

Abstract
In this study, we probe the folding of KvAP, a voltage gated K+ (Kv) channel. The KvAP channel though of archaebacterial origin is structurally and functionally similar to eukaryotic Kv channels. An advantage of the KvAP channel is that it can be folded in vitro from an extensively unfolded state and the folding can be controlled by temperature. We utilize these properties of the KvAP channel to separately study the membrane insertion and the tetramerization stages during folding. We use two quantitative assays: a Cys PEGylation assay to monitor membrane insertion and a cross-linking assay to monitor tetramerization. We show that during folding the KvAP polypeptide is rapidly inserted into the lipid bilayer with a “native-like” topology. We identify a segment at the C-terminus that is important for multimerization of the KvAP channel. We show that this C-terminal domain forms a dimer, which raises the possibility that the tetramerization of the KvAP channel proceeds through a dimer of dimers pathway. Our studies show that the in vitro folding of the KvAP channel mirrors aspects of the cellular assembly pathway for voltage gated K+ channels and therefore suggest that evolutionarily distinct Kv channels share a common folding pathway. The pathway for the folding and assembly of a Kv channel is of central importance as defects in this pathway have been implicated in the etiology of several disease states. Our studies indicate that the KvAP channel provides an experimentally tractable system to elucidate the folding mechanism of Kv channels.
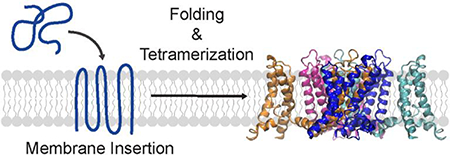
Graphical Abstract
INTRODUCTION
Voltage-gated K+ (Kv) channels are found in all kingdoms of life and are critical for the generation of electrical impulses by excitable cells.1, 2 Kv channels form tetramers in the cell membrane. Each monomer consists of six transmembrane segments (S1-S6) that are arranged in two distinct domains; the voltage-sensing domain (VSD, S1-S4) responsible for sensing the membrane potential and the pore domain (PD, S5-S6) responsible for selectively translocating K+ across the membrane (Figure 1A, B).3, 4 Additionally, Kv channel subunits can contain cytoplasmic domains that are involved in various activities such as interacting with proteins that modify channel function or assisting in the assembly of the channel.5–10
Figure 1. Structure and folding of the KvAP channel.

(A) Top view of the tetrameric KvAP channel. The structural model of the KvAP channel previously described31 is shown. The structure illustrates the domain swap between the pore and voltage sensor domains of adjacent subunits in the KvAP channel. (B) Topology map illustrating the domains and the structural features of a KvAP channel subunit. Dashed lines are membrane boundaries. (C) The folding pathway of the KvAP channel involves the stages of membrane insertion, secondary/tertiary folding and tetramerization. Structure figures were generated with VMD.60
In humans, genetic defects in Kv channels have been linked to several disease states including ataxia, epilepsy, diabetes, and heart arrhythmias such as Short-QT and Long-QT syndrome.11 While the mechanisms underlying the disease causing mutations could be altered voltage gating, ion permeation or abnormal mRNA transcription/translation, it is increasingly being recognized that the overwhelming majority of disease causing mutations are linked to defects in the assembly and trafficking of Kv channels.12–17 For example, the majority of point mutations (88% of 167 mutations tested) in the Kv11.1 channel (also known as hERG) that cause Long-QT syndrome show an assembly/trafficking deficient phenotype.12 This observation points to a central role for the folding/assembly of Kv channel in the etiology of disease states. In spite of this importance, very little is known about the folding process of Kv channels.
Our present knowledge on the folding and assembly of Kv channels comes primarily from in vivo studies on the Shaker K+ channel (mammalian homologs are Kv1.x).18 Membrane protein folding is proposed to take place in two stages: insertion of the polypeptide into the membrane and the folding of the polypeptide in the membrane (Figure 1C).19–21 In certain cases, the process of folding can precede the insertion of the polypeptide into the lipid bilayer.22 In the folding of Kv channels, an additional step of tetramerization is required to produce the functional channel. Folding studies on the Shaker channel family have probed the membrane insertion of specific transmembrane segments and the role of the cytoplasmic domain, referred to as the T1 domain, in tetramer assembly and channel subtype segregation.8, 23–25 The studies on insertion of the transmembrane segments have been carried out using in vitro translation systems while the tetramer assembly studies have been carried out by channel expression in Xenopus oocytes or HEK cells. Cellular expression systems such as oocytes or HEK cells provide a challenging venue to probe the mechanism of channel folding and assembly as the tools that can be used to probe membrane protein folding in a cellular context are limited. The use of an in vitro system is advantageous in this regard, however the efficiency of folding and assembly of the Shaker family of channels in an in vitro system is very low.26 Due to these limitations, there are a number of essential questions regarding the folding of Kv channels that are yet to be answered.
