastins 1–4.
| Protein | Sequence | #aa | hyd | Tc(°C)a |
|---|---|---|---|---|
| 20’ | (VPGVGG)5 | 30 | 0.9 | ND |
| 24’ | (APGVGV)7 | 42 | 1.3 | ND |
| 2123 | EAQA5KA2KYGVGTPA5KA3KAAQFG | 33 | 0.2 | NC |
| X’ | DA5KA2KF | 11 | 0.4 | NC |
| 1 | 20’−2123–24’−2123–24’ | 182 | 0.8 | 52 |
| 2 | 20’−20’−2123–24’−2123–24’−24’ | 254 | 0.9 | 32 |
| 3 | 20’-X’−24’-X’−24’ | 138 | 1.0 | 44 |
| 4 | 24’-X’−24’-X’−24’-X’−24’ | 203 | 1.1 | 36 |
Tc values were determined spectrophotometrically by the onset of turbidity at 440 nm under standard conditions: 25 μM protein in pH 7, 50 mM Tris with 1.5 M NaCl and 1 mM CaCl2. For easy detection, constructs 1–4 contain tryptophan residues at the N- and C- termini.
Coacervation Temperatures, Tc.
The inverse transition, coacervation, observed in tropoelastin, minielastins and elastin-like proteins, is fundamental to the formation of an elastic matrix. It is generally assumed to be an entropy driven, hydrophobic collapse.37 Transition temperatures of 1 -4, Table 1, have an order consistent with the overall hydrophobicity of the construct and the number of residues in the hydrophobic modules. Relative to 1, constructs 2 and 3 have lower Tc’s by 20 °C and 8 °C, respectively. Thus, doubling the length of the terminal hydrophobic modules (2) has a greater effect on Tc than decreasing the length of the less hydrophilic cross-linker (3) by approximately the same amount. Similarly, the molecular weight and the number of residues in the hydrophobic modules of 4 is intermediate between 2 and 3 and its Tc is intermediate between 2 and 3 even though 4 is more hydrophobic than 3. A similar lowering of Tc with the molecular weight of elastin-like proteins was reported by Chilkoti and coworkers.38 Compared to the closely related minielastin 202424, Tc = 29 °C, the transition temperature of 1 is significantly higher, 52 °C.31,35 The increase in Tc is the result of the modestly shorter lengths of 20’ and 24’ compared to 20 and 24 (30 and 45 residues compared to 35 and 49 residues), and the lower hydrophobicity of 20’ compared to 20 (1.2 and 0.9).
Circular dichroism spectroscopy.
CD spectra of minielastins 1–4 and constituent modules, Figure 1, are characteristic of CD spectra from other minielastins, tropoelastin and many intrinsically disordered proteins.31,39,40 These spectra have two features: an intense negative peak near 200 nm and a less intense, negative shoulder centered near 222 nm. For example, the spectrum of 1 has values of θ200 and θ222 that are closely similar to those reported for the minielastin 202424 31 and to tropoelastin, albeit θ200 and θ222 are somewhat less negative. Also like tropoelastin, θ200 values decrease in magnitude when T is increased, Figures 1c,d.
Figure 1.
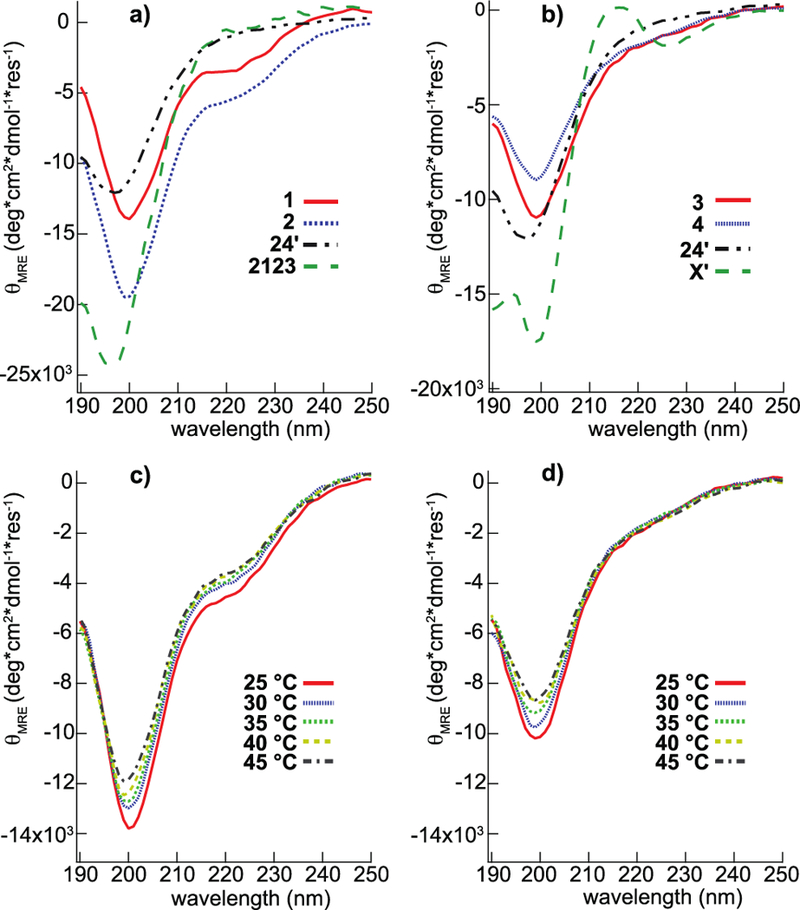
Circular dichroism analysis of elastin-like proteins and their temperature invariance. Spectra of elastin-like constructs at 25 °C: 1, 2, 24’ and 2123 (a); 3, 4, 24’ and X’ (b). Temperature dependence of the CD spectra of 2 and 4 are shown in (c) and (d), respectively.
In favorable cases, protein CD spectra can be deconvoluted into the percentages of helical, sheet, turn and unordered secondary structures present in the protein. We have used two algorithms with basis sets that include spectra of intrinsically disordered proteins.41–43 Averaged over 1–4, we find 7 ± 5% helix, 22 ± 8% sheet, 23 ± 7% turn and 52 ± 10% unordered. Differences among the four minielastins are obscured by the range of results obtained using different algorithm/basis set combinations and this uncertainty is similar to the wide range of percentages reported from CD studies of tropoelastin, 3–10% helix, 30–45% sheet, 17–26% turn and 21–55% unordered.40,44–46 Thus, reliable interpretation of CD spectra of these proteins is difficult. Note also that CD spectroscopy operates on a time-scale that is fast compared to protein dynamics, i.e., spectra of transient secondary structures are captured. In one case, CD analysis of a dynamically disordered elastic protein significantly overestimated the amount of secondary structure measured by other techniques.47
Uversky proposed an alternative analysis.39 Intrinsically disordered proteins are categorized as “mostly unfolded” or “showing signs of residual secondary structure” based on the following intensities of the θ200 and θ222 features: The former have θ200 (deg/dmole) from −21700 to −16100 and θ222 from −2400 to −1000 while the latter have θ200 from 12000 to −19400 and θ222 from −5000 to −2800.39 By these criteria, the isolated modules, 2123 and X’ are unfolded and minielastins 1, 3 and 4 show signs of residual helical secondary structure. We emphasize that “showing signs of residual helical secondary structure” could indicate small regions with stable helical secondary structure or a dynamic protein with an ensemble of structures different from that found in a completely unfolded state. This underlines the need for a technique that probes structure and dynamics at the residue level and distinguishes between transient and stable structures.
NMR Structural Analysis – hydrophobic domains.
We have used a combination of 2D and 3D experiments to obtain nearly complete assignments in the isolated modules 24’, 2123 and X’ and in minielastins 1–4. Missing assignments are in residues at or near module junctions and a few of the alanyl residues in 2123 cross-link modules. Chemical shift assignments are in the Supporting Information Table S1–S4 and representative 15N HSQC spectra are shown in Figure 2. Superimposed on the spectrum of 4 are spectra of the isolated hydrophobic module, 24’ (blue), and the isolated cross-link modules, X’ (red), contained in 4.
Figure 2.
