

Abstract
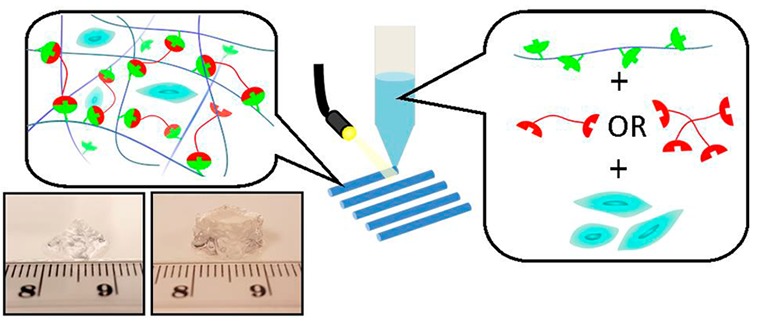
Bioprinting is a powerful technique that allows precise and controlled 3D deposition of biomaterials in a predesigned, customizable, and reproducible manner. Cell-laden hydrogel (“bioink”) bioprinting is especially advantageous for tissue engineering applications as multiple cells and biomaterial compositions can be selectively dispensed to create spatially well-defined architectures. Despite this promise, few hydrogel systems are easily available and suitable as bioinks, with even fewer systems allowing for molecular design of mechanical and biological properties. In this study, we report the development of a norbornene functionalized alginate system as a cell-laden bioink for extrusion-based bioprinting, with a rapid UV-induced thiol–ene cross-linking mechanism that avoids acrylate kinetic chain formation. The mechanical and swelling properties of the hydrogels are tunable by varying the concentration, length, and structure of dithiol PEG cross-linkers and can be further modified by postprinting secondary cross-linking with divalent ions such as calcium. The low concentrations of alginate needed (<2 wt %), coupled with their rapid in situ gelation, allow both the maintenance of high cell viability and the ability to fabricate large multilayer or multibioink constructs with identical bioprinting conditions. The modularity of this bioink platform design enables not only the rational design of materials properties but also the gel’s biofunctionality (as shown via RGD attachment) for the expected tissue-engineering application. This modularity enables the creation of multizonal and multicellular constructs utilizing a chemically similar bioink platform. Such tailorable bioink platforms will enable increased complexity in 3D bioprinted constructs.
Introduction
Additive manufacturing is an increasingly powerful technique used for biofabrication of three-dimensional (3D) constructs in tissue engineering and regenerative medicine.1−4 Bioprinting offers controlled deposition and patterning of polymers, composites, or hydrogels to form well-defined scaffolds with the ability to combine multiple material compositions.5 The bioprinting of cell-laden biomaterials, termed bioinks, allows for the deposition of cells encapsulated in a defined 3D construct and provides a method for the development of complex synthetic biological systems and tissue-engineered constructs.6,7 However, the number of readily accessible materials allowing printability, high cell viability, and user defined customization remains low.
A number of available bioprinters have been developed, including inkjet or droplet-on-demand, continuous extrusion or pressure-based, and laser-assisted bioprinting.8,9 All of these techniques allow spatial control over cell deposition, as well as strategic placement of bioinks with different formulations; however, extrusion bioprinting10,11 is one of the most commonly used setups due to its ease of operation, affordability, and ability to bioprint high cell densities.8 This technique allows the layer-by-layer deposition of fibers in a controlled manner and can facilitate bioprinting of a wide range of bioinks, where the viscosity of the chosen bioinks often plays a vital role in determining the bioprintability of the materials.12 High viscosity materials (high polymeric concentrations) provide structures with high integrity and the ability to support their own weight but upon gelation limit the mobility of encapsulated cells and their capacity to restructure their surrounding matrix. On the contrary, lower viscosity materials provide a less crowded and more remodelable environment for cells but significantly lack printability or structural integrity. Therefore, there are many efforts toward bridging this incompatibility gap, where systems not only meet the demands for good printability but also provide a suitable environment for cells.12−14
Various hydrogel systems have been developed as bioinks for bioprinting,15−18 including natural polymers like gelatin19−22 and collagen23 or synthetic polymers such as polyethylene glycol,24 pluronic,25 and polyvinylpyrrolidone.26 Depending on the choice of polymer, different chemistries can be employed for the cross-linking mechanism, with photoinitiated reactions becoming a popular choice due to spatial and temporal control of cross-linking.27 Free radical polymerization of (meth)acrylates has been widely used in the design of photoreactive bioinks.5,20−22 However, such free-radical chain-growth cross-linking events inherently produce nonbiodegradable backbones, only allow tailorability of mechanical properties via degree of (meth)acrylation or concentration, and generally require low oxygen environments for efficient cross-linking. Photoinitiated thiol–ene chemistry is well-suited as an alternative and recently has found utility in biofabrication.5,19,28 The photoinitiated thiol–ene can be highly efficient, orthogonal to other chemistries, and tolerant to oxygen and can form more homogeneous (step-growth) hydrogel networks compared to free radical polymerized diacrylates.29,30 In addition, functionality is straightforward to incorporate in the hydrogel design as many biomolecules containing thiols can be conjugated to the network, and dithiol cross-linkers can be biodegradable or biomolecules, themselves. Notable examples of thiol–ene hydrogels for fabrication include the cell-laden hyaluronic acid systems functionalized with both methacrylates and norbornenes allowing for dual cross-linking (via photoinitiated radical chain and step-growth thiol–ene reactions, respectively)5 and fully synthetic allylated and thiolated poly(glycidol) systems that allow for cell-laden bioink bioprinting via cross-linking upon exposure to UV (mixed mechanism).28,31
Alginate is a naturally derived polysaccharide made up of β-d-mannuronic acid (M units) and α-l-guluronic acid (G units), which can easily cross-link via its G-blocks to form hydrogels in the presence of multivalent ions, especially Ca2+ ions have been extensively used to prepare hydrogel biomaterials and as synthetic extracellular matrices (ECM) for cell encapsulation.32 There are also free hydroxyl and carboxyl groups on the alginate backbone, which are available for chemical modification, either to alter physical properties of the native alginate, allow for cross-linking, or to introduce bioactivity to the polymer scaffold.35,36 Due to the stiffness of its chains, alginate has high solution viscosities at relatively low weight concentrations and is known to possess shear-thinning properties.33,34
Alginate has been widely used to explore bioprinting, and the results reiterate the difficulty of balancing gels with beneficial mechanical properties for printing (high polymer concentration and high modulus) and desirable properties for 3D cell culture (low polymer concentration and low modulus). One of the earliest reports of alginate bioprinting was with low sodium alginate concentrations (1 w/v % to 2 w/v %) loaded with endothelial cells and cross-linked by bioprinting into a calcium containing bath.37 With low alginate concentration, the cell viability remained high, yet the elastic modulus and the structural integrity of the gels significantly deteriorated over time. Further studies with bioprinting of fibroblast-laden alginate via extrusion printing has shown good printability (5 layers) at high concentration (10 wt %), while only a single layer was possible at concentrations relevant for longer term cell culture (2 wt %).38 In order to circumvent this problem, some reports utilize rheological modifiers in order to allow high viscosity at rest with a lower network concentration.41,42 In our lab, we have also attempted bioprinting cell-laden alginate solutions at different concentrations and have observed significant decreases in cell viability at concentrations above 4 w/v % for various cell lines, including islets and beta-cell lines.39 The cell death observed has been attributed to the high viscosity of the polymer solutions, resulting in high shear stresses on cells.40 While one should be aware that differences between alginate polymer architectures can confound the direct comparison of literature results, nevertheless, the general trend of alginate inks (and bioinks in general) with desirable fabrication parameters leading to decreased cell viability remains a significant hurdle for 3D printing of complex cell-laden constructs.
In this work, we present our modular alginate-based bioinks, cross-linked via the photoinitiated thiol–ene reaction. Alginate was chosen as the polymeric foundation of this bioink due to its low-cost and availability, popularity in biomaterials use, and reputation as a “blank slate” hydrogel scaffold (due to its antifouling nature and lack of cell-specific interactions). The photoinitiated thiol–ene reaction was chosen to introduce spatial and temporal control in the cross-linking chemistry, which is lacking in the typical ionotropic cross-linking chemistry. Alginate is first functionalized with norbornene, one of the most reactive substrates for the radical thiol–ene reaction,43 allowing ultrafast light-triggered thiol–ene cross-linking with precise spatial and temporal control. Furthermore, norbornenes undergo purely step-growth polymerization (little to no homopolymerization or chain growth),44 allowing one to potentially engineer a biodegradable network and circumventing the presence of nondegradable (meth)acrylate networks normally associated with light based gelation techniques. The modularity of this bioink design allows tuning of the mechanical and bioactive properties of the hydrogels, rendering them applicable for various tissue engineering applications. Figure 1 depicts the overall strategy in the current work, from the modification of alginate to the bioprinting process.