Here, we investigate the folding of the Kv channel, KvAP. The KvAP channel is derived from Aeropyrum pernix, an hyperthermophilic archaeon that has an optimal growth temperature of 90 −95 °C.27 The KvAP channel, though archaebacterial in origin, shares extensive structural and functional similarity to eukaryotic Kv channels like Shaker.27–29 We have previously reported the in vitro folding of the KvAP channel and established by biochemical, spectroscopic and functional studies that the KvAP channel obtained by in vitro folding is identical to the native channel.30 The ability to carry out in vitro folding makes the KvAP channel a good model system to probe the folding of a Kv channel.
In this study, we use two quantitative assays to probe specific stages during the in vitro folding of the KvAP channel. We use a Cys PEGylation assay to monitor the insertion of the TM segments into the lipid bilayer and a chemical crosslinking assay to follow the tetramerization of the KvAP channel. We find that the membrane insertion of the KvAP polypeptide is rapid and takes place with the native-like topology. We also identify a protein segment at the C-terminus that is important for multimerization of the KvAP channel. Our findings on the folding pathway of the KvAP channel mirror the findings on the cellular assembly of eukaryotic Kv channels. Our studies therefore suggest that these evolutionarily distant channels potentially share a common folding pathway. Importantly, our studies establish the KvAP channel as an important model system to elucidate the folding mechanism of Kv channels.
EXPERIMENTAL PROCEDURES
Expression and purification of KvAP constructs
A His6 tagged WT KvAP in the pQE60 plasmid was kindly provided by R. MacKinnon (The Rockefeller University).27 The single cysteine residue (C247) in the WT KvAP channel was substituted by Ser (KvAP-C247S). The Cys variants used in the PEGylation experiments were generated in the KvAP C247S background using Quickchange mutagenesis (Agilent). The KvAPΔCTD construct has a deletion of residues C247-K282.31 The KvAP+Lys construct has a C247K and a P249K substitution. The KvAPΔCTD+Lys construct has the C247K and P249K substitutions along with a deletion of residues L252-K282.
Protein expression and membrane preparation for the KvAP channel constructs was carried out as previously described.30 For protein purification, the membranes were solubilized by decyl-β-D-maltoside (DM, 2% w/v) and the KvAP protein was purified by metal affinity chromatography followed by size exclusion chromatography (SEC). SEC was carried out using a Superdex S200 column using 50 mM HEPES-KOH, 150 mM KCl and 0.25 % DM as the running buffer. The KvAP channel tetramer was unfolded by trichloroacetic acid precipitation as previously described30 to provide the unfolded protein that was used for the in vitro folding and the PEGylation experiments.
In vitro folding of KvAP
In vitro folding of KvAP was carried out using large unilamellar vesicles (LUVs) composed of either 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) or Asolecitin (Aso, 20% L-α-Phosphatidylcholine from Soybean). POPC was purchased from Avanti Polar Lipids in chloroform, aliquoted into glass vials, dried under a gentle stream of Argon gas and residual solvent was removed by lyophilization overnight. Aso was purchased from Avanti Polar Lipids in granule form, dissolved in cyclohexane, frozen with liquid Nitrogen and lyophilized overnight. Lipids were stored under Argon at −80 °C prior to use.
A fresh stock of lipids were prepared for each experiment by solubilizing the lipids with Lipid buffer (100 mM sodium phosphate pH 7.0, 150 mM KCl, and 1 mM EDTA) for 30 minutes with gentle agitation. LUVs were prepared by extrusion at a concentration of 10 mg mL−1 using 400 nm filters. Unfolded KvAP-C247S was dissolved in SDS-Protein buffer (100 mM sodium phosphate pH 7.0, 1% (w/v) SDS, and 1 mM EDTA) and A280 was measured with a Nanodrop (ThermoFisher) to determine protein concentration. The protein was diluted to 150 μM with SDS-Protein buffer for in vitro folding experiments. The unfolded protein stock solution was prepared fresh prior to each experiment.
For in vitro folding, unfolded KvAP was diluted 10 fold into LUVs giving final concentrations of 15 μM protein and 11.84 mM POPC; a molar lipid: monomer ratio of 789:1. Because Aso is a heterogeneous mixture, the molar concentration and ratio cannot be determined. Protein and lipid were mixed at room temperature (about 20 °C). The presence of KCl in the lipid buffer causes precipitation of SDS.19 The zero time point was taken at room temperature and in vitro folding was initiated by heating the sample with a thermal cycler (Applied Biosystems). At time points, the folding reaction was quenched by the addition of 2% (w/v) dodecylphosphocholine (Fos-12, Anatrace). Samples were incubated in Fos-12 at room temperature for 1 minute and then crosslinked with 0.1% (w/v) glutaraldehyde for 30 minutes. Crosslinking was quenched by the addition of 5X SDS loading buffer (250 mM Tris-HCl pH 6.8, 8% SDS, 0.1% bromophenol blue, 40% glycerol and 100 mM DTT).