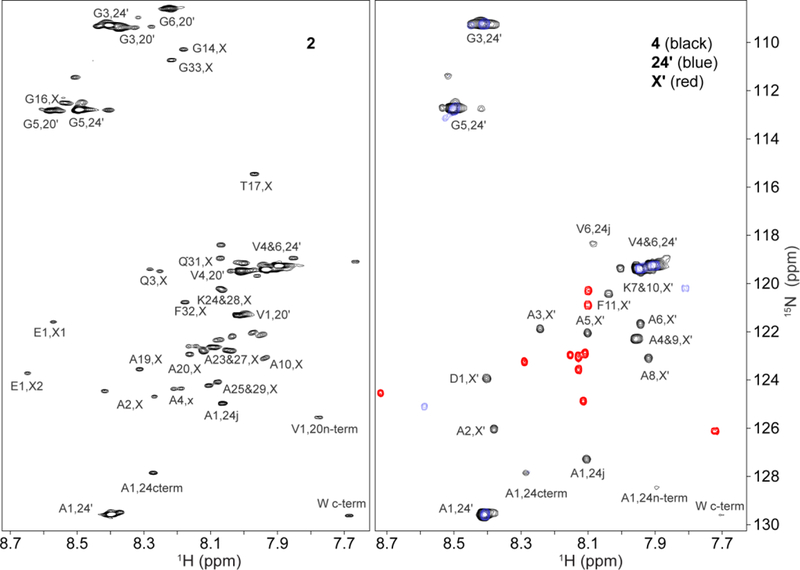
NMR spectral assignments of designed elastin proteins 2 and 4 and their component domains. 15N HSQC peaks from the isolated hydrophobic module, 24’ (blue), and cross-linker, X’ (red), are superimposed on spectra (black peaks) of minielastins 2 (left) and 4 (right) that contain them. Assignments indicate the module and the residue position within the module. For example, A1,24 is the first residue in the APGVGV repeat of the 24’ hydrophobic module and A4,X or A4,X’ are the fourth residues in the 2123 or X’ cross-link modules, respectively.
Chemical shifts are, in most cases, well dispersed and resonances are labeled according to position in the hydrophobic repeats or the position in the crosslink module, Figure 2. Prolyl residues, not observed in 15N HSQC spectra, were assigned using 3D experiments (see materials and methods). Assigned chemical shifts are diagnostic of local secondary structure and NOEs from 3D 15N-edited NOESY HSQC spectra provide qualitative interatomic distances. Since up to six backbone atoms (HN, N, Cα, Hα, Cβ and CO) can be observed, the NMR data is both redundant and residue-specific. Moreover, since the relevant NMR time scale is slow (< 103 s−1) compared to typical conformational dynamics in proteins, we unambiguously observe chemical shifts and NOEs that are averaged over accessible conformations.
In Figure 2, it is seen that the spectrum of the isolated 24’ module superimposes on the peaks assigned to the same residues in minielastin 4. More generally, we find that Hα, HN, Cα, Cβ, CO and N shifts in the APGVGV repeat are essentially equivalent in the minielastins 1-4 and the isolated module 24’. Similarly, Hα, Cα, Cβ and CO shifts are invariant among the different proteins as are chemical shifts for residues in the 20’ repeat, VPGVGG, in the three constructs that contain this module, 1, 2 and 3. For the constructs studied here, we conclude that structural properties of the hydrophobic modules are independent of whether or not they are flanked by a cross-link module or by the sequence of the cross-linker.
Unlike the hydrophobic modules, all peaks from the isolated crosslink modules do not superimpose on the spectra of the relevant minielastin, Figure 2. This is evident for alanyl residues in the A5KA2/3K motifs. Note also that signals from the first 4 residues, EAQA, of the 2123 cross-linker are doubled in minielastin 2, i.e., their shifts depend on the preceding hydrophobic modules, 20’ or 24’. Insofar as random coil chemical shifts for these residues can be accounted for by no more than the two preceding residues18, the observation that four residues are affected indicates some level of non-random structure at the beginning of the cross-link module.
To assess secondary structure in these proteins, we have compared observed with calculated secondary shifts, Figure 3. Secondary shifts are referenced to random coil shifts that, as the name indicates, vary in a systematic way with secondary structure. We have used the sequence-corrected random coil shifts of Poulsen18 and calculated shifts from the homology based program Sparta.48 For simplicity, the calculated chemical shifts for helical and extended sheet structures are averaged over the sequence. Another method for determining random coil shifts, that of Vendruscolo and coworkers49, gives similar results with RMS differences between the two methods of 0.11 ppm for HN, 1.15 ppm for N, 0.41 ppm for Cα, 0.08 ppm for Hα, 0.35 ppm for Cβ and 0.56 ppm for CO. Based on this, we use ±0.1 ppm for H’s and ±0.5 ppm for heteronuclei as reasonable error limits for interpreting secondary shifts. Amide secondary shifts are not discussed further in this context since the standard deviation for amide nitrogen shifts is larger.
Figure 3.
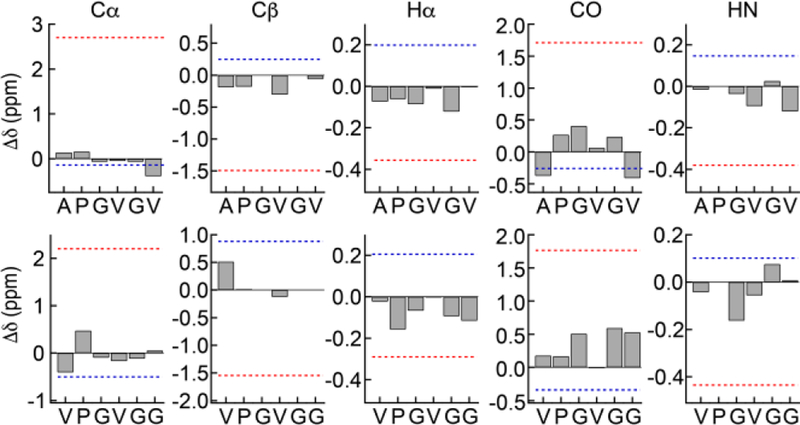
Disorder in elastin hydrophobic domains. Observed and calculated secondary shifts in the APGVGV and VPGVGG repeats of minielastin 2. Observed secondary shift deviations from random coil values are depicted as gray bars and the average of calculated secondary shifts for the sequence constrained to α−helix (red) and sheet (blue) structures are indicated by dotted lines.
Within these error limits, all secondary shifts are small and comparable to random coil values for all residues in both the APGVGV and VPGVGG repeats. All observed secondary shifts exclude helical structure in the hydrophobic modules. However, sheet and other extended structures like ppII have secondary shifts that are in some cases close to random coil values. A different structural model proposed for ELP’s has a type II β−turn at the first three residues.12 With this feature incorporated into the repeats studied here, calculated secondary chemical shifts are small only at the prolyl residue. i.e., the chemical shift data does not support this model for these minielastins. Note that the large downfield shifts of V1,20 and A1,24 or the upfield shifts of G3,20 and G3,24 relative to other alanine, valine or glycine residues in these proteins, Figure 2, is accounted for by their position in the sequence, before or after proline, and do not indicate a conformational preference different from that in short, unstructured peptides.
NOE intensities, related to the distance between proton pairs, are complementary to the chemical shift results. Compared to spectra of proteins with unique structures, the number of NOEs observed in 15N-edited NOESY HSQC spectra of these proteins is small. This indicates significant conformational averaging, i.e., a dynamic ensemble of structures. Since a small number of NOE constraints are observed and a potentially large number of structures are to be determined, simplifying assumptions must be made to understand the available data. We assume that the NOE data provides qualitative populations of the conformations associated with secondary structures found in known proteins.50–52 These are conveniently visualized as localized regions in Ramachandran plots. Proline aside (it has no amide proton), three residue types need to be considered; glycine, residues before proline and all other residue types.50,51 Glycine with the greatest conformational variability is found in three secondary conformations: extended, right-handed α−helix and left-handed α−helix. Extended conformation includes β−sheet and ppII since they are similar and not easily distinguished by a small number of NOE distances. Also, since right-and left-handed α−helices are mirror images, a glycyl residue in either of these secondary structures will have the same NOEs. Residues before proline are predominantly found in an expanded sheet/ppII region and in less populated helix conformation. All other residues are found primarily in the sheet/ppII and helix regions. Thus, a feasible approach is to compare observed NOEs to calculated NOEs for residues constrained to sheet and helix conformations.