Figure 1.

Schematic overview of the strategy employed to develop photoactive alginate bioink (Alg-norb) for bioprinting of hydrogels reported in the current work.
Materials
All materials were obtained from the supplier indicated and used without further purification unless otherwise noted. 1-(3-(Dimethylamino)propyl)-3-ethylcarbodiimide hydrochloride (98+%, VWR, EDC-HCl), N-hydroxysulfosuccinimide sodium salt (≥98%, Sigma-Aldrich, sulfo-NHS), 5-norbornene-2-methylamine (mixture of isomers, TCI Chemicals), O-(2-mercaptoethyl)-O′-methylpolyethylene glycol (≥95%, Aldrich, mPEG thiol), lithium phenyl(2,4,6-trimethylbenzoyl)phosphinate (>98.0%, TCI Chemicals, LAP), N,N-dimethylformamide (anhydrous, Sigma-Aldrich, 99.5%), deuterium oxide (Aldrich, 99.9 atom % D), poly(ethylene glycol) dithiol (PEG dithiol 1500, 1500 Da, Aldrich), PEG dithiol 5000 Da (Laysan Bio), 4arm polyethylene glycol thiol (4arm PEG thiol 5000, 5000 Da, >90% substitution, JenKem Technology USA), CGGGRGDS (Chinapeptides), and Dulbecco’s phosphate buffer saline (DPBS, no calcium, no magnesium, ThermoFisher Scientific) were all used as obtained from the suppliers. Alginate (Manugel GMB, FMC, Lot No. G9402001) was purified before use (vide infra). 2-(N-Morpholino)ethanesulfonic acid (MES) buffer was prepared by dissolving 4.88 g of MES hydrate (≥99.5%, Sigma) and 4.38 g of NaCl (Bioxtra, Sigma-Aldrich) in 250 mL of deionized water (0.100 M MES, 0.300 M NaCl).
Methods
Purification of Alginate
Alginate (Manugel GMB, FMC, Lot No. G9402001) was purified before use. A 1 wt % alginate solution in deionized water was stirred with activated charcoal Norit (Sigma-Aldrich) for 1 h, followed by filtration with 11 μm, 1.2 μm, 0.45 μm, and 0.2 μm Whatman membrane filters to remove particulate matter. Water was removed by lyophilization to yield white fibrous alginate. Alginate was characterized via NMR to determine the M/G block composition of the polymer. See the Supporting Information for details.
General Procedure for Functionalization of Alginate with Norbornene Methylamine
Alginate (0.0506 g, 2.56 × 10–4 mol COOH groups) was weighed into a glass vial and dissolved with 5 mL of MES buffer. pH was adjusted to 6.5 with NaOH. EDC-HCl (0.0446 g, 2.33 × 10–4 mol) and sulfo-NHS (0.0360 g, 1.66 × 10–4 mol) were weighed and added to the vial in one portion before being left to stir for 30 min. Then, the pH of the reaction solution was adjusted with a pH paper to approximately 8 using NaOH (1 M). 5-Norbornene-2-methylamine was then added, and reaction was left to stir for 18 h at room temperature (rt). The reaction solution was then transferred to a 10 kDa MWCO dialysis tube and dialyzed against NaCl solutions, starting from 100 mM, 50 mM, 25 mM, and finally deionized water, with change of dialysate every 10 to 18 h. Water was then removed via lyophilization to yield functionalized alginate (Alg-norb). Degree of functionalization was determined via NMR using dimethylformamide as an internal standard (see the Supporting Information for details). Molecular weight and molar mass dispersity were measured via aqueous gel permeation chromatography (GPC) (data included in the Supporting Information). This modification was also successfully carried out in 0.5 and 1 g scales.
Representative Conjugation of Thiol-RGD to Alg-norb
Alg-norb (0.0506 g, 12% functionalized, 3.03 × 10–5 mol of norbornene) was weighed into a glass vial, and 2.5 mL of water was added (2 w/v % solution) and stirred until Alg-norb was dissolved completely. LAP (1.62 mg, 5.50 × 10–6 mol, 2.2 mM) and CGGGRGDS (0.360 mg, 5.09 × 10–7 mol, 0.2 mM) were added to the Alg-norb solution. The mixture was then placed in UV oven (365 nm, 10 mW/cm2) for 30 s. The reaction solution was transferred to a 10 kDa MWCO tube and dialyzed against deionized water for 2 days with change of dialysate every 10 to 18 h. Purified polymer solution was then lyophilized to yield RGD-Alg-norb as a white solid (0.043 g, 85%).
Swelling Experiments
Polydimethylsiloxane (PDMS) molds in disc geometries were prepared in a diameter of 6 mm with thickness of 2 mm for swelling experiments. Stock solutions in PBS of 2.5 wt % Alg-norb, 169.95 mM LAP, and 100 mM of cross-linker (PEG dithiol 1500, PEG dithiol 5000, and 4-arm PEG 5000) were prepared. Alg-norb solutions with the different cross-linkers were then prepared by mixing the stock solutions homogeneously in Eppendorf tubes at appropriate dilution ratios. Final concentrations of Alg-norb and LAP were 2 wt % and 2 mM, respectively. Details on the concentration of cross-linkers used are tabulated in Table S1 (Supporting Information). 60 μL of polymer solution was added to each mold, and hydrogels were prepared at 10 mW/cm2 for 60 s using a 365 nm LED. Four hydrogels were prepared for each reaction condition. Mass of swollen gels at equilibrium was measured after hydrogels were swollen to a constant mass in a deionized water bath (with occasional water replacement) at rt. Hydrogels were then dried to constant mass, first at rt for 24 h, followed by under vacuum at 60 °C. The mass swelling ratio (q) was defined as the mass ratio of hydrogels at equilibrium to the dried gels.
Cell Viability Assays in 2D Culture
L929 fibroblast cell line from mouse (passage 3) was suspended in cell culture medium (Dulbecco’s Modified Eagle’s Medium (DMEM, ThermoFisher) with glutamax) supplemented with 1% penicillin–streptomycin and 10% fetal bovine serum (FBS) at a concentration of 7500 cells/mL. 200 μL of the cell suspension was cultured in 15 wells of a 96 black well clear bottom tissue culture treated plate. Plate was incubated for 24 h at 37 °C with 5% CO2, and then medium was aspirated and replaced with 100 μL of medium. Cells were then exposed to 10 mW/cm2 of 365 nm LED for 0, 30, 60, 120, and 300 s.
Live/Dead Assay of Hydrogels
The viability of the cells exposed to the bioprinting conditions was evaluated using a LIVE/DEAD viability/cytotoxicity kit (Thermofisher). Stock solutions of the assay, ethidium homodimer-1 (0.036 μM) and calcein AM (1 μM), were prepared in 10 mL of PBS without calcium and magnesium. One mL of calcein stock solution was added to each scaffold and incubated for 20 min at 37 °C. After that, 1 mL of the ethidium-1 stock solution was added to the wells and incubated for an additional 10 min at 37 °C. The dye solutions were then aspirated from the wells, and 1 mL of culture medium without phenol red was added to wells before imaging. Calcein AM produced a green fluorescence in live cells (ex/em = 495/515 nm) and ethidium homodimer-1 binds to nucleic acids of cells with damaged membranes to produce red fluorescence (ex/em = 495/635 nm). A live cell imaging Nikon TI-E with environmental control with a 10× objective (WD = 15, NA = 0.3) was used for cell imaging. Images were typically acquired via 1024 μm × 1024 μm scans with Z stacks of 5 to 10 μm at three different regions of a hydrogel. Cell viability was estimated using CellProfiler 3.0.0. through quantification of the number of live cells over total number of cells.