Samples were then subjected to SDS-PAGE on 12% acrylamide gels.32 Gels were stained with Coomassie blue, imaged (Biorad imager) and the densities of bands were quantitated by ImageLab (Biorad) using a 50 mm disk background subtraction. The percent tetramer was calculated as the density of the tetrameric species divided by the sum of all species in each lane. Each gel consisted of a control lane in which the protein was not crosslinked with glutaraldehyde.
PEGylation assay
The unfolded KvAP polypeptides were suspended in the SDS-Protein buffer and diluted into POPC vesicles at room temperature similar to the procedure used for in vitro folding except that the SDS-Protein and Lipid buffers were supplemented with 5 and 1 mM DTT respectively. Immediately following dilution, the protein/lipid mixture was diluted 1.25X with water and then reacted with 3 mM 2 kDa PEG-maleimide (PEG-mal, Creative PEGWorks). Reducing conditions were chosen to balance the reactivity of the PEG-mal reagent (3 mM) with the final concentration of DTT (1.1 mM) as DTT will react with the PEG-mal. Using higher concentrations of PEG-mal (>3 mM) resulted in non-specific reactivity. The maximum time between the addition of the KvAP polypeptide to the POPC vesicles and the start of the incubation with PEG-mal was 30 seconds. A negative control was carried out without the addition of PEG-mal and a positive control was carried out by reacting with PEG-mal in the presence of SDS (by diluting the protein lipid mixture 1.25X using 10% SDS for a final SDS concentration of 2%). All reactions were carried out at room temperature for 30 minutes and quenched with 5X SDS loading buffer. Lack of tetramerization of the KvAP channel during the incubation with PEG-mal was verified by glutaraldehyde crosslinking. Samples were subjected to SDS-PAGE on 12% acrylamide gels, imaged and quantitated as described. The PEG-mal reactivity was quantitated as the reacted density (higher gel-shift band) divided by the sum of the reacted and unreacted bands. We observed that a fraction of the cysteines were unreactive to PEG-mal as judged by the absence of full reactivity in the SDS control. To account for this, the reactivity was normalized as:
Expression and analysis of SUMO-CTD and SUMO-GCN4
The SUMO-CTD fusion protein consisted of KvAP channel residues L239-K282 with a C247S substitution that was cloned after G98 of the yeast SUMO protein.33 In the SUMO-GCN4 fusion protein, residues G250-K282 of the SUMO-CTD fusion protein were replaced with a Gly3 linker and the tetrameric GCN4 sequence (GGGRMKQIEDKLEEILSKLYHIENELARIKKLLGER).34 The SUMO fusion proteins also consisted of a C-terminal thrombin site followed by a His6 tag. The SUMO fusion proteins were expressed in Escherichia coli Rosetta2 (DE3) cell using the high-density expression protocol.35 Purification of the SUMO fusion proteins using metal affinity chromatography was carried out as previously described.33 The SUMO protein was obtained by cleavage of the SUMO-CTD fusion protein using the SUMO protease as previously described.33
SEC analysis of the SUMO fusion proteins was carried out using a Superdex S200 column (GE Healthcare) in 50 mM HEPES-NaOH pH 7.5, 150 mM NaCl, 1 mM EDTA. The peak fractions obtained were crosslinked using 0.1% glutaraldehyde for 30 minutes at room temperature. As a control, glutaraldehyde crosslinking was also carried out in the presence of 2% SDS. The crosslinked samples were analyzed by SDS-PAGE.
To test the temperature dependence of CTD dimerization, the SUMO-CTD fusion protein in 50 mM HEPES-NaOH pH 7.5, 150 mM NaCl was dissociated by treatment with 0.35% SDS. Dissociation was confirmed by crosslinking using 0.1% glutaraldehyde. For re-association, the SDS sample was diluted 10 fold in 50 mM HEPES-NaOH pH 7.5, 150 mM NaCl and incubated at the desired temperature for 10 minutes. The extent of re-association was then determined by crosslinking with 0.1% glutaraldehyde followed by SDS-PAGE.
Expression and purification of S6 and S6+CTD peptides
The S6 and the S6+CTD peptides were recombinantly expressed as SUMO fusion proteins (Table 1). The SUMO fusion protein used for the expression of the S6+CTD peptide consisted of the KvAP channel residues 191–282 (with a V191C and a C247S substitution) cloned after Gly98 of the yeast SUMO protein.33 The SUMO-S6+CTD fusion protein also consisted of an N-terminal His6 tag and a C-terminal Strep tag. The SUMO-S6 fusion protein consisted of KvAP channel residues 191–246 (with a V191C substitution). The SUMO-S6 and the SUMO-S6+CTD fusion proteins were expressed in Escherichia coli Rosetta2 (DE3) cells using the high-density expression protocol, purified and proteolysed using the SUMO protease as previously described.33 Following proteolysis, the peptides were purified by RP-HPLC on a C4 column using a 50–100% gradient of Buffer C [6: 3: 1, isopropanol: acetonitrile: H2O + 0.1% trifluoroacetic acid] and confirmed by Electrospray Ionisation Mass Spectrometry (S6 peptide: observed mass = 5701.46 ± 1.86, expected mass = 5701.78; S6+CTD peptide: observed mass = 11015.78 ± 1.44, expected mass = 11015.78). Purified peptides were lyophilized and stored at −80 °C.