Strips from the 3D 15N-edited NOESY HSQC spectrum, Figure 4a, depict backbone NOEs and longer-range interactions to side chain protons in the hydrophobic modules. Peaks in the strip plots are color-coded for the intra-residue αN(i,i) NOE (NOEint ,red), the sequential αN(i-1,i) NOE (NOEseq, green) and blue or black for long range NOEs. Also observed, but not shown in Figure 4a, are NOEs between HN’s of adjacent residues, NOENN. The ratios NOEseq/NOEint and NOENN/NOEint are bar graphed in Figures 4b–e and compared with “benchmark” calculations for the hydrophobic repeats constrained to helix and sheet/ppII (extended) structures. Ratios are used because factors other than inter-proton distances approximately cancel. Details of the “benchmark” calculations and the distances used that account for both Hα’s in glycine residues are given in the Supporting Information, Table S5. The ratios NOEseq/NOEint and NOENN/NOEint readily distinguish between helix and extended conformations and have been used to confirm extended structure in polyalanine peptides.53 NOEseq/NOEint is large for extended and small for helix and vice versa for NOENN/NOEint. While secondary shifts do not exclude the possibility of extended structure across the hydrophobic repeats, the NOE results eliminate this possibility since a predominantly extended conformation is indicated at a single residue, alanine before proline in 24’. Moreover, a stable structure like the type II β-turn model54 proposed for the structure of (VPGVG)n is inconsistent with both the chemical shift and NOE data. Spectra of stable structures would have greater secondary shifts and more NOEs. NOEs for all glycine residues in the both repeats are approximately centered between the helix and sheet/ppII values indicating high disorder, i.e., dynamic sampling of both helix and extended conformations in approximately equal proportion. The smaller preference for extended conformations, i.e., greater flexibility, in the valine before proline combined with three rather than two glycine residues per repeat indicates that 20’ is more flexible than 24’. Modeling shows that the long-range NOEs to the G3 HN from A1 and V6 methyls (Figure 4 strip plots) are consistent with both helix and extended structures.
Figure 4.
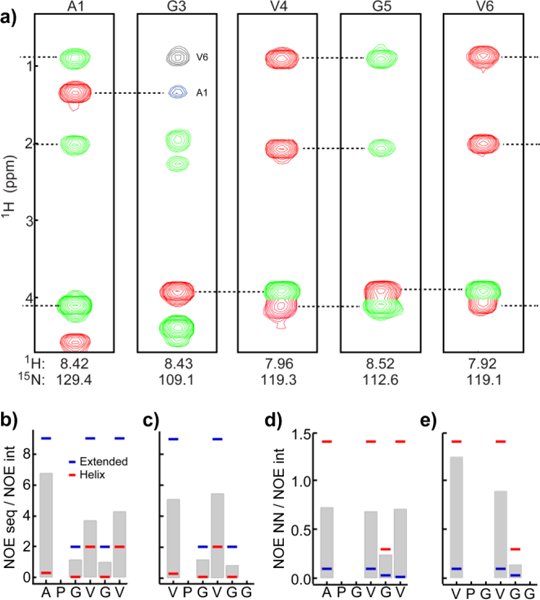
Average interproton distance in hydrophobic domains. (a) Strip plots from the 3D 15N-edited NOESY HSQC spectrum of 2 at selected residues. Intra-residue NOEs (red) were identified in the 3D 15N-edited TOCSY HSQC spectrum. Sequential NOEs from the residue before are colored green and longer-range correlations are colored blue or black. Observed NOE ratios NOEseq/NOEint (b, d) and NOENN/NOEint (c, e) are gray bars. Benchmark NOE ratios are indicated for α−helix (red) and β−sheet (blue).
Given the high proline content of these proteins, cis X-pro linkages are potentially important since they cause local straightening of the protein backbone.52 Cis linkages were identified by their characteristic downfield shifted 13Cβ signals (34.3 ppm) and upfield shifted 13Cγ signals (24.5 ppm) relative to the trans prolyl conformation, 32.1 ppm and 27.3 ppm, respectively (Supporting Information, Figure S1). Peak heights indicate 6–7% cis linkages in all constructs; a value that is consistent with most proline-containing peptides. Thus, minielastins 1–4 have 1–2 slowly interconverting cis X-pro linkages each.
NMR Structural Analysis – crosslinking domains.
Secondary shifts for the 2123 and X’ cross-link modules with and without flanking modules are shown in Figure 5. Unlike the hydrophobic modules, secondary shifts are significantly affected by the presence of flanking modules. When flanked by 24’, secondary shifts at all backbone atoms aside from residues at the center of the 2123 sequence deviate from random coil values in the pattern characteristic of helical structure: Cα and CO (not shown) shifts are upfield while Cβ, Hα and HN (not shown) shifts are downfield. Although the secondary shifts are less than those calculated for a stable, fully formed helix, this characteristic shift pattern is systematically observed. We conclude that the cross-link motif, A5KA2/3K, lacks significant secondary structure when not flanked by hydrophobic modules, but when flanked by hydrophobic modules, it is partially helical. The central YGVGTP sequence in 2123, similar to a hydrophobic repeat, is disordered with or without flanking hydrophobic modules.
Figure 5.
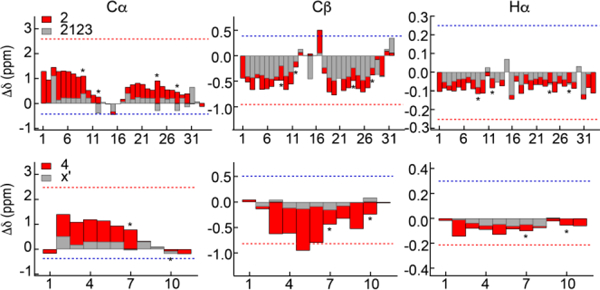
Helical structure in elastin crosslinking domains. Secondary shifts for the cross-link modules, 2123 and X’, with (2 and 4, red bars) and without (2123 and X’, gray bars) flanking 24’ hydrophobic modules. Gray bars are superimposed on red bars and asterisks indicate positions of lysine residues. Numbers refer to sequences in Table 1. Dotted lines indicate an average of the calculated secondary shifts for the sequences constrained to helix (red) and extended (blue) structures.
Nascent helicity in the cross-link modules is also indicated by backbone NOEs between amide protons. Specifically, sequential NOEs (NOENN) were observed with a mix time of 100 ms for residues A2,X’–A6,X’ in minielastin 4 (Supporting Information, Figure S2). Consistent with the formation of a more compact helix-like structure, NOENN values are ~5-fold larger than in the hydrophobic domains on a per residue basis, i.e., adjacent amide protons are ~30% closer in the cross-linker. At a longer mix time, 250 ms, the HN(i):HN(i+2) NOEs were also observed. NOEseq and NOEint were not resolved due to the small differences in Hα chemical shifts.
Temperature dependence of structure and H-bonding.
Changes in Cα, Hα and Cβ chemical shifts are good indicators of structural transitions and the temperature coefficients of amide proton chemical shifts are useful for identifying intramolecular hydrogen bonds 55,56 insofar as significant structural transitions are absent.57 Superimposed in Figure 6a are 13C HSQC spectra of 4 obtained at 20 °C, 30 °C and 40 °C. The absence of significant structural transitions from 20 °C to 40 °C is indicated by the invariance of Cα, Hα and Cβ chemical shifts over this range of temperature. Amide proton chemical shifts are, however, temperature dependent, Figure 6b (Supporting Information, Figure S3). Amide protons with intramolecular H-bonds are less susceptible to temperature-induced chemical shift perturbation than exposed amides H-bonded with water. This correlation is improved by including HN secondary shifts in the analysis.57 For the proteins studied here, secondary shifts at 25 °C are negligible for residues in hydrophobic modules or −0.25 ± 0.05 ppm for residues proximal to lysyl residues in crosslink modules (Supporting Information, Figure S6). With these secondary shifts, intramolecular H-bonding is likely only in residues with ΔδHN/ΔT > −4.6 ppb/K.58 From temperature coefficients for 4, Figure 6c, intramolecular H-bonding is likely absent in the hydrophobic modules (−9.6 < ΔδHN/ΔT < −7.1) and likely present only in the AKAAK sequence in the cross-linker (−4.5 < ΔδHN/ΔT < −3).