Staining Cells with Fluorescent Probes
L929 cells were stained with CellTracker green CMFDA and CellTracker red CMTPX dyes. Stock solutions of 10 mM of CellTracker green CMFDA and red CMTPX were prepared by dissolving 50 μg of the stock powder in anhydrous dimethylfsulfoxide. The CMFDA and CMTPX stock solutions were then diluted with warm DMEM to prepare concentrations of 8.5 μM and 5 μM, respectively. Dye solutions were added to L929 pellets, resuspended, and incubated for 15 min at 37 °C. Cells were centrifuged, and dye solutions were removed. Warm medium was added, and cell pellets were resuspended before incubation for another 15 min. Cells were then transferred to Alg-norb solutions and mixed homogeneously for bioprinting.
Printer Parameters
Alg-norb with specific formulations of photoinitiator and cross-linker (3 million cells/mL) was loaded into amber syringes, which were loaded into a custom holder, designed to hold a cartridge and LED light source (4-wavelength high-Power LED source, Thorlabs, 365 nm, approximately 10 mW/cm2). Bioprinting was carried out with a metal G25 needle on a BioScaffolder (GeSiM - Gesellschaft für Silizium-Mikrosysteme mbH, Germany) controlled through proprietary software. In general, scaffold geometries and settings were set to a polygon of four corners, comprising of 13 meandered strands placed at a distance of 0.53 mm apart. The angle of deposition was turned 90° after each layer. Height of each layer was set to 0.2 mm, and the number of layers was varied according to experiment requirements.
NMR
Spectra were acquired on a 700 MHz Bruker spectrometer at 325 K. NMR samples were prepared between 2 and 3 mg in 0.5 mL of D2O. Water suppression pulse sequence was also applied to spectra.
Rheology
Measurements were carried out on MCR 302 rheometer (Anton Paar) fitted with a 10 mm diameter parallel plate geometry and a quartz glass bottom with fiber optic light guide. Prepared solutions (30 μL) of Alg-norb with specific formulations of photoinitiator and cross-linker was pipetted onto the middle of the sample holder, which is between the glass surface (above UV source) and parallel plate geometry. To compare the rate of network formation for the different cross-linkers, UV was applied (1 W/cm2 at 365 nm) using a Bluepoint 4 light source (Hönle UV technology) for 60 s during oscillatory time sweeps (1.0 Hz, 1.0% strain). Rheological properties of the hydrogels were also studied via frequency sweeps performed from 0.5 to 50 Hz (1% strain) and strain sweeps from 0.2 to 500% shear strain (1 Hz). All measurements were carried out in duplicate.
Results and Discussion
Synthesis of Alg-norb and Thiol–Ene Reactions
Alginate was modified via aqueous EDC (1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide)/NHS (N-hydroxysuccinimide) coupling of an amine to the carboxylic acids on the alginate backbone. The alginate carboxylic acid groups are first activated by EDC, and then a more stable activated ester is formed with NHS, forming amine reactive NHS esters. Subsequent treatment of the semistable NHS ester with norbornene methylamine enables amide bond formation and attachment of the norbornene to the alginate backbone (Scheme 1a).
Scheme 1. Reaction Schemes of a) the Carbodiimide Reaction between Alginate and Norbornene Methylamine and Photoinitiated Thiol–Ene Reactions of Alg-norb with b) RGD Peptide Sequence (CGGGRGDS).

Amidation reactions on alginate using the EDC/NHS coupling mechanism have been reported in various reaction conditions, which have resulted in varying degrees of yield,45,46 yet there remains little documentation in the literature on optimization, reproducibility, and control of this reaction. Consequently, in this work several different reaction conditions were briefly explored to optimize the modification and to ensure reproducibility. Reactions were attempted in both phosphate and MES buffer at pH 6.5.45,46 Our results showed more efficient functionalization in MES buffer (∼40 mol % yield, vide infra), while very poor efficiency was achieved in phosphate buffer (4.3 mol % yield), in line with the documented instability of the acylurea intermediate in phosphate buffer.47 Ultimately, pH 6.5 was utilized, within the range of pH (pH 4.5 to 7.2) reported feasible for the formation of the activated NHS ester. In the second step of the reaction, one can aminolyze the activated NHS ester with norbornene methylamine either at the pH used for NHS ester formation (pH 6.5) or raise the pH in order to increase the nucleophilicity of the amine coupling partner (pH 8.0). While only a slightly improved yield (∼+2%) at the higher pH (pH 8.0) was observed, subsequent reactions were carried out after an adjustment of pH with the addition of NaOH in accordance with the reaction mechanism.
Due to complexities in the NMR of alginate, a more reliable method for quantifying the amount of norbornene functionalization on alginate was developed utilizing 1H NMR with anhydrous DMF as an internal standard. An exemplar NMR spectrum of a norbornene-functionalized alginate (Figure S3) and details on the calculation steps are included in the Supporting Information.
In order to check the control and reproducibility of our functionalization approach, we investigated the reproducibility of the reaction and the ability to tailor the degree of norbornene functionalization. By simply adding different stoichiometric amounts of norbornene to the reaction, while proportionally altering the equivalents of EDC and NHS, we were able to straightforwardly change the degree of functionalization from 4% to 12% with good control (Table 1). The experimental yield of functionalization was relatively consistent at approximately 40% for all equivalents of norbornene tried.
Table 1. Comparison of Percentage Norbornene Functionalization and Yield of Reaction with Different Equivalents of Norbornene, EDC, HCl, and NHS in MES Buffer.
| Norbornene equiva | EDC equiv | NHS equiv | % mol norbornene functionalization | % yield |
|---|---|---|---|---|
| 0.3 | 2.4 | 0.9 | 12.0 | 40 |
| 0.2 | 1.6 | 0.6 | 7.1 | 36 |
| 0.1 | 0.8 | 0.3 | 4.2 | 42 |
Calculated according to mol equivalents of COOH groups of alginate.
Having control over the alginate functionalization, next a model photoinitiated thiol–ene reaction with a 800 Da SH-PEG-OMe was employed to investigate the fidelity of the norbornene groups attached to alginate and the reaction efficiency in a solution phase model reaction. LAP was utilized as the photoinitiator because it is water-soluble, highly efficient at 365 nm, and has shown good biocompatibility for fabrication of cell-laden hydrogels (up to certain concentrations).48 Following the model reaction, the reacted Alg-norb sample showed peaks attributed to the main chain and methyl protons of PEG-OMe at 3.8 and 3.4 ppm, respectively (as shown in Figure S5 in the Supporting Information), with an estimated 90% functionalization of the available PEG-OMe now on the alginate backbone.
Due to alginate’s lack of bioactivity for cell adhesion, a similar reaction was carried out with cystine (thiol) terminated RGD peptide (CGGGRGDS) (Scheme 1b). RGD is a common tripeptide motif present in many ECM proteins and therefore has been widely employed for promoting cell adhesion on biologically inert synthetic and natural biomaterials.49,50 Success of the reaction was verified via NMR, as shown in Figure 2, with a decrease in the peaks corresponding to the norbornene double bonds at 6.2 ppm and the appearance of peaks attributed to the methylene groups of the peptide at 1.8 ppm.
Figure 2.
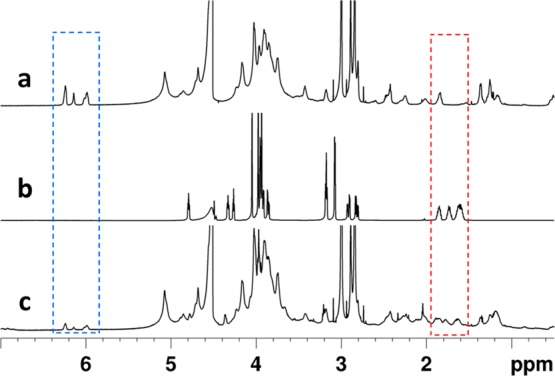
1H NMR spectra (in D2O) of a) Alg-norb, b) CGGGRGDS, and c) Alg-norb reacted with 1 mM CGGGRGDS. The observed decrease in intensity of the double bonds of the norbornene groups after the reaction (highlighted in the blue box at 6.2 ppm) and the appearance of the peaks corresponding to the peptide sequence are highlighted in the red box at 1.7 ppm.