Table 1.
Sequences of peptides used in the folding competition assay.
| Peptide | Sequencea |
|---|---|
| S6 | C191VTATTVGYGDVVPATPIGKVIGIAVMLTGISALTLLIGTVSNMFQKILVGEPEPS246G |
| S6-CTD | C191VTATTVGYGDVVPATPIGKVIGIAVMLTGISALTLLIGTVSNMFQKILVGEPEPSSSPAKLAEMVSSMSEEEFEEFVRTLKNLRRLENSMK282GSWSHPQFEK |
The sequence in red are residues in the CTD of KvAP. The transmembrane segment (residues 207–237) of the KvAP channel is indicated in bold. In addition, the peptides contain the selectivity filter (residues 196–201) and the C-terminal end of the pore helix. The peptides also carry a V191C and a C247S (in S6-CTD) substitution. The underlined region of S6-CTD is a strep-tag.
The CTD competition assay
Unfolded KvAP C247S and the competitor peptides (S6 and S6 + CTD) were rehydrated in SDS-Protein buffer supplemented with 100 mM DTT and 1 mM TCEP at concentrations of 400 μM (KvAP) or 2 mM (competitor peptides). The S6 and S6+CTD peptide concentrations were determined by A280. We confirmed the relative concentrations of the competitor peptides by RP-HPLC. In the competition assay, 2.5 μL of the KvAP (400 μM) was mixed with 2.5 μL of the competitor peptide (2 mM) or 2.5 μL protein buffer and then mixed with Aso liposomes (45 μL, 10 mg/mL). The folding reaction was allowed to proceed for 90 minutes at 50 °C with intermittent mixing. At the end of the incubation period, 2% Fos-12 was added and the reaction was crosslinked using 0.1% glutaraldehyde for 30 minutes at room temperature. The crosslinking reaction was quenched with 5X SDS loading buffer. In control reactions for each competitor, the competitor was added after the folding step at 50 °C but before glutaraldehyde crosslinking was carried out. Equal amounts of the crosslinking reactions were carefully loaded and electrophoresed on a 12% SDS-PAGE gel. The SDS-PAGE gel was stained and analyzed as previously described. Percent tetramer was determined by dividing the density of the KvAP tetramer band in each lane by the total density in the lane containing KvAP cross-linked with no competitor peptide. Percent inhibition for each competitor peptide was then determined as:
where the “with competitor” condition was when the competitor peptide was added at the beginning of the folding reaction, and the “control” condition was when the competitor peptide was added after the folding reaction but before the glutaraldehyde cross-linking was carried out. This analysis requires the presence of the same amounts of the KvAP polypeptide being present in each of the lanes and therefore great care was taken to ensure that equal amounts of the crosslinking reaction were loaded on the SDS-PAGE gel in each case.
RESULTS
Temperature dependent folding of the KvAP channel
We used glutaraldehyde cross-linking to monitor the folding of the KvAP channel.30 Glutaraldehyde cross-linking of the native KvAP channel gives a protein band that corresponds to a tetramer on SDS-PAGE, while crosslinking of the unfolded KvAP polypeptide results in only a monomeric species. In vitro folding of the KvAP channel is carried out using lipid vesicles.30 Briefly, the unfolded KvAP polypeptide is solubilized in SDS and then diluted into POPC lipid vesicles. The folding is quenched at various time points by the addition of the detergent Fos-12, which disrupts the lipid vesicles, and cross-linked by glutaraldehyde (Figure 2A, B). The oligomeric species obtained on cross-linking are separated by SDS-PAGE and quantified. The folding yields are calculated from the density of the tetrameric species divided by the total protein density. As previously reported, the folding reaction is extremely slow at room temperature with only a ~4% tetramer yield at the 2 hour time point but the extent of folding increases with temperature30 and a 75% yield of tetramer is observed at the 2 hour time point at 80 °C (Figure 2C). Elevated temperature is required through-out the folding reaction. Figure 2D shows the effects of a downshift in temperature during the folding reaction. Here, aliquots of the in vitro folding reaction of the KvAP channel at 50 °C were taken at different time points, the temperature was dropped to 20 °C and the progress of the folding was monitored. We observed that following the drop in temperature to 20 °C, no additional folding took place and the extent of tetramer stayed steady through-out the time course. These studies indicate the requirement for higher temperature for tetramer formation and point to an unique feature of the in vitro folding of the KvAP channel, that it can be controlled by a change in temperature.
Figure 2. In vitro folding of the KvAP channel is dependent on temperature.