Figure 6.
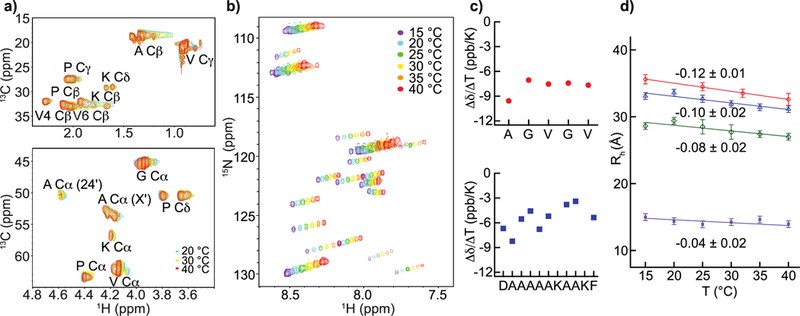
Temperature invariance of the disordered hydrophobic domains and temperature-driven compaction of designed elastins. (a) Superimposed 13C HSQC spectra of minielastin 4 obtained at 20 °C (green), 30 °C (yellow) and 40 °C (red). None of the resonances significantly changes chemical shift. (b) Superimposed 15N HSQC spectra of minielastin 4 obtained at 15–40 °C (c) Graphs of HN chemical shift temperature coefficients obtained from (b) for the residues in the hydrophobic module (red) and the cross-link module (blue) of minielastin 4. (d) Temperature dependent slopes of the hydrodynamic radii, Rh, of minielastins 1 (blue), 3 (green) and 4 (red) and module 24’ (purple) determined by NMR.
Molecular volume.
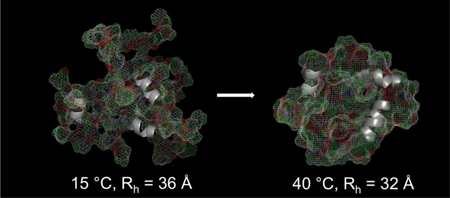
The hydrodynamic radius, Rh, is a signature property related to the compactness of a protein’s structure. Folded proteins are tightly packed and the volume varies linearly with the number of residues, N, so Rh ≈ N1/3. Disordered proteins are not well packed and the chain trajectory is approximately a random walk so Rh ≈ N½.59 These features are well represented by empirical scaling equations of the form Rh ≈ aNb with parameters (a,b) determined from databases of proteins with known size and chain length.60,61–63 Molecular weights determined by equilibrium and velocity AUC, Table 2, compare well with those calculated from the sequences confirming that the proteins are monomeric at 20 °C and pH 6 phosphate buffer. Hydrodynamic radii of 1–4 determined by AUC at a protein concentration of ~30 μM and gradient NMR 64 at ~10-fold higher concentration are in good agreement. Frictional ratios are greater than 1 and indicate that the proteins are highly elongated and/or dynamically disordered. Moreover, the observed values, ~2, are in the lower part of the range found in disordered proteins, 1.6–2.865 and somewhat larger than tropoelastin, 1.4.66 Consistent with the CD and NMR results, the hydrodynamic radii are significantly larger than those for well-folded proteins. However, these minielastins are smaller than other IDP’s with the same chain length, proline content and charge. To further investigate the apparent compactness of these proteins, we used gradient NMR to see the effects of temperature on the hydrodynamic radii of 3 and 4. Elevated temperature promotes coacervation in natural elastins, the poorly understood inverse transition that is required for assembly of the elastic matrix. While we have not followed this property through the phase transition, we find that Rh linearly decreases by 7–8% over a temperature increase from 15–40 °C, Figure 6d. While the decrease in radius is not large, it is reversible and correspond to a statistically significant volume decrease of 25%.
Table 2.
MW and MWAUC are molecular weights (kDa) calculated from the sequence and measured by analytical ultracentrifugation (AUC), respectively.
DISCUSSION
We have described the design and solution properties of minielastins adapted for NMR studies. Like natural elastins, hydrophobic modules are alternated with cross-link modules. The hydrophobic modules are repeats of prominent 6-residue sequences found in natural elastins, APGVGV or VPGVGG, and the cross-link modules contain one or two A5KA2/3K motifs, also prominent in natural elastins. Importantly, these minielastins undergo the signature inverse phase transition found in natural elastin, coacervation. In the constructs studied here, the temperatures of the inverse transition, Tc, varies systematically. Increasing the length of the hydrophobic modules or decreasing the number of A5KA2/3K motifs in the cross-link module decreases Tc.
These minielastins yield well-resolved solution NMR spectra that were assigned using standard 3D NMR techniques. Assignments are complete aside from some residues in the hydrophobic modules at junctions with cross-link modules and some alanyl residues in the larger cross-link module. This has allowed us to probe structure and dynamics at the residue level in proteins that share key features with natural elastins. We expect that chemical shift assignments are transferable to other minielastins built with the same modules. This greatly simplifies NMR studies.
Disorder in the hydrophobic domains.
Chemical shifts of the APGVGV and VPGVGG repeats are equivalent in all four minielastins. In addition, chemical shifts are equivalent to those in the isolated, hydrophobic module. Thus, flanking cross-link modules have little effect on structure and dynamics in the hydrophobic modules. Secondary shifts for all residues in the hydrophobic modules at Cα, Hα, HN, CO and Cβ are close to random coil values and this is sufficient to exclude stable α-helices and type II β−turns in hydrophobic modules. Stable extended structures like sheet and ppII are excluded by comparing the NOE ratios, NOEseq/NOEint and NOENN/NOEint, with calculated values for helix and extended structures. Only the alanine in APGVGV has an apparent preference for extended structure. Glycyl residues in both repeats show NOE ratios that are intermediate between compact helix-like and extended structures indicating dynamical averaging with approximately equal sampling of extended and helical conformations. Valine residues, are somewhat less disordered with a small preference for the more compact helix conformations; presumably due to the large hydrophobic side chain. With greater glycine content and a smaller preference for extended conformation in the residue before proline, our results indicate that 20’ is somewhat more flexible than 24’. Combined, the results from secondary chemical shifts, backbone NOEs and HN temperature coefficients all indicate that the hydrophobic modules are dynamically disordered and devoid of stable secondary structure.
Helical structure in the cross-linking domains.
Chemical shifts of the cross-linkers 2123 and X’ are essentially the same among the minielastins (aside from four residues at the N-terminal junction) but are different from shifts in the isolated cross-linker. Thus, flanking modules affect the cross-link modules. Comparison of secondary shifts in the A5KA2/3K motifs with calculated secondary shifts indicates that this motif is random coil without and partially helical with flanking hydrophobic modules. Previously, Tamburro and coworkers studied natural elastin cross-link domains by CD and 1H NMR.7 They found predominantly random coil structure in aqueous buffer and partial helicity with added trifluoroethanol, a helix promoting solvent.7 Here we find that flanking the cross-linker with hydrophobic modules has the same effect, albeit to a lesser degree.
A simple picture summarizes the results presented above. Hydrophobic modules form collapsed “molten globules” in which the protein backbone is highly disordered and dynamic. Consistent secondary structure encompassing even a few residues is absent and this is unaffected by flanking cross-link modules. The hydrophobic modules act as helical endcaps promoting helix formation in the short crosslinking domain peptides which are completely unfolded in their absence.67 In their presence, these cross-link modules form dynamic α-helices with helicity greatest near the lysyl residues. Helix formation brings the i and i+¾ lysine side chains to close proximity, positioning them to form desmosine crosslinks. A recent paper describes a different minielastin in which similarly short crosslinking domains flank the hydrophobic domain, but the crosslinking domains did not adapt a partially helical conformation.36 However, alanine-rich helices less than two heptads in length do not adopt helical conformations without two flanking helical endcaps,68 so a short crosslinking domain with a single endcap would not be expected to even partially form a helix. Additionally, the environment provided by flanking hydrophobic domains can be expected to promote helix formation.53 We note that natural elastin sequences always have the crosslinking domain flanked on both sides by hydrophobic domains.