Formation of 3D Hydrogel Networks
Due to the step growth nature of norbornene thiol–ene cross-linking,44 the choice of cross-linker has a major influence on the final network properties, including mesh size and mechanical properties. We chose to use a small series of thiol end group functionalized polyethylene glycol (PEG) chains of different molecular weights (1500 and 5000) and number of arms (2 and 4) to investigate the tailorability of the hydrogel system. The smallest PEG linker used was 1500 Da, due to the similarity in molecular weight to many matrix metalloproteinase (MMP) enzyme cleavable peptide linkers to be incorporated in later generations of these bioinks. The different PEG cross-linkers and concentrations used for formation of hydrogels are shown in Table 2. In all cases rapid and reliable gelation within minutes could be effected by irradiation at 365 nm of the pregel formulation, which is further discussed later in this section.
Table 2. Chemical Compositions and Mass Swelling of Hydrogel Samples.
| Water, rt | |||||
|---|---|---|---|---|---|
| Sample | Cross-linker | Molecular Mass (g/mol) | Ratio of SH:norbornene | Concentration of cross-linker (mol %) | Average swelling ratioa |
| Alg-norb10-1 | PEG dithiol 1500 | 1500 | 0.9:1 | 10 | 254.6 ± 14.8 |
| Alg-norb10-2 | PEG dithiol 1500 | 1500 | 0.5:1 | 5 | 1199.0 ± 114.3 |
| Alg-norb10-3 | PEG dithiol 5000 | 5000 | 0.9:1 | 10 | 337.7 ± 23.7 |
| Alg-norb10-4 | 4arm PEG 5000 | 5000 | 0.9:1 | 10 | 161.7 ± 16.3 |
Mass swelling ratios (swollen/dried) of Alg-norb hydrogels (11 mol % norbornene functionalization) cross-linked with different cross-linkers in deionized water. Four samples were measured for each condition.
The water uptake capacity of the hydrogels was measured via the mass swelling ratio to provide an indication on the density of the network structure (Table 2). With the 10 mol % PEG dithiol 1500 cross-linker (Alg-norb10-1) a swelling ratio of 254.6 ± 14.8 was measured, and with a decrease in cross-linker added (5 mol %) (Alg-norb10-2), a higher swelling ratio was observed (1199 ± 114.3). This is expected as less cross-linkable units would decrease the cross-linking density of the hydrogel, therefore forming less compact network structures. By increasing the molecular weight of the PEG cross-linker from 1500 to 5000 Da (Alg-norb10-3), which inherently increases the distance between cross-linking points, a slight increase in water uptake was observed (337.7 ± 23.70, both at 10 mol %). A tighter and more compact network could be synthesized by switching out the bifunctional cross-linkers for multiarm cross-linkers; the use of a 4-arm PEG thiol 5000 Da cross-linker (Alg-norb10-4) showed a decrease in swelling behavior of the hydrogels (161.7 ± 16.30). The swelling properties of hydrogels were also studied in PBS and cell culture medium, DMEM, at room temperature and 37 °C. As shown in Figure 3, no significant changes were observed in the size of the hydrogel when placed in different baths at room temperature or 37 °C. These observations showed that these hydrogels are not likely to undergo any drastic changes during the course of fabrication and cell culture.
Figure 3.
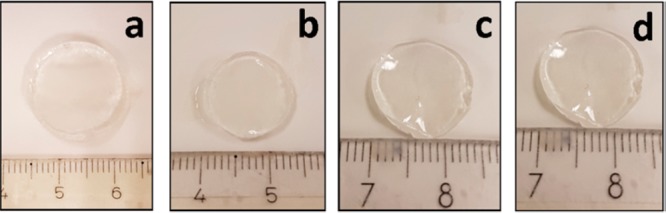
Alg-norb hydrogel (2 w/v%) cross-linked with 10 mol % of 1500 Da PEG dithiol (Alg-norb10-1) swelled in a) water at 23 °C, b) PBS at 23 °C, c) PBS at 37 °C, and d) DMEM at 37 °C.
Gelation kinetics of printed formulations are also inherently important design parameters in the evaluation of bioink candidates. The gelation kinetics of these formulations were monitored via in situ gelation on a photorheometer setup equipped with a transparent window and UV light source. It should be noted that the 2 w/v % Alg-norb (11 mol % norbornene functionalization) employed for our gelation experiments exhibited shear-thinning behavior with a viscosity of 0.16 Pa·s (detailed data included in the SI). Gelation rates of Alg-norb 2 w/v% using the different PEG cross-linkers were measured and compared as shown in Figure 4a. All ink formulations rapidly built network strength, with gelation occurring on subsecond time scales and final network properties achieved within 3 s, although it should be noted that the light intensity on the photorheometer is higher (1 W/cm2) compared to the lamp used for bioprinting (10 mW/cm2). Starting as a liquid (G″ > G′), upon UV exposure a buildup in molecular weight before cross-linking is seen by an initial increase in the viscous loss modulus (G″), followed by a sharp transition and gelation upon true cross-linking of the network. When the material passes the gel point, the hydrogel changes rapidly from a viscous to an elastic material with a large increase in G′ and a concomitant decrease in G″ (decrease in the viscous/flowing component of the material due to completed cross-linking). The rapid rates of gelation observed aid in the formation of 3D structures as polymer solutions are extruded out of the needle and can minimize exposure of encapsulated cells to UV light.
Figure 4.

Shear storage (G′) and loss (G″) moduli, G′, measured as a function of a) UV illumination time, b) frequency, and c) strain for Alg-norb with different PEG cross-linkers (1 = Alg-norb10-1; 2 = Alg-norb10-2; 3 = Alg-norb10-3; 4 = Alg-norb10-4). d) Strain sweep of Alg-norb10-1 (blue lines) and after addition of 100 mM CaCl2 for 3 min (red lines). UV intensity is 1 W/cm2 for all rheological measurements.
Matrix elasticity plays a crucial role in the regulation of cell functions such as differentiation, proliferation, and viability.51 Therefore, to design and develop synthetic hydrogel networks to function as a cellular environment, it is essential to understand and tune the mechanical properties of the hydrogels for tissue specific applications. We again turned to a photorheometer to investigate the effect of the cross-linker and cross-linking density on gel mechanical properties. Frequency sweeps (Figure 4b) for the different gels (Alg-norb10-1 to 4) showed typical frequency independent storage moduli and a large linear elastic regime, characteristic of covalently cross-linked gels. The shear storage moduli (G′) from the strain sweeps of the hydrogels were also measured on a photorheometer (Figure 4c). Hydrogels (Alg-norb 2 w/v %) cross-linked with 10 mol % of PEG dithiol 1500 (Alg-norb10-1) had storage modulus approximately 1.6 kPa, with a drop in modulus (to approximately 600 Pa) when the amount of cross-linker was halved (5 mol %). A slight increase (2.4 kPa) in modulus was observed when cross-linking was carried out with the longer PEG dithiol 5000. The use of a multiarm cross-linker, 4-arm PEG thiol 5000 Da, proceeded to form stiffer network structures with an observed storage modulus of 6 kPa. The rheological properties of Alg-norb with much lower concentrations of norbornene groups (0.24 mol %) cross-linked with excess of the PEG cross-linkers (10 mol %) showed softer gels with similar trends (see the SI for more information). Within the strain sweep measurements, these hydrogels all showed strains at break above 100%, often with significant deviations from linearity around 50% (4-arm PEG 5000 around 10%). Notably, most of these hydrogels also show some strain stiffening52 behavior upon deformation (and before failure), with the exception of 4-arm PEG 5000 and the Ca2+ cross-linked materials (see below). Full strain sweep curves are included in the Supporting Information (Figures S7 and S8).
Other than altering the cross-linker topology and cross-linking density, the mechanical properties of these alginate hydrogels can also be altered via addition of multivalent ions such as calcium. As an example shown in Figure 4d, 100 mM of CaCl2 solution was introduced to the hydrogel formed with the PEG thiol 1500 (Alg-norb10-1) for 3 min and showed an increase in modulus from approximately 1.6 to 6 kPa, albeit with a much reduced strain at which the network begins to lose performance (60% vs 2.5%). Furthermore, a weak gel with an initial modulus between 60 and 90 Pa can have its modulus increased up to over 10 kPa (again with reduced strain at break) when treated similarly with CaCl2 (Figure S8 in the Supporting Information). This reduction in the strain at which the network yields is hypothesized to be a consequence of the tighter and more rigid network formed due to the addition of the divalent ions. Based on the shear moduli measurements, the average mesh sizes for hydrogels were also estimated to be between approximately 11 nm (Alg-norb10-4) to 23 nm (Alg-norb10-1). The addition of calcium ions further decreased the average mesh size by approximately half. As for the weak hydrogels with low norbornene functionalization (0.24 mol %), mesh sizes were between 34 and 44 nm with a significant decrease to approximately 9 nm when calcium ions were introduced to the PEG dithiol 1500 network. More details on the derived values are included in Table S1.