(A, B) Time course of the in vitro folding of the KvAP channel at 20 °C (A) and at 80 °C (B). Representative SDS-PAGE gels showing glutaraldehyde crosslinking of the in vitro folding reaction at various time points over 2 hours is shown. The last lane is a time point at 2 hours without glutaraldehyde added (−). The oligomeric nature of the crosslinked band is indicated. (C) Tetramerization kinetics of the KvAP channel at different temperatures are plotted. Data have been fitted with either a single (40 °C and 80 °C) or a double exponential (50 °C). Data at 20 °C could not be fitted to an exponential. (D) Temperature downshift arrests folding of KvAP. The in vitro folding reaction for the KvAP channel was carried out at 50 °C. After incubation for 2, 5 or 16 minutes, the samples were transferred from 50 °C to 20 °C and the tetramerization kinetics were followed. No additional tetramerization was observed at 20 °C. The data points following the downshift are fitted with a straight line. Error bars shown in C and D are standard deviations for n =3 using at least 2 independent protein preparations.
Membrane insertion is not rate limiting in the folding of KvAP
We used a quantitative Cys PEGylation assay 30, 36 to determine the membrane insertion of the various segments of the KvAP polypeptide during in vitro folding. The Cys-PEGylation assay has been previously used to monitor membrane protein topology.37–42 The assay measures the labelling of a Cys residue in the KvAP channel by PEG-2K-maleimide (PEG-mal) in the presence of lipid vesicles. If the Cys residue is within the hydrophobic core of the membrane, then it is not accessible to the PEG-mal reagent, whereas a Cys that is present outside the lipid bilayer is accessible to labelling. Labelling by PEG-mal is determined by a 2 kDa shift in the mobility on SDS-PAGE, and the extent of labelling provides the degree of membrane insertion of the segment of the KvAP channel with the Cys substitution.
In the assay (Figure 3A), Cys mutants of the KvAP channel were unfolded, solubilized in SDS and then diluted 10-fold into POPC vesicles at room temperature. Immediately following dilution (< 30 seconds), an aliquot was withdrawn and reacted with PEG-mal for 30 min. A negative control without PEG-mal and a positive control with labelling in the presence of SDS was also performed. To test the assay we used two Cys mutants, 37C (Cys substitution at residue 37) in transmembrane helix S1 and 143C in the loop region between transmembrane helices S4 and S5 (Figure 3B). Using the Cys PEGylation assay, we observed that 37C does not react with PEG-mal in the presence of lipid vesicles but is labelled when the lipid vesicles are disrupted by SDS (Figure 3C). 143C reacts with PEG-mal in the presence of lipid vesicles but does not show complete modification. The extent of modification of 143C observed in the presence of lipid vesicles was similar to the modification observed when the reaction was carried out in the presence of SDS. The lack of complete modification in the presence of SDS indicates that a fraction of the 143C was unreactive, probably due to some spurious reaction during protein unfolding. The unfolding procedure used involved treatment with strong acids, organic solvents and lyophilization to ensure extensive unfolding.30 We expect that under the harsh conditions used, a fraction of Cys undergo some spurious reaction and becomes unreactive to PEG-mal. As a result, we do not observe complete reactivity to PEG-mal in SDS. Complete reactivity to PEG-mal in SDS was also not observed for 37C, albeit the fraction unreactive was small. The fraction unreactive to PEG-mal varied depending on the Cys mutant and the protein preparation. To account for the unreactive fraction, the reactivity to PEG-mal in lipid was normalized to the reactivity observed when the lipid vesicles were disrupted by SDS (Figure 3D). From the normalized data, we can conclude that 37C is buried within the lipid bilayer while 143C is solvent exposed.
Figure 3. The Cys-PEGylation assay used to assess the membrane insertion of KvAP.

(A) The reaction scheme used in Cys-PEGylation assay is shown. (B) Topology map of the VSD of the KvAP channel illustrating the cysteines used to demonstrate the methodology. 37C is transmembrane and 143C is in a loop outside of the membrane. (C) SDS-PAGE gels showing the results of the Cys-PEGylation for 37C and 143C. (D) PEGylation results for 37C and D143C were quantified and plotted as bar graphs. Error bars are standard deviations from n=3 experiments using at least 2 independent protein preparations.
We generated Cys mutants of the KvAP channel with individual Cys substitutions introduced into each of the transmembrane helices and the loops to cover the topology as shown in Figure 4A. Additional Cys substitutions, two in S4 and four in the pore helix, were chosen to monitor the membrane insertion of these regions more closely. As shown in Figure 4B, all the loop Cys showed high reactivity with PEG-mal indicating solvent accessibility. Loops sites predicted to be on either side of the lipid vesicle reacted similarly indicating that the PEG-mal reagent can readily permeate into the interior of the lipid vesicle as previously proposed.30 While the presence of KCl in the assay buffer precipitates most of the SDS (which is used to keep the unfolded KvAP in solution), we anticipate that a small fraction of the SDS is not precipitated and incorporates into lipid vesicles. The incorporation of SDS into the lipid bilayers makes the vesicles leaky and allows permeability of PEG-mal into the vesicles thereby accounting for the ability of the PEG-mal reagent to react with Cys on either side of the vesicle. In contrast to the Cys residues in the loop regions, the transmembrane Cys showed no or very low reactivity indicating that the transmembrane helices are incorporated into the lipid bilayer. Both the sites in S4, 122C in the middle of the S4 segment and 127C predicted to be deeper within the membrane showed no accessibility to the PEG-mal reagent in the presence of lipid vesicles indicating membrane insertion of the S4 segment. For the pore helix Cys, we saw a gradient of reactivity, with decreased reactivity as the sites moved down the pore helix, into the membrane (Figure 4B). Data on the modification of the pore helix Cys residues is presented in Supplementary figure 1. The pattern of reactivity observed is the expected result if the pore helix is inserted into the membrane as the side chain becomes increasingly shielded from the solvent as it moves towards the center of the bilayer. The pore helix is therefore also inserted into the bilayer as observed in the native structure of the KvAP channel pore. Similar results have been previously reported for the biogenesis of the pore of the Kv1.3 channel.43, 44
Figure 4. Membrane insertion of KvAP during folding.