Compact disordered structure.
The principle manner in which the hydrophobic domains differ from truly random coil structure is in their degree of condensation – both their Rh and their frictional ratios lie between those typical of folded proteins and non-elastin intrinsically disordered proteins of the same chain length. Marsh et al. performed an analysis of a large number of natural intrinsically disordered proteins and used this data to create an empirical equation which predicts the Rh of a protein as a function of its amino acid composition.61 As Table 2 demonstrates, this equation significantly overestimates the hydrodynamic radius of our elastin-like proteins. However, elastin differs from the proteins in their analysis in that it has a significantly lower fractional charged residue content, a higher proline content, and a significantly higher fractional content of hydrophobic residues. The hydrophobic content in particular offers an explanation of both the decreased hydrodynamic radius and, as described below, the temperature-dependent compaction of these proteins.
Temperature-driven compaction.
As the temperature is raised to 40 °C, the proteins become even more compact, with the hydrodynamic radius decreasing linearly over this range. This behavior was first demonstrated in dilute polymers dissolved in hydrophobic solvents in 197669 and theoretically predicted to occur in some biopolymers by Flory in 1986.70 Temperature-driven compaction has been observed in the disordered N-terminal region of the p53 protein and attributed to the melting of polyprotein II structure in that domain.53,63,71 However, the NOE data (figure 4), indicates that dynamic conformations in these minielastins at 25 °C is not significantly biased toward extended structures (sheet and ppII) relative to compact structures (helix). Also, as temperature is increased to 40 °C, the CD spectra (figure 1c,d) of constructs 2 and 4 is opposite to that expected for a decrease in sampling of ppII and backbone chemical shifts (figure 6a) are essentially constant so compaction via this mechanism is unlikely. An alternative explanation is compaction driven by the entropic hydrophobic effect, which increases as the temperature is elevated. While the role of hydrophobic side chain surface area was shown not to play a role in room temperature compaction in the natural intrinsically disordered protein dataset of Marsh et al.,61 we believe that the exceptional hydrophobicity of the elastin-like proteins studied here and the absence of charged side chains in the hydrophobic domains enables these proteins to exhibit the classical temperature compaction behavior predicted by Flory more than 30 years ago and not seen in most natural intrinsically disordered proteins - as the temperature increases, the strength of the hydrophobic effect increases, and water-filled cavities in the protein are increasingly destabilized. This hypothesis is supported by recent MD simulation72 showing a gradual reduction of the radius of hydration with expulsion of water as the temperature is increased. This property also seems likely connected to the coacervation behavior which these proteins exhibit – as the temperature increases, the protein contracts while expelling water, and eventually the association of separate chains is needed to accommodate the increasing need to reduce the solvent-exposed hydrophobic surface area. This was seen in the MD simulations of (GVGVP)3.72 The temperature induced compaction observed here in minielastins, 25%, is the same as the volume decrease in natural elastin material over the temperature change, 15–40 °C,8 and the latter is directly related to recoil force in an entropic elastomer. In addition to the connection of hydrophobicity to coacervation, our results also connect Rh to hydrophobicity and, in turn, to recoil. Until now, this connection is based on thermochemical, i.e., macroscopic rather than molecular based experiments.10,11 Thus, a common physical basis for coacervation and, at least in part, recoil is suggested and this is an inherent feature of the monomeric protein.
MATERIALS AND METHODS
Protein expression.
Genes containing an N-terminal TEV protease cut site were synthesized by Integrated DNA Technologies (Corralville, IA) and inserted in expression vector pET32a(+) (Novagen, Inc., Madison, WI) as described earlier.73 Genes were transformed into NiCo21(DE3) competent E.coli cells. For unlabeled growths, cells were initially grown in 5 mL of LB (per liter: 10 g bactotyptone, 5 g of yeast extract, 10 g NaCl, pH 7.5) with ampicillin for 8 hours, then transferred into 100 mL LB with ampicillin overnight. 50 mL of LB from the overnight growths was added to 1 L of TPP (per liter: 20 g bactotryptone, 15 g yeast extract, 8 g NaCl, 4 g Na2HPO4, 2 g KH2PO4, pH 7.5, after autoclaving: 25 mL of 40% sterile glucose) with ampicillin. For unlabeled growths, cells were initially grown in 5 mL of LB (per liter: 10 g bactotyptone, 5 g of yeast extract, 10 g NaCl, pH 7.5) with ampicillin for 8 hours, then transferred into 100 mL LB with ampicillin overnight. 50 mL of LB from the overnight growths was added to 1 L of TPP (per liter: 20 g bactotryptone, 15 g yeast extract, 8 g NaCl, 4 g Na2HPO4, 2 g KH2PO4, pH 7.5, after autoclaving: 25 ml of 40% sterile glucose) with ampicillin.
For labeled growths, cells were grown overnight in 5 mL of M9 minimal media (per liter: 12 g Na2HPO4, 6 g KH2PO4, 0.5 g NaCl, 1 g 15N NH4Cl, pH 7.4, after autoclaving: 25 mL 40% glucose (13C glucose for carbon labeled samples), 2 mL of trace metal solution (per 1 L: 500 mL EtOH, 0.8 mL concentrated HCl, 0.5 g FeCl2·4H2O, 18.4 mg CaCl2·2H2O, 6.4 mg H3BO3, 4 mg MnCl2·4H2O, 1.8 mg CoCl2·6H2O, 0.4 mg CuCl2·2H2O, 34 mg ZnCl2, 60.5 mg Na2MoO4·2H2O, 26.8 g MgCl2·6H2O), 1 mL vitamin solution (per 500 mL: 1.1 mg biotin, 1.1 mg folic acid, 110 mg para-aminobenzoic acid, 110 mg riboflavin, 220 mg pyridoxine HCl, 220 mg thiamine HCl, 220 mg niacinamide)) with carbenicillin. Cells were then transferred to 1 L of M9 minimal media with carbenicillin.
1 L growths were placed in a shaker at 37 °C until reaching an OD600 of between 0.7 and 1, then induced with 200 mg of isopropyl-β-D-thiogalactopyranoside (IPTG) overnight at 18 °C.
Protein Purification.
Purification followed a previously used procedure.74 Cells were pelleted via centrifugation at 9000 RPM for 15 minutes, resuspended in 20 mL of wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, 3.1 mM NaN3, pH8) with an added microspatula tip of Pefabloc and 125 μL of DNase solution (20 mg DNase I in 10 mL of 20% glycerol, 75 mM NaCl), lysed with a French press, and purified via Ni-NTA column. Lysate was poured over the Ni-NTA column, rinsed with wash buffer and eluted with 25 mL of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, 3.1 mM NaN3, pH 8.0). The affinity tag was cut with TEV protease and removed via a second Ni-NTA column. Final purification was carried out via HPLC using a 250 × 20 mm Higgins Analytical PROTO 3000 C18 10 μM column, with a gradient of 20%−80% acetonitrile with 0.1% TFA over 60 minutes.
Coacervation.
Lyophilized proteins were dissolved in the coacervation buffer 50 mM Tris, 1.5 M NaCl, 1 mM CaCl2 at pH 7.5 to a final concentration of 25 μM unless otherwise stated. Coacervation studies were performed using an HP 8452A diode array spectrophotometer running OLIS SpectralWorks software version 4.9.6 connected to a Quantum Northwest TC125 temperature controller. Solutions were placed in a quartz cuvette and were equilibrated at initial temperature for at least five minutes. Absorbance at 440 nm was monitored with increasing temperature and a heating rate of 1 °C/min with gentle stirring. Coacervation temperatures indicated are the onset temperature of turbidity observed as an increase in absorbance.
NMR Spectroscopy.
Solutions were prepared by dissolving the protein in 350 μL of pH 6, 50 mM phosphate buffer with 10% D2O and an internal reference, DSS, to a final protein concentration of approximately 300 μM. NMR spectra were obtained on a 700 MHz Varian NMR spectrometer with an HCN cryoprobe at 25°C. Spectra were processed and visualized with NMRPipe75 and NMRFAM-Sparky.76,77 Optimal experimental conditions were determined and sample integrity was monitored using 15N HSQC spectroscopy. Assignments were made with standard 3D experiments,78 primarily the CBCANH and CBCA(CO)NH pair of experiments combined with HACAN and HACA(CO)N to observe prolyl residues. Additional assignments, for example the alanyl residues in the X’ cross-link module (Supporting Information, Figure S4), were obtained or confirmed with HNCO and HN(CA)CO. The HNN experiment was also useful for assignments in cross-link modules.