Through the concentration of cross-linkable units, choice of cross-linker, and adjustment of cross-linking density, it was possible to prepare gels of 2 wt % alginate (good for both printing and cell viability, see below) with shear moduli (G) ranging from approximately 0.05 to 10 kPa (E ≈ 0.15 to 30 kPa). This moduli range is useful for tuning the elasticities of microenvironments for different tissues and can be manipulated to direct lineage of stem cells.53 For example, soft alginate gels as opposed to stiff gels have proven to be better matrices to support neurite growth,54 attributed to the similarity in elastic modulus of brain tissues;55−57 stiffer gels, which mimic the elasticity of muscle tissues, can potentially be used as matrices for differentiation of stem cells to myoblasts.58
Bioprinting and Biocompatibility of Alg-norb
To determine the suitability of our alginate bioink platform for bioprinting, we adopted a number of strategies toward producing stable and well-defined structures. From bioprinting in air directly on tissue culture plates, to the use of nondesirable rheological modifiers, to the use of nonreactive baths to deposit into (see the SI for details), we found the most desirable performance when bioprinting directly into cell-culture medium. Bioprinting in cell culture medium allowed the formation of defined structures and was straightforward in its implementation. It should be noted that medium without phenol red was utilized to avoid possible detrimental effects to cells, and all further printing of cell-laden structures was performed in a culture medium bath.
Using a 10 mW/cm2 365 nm LED, test printing of scaffolds was used to determine the optimized conditions for the Alg-norb system. Figures 5a and b are example images of a two-layered bioprinted construct produced by cell-laden Alg-norb hydrogels. At day 0, immediately after bioprinting, the fibers and the pores of the scaffolds were visible (approximately 400 and 200 μm, respectively, Figure 5a). The hydrogels undergo slight swelling over time, and at day 7 the pores were no longer visible under high magnification (Figure 5b). We have also conducted mass swelling experiments of similar hydrogel slabs incubated in cell culture medium at 37 °C immediately after gelation. The mass of hydrogels increased up to 1.6 times before reaching equilibrium after approximately 24 h (Table S2 in the SI). The ability of the hydrogels to self-sustain and maintain the integrity of the bioprinted structures was shown via the selective bioprinting of not only two differently labeled cell populations within a similar bioink, but also two distinct materials formulations with distinct cell types. Initially, two populations of L929 were stained with green and red dyes, which were then extruded in a controlled manner to produce alternating fibers in the X-Y and Z directions. Direct visualization of the two different bioinks to showcase control in the X-Y and Z directions is shown in Figures 5c and d. Going one step further, this platform also allowed the printing of two distinctly different bioinks – L929 in 2 wt % Alg-norb10-1 and ATDC5 in 2 wt % Alg-norb10-4 – with distinctly different mechanical properties (Figure S19).
Figure 5.
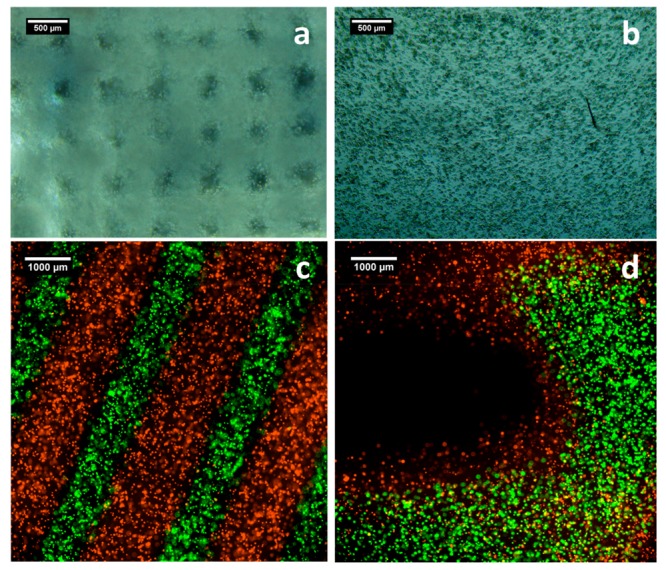
Images of 3D bioprinted hydrogels loaded with cells at a) day 0 and b) day 7. Green and red cell tracker labeled L929 as two different bioinks printed as alternating fibers c) in the X-Y plane and d) in the Z direction.
Utilizing optimized printing conditions (see below), multilayer architectures like a simple pyramid and a cubic structure were able to be recreated with sufficient integrity over 26 layers of bioprinting. As shown in Figure 6, the structural integrity and initial 3D geometries of both structures were well-preserved postprinting. While some equilibrative swelling does decrease the porosity of the printed constructs, porous-like structures were maintained and observed in the X-Y and Z planes as shown in Figures 6d and e. The stability and robustness of the bioprinted structures over time have been maintained in PBS for over two months.
Figure 6.

Scaffolds bioprinted in a) the geometry of a pyramid. b) and c) the geometry of a cube. Porous-like structures can be seen in the cube scaffold shown in d) X-Y and e) Z planes when imaged between two glass coverslips. Of note, the bioprinting conditions used to produce these scaffolds match those optimized for high-cell viability. These scaffolds have shown stability in PBS for over two months. Theoretical side length = 6.9 mm (13 strands, 0.53 mm between strands), total height = 5.2 mm (200 μm/layer, 26 layers).
As bioinks, the developed polymer platform should not only show printability but also the ability to support cell viability throughout and after the bioprinting process. Among the parameters that potentially influence cell viability in extrusion-based bioprinting are the period of exposure to UV, concentration of the bioink, pressure applied for extrusion, and intensity of UV. For these cell viability experiments, we chose to work with a mouse fibroblast, L929, as it is a well characterized cell line commonly used for cytotoxicity testing in biomaterials59,60 and is recommended by ASTM medical device standard F813. The effect of UV irradiation on the viability of cells was first studied in 2D on tissue culture treated plates, where we found high viability (96%) up to 120 s of irradiation time, indicating reasonable UV tolerance of these cells (Figures S10 and S11 in the Supporting Information). As we have shown that bioactivation via conjugation of peptides with cysteines was feasible in our hydrogel system, it was also important to determine the functionality of the conjugated peptides. 2D seeding of L929 cells on Alg-norb10-1 hydrogel slabs with 2 mM of RGD covalently attached showed more cell attachment with elongated morphologies as compared to hydrogels with lower (0.2 mM) or no concentrations of RGD (Figure S17). For cell viability experiments with cells encapsulated in hydrogels, limited observation of cellular spreading in the 3D hydrogels was observed (as compared to 2D) within the time frame of our experiments with or without the presence of RGD sequences as adhesive sites (example shown in Figure S18 in the Supporting Information). The interaction of cells, which dictate their morphology, migration, and adhesion, have been reported to differ significantly in a 2D and a 3D environment.61
Next, the effect of the viscosity of the polymer and bioprinting parameters such as extrusion pressure and printing speed on cell viability was studied. Alg-norb solutions of 1 and 2 w/v % were investigated and compared to determine if viscosity was a contributing factor to printability and cell viability. Lower extrusion pressure ranges (15 to 30 kPa) had to be applied during bioprinting for the 1 w/v % Alg-norb solutions due to its lower viscosity, while extrusion pressures of 30 to 40 kPa were tested for the 2 w/v % solutions. At the overlapping extrusion pressure of 30 kPa, we observed higher cell viability in the 1 w/v % Alg-norb (91%) compared to 2 w/v % (79%) on day 1 (Figures S14 and S15 in the Supporting Information). In both polymer concentrations, better cell viability was observed when pressures were kept low. The comparison of bioprinting speeds of 5 and 10 mm/s showed lower viability of cells at the slower bioprinting speed, attributed to the longer exposure times to the UV light (Figures S13 and S15). Based on the studies conducted, the optimal conditions to produce scaffolds with defined structures bioprinted with L929 were at a bioprinting speed of 10 mm/s with an extrusion pressure of 30 kPa. Cell viability for L929 under these conditions for day 1 (81 ± 4.4%) and day 7 (87 ± 5.5%) are shown in Figure 7.