(A) Topology map of the KvAP channel showing the location of the Cys substitutions used for the PEGylation assays. (B) Bar graph showing the PEGylation results for the Cys substitutions. Sites colored in gold are transmembrane, blue are in loops outside of the membrane and red are sites in the pore helix. Error bars are standard deviations from n=3 experiments using at least 2 independent protein preparations. Some error bars are zero because the measured reactivity was zero.
Thiol maleimide reactions are fast; initial rates measured using a test maleimide (mol. wt. ~500) and a thiol containing peptide have been measured with an average of 734 M−1s−1 at pH 7.4.45 Reactions using the larger PEG-mal are expected to be slower. However, we use an excess of the PEG-mal reagent in the assay and therefore we expect the Cys residues to be rapidly labelled. The lack of labelling of the Cys residues in the transmembrane regions during in vitro folding indicates that the KvAP polypeptide is rapidly inserted into the lipid bilayer while the pattern of labeling indicates that the KvAP polypeptide is inserted with a native-like topology. Further, the PEG-mal assay is carried out at 20 °C at which tetramer formation is very slow, which suggests that the correct membrane topology of the KvAP channel during in vitro folding, in particular the topology of the pore helix, is not dependent on tetramer formation, similar to the results previously reported for the Kv1.3 channel.43 Our data indicates that the formation of the KvAP channel tetramers takes place through the association of subunits in the lipid bilayer with the native topology.
The CTD is required for efficient in vitro folding of the KvAP channel
A key step in the folding of a Kv channel is the formation of the channel tetramer. In the Shaker Kv channel family, this process of tetramerization is facilitated by an N-terminal domain referred to as the tetramerization (or T1) domain while in other members of the voltage gated channel superfamily, tetramerization is driven by a coiled coil region present at the C-terminus. 8, 25, 46–49 Influenced by these precedents, we investigated whether the extracellular domains of the KvAP channel play a role in channel tetramerization. We focused on the C-terminal domain (CTD), which is 44 amino acid long and was not visualized in the crystal structure (Figure 5A). A protein BLAST optimized for short sequences did not yield any hits indicating that the sequence of the KvAP CTD is unique (Supplementary figure 2).50 Based on the sequence, the CTD is predicted to be α-helical but does not have a propensity to form a coiled coil as previously observed for the C-terminal association domains seen in certain members of the voltage gated channel superfamily.46–49
Figure 5. Deletion of the CTD abolishes tetramerization of KvAP in POPC lipids.

(A) The C-terminal sequences of the KvAP channel constructs used are shown. All constructs contain the wild type KvAP channel residues 1 – 238. The S6 transmembrane segment in the KvAP channel ends at residue 237. The Lys residues introduced to enhance glutaraldehyde crosslinking are indicated in red. (B) Time course of in vitro folding of the KvAPΔCTD+ Lys channel and the full length control in POPC lipid vesicles at 50 °C. Shown to the right are schematic illustrations of the constructs used. Data for the full-length KvAP channel is from Figure 2. Tetramerization kinetics were quantitated from n=3 experiments with at least 2 protein preparations. Error bars are standard deviations. Tetramerization kinetics for KvAPΔCTD+Lys could not be fitted with an exponential equation.
To test the influence of the CTD on channel tetramerization, we generated a KvAP channel construct with a deletion of residues 247–282 at the C-terminus (KvAP-ΔCTD, Figure 5A). The S6 transmembrane segment extends to Lys237 and the KvAP-ΔCTD construct therefore consists of only nine residues at the C-terminus compared to the native channel. We observed that KvAP-ΔCTD on native expression in E. coli gave lower yields and the protein migrated on size exclusion chromatography (SEC) as a mixture of a tetramer and monomeric/dimeric species (Supplementary figure 3). The lower protein yields and incomplete tetramer formation compared to the full–length KvAP suggests a role for the CTD in tetramer assembly in the cell. Next, we investigated if the CTD has an effect on the in vitro folding of the KvAP channel. A challenge in evaluating the in vitro folding of the ΔCTD channel was that the KvAP-ΔCTD tetramer did not cross-link efficiently with glutaraldehyde (Supplementary figure 3). We speculated that the inefficient cross-linking of the KvAP-ΔCTD was because it lacked three Lys residues compared to the full length channel (Figure 5A). In an attempt to increase the cross-linking efficiency, we introduced additional Lys residues to generate a KvAP-ΔCTD+Lys construct (Figure 5A). Crosslinking of the KvAP-ΔCTD+Lys tetramer with glutaraldehyde, following purification by SEC, was close to 100% (Supplementary Figure 3). As a control, we introduced Lys residues at equivalent positions into full length KvAP and observed that crosslinking of the KvAP+ Lys was similar to the KvAP C247S protein (Supplementary figure 3). Importantly, the kinetics of tetramerization of the KvAP+Lys was similar to the KvAP C247S protein indicating that the introduction of the Lys residues did not alter this process (Supplementary figure 3).