NOEs were measured in 15N-edited NOESY HSQC spectra with 100 ms or 250 ms mix times as noted. Intra-residue and sequential NOEs were assigned by comparison with the 15N-edited TOCSY HSQC spectrum obtained with a 70 ms mix.
Diffusion constants and, in turn, hydrodynamic radii were measured by pulsed-gradient NMR with the bipolar pair pulse stimulated echo (dbppste) experiment in the Varian NMR DOSY package. Gradients were calibrated against the known diffusion constants of three samples, 1% H2O in 2H2O,79 ubiquitin80 and lysozyme.81 Signal decay, S, as a function of gradient strength, g, was fit to a Gaussian function, S = S0exp(−g2/w2) (Supporting Information, Figure S5) and the diffusion constant was determined from the standard relation w−2 = Dγ2δ2(Δ−δ/3−τ/2).64 The diffusion constant was determined twice at each temperature to confirm thermal equilibration and measured again at the starting temperature, 25 °C, following each temperature change to confirm reversibility. The standard deviation of the multiple determinations at 25 °C was used as the experimental uncertainty in the diffusion constants. Hydrodynamic radii, Rh, were calculated using the Stokes-Einstein relation, D = kBT/6πηRh, with the known viscosity of water at each temperature.82 Hydrodynamic radii determined by NMR at 20 °C were confirmed by analytical ultracentrifugation.
Analytical Ultracentrifugation.
Sedimentation velocity and equilibrium experiments were carried out in a Beckman Coulter ProteomeLab XL-A analytical ultracentrifuge (Beckman Coulter Inc., Brea, CA) at 20°C in standard 2 sector cells. Buffer density was determined on a Mettler/Paar Calculating Density Meter DMA 55A at 20.0 °C and viscosity was measured using an Anton Parr AMVn Automated Microviscometer at 20 °C. Sedimentation velocity data were analyzed with the program Sedfit 83 (www.analyticalultracentrifugation.com) using the continuous c(s) distribution model. The partial specific volumes of peptides were calculated from the amino acid composition using the Protparam tool in ExPASy (expasy.org). Rh and f/f0 were calculated by Sedfit. Errors in Rh are in the range of ± 10%. Experimental sedimentation coefficients were corrected to s20,w using the corrections based on the measured density and viscosity. Sedimentation equilibrium experiments were carried out in standard 2 sector cells using 150 μL of buffer in the reference sector and 130 μL of sample in the sample sector. Three different rotor speeds were used and equilibrium was confirmed by observing the identical distribution of 280 nm absorbance in 3 successive scans at two hour intervals for each rotor speed. Data were analyzed using the program Sedphat (www.analyticalultracentrifugation.com). Global analysis was carried out using the single species model and allowing the values of molecular weight, cell bottom and meniscus to float. Error analysis was performed using the Monte-Carlo error analysis feature of Sedphat with 1000 iterations. Molecular weights of peptides are reported as the average of sedimentation equilibrium and velocity experiments. AUC-determined molecular weights generally are in the range of ± 5% error.84
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge support from the NSF (DMR-1410678 ) to RJW. RLK gratefully acknowledges support via National Institutes of Health grant 1R01-GM111932. Program and infrastructure support from the National Institutes of Health National Center for Research Resources to the City College of New York (5G12MD007603–30). RLK is a member of the New York Structural Biology Center (NYSBC). NMR data collected at NYSBC was made possible by a grant from NYSTAR and ORIP/NIH facility improvement grant CO6RR015495. JMP was the recipient of a fellowship award from the U.S. Department of Education Graduate Assistance in Areas of National Need (GAANN) Program in Biochemistry, Biophysics, and Biodesign at The City College of New York (PA200A150068).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Chemical shift assignments for proteins 1–4, distance and calculated NOE, temperature coefficients of amide protons, HNCO/HN(CA)CO spectra of cross-link in 24’, Gradient decay from diffusion experiment (PDF)
REFERENCES
- (1).Keeley FW The Evolution of Elastin In Evolution of Extracellular Matrix, Keeley FW; Mecham RP, Eds. Springer Berlin Heidelberg: Berlin, Heidelberg, 2013; pp 73–119. [Google Scholar]
- (2).Mithieux SM; Weiss AS Elastin In Fibrous Proteins: Coiled-Coils, Collagen and Elastomers, Parry DAD; Squire JM, Eds. 2005; Vol. 70, pp 437-+. [Google Scholar]
- (3).Yeo GC; Keeley FW; Weiss AS Coacervation of Tropoelastin. Adv. Colloid Interface Sci. 2011, 167 (1–2), 94–103. [DOI] [PubMed] [Google Scholar]
- (4).Heinz A; Ruttkies CKH; Jahreis G; Schrader CU; Wichapong K; Sippl W; Keeley FW; Neubert RHH; Schmelzer CEH In vitro Cross-linking of Elastin Peptides and Molecular Characterization of the Resultant Biomaterials. Biochim. Biophys. Acta, Gen. Subj. 2013, 1830 (4), 2994–3004. [DOI] [PubMed] [Google Scholar]
- (5).He D; Chung M; Chan E; Alleyne T; Ha KCH; Miao M; Stahl RJ; Keeley FW; Parkinson J Comparative Genomics of Elastin: Sequence Analysis of a Highly Repetitive Protein. Matrix Biol. 2007, 26 (7), 524–540. [DOI] [PubMed] [Google Scholar]
- (6).Tamburro AM; Bochicchio B; Pepe A Dissection of Human Tropoelastin: Exon-by-exon Chemical Synthesis and Related Conformational Studies. Biochemistry 2003, 42 (45), 13347–13362. [DOI] [PubMed] [Google Scholar]
- (7).Tamburro AM; Pepe A; Bochicchio B Localizing Alpha-Helices in Human Tropoelastin: Assembly of the Elastin “Puzzle”. Biochemistry 2006, 45 (31), 9518–9530. [DOI] [PubMed] [Google Scholar]
- (8).Gosline JM Temperature-dependent Swelling of Elastin. Biopolymers 1978, 17 (3), 697–707. [DOI] [PubMed] [Google Scholar]
- (9).Lillie MA; Gosline JM The viscoelastic basis for the tensile strength of elastin. Int. J. Biol. Macromol. 2002, 30 (2), 119–127. [DOI] [PubMed] [Google Scholar]
- (10).Weis-Fogh T Andersen, S. O. New Molecular Model for the Long-range Elasticity of Elastin. Nature 1970, 227, 4. [DOI] [PubMed] [Google Scholar]
- (11).Gosline JM Hydrophobic Interaction and a Model for Elasticity of Elastin. Biopolymers 1978, 17 (3), 677–695. [DOI] [PubMed] [Google Scholar]
- (12).Urry DW; Hugel T; Seitz M; Gaub HE; Sheiba L; Dea J; Xu J; Parker T Elastin: a Representative Ideal Protein Elastomer. Philos. Trans. R. Soc., B 2002, 357 (1418), 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pometun MS; Chekmenev EY; Wittebort RJ Quantitative Observation of Backbone Disorder in Native Elastin. J. Biol. Chem. 2004, 279 (9), 7982–7987. [DOI] [PubMed] [Google Scholar]
- (14).Muiznieks LD; Weiss AS; Keeley FW Structural Disorder and Dynamics of Elastin. Biochem. Cell Biol. 2010, 88 (2), 239–250. [DOI] [PubMed] [Google Scholar]
- (15).Roberts S; Dzuricky M; Chilkoti A Elastin-like Polypeptides as Models of Intrinsically Disordered Proteins. FEBS Lett. 2015, 589 (19), 2477–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kumashiro KK; Ohgo K; Elliott DW; Kagawa TF; Niemczura WP Backbone Motion in Elastin’s Hydrophobic Domains as Detected by 2H NMR Spectroscopy. Biopolymers 2012, 97 (11), 882–888. [DOI] [PubMed] [Google Scholar]