Figure 7.
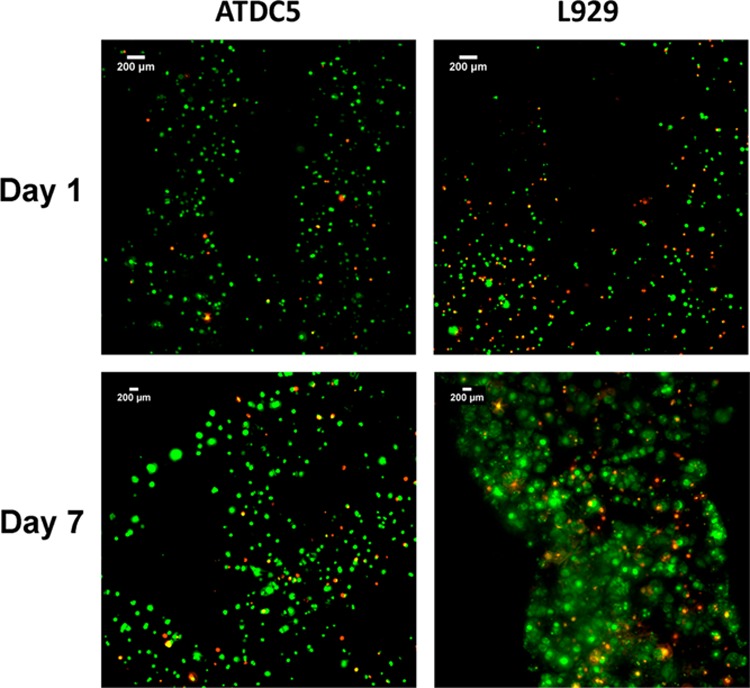
Viability of ATDC5 and L929 cells in bioprinted scaffolds over day 1 and 7. Green stain represents live cells and red stain represents dead cells. ATDC5 scaffolds were bioprinted at 5 mm/s with 30 kPa pressure, and L929 scaffolds were bioprinted at 10 mm/s with 30 kPa pressure.
While L929 cells provide a basis for the optimization of bioprinting parameters, we also investigated the use of a chondrogenic cell line (ATDC 5) more relevant to study cartilage tissue regeneration. When compared to the L929 scaffolds, ATDC5 showed better cell viability under identical conditions (Figure S14 in the Supporting Information), suggesting a resistance to the shear forces from printing. Additionally, ATDC5 could be deposited at 5 mm/s while maintaining cell viability, suggesting that these cells were also less sensitive to UV irradiation. For ATDC5, scaffolds with defined structures could be formed at 30 kPa at a lower extrusion speed of 5 mm/s while still maintaining high cell viability for day 1 (93 ± 2.8%) and day 7 (86 ± 4.6%). Figure 7 depicts the live/dead assays for bioprinted L929 and ATDC5 in Alg-norb hydrogels in their optimized conditions.
Conclusions
In this work, we have demonstrated the design of a modular alginate-based hydrogel system that facilitates the 3D bioprinting of multiple cell types and allows for the tailorability of mechanical properties (via cross-linker and density) and the chemical functionality (via RGD attachement). This modified bioink has extended the biofabrication window of alginate, allowing for printability at a lower concentration (2 wt %) with high cell survivability (>80%) and the creation of stable 3-dimensional constructs. Rapid UV-induced thiol–ene gelation not only created inks with a range mechanical properties (G′ from 0.05 to 10 kPa, and secondary Ca2+ cross-linking) but also allowed the straightforward incorporation of a thiol containing adhesive peptide (HS-RGD). Optimization of printing parameters has shown the susceptibility of L929 to shear stress and resulted in optimized bioprinting conditions for ATDC5 and L929 at 30 kPa pressure at 5 mm/s and 30 kPa at 10 mm/s, respectively. More complex multi-ink geometries have been created using this platform, including multimaterial and multicell inks. Via judicious choice of ink concentration, cross-linker, and bioactive molecules, this alginate ink platform allows for a high degree of tailorability for different tissue engineering cell types and tissue targets. While these current ink formulations do not allow for the printing of highly porous and highly accurate gel grid structures (akin to solid 3D printed constructs), their utility in the creation of well-defined 3D multicellular constructs remains of high interest. Future research with this platform will include the creation of complex tissue relevant 3D geometries with multimaterial and multicell inks, the inclusion of passive or active degradation mechanisms within the ink to allow resorption, and the exploration of cell-responsive elements51 within the ink formation.
Acknowledgments
The authors would like to thank Joost van Dongen for aqueous GPC measurements of the produced alginate polymers and Dr. Hoon Suk Rho for capturing images of the bioprinted scaffolds. H.W.O., C.A.M., A.C., L.M., and M.B.B. would all like to acknowledge the Province of Limburg for funding. A.C. and L.M. would also like to thank Brightlands Materials Center for funding.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.8b00696.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Mota C.; Puppi D.; Chiellini F.; Chiellini E. Additive Manufacturing Techniques for the Production of Tissue Engineering Constructs. J. Tissue Eng. Regener. Med. 2015, 9, 174–190. 10.1002/term.1635. [DOI] [PubMed] [Google Scholar]
- Mironov V.; Reis N.; Derby B. Bioprinting: A Beginning. Tissue Eng. 2006, 12 (4), 631–634. 10.1089/ten.2006.12.631. [DOI] [PubMed] [Google Scholar]
- Jose R. R.; Rodriguez M. J.; Dixon T. A.; Omenetto F.; Kaplan D. L. Evolution of Bioinks and Additive Manufacturing Technologies for 3D Bioprinting. ACS Biomater. Sci. Eng. 2016, 2 (10), 1662–1678. 10.1021/acsbiomaterials.6b00088. [DOI] [PubMed] [Google Scholar]
- Melchels F. P. W.; Domingos M. A. N.; Klein T. J.; Malda J.; Bartolo P. J.; Hutmacher D. W. Additive Manufacturing of Tissues and Organs. Prog. Polym. Sci. 2012, 37 (8), 1079–1104. 10.1016/j.progpolymsci.2011.11.007. [DOI] [Google Scholar]
- Ouyang L.; Highley C. B.; Sun W.; Burdick J. A. A Generalizable Strategy for the 3D Bioprinting of Hydrogels from Nonviscous Photo-Crosslinkable Inks. Adv. Mater. 2017, 29, 1604983. 10.1002/adma.201604983. [DOI] [PubMed] [Google Scholar]
- Moroni L.; Boland T.; Burdick J. A.; De Maria C.; Derby B.; Forgacs G.; Groll J.; Li Q.; Malda J.; Mironov V. A.; Mota C.; Nakamura M.; Shu W.; Takeuchi S.; Woodfield T. B. F.; Xu T.; Yoo J. J.; Vozzi G. Biofabrication: A Guide to Technology and Terminology. Trends Biotechnol. 2018, 36 (4), 384–402. 10.1016/j.tibtech.2017.10.015. [DOI] [PubMed] [Google Scholar]
- Groll J.; Boland T.; Blunk T.; Burdick J. A.; Cho D. W.; Dalton P. D.; Derby B.; Forgacs G.; Li Q.; Mironov V. A.; Moroni L.; Nakamura M.; Shu W.; Takeuchi S.; Vozzi G.; Woodfield T. B. F.; Xu T.; Yoo J. J.; Malda J. Biofabrication: Reappraising the Definition of an Evolving Field. Biofabrication 2016, 8, 013001. 10.1088/1758-5090/8/1/013001. [DOI] [PubMed] [Google Scholar]
- Murphy S. V.; Atala A. 3D Bioprinting of Tissues and Organs. Nat. Biotechnol. 2014, 32 (8), 773–785. 10.1038/nbt.2958. [DOI] [PubMed] [Google Scholar]
- Hölzl K.; Lin S.; Tytgat L.; Van Vlierberghe S.; Gu L.; Ovsianikov A. Bioink Properties Before, During and After 3D Bioprinting. Biofabrication 2016, 8, 032002. 10.1088/1758-5090/8/3/032002. [DOI] [PubMed] [Google Scholar]
- You F.; Eames B. F.; Chen X. Application of Extrusion-Based Hydrogel Bioprinting for Cartilage Tissue Engineering. Int. J. Mol. Sci. 2017, 18 (7), 1597. 10.3390/ijms18071597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbolat I. T.; Hospodiuk M. Current Advances and Future Perspectives in Extrusion-Based Bioprinting. Biomaterials 2016, 76, 321–343. 10.1016/j.biomaterials.2015.10.076. [DOI] [PubMed] [Google Scholar]