With the KvAP-ΔCTD+Lys construct, we evaluated the effect of deletion of the CTD on in vitro folding. We found that the extent of tetramer formation in POPC LUVs at 50 °C was very low (about 4% tetramer at 2 hours) compared to 75% for the full length protein (Figure 5B). When we used Aso lipids instead of POPC, we observed that tetramer assembly for the KvAP-ΔCTD+Lys did take place though only to approximately half the yield observed for the full length protein (Supplementary Figure 4). Together these results indicate that the CTD increases the efficiency of the tetramerization process and that the effect of the CTD on the tetramerization, is dependent on the lipid environment used for the in vitro folding reaction.
The CTD is dimeric in solution
A potential role for the CTD in the folding process could be to act as a tetrameric association domain. To evaluate this possibility, we investigated the oligomeric state of the CTD in solution. For this purpose, we fused the CTD (residues L239-K282 of KvAP) with SUMO (Small Ubiquitin-like Modifier protein SMT3 from yeast) to obtain a fusion protein with a convenient size for SDS-PAGE and for high level expression in Escherichia coli (Figure 6A, Supplementary Table 1).51 We substituted the Cys residue in the sequence (C247) with serine to prevent complications due to disulfide bond formation. As a control, we generated a SUMO fusion to a variant of the GCN4 peptide that forms a tetrameric coiled coil and is of a similar size to the CTD (Figure 6A, Supplementary Table 1).34 We observed through SEC and crosslinking of the purified fusion proteins that the SUMO-CTD fusion was dimeric in solution while the control SUMO-GCN4 was tetrameric and the SUMO protein was monomeric (Figure 6B, C). We tested a SUMO CTD fusion with the native C247 residue and observed that it readily forms a disulfide cross-linked dimer (Supplementary figure 5). These results indicate that the CTD peptides are dimeric in solution and associate through the formation of a parallel dimer.
Figure 6. The CTD of KvAP is a dimer.

(A) Schematic illustration of the SUMO-GCN4 and SUMO-KvAP CTD constructs used. (B) SEC of the SUMO-KvAP-CTD, SUMO-GCN4 and SUMO proteins show that the SUMO- KvAP-CTD is a dimer, SUMO-GCN4 is a tetramer while the SUMO protein migrates as a monomer. (C) SDS-PAGE gel showing the glutaraldehyde cross-linking of the SUMO- KvAP-CTD and SUMO-GCN4 proteins confirms that the SUMO- KvAP-CTD is a dimer while SUMO-GCN4 is a tetramer.
The native KvAP channel purifies with the subunits linked by a C247 disulfide bond. Disulfide bond formation indicates that the CTD of the neighboring subunits are in close proximity in the native channel. The C247 disulfide bond is however not functionally important as deletion of the disulfide bond by a C247S substitution does not alter channel assembly or function.30, 52
The isolated CTD inhibits KvAP tetramerization
Next, we investigated whether CTD dimerization is an important step in the folding of the KvAP channel. For this purpose, we devised a competition assay (Figure 7A). In this assay we tested whether the presence of excess CTD can inhibit the tetramerization of the KvAP channel. The CTD is highly water soluble while the folding of the KvAP channel takes place in lipid bilayers and therefore to localize the CTD to the bilayer surface, we used the S6-CTD peptide (residues 191–282, Table 1). To test for inhibition, we carried out in vitro folding of the KvAP channel in the presence of a five-fold molar excess of the purified S6-CTD peptide. After folding for 90 mins at 50 °C, the extent of folding was determined by glutaraldehyde crosslinking (Figure 7B). We observed that presence of the S6+CTD peptide resulted in a 60% decrease in the tetramer yield (Figure 7C). As a control, we also tested the S6 peptide (residues 191–246, Table 1) without the CTD and did not see an inhibition of tetramer formation.
Figure 7. The CTD inhibits in vitro folding of KvAP.