- (17).Ohgo K; Niemczura WP; Seacat BC; Wise SG; Weiss AS; Kumashiro KK Resolving Nitrogen-15 and Proton Chemical Shifts for Mobile Segments of Elastin with Two-dimensional NMR Spectroscopy. J. Biol. Chem. 2012, 287 (22), 18201–18209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kjaergaard M; Poulsen FM Sequence Correction of Random Coil Chemical Shifts: Correlation between Neighbor Correction Factors and Changes in the Ramachandran Distribution. J. Biomol. NMR 2011, 50 (2), 157–165. [DOI] [PubMed] [Google Scholar]
- (19).Krivokhizhina TV; Wittebort RJ 2Q NMR of (H2O)-H-2 Ordering at Solid Interfaces. J. Magn. Reson. 2014, 243, 33–39. [DOI] [PubMed] [Google Scholar]
- (20).Baker PJ; Haghpanah JS; Montclare JK, Elastin-Based Protein Polymers. In Polymer Biocatalysis and Biomaterials II, Cheng HN; Gross RA, Eds. 2008; Vol. 999, pp 37–51. [Google Scholar]
- (21).Hassouneh W; Zhulina EB; Chilkoti A; Rubinstein M Elastin-like Polypeptide Diblock Copolymers Self-Assemble into Weak Micelles. Macromolecules 2015, 48 (12), 4183–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Le DHT; Hanamura R; Dieu-Huong P; Kato M; Tirrell DA; Okubo T; Sugawara-Narutaki A Self-Assembly of Elastin-Mimetic Double Hydrophobic Polypeptides. Biomacromolecules 2013, 14 (4), 1028–1034. [DOI] [PubMed] [Google Scholar]
- (23).Sallach RE; Cui W; Wen J; Martinez A; Conticello VP; Chaikof EL Elastin-mimetic Protein Polymers Capable of Physical and Chemical Crosslinking. Biomaterials 2009, 30 (3), 409–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sun F; Zhang WB; Mahdavi A; Arnold FH; Tirrell DA Synthesis of Bioactive Protein Hydrogels by Genetically Encoded SpyTag-SpyCatcher Chemistry Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (31), 11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wu X; Sallach RE; Caves JM; Conticello VP; Chaikof EL Deformation Responses of a Physically Cross-linked High Molecular Weight Elastin-like Protein Polymer. Biomacromolecules 2008, 9 (7), 1787–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yuvienco C; More HT; Haghpanah JS; Tu RS; Montclare JK Modulating Supramolecular Assemblies and Mechanical Properties of Engineered Protein Materials by Fluorinated Amino Acids. Biomacromolecules 2012, 13 (8), 2273–2278. [DOI] [PubMed] [Google Scholar]
- (27).Huang L; McMillan RA; Apkarian RP; Pourdeyhimi B; Conticello VP; Chaikof EL Generation of Synthetic Elastin-mimetic Small Diameter Fibers and Fiber Networks. Macromolecules 2000, 33 (8), 2989–2997. [Google Scholar]
- (28).McMillan RA; Conticello VP Synthesis and Characterization of Elastin-mimetic Protein Gels Derived from a Well-defined Polypeptide Precursor. Macromolecules 2000, 33 (13), 4809–4821. [Google Scholar]
- (29).Cirulis JT; Keeley FW Kinetics and Morphology of Self-Assembly of an Elastin-like Polypeptide Based on the Alternating Domain Arrangement of Human Tropoelastin. Biochemistry 2010, 49 (27), 5726–5733. [DOI] [PubMed] [Google Scholar]
- (30).Miao M; Cirulis JT; Lee S; Keeley FW Structural Determinants of Cross-linking and Hydrophobic Domains for Self-assembly of Elastin-like Polypeptides. Biochemistry 2005, 44 (43), 14367–14375. [DOI] [PubMed] [Google Scholar]
- (31).Miao M; Bellingham CM; Stahl RJ; Sitarz EE; Lane CJ; Keeley FW Sequence and Structure Determinants for the Self-aggregation of Recombinant Polypeptides Modeled after Human Elastin. J. Biol. Chem. 2003, 278 (49), 48553–48562. [DOI] [PubMed] [Google Scholar]
- (32).Bellingham CM; Lillie MA; Gosline JM; Wright GM; Starcher BC; Bailey AJ; Woodhouse KA; Keeley FW Recombinant Human Elastin Polypeptides Self-assemble into Biomaterials with Elastin-like Properties. Biopolymers 2003, 70 (4), 445–455. [DOI] [PubMed] [Google Scholar]
- (33).Bellingham CM; Keeley FW Self-ordered Polymerization of Elastin-based Biomaterials. Curr. Opin. Solid State Mater. Sci. 2004, 8 (2), 135–139. [Google Scholar]
- (34).Kyte J; Doolittle RF A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157 (1), 105–132. [DOI] [PubMed] [Google Scholar]
- (35).Bellingham CM; Woodhouse KA; Robson P; Rothstein SJ; Keeley FW Self-aggregation Characteristics of Recombinantly Expressed Human Elastin Polypeptides. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 2001, 1550 (1), 6–19. [DOI] [PubMed] [Google Scholar]
- (36).Reichheld SE; Muiznieks LD; Keeley FW; Sharpe S Direct Observation of Structure and Dynamics During Phase Separation of an Elastomeric Protein. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (22), E4408–E4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Urry DW; Luan CH; Parker TM; Gowda DC; Prasad KU; Reid MC; Safavy A Temperature of Polypeptide Inverse Temperature Transition Depends on Mean Residue Hydrophobicity. J. Am. Chem. Soc. 1991, 113 (11), 4346–4348. [Google Scholar]
- (38).Meyer DE; Chilkoti A Quantification of the Effects of Chain Length and Concentration on the Thermal Behavior of Elastin-like Polypeptides. Biomacromolecules 2004, 5 (3), 846–851. [DOI] [PubMed] [Google Scholar]
- (39).Uversky VN Natively Unfolded Proteins: A Point where Biology Waits for Physics. Protein Sci. 2002, 11 (4), 739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Vrhovski B; Jensen S; Weiss AS Coacervation Characteristics of Recombinant Human Tropoelastin. Eur. J. Biochem. 1997, 250 (1), 92–98. [DOI] [PubMed] [Google Scholar]
- (41).Whitmore L; Wallace BA Protein Secondary Structure Analyses from Circular Dichroism Spectroscopy: Methods and Reference Databases. Biopolymers 2008, 89 (5), 392–400. [DOI] [PubMed] [Google Scholar]
- (42).Sreerama N; Woody RW Estimation of Protein Secondary Structure from Circular Dichroism Spectra: Comparison of CONTIN, SELCON, and CDSSTR Methods with an Expanded Reference Set. Anal. Biochem. 2000, 287 (2), 252–260. [DOI] [PubMed] [Google Scholar]
- (43).Provencher SW A Constrained Regularization Method for Inverting Data Represented by Linear Algebraic or Integral-Equations. Comput. Phys. Commun. 1982, 27 (3), 213–227. [Google Scholar]
- (44).Debelle L; Alix AJP; Wei SM; Jacob MP; Huvenne JP; Berjot M; Legrand P The Secondary Structure and Architecture of Human Elastin. Eur. J. Biochem. 1998, 258 (2), 533–539. [DOI] [PubMed] [Google Scholar]
- (45).Debelle L; Alix AJP; Jacob MP; Huvenne JP; Berjot M; Sombret B; Legrand P Bovine Elastin and Kappa-Elastin Secondary Structure Determination by Optical Spectroscopies. J. Biol. Chem. 1995, 270 (44), 26099–26103. [DOI] [PubMed] [Google Scholar]
- (46).Debelle L; Alix AJP Optical Spectroscopic Determination of Bovine Tropoelastin Molecular-Model. J. Mol. Struct. 1995, 348, 321–324. [Google Scholar]
- (47).Balu R; Knott R; Cowieson NP; Elvin CM; Hill AJ; Choudhury NR; Dutta NK Structural Ensembles Reveal Intrinsic Disorder for the Multi-Stimuli Responsive Bio-mimetic Protein Rec1-resilin. Sci. Rep. 2015, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Shen Y; Bax A SPARTA Plus : a Modest Improvement in Empirical NMR Chemical Shift Prediction by Means of an Artificial Neural Network.J. Biomol. NMR 2010, 48 (1), 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).De Simone A; Cavalli A; Hsu STD; Vranken W; Vendruscolo M Accurate Random Coil Chemical Shifts from an Analysis of Loop Regions in Native States of Proteins. J. Am. Chem. Soc. 2009, 131 (45), 16332-+. [DOI] [PubMed] [Google Scholar]