- Malda J.; Visser J.; Melchels F. P.; Jüngst T.; Hennink W. E.; Dhert W. J. A.; Groll J.; Hutmacher D. W. 25th Anniversary Article: Engineering Hydrogels for Biofabrication. Adv. Mater. 2013, 25 (36), 5011–5028. 10.1002/adma.201302042. [DOI] [PubMed] [Google Scholar]
- Wang L. L.; Highley C. B.; Yeh Y.-C.; Galarraga J. H.; Uman S.; Burdick J. A. Three-Dimensional Extrusion Bioprinting of Single- and Double-Network Hydrogels Containing Dynamic Covalent Crosslinks. J. Biomed. Mater. Res., Part A 2018, 106, 865–875. 10.1002/jbm.a.36323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highley C. B.; Rodell C. B.; Burdick J. A. Direct 3D Printing of Shear-Thinning Hydrogels into Self-Healing Hydrogels. Adv. Mater. 2015, 27 (34), 5075–5079. 10.1002/adma.201501234. [DOI] [PubMed] [Google Scholar]
- Donderwinkel I.; van Hest J. C. M.; Cameron N. R. Bio-Inks for 3D Bioprinting: Recent Advances and Future Prospects. Polym. Chem. 2017, 8, 4451–4471. 10.1039/C7PY00826K. [DOI] [Google Scholar]
- Jungst T.; Smolan W.; Schacht K.; Scheibel T.; Groll J. Strategies and Molecular Design Criteria for 3D Printable Hydrogels. Chem. Rev. 2016, 116 (3), 1496–1539. 10.1021/acs.chemrev.5b00303. [DOI] [PubMed] [Google Scholar]
- Guvendiren M.; Molde J.; Soares R. M. D.; Kohn J. Designing Biomaterials for 3D Printing. ACS Biomater. Sci. Eng. 2016, 2 (10), 1679–1693. 10.1021/acsbiomaterials.6b00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospodiuk M.; Dey M.; Sosnoski D.; Ozbolat I. T. The Bioink: A Comprehensive Review on Bioprintable Materials. Biotechnol. Adv. 2017, 35 (2), 217–239. 10.1016/j.biotechadv.2016.12.006. [DOI] [PubMed] [Google Scholar]
- Bertlein S.; Brown G.; Lim K. S.; Jungst T.; Boeck T.; Blunk T.; Tessmar J.; Hooper G. J.; Woodfield T. B. F.; Groll J. Thiol-Ene Clickable Gelatin: A Platform Bioink for Multiple 3D Biofabrication Technologies. Adv. Mater. 2017, 29, 1703404. 10.1002/adma.201703404. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Jin X.; Dai R.; Holzman J. F.; Kim K. An Ultrafast Hydrogel Photocrosslinking Method for Direct Laser Bioprinting. RSC Adv. 2016, 6, 21099–21104. 10.1039/C5RA24910D. [DOI] [Google Scholar]
- Yin J.; Yan M.; Wang Y.; Fu J.; Suo H. 3D Bioprinting of Low Concentration Cell-Laden Gelatin Methacrylate (GelMA) Bioinks with Two-Step Crosslinking Strategy. ACS Appl. Mater. Interfaces 2018, 10 (8), 6849–6857. 10.1021/acsami.7b16059. [DOI] [PubMed] [Google Scholar]
- Lim K. S.; Schon B. S.; Mekhileri N. V.; Brown G. C. J.; Chia C. M.; Prabakar S.; Hooper G. J.; Woodfield T. B. F. New Visible-Light Photoinitiating System for Improved Print Fidelity in Gelatin-Based Bioinks. ACS Biomater. Sci. Eng. 2016, 2, 1752–1762. 10.1021/acsbiomaterials.6b00149. [DOI] [PubMed] [Google Scholar]
- Rhee S.; Puetzer J. L.; Mason B. N.; Reinhart-King C. A.; Bonassar L. J. 3D Bioprinting of Spatially Heterogeneous Collagen Constructs for Cartilage Tissue Engineering. ACS Biomater. Sci. Eng. 2016, 2, 1800–1805. 10.1021/acsbiomaterials.6b00288. [DOI] [PubMed] [Google Scholar]
- Gao G.; Yonezawa T.; Hubbell K.; Dai G.; Cui X. Inkjet-Bioprinted Acrylated Peptides and PEG Hydrogel with Human Mesenchymal Stem Cells Promote Robust Bone and Cartilage Formation with Minimal Printhead Clogging. Biotechnol. J. 2015, 10, 1568–1577. 10.1002/biot.201400635. [DOI] [PubMed] [Google Scholar]
- Müller M.; Becher J.; Schnabelrauch M.; Zenobi-Wong M. Nanostructured Pluronic Hydrogels as Bioinks for 3D Bioprinting. Biofabrication 2015, 7, 035006. 10.1088/1758-5090/7/3/035006. [DOI] [PubMed] [Google Scholar]
- Ng W. L.; Yeong W. Y.; Naing M. W. Polyvinylpyrrolidone-Based Bio-Ink Improves Cell Viability and Homogeneity during Drop-on-Demand Printing. Materials 2017, 10, 190. 10.3390/ma10020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai S.; Kamei H.; Mori T.; Hotta T.; Ohi H.; Nakahata M.; Taya M. Visible Light-Induced Hydrogelation of an Alginate Derivative and Application to Stereolithographic Bioprinting Using a Visible Light Projector and Acid Red. Biomacromolecules 2018, 19 (2), 672–679. 10.1021/acs.biomac.7b01827. [DOI] [PubMed] [Google Scholar]
- Stichler S.; Jungst T.; Schamel M.; Zilkowski I.; Kuhlmann M.; Böck T.; Blunk T.; Teßmar J.; Groll J. Thiol-Ene Clickable Poly(glycidol) Hydrogels for Biofabrication. Ann. Biomed. Eng. 2017, 45 (1), 273–285. 10.1007/s10439-016-1633-3. [DOI] [PubMed] [Google Scholar]
- Senyurt A. F.; Wei H. Y.; Hoyle C. E.; Piland S. G.; Gould T. E. Ternary Thiol-Ene/acrylate Photopolymers: Effect of Acrylate Structure on Mechanical Properties. Macromolecules 2007, 40, 4901–4909. 10.1021/ma062534b. [DOI] [Google Scholar]
- Hoyle C. E.; Lee T. Y.; Roper T. Thiol-Enes: Chemistry of the Past with Promise for the Future. J. Polym. Sci., Part A: Polym. Chem. 2004, 42, 5301–5338. 10.1002/pola.20366. [DOI] [Google Scholar]
- Stichler S.; Bertlein S.; Tessmar J.; Jüngst T.; Groll J. Thiol-Ene Cross-Linkable Hydrogels as Bioinks for Biofabrication. Macromol. Symp. 2017, 372, 102–107. 10.1002/masy.201600173. [DOI] [Google Scholar]
- Smidsrød O.; Skjåk-Brłk G. Alginate as Immobilization Matrix for Cells. Trends Biotechnol. 1990, 8, 71–78. 10.1016/0167-7799(90)90139-O. [DOI] [PubMed] [Google Scholar]
- Morris E. R.; Cutler A. N.; Ross-Murphy S. B.; Rees D. A.; Price J. Concentration and Shear Rate Dependence of Viscosity in Random Coil Polysaccharide Solutions. Carbohydr. Polym. 1981, 1, 5–21. 10.1016/0144-8617(81)90011-4. [DOI] [Google Scholar]
- Rezende R. A.; Bártolo P. J.; Mendes A.; Filho R. M. Rheological Behavior of Alginate Solutions for Biomanufacturing. J. Appl. Polym. Sci. 2009, 113, 3866–3871. 10.1002/app.30170. [DOI] [Google Scholar]
- Yang J. S.; Xie Y. J.; He W. Research Progress on Chemical Modification of Alginate: A Review. Carbohydr. Polym. 2011, 84 (1), 33–39. 10.1016/j.carbpol.2010.11.048. [DOI] [Google Scholar]
- Pawar S. N.; Edgar K. J. Alginate Derivatization: A Review of Chemistry, Properties and Applications. Biomaterials 2012, 33 (11), 3279–3305. 10.1016/j.biomaterials.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Khalil S.; Sun W. Bioprinting Endothelial Cells With Alginate for 3D Tissue Constructs. J. Biomech. Eng. 2009, 131 (11), 111002. 10.1115/1.3128729. [DOI] [PubMed] [Google Scholar]