- (50).Ho BK; Brasseur R The Ramachandran Plots of Glycine and Pre-proline. BMC Struct. Biol. 2005, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ho BK; Thomas A; Brasseur R Revisiting the Ramachandran Plot: Hard-Sphere Repulsion, Electrostatics, and H-bonding in the Alpha-Helix. Protein Sci. 2003, 12 (11), 2508–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Macarthur MW; Thornton JM Influence of Proline Residues on Protein Conformation. J. Mol. Biol. 1991, 218 (2), 397–412. [DOI] [PubMed] [Google Scholar]
- (53).Chen K; Liu ZG; Kallenbach NR The Polyproline II Conformation in Short Alanine Peptides is Noncooperative. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (43), 15352–15357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Luan CH; Chang DK; Parker TM; Krishna NR; Urry DW Beta-Spiral Conformations of the Elastomeric Polytetrapeptides, (VPGG)n and (IPGG)n, by 2D NMR and Molecular Mechanics Studies. Int. J. Quantum Chem 1991, 40, 183–198. [Google Scholar]
- (55).Merutka G; Dyson HJ; Wright PE Random Coil H-1 Chemical-Shifts Obtained as a Function of Temperature and Trifluoroethanol Concentration for the Peptide Series GGXGG. J. Biomol. NMR 1995, 5 (1), 14–24. [DOI] [PubMed] [Google Scholar]
- (56).Baxter NJ; Williamson MP Temperature Dependence of H-1 Chemical Shifts in Proteins. J. Biomol. NMR 1997, 9 (4), 359–369. [DOI] [PubMed] [Google Scholar]
- (57).Andersen NH; Neidigh JW; Harris SM; Lee GM; Liu ZH; Tong H Extracting Information from the Temperature Gradients of Polypeptide NH Chemical Shifts .1. The Importance of Conformational Averaging. J. Am. Chem. Soc. 1997, 119 (36), 8547–8561. [Google Scholar]
- (58).Cierpicki T; Otlewski J Amide Proton Temperature Coefficients as Hydrogen Bond Indicators in Proteins. J. Biomol. NMR 2001, 21 (3), 249–261. [DOI] [PubMed] [Google Scholar]
- (59).Tanford C, Physical Chemistry of Macromolecules. Wiley: 1961. [Google Scholar]
- (60).Wilkins DK; Grimshaw SB; Receveur V; Dobson CM; Jones JA; Smith LJ Hydrodynamic Radii of Native and Denatured Proteins Measured by Pulse Field Gradient NMR Techniques. Biochemistry 1999, 38 (50), 16424–16431. [DOI] [PubMed] [Google Scholar]
- (61).Marsh JA; Forman-Kay JD Sequence Determinants of Compaction in Intrinsically Disordered Proteins. Biophys. J. 2010, 98 (10), 2383–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Tomasso ME; Tarver MJ; Devarajan D; Whitten ST Hydrodynamic Radii of Intrinsically Disordered Proteins Determined from Experimental Polyproline II Propensities. PLoS Comput. Biol. 2016, 12 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Langridge TD; Tarver MJ; Whitten ST Temperature Effects on the Hydrodynamic Radius of the Intrinsically Disordered N-terminal Region of the p53 Protein. Proteins: Struct., Funct., Bioinf. 2014, 82 (4), 668–678. [DOI] [PubMed] [Google Scholar]
- (64).Johnson CS Diffusion Ordered Nuclear Magnetic Resonance Spectroscopy: Principles and Applications. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34 (3–4), 203–256. [Google Scholar]
- (65).Scott DJ; Winzor DJ Characterization of Intrinsically Disordered Proteins by Analytical Ultracentrifugation In Analytical Ultracentrifugation, Cole JL, Ed. 2015; Vol. 562, pp 225–239. [DOI] [PubMed] [Google Scholar]
- (66).Toonkool P; Regan DG; Kuchel PW; Morris MB; Weiss AS Thermodynamic and Hydrodynamic Properties of Human Tropoelastin - Analytical Ultracentrifuge and Pulsed Field-Gradient Spin-Echo NMR Studies. J. Biol. Chem. 2001, 276 (30), 28042–28050. [DOI] [PubMed] [Google Scholar]
- (67).Aurora R; Rose GD Helix Capping. Protein Sci. 1998, 7 (1), 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Henchey LK; Jochim AL; Arora PS Contemporary Strategies for the Stabilization of Peptides in the Alpha-Helical Conformation. Curr. Opin. Chem. Biol. 2008, 12 (6), 692–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Slagowski E; Tsai B; McIntyre D Dimensions of Polystyrene Near and Below Theta Temperature. Macromolecules 1976, 9 (4), 687–688. [Google Scholar]
- (70).Erman B; Flory PJ Critical Phenomena and Transitions in Swollen Polymer Networks and in Linear Macromolecules. Macromolecules 1986, 19 (9), 2342–2353. [Google Scholar]
- (71).Shi ZS; Chen K; Liu ZG; Ng A; Bracken WC; Kallenbach NR, Polyproline II propensities from GGXGG peptides reveal an anticorrelation with beta-sheet scales. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (50), 17964–17968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Tarakanova A; Huang W; Weiss AS; Kaplan DL; Buehler MJ Computational Smart Polymer Design Based on Elastin Protein Mutability. Biomaterials 2017, 127, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Zhang L; Andersen EME; Khajo A; Maggliozzo RS; Koder RL Dynamic Factors Affecting Gaseous Ligand Binding in an Artificial Oxygen Transport Protein. Biochemistry 2013, 52, 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Zhang L; Anderson JLR; Ahmed I; Norman JA; Negron C; Mutter AC; Dutton PL; Koder RL Manipulating Cofactor Binding Thermodynamics in an Artificial Oxygen Transport Protein. Biochemistry 2011, 50 (47), 10254–10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A Nmrpipe - a Multidimensional Spectral Processing System Based on Unix Pipes. J. Biomol. NMR 1995, 6 (3), 277–293. [DOI] [PubMed] [Google Scholar]
- (76).Lee W; Tonelli M; Markley JL NMRFAM-SPARKY: Enhanced Software for Biomolecular NMR Spectroscopy. Bioinformatics 2015, 31 (8), 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Goddard TD; Kneller DG, SPARKY 3. University of California, San Francisco, 2008. [Google Scholar]
- (78).Cavanagh J; Fairbrother WJ; Palmer AG; Skelton NJ; Rance M, Protein NMR Spectroscopy: Principles and Practice. Elsevier Science: 2010. [Google Scholar]
- (79).Wang JH Self-Diffusion Coefficients of Water. J. Phys. Chem. 1965, 69 (12), 1. [Google Scholar]
- (80).Altieri AS; Hinton DP; Byrd RA Association of Biomolecular Systems Via Pulsed-Field Gradient Nmr Self-Diffusion Measurements. J. Am. Chem. Soc. 1995, 117 (28), 7566–7567. [Google Scholar]
- (81).Price WS; Tsuchiya F; Arata Y Lysozyme Aggregation and Solution Properties Studied Using PGSE NMR Diffusion Measurements. J. Am. Chem. Soc. 1999, 121 (49), 11503–11512. [Google Scholar]
- (82).Huber ML; Perkins RA; Laesecke A; Friend DG; Sengers JV; Assael MJ; Metaxa IN; Vogel E; Mares R; Miyagawa K New International Formulation for the Viscosity of H2O. J. Phys. Chem. Ref. Data 2009, 38 (2), 101–125. [Google Scholar]
- (83).Schuck P Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 2000, 78 (3), 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Zhao HY; Ghirlando R; Alfonso C; Arisaka F; Attali I; Bain DL; Bakhtina MM; Becker DF; Bedwell GJ; Bekdemir A; et al. A Multilaboratory Comparison of Calibration Accuracy and the Performance of External References in Analytical Ultracentrifugation. Plos One 2015, 10 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.