- Shi P.; Laude A.; Yeong W. Y. Investigation of Cell Viability and Morphology in 3D Bio-Printed Alginate Constructs with Tunable Stiffness. J. Biomed. Mater. Res., Part A 2017, 105, 1009–1018. 10.1002/jbm.a.35971. [DOI] [PubMed] [Google Scholar]
- Marchioli G.; van Gurp L.; van Krieken P. P.; Stamatialis D.; Engelse M.; van Blitterswijk C. A.; Karperien M. B. J.; de Koning E.; Alblas J.; Moroni L.; van Apeldoorn A. A. Fabrication of Three-Dimensional Bioplotted Hydrogel Scaffolds for Islets of Langerhans Transplantation. Biofabrication 2015, 7 (2), 025009. 10.1088/1758-5090/7/2/025009. [DOI] [PubMed] [Google Scholar]
- Ning L.; Guillemot A.; Zhao J.; Kipouros G.; Chen X. Influence of Flow Behavior of Alginate–Cell Suspensions on Cell Viability and Proliferation. Tissue Eng., Part C 2016, 22 (7), 652–662. 10.1089/ten.tec.2016.0011. [DOI] [PubMed] [Google Scholar]
- Markstedt K.; Mantas A.; Tournier I.; Martínez Ávila H.; Hägg D.; Gatenholm P. 3D Bioprinting Human Chondrocytes with Nanocellulose-Alginate Bioink for Cartilage Tissue Engineering Applications. Biomacromolecules 2015, 16, 1489–1496. 10.1021/acs.biomac.5b00188. [DOI] [PubMed] [Google Scholar]
- Leppiniemi J.; Lahtinen P.; Paajanen A.; Mahlberg R.; Metsä-Kortelainen S.; Pinomaa T.; Pajari H.; Vikholm-Lundin I.; Pursula P.; Hytönen V. P. 3D-Printable Bioactivated Nanocellulose-Alginate Hydrogels. ACS Appl. Mater. Interfaces 2017, 9, 21959–21970. 10.1021/acsami.7b02756. [DOI] [PubMed] [Google Scholar]
- Cramer N. B.; Reddy S. K.; Cole M.; Hoyle C.; Bowman C. N. Initiation and Kinetics of Thiol-Ene Photopolymerizations without Photoinitiators. J. Polym. Sci., Part A: Polym. Chem. 2004, 42, 5817–5826. 10.1002/pola.20419. [DOI] [Google Scholar]
- Hoyle C. E.; Bowman C. N. Thiol–Ene Click Chemistry. Angew. Chem., Int. Ed. 2010, 49 (9), 1540–1573. 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- Desai R. M.; Koshy S. T.; Hilderbrand S. A.; Mooney D. J.; Joshi N. S. Versatile Click Alginate Hydrogels Crosslinked via Tetrazineenorbornene Chemistry. Biomaterials 2015, 50 (1), 30–37. 10.1016/j.biomaterials.2015.01.048. [DOI] [PubMed] [Google Scholar]
- Boekhoven J.; Rubertpérez C. M.; Sur S.; Worthy A.; Stupp S. I. Dynamic Display of Bioactivity through Host-Guest Chemistry. Angew. Chem., Int. Ed. 2013, 52 (46), 12077–12080. 10.1002/anie.201306278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal D.; Vijay I. K. A Method for the High Efficiency of Water-Soluble Carbodiimide-Mediated Amidation. Anal. Biochem. 1994, 218, 87–91. 10.1006/abio.1994.1144. [DOI] [PubMed] [Google Scholar]
- Fairbanks B. D.; Schwartz M. P.; Bowman C. N.; Anseth K. S. Photoinitiated Polymerization of PEG-Diacrylate with Lithium Phenyl-2,4,6-Trimethylbenzoylphosphinate: Polymerization Rate and Cytocompatibility. Biomaterials 2009, 30 (35), 6702–6707. 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellis S. L. Advantages of RGD Peptides for Directing Cell Association with Biomaterials. Biomaterials 2011, 32 (18), 4205–4210. 10.1016/j.biomaterials.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersel U.; Dahmen C.; Kessler H. RGD Modified Polymers: Biomaterials for Stimulated Cell Adhesion and beyond. Biomaterials 2003, 24, 4385–4415. 10.1016/S0142-9612(03)00343-0. [DOI] [PubMed] [Google Scholar]
- Ooi H. W.; Hafeez S.; van Blitterswijk C. A.; Moroni L.; Baker M. B. Hydrogels That Listen to Cells: A Review of Cell-Responsive Strategies in Biomaterial Design for Tissue Regeneration. Mater. Horiz. 2017, 4, 1020–1040. 10.1039/C7MH00373K. [DOI] [Google Scholar]
- Kouwer P. H. J.; Koepf M.; Le Sage V. A. A.; Jaspers M.; van Buul A. M.; Eksteen-Akeroyd Z. H.; Woltinge T.; Schwartz E.; Kitto H. J.; Hoogenboom R.; Picken S. J.; Nolte R. J. M.; Mendes E.; Rowan A. E. Responsive Biomimetic Networks from Polyisocyanopeptide Hydrogels. Nature 2013, 493 (7434), 651–655. 10.1038/nature11839. [DOI] [PubMed] [Google Scholar]
- Engler A. J.; Sen S.; Sweeney H. L.; Discher D. E. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 2006, 126, 677–689. 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- Matyash M.; Despang F.; Mandal R.; Fiore D.; Gelinsky M.; Ikonomidou C. Novel Soft Alginate Hydrogel Strongly Supports Neurite Growth and Protects Neurons Against Oxidative Stress. Tissue Eng., Part A 2012, 18 (1–2), 55–66. 10.1089/ten.tea.2011.0097. [DOI] [PubMed] [Google Scholar]
- Chatelin S.; Constantinesco A.; Willinger R. Fifty Years of Brain Tissue Mechanical Testing: From in Vitro to in Vivo Investigations. Biorheology 2010, 47, 255–276. 10.3233/BIR-2010-0576. [DOI] [PubMed] [Google Scholar]
- Buxboim A.; Ivanovska I. L.; Discher D. E. Matrix Elasticity, Cytoskeletal Forces and Physics of the Nucleus: How Deeply Do Cells “Feel” Outside and In?. J. Cell Sci. 2010, 123 (3), 297–308. 10.1242/jcs.041186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges P. C.; Miller W. J.; Meaney D. F.; Sawyer E. S.; Janmey P. A. Matrices with Compliance Comparable to that of Brain Tissue Select Neuronal over Glial Growth in Mixed Cortical Cultures. Biophys. J. 2006, 90, 3012–3018. 10.1529/biophysj.105.073114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler A. J.; Richert L.; Wong J. Y.; Picart C.; Discher D. E. Surface Probe Measurements of the Elasticity of Sectioned Tissue, Thin Gels and Polyelectrolyte Multilayer Films: Correlations between Substrate Stiffness and Cell Adhesion. Surf. Sci. 2004, 570 (1–2), 142–154. 10.1016/j.susc.2004.06.179. [DOI] [Google Scholar]
- Liu X.; Tang M.; Zhang T.; Hu Y.; Zhang S.; Kong L.; Xue Y. Determination of a Threshold Dose to Reduce or Eliminate CdTe-Induced Toxicity in L929 Cells by Controlling the Exposure Dose. PLoS One 2013, 8 (4), e59359. 10.1371/journal.pone.0059359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson R. R.; Miller J. A.; Reichert W. M. Polyimides as Biomaterials: Preliminary Biocompatibility Testing. Biomaterials 1993, 14 (8), 627–635. 10.1016/0142-9612(93)90183-3. [DOI] [PubMed] [Google Scholar]
- Hakkinen K. M.; Harunaga J. S.; Doyle A. D.; Yamada K. M. Direct Comparisons of the Morphology, Migration, Cell Adhesions, and Actin Cytoskeleton of Fibroblasts in Four Different Three-Dimensional Extracellular Matrices. Tissue Eng., Part A 2011, 17 (5–6), 713–724. 10.1089/ten.tea.2010.0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.