(A) The reaction scheme used in the competition assay is shown. The competitors used are the S6-CTD or the S6 peptide. The schematic representation of the proteins used in the assay are shown. (B) SDS-PAGE gel showing the glutaraldehyde crosslinking of the KvAP folding reaction carried out in the presence of the S6+CTD or the S6 peptide. The identity of the protein bands observed on glutaraldehyde crosslinking are indicated. The protein bands indicated by the asterisk potentially corresponds to a KvAP+S6-CTD dimer and a S6-CTD tetramer based on their molecular weights. Control lanes where the competitor was added after completion of folding are marked with a C. A five-fold molar ratio of competitor to unfolded KvAP is used in all lanes. Monomers of S6-CTD and the S6 peptide are not visible on this gel because of their low molecular weight. A decrease in the density of the KvAP tetramer band is seen in the case of folding in the presence of S6-CTD but not in the presence of the S6 peptide. (C) Inhibition of the in vitro folding reaction. Bar graph showing the % of tetramer inhibition observed in the presence of the S6+CTD and the S6 peptide. Error bars correspond to the standard deviation for n = 3 from at least 2 protein preparations.
As the unfolded KvAP polypeptide has a strong tendency to aggregate, the glutaraldehyde crosslinking reaction to evaluate the in vitro folding of KvAP is carried out in the presence of Fos-12.30 The addition of Fos-12 dissociates the non-specific aggregates without affecting the native KvAP channel tetramers. However, Fos-12 dissociates the CTD dimers (Supplementary figure 6) and we expect a similar effect of Fos-12 on the S6-CTD dimer and the heterodimer of the S6-CTD with the KvAP polypeptide. Due to these effects of Fos-12, glutaraldehyde crosslinking was not able to efficiently trap these species and therefore, only the level of the KvAP tetramer was used for the analysis. The assay therefore indicates that the S6-CTD peptide can inhibit KvAP tetramer formation while the S6 peptide lacking the CTD cannot. As the CTD readily forms dimers, our data suggests that dimerization through the CTD is potentially a critical step in the assembly process of the KvAP tetramer.
Dimerization of the CTD is not temperature dependent
A key aspect of the KvAP folding reaction is that it takes place efficiently only at higher temperatures. During the folding reaction, membrane insertion of the KvAP polypeptide takes place rapidly at 20 °C and therefore not the origin of the temperature dependence. Our studies have shown that dimerization of the CTD is an important step in the in vitro folding of the KvAP and therefore we investigated if CTD dimerization is the temperature dependent step. We observed that the SUMO-CTD dimer can be dissociated by SDS and re-associated by dilution of the detergent. We tested the temperature dependence of the CTD dimerization by using this assay (Figure 8A). We observed that the dimerization of the CTD was not temperature dependent as the extent of dimerization observed on dilution of SDS was independent of temperature (Figure 8B,C). Importantly, the extent of dimerization observed at 20 °C, a temperature at which tetramerization was not observed, was similar to the extent observed at 80 °C at which tetramerization was efficient. We therefore conclude that the temperature dependence of the in vitro folding of KvAP does not originate from a requirement for the dimerization of the CTD.
Figure 8: Dimerization of the CTD is not dependent on temperature.

(A) The reaction scheme used for the CTD dimerization assay. The SUMO-CTD construct is used in this assay for easy visualization on an SDS-PAGE gel. (B) SDS-PAGE gel showing the extent of dimerization of SUMO-CTD as determined by glutaraldehyde crosslinking. The dimeric SUMO-CTD is dissociated by treatment with 0.35 % SDS, re-associated at the indicated temperature and the extent of re-association is determined by glutaraldehyde crosslinking. Glutaraldehyde crosslinking of the native SUMO-CTD before (−) and after treatment (+) with 0.35% SDS is also shown. Glutaraldehyde crosslinking of the monomeric and the dimeric SUMO-CTD proteins results in an additional band that is indicated by an asterisk. (C) The extent of SUMO-CTD dimerization at the different temperatures were quantified and plotted as a bar graph. The error bars are standard deviations from three experiments with two different protein preparations.
DISCUSSION
The folding of membrane proteins can be considered in two stages with insertion of the polypeptide into the lipid bilayer in stage I and folding within the lipid bilayer in stage II.20, 21 In certain cases, the folding can precede the insertion of the polypeptide into the lipid bilayer.22 For multimeric proteins, there is an additional stage of association of the subunits for the formation of the native multimeric protein that also has to be considered. Multimerization can take place in an independent step or in conjunction with the folding in stage II. In this study we investigate these stages in the in vitro folding of the KvAP channel. We show that elevated temperature is required for the in vitro folding of the KvAP channel, which is in keeping with its hyperthermophilic origin. We utilize the unique temperature dependence of the in vitro folding of the KvAP channel to separately study the membrane insertion and the tetramerization stages in the in vitro folding of the KvAP channel.
We use a Cys-PEGylation assay to probe the membrane insertion and our assays indicate that the KvAP polypeptide rapidly inserts into the lipid bilayer with the correct membrane topology. We specifically show that S4, despite having four positive charges, is able to insert into the membrane in the context of the whole protein. Previous studies have investigated the membrane insertion of S4 from the KvAP, Shaker, Kv1.3 and KAT1 channels by using a translocon-assisted insertion assay.23, 24, 53–55 These studies showed that the membrane insertion of the isolated S4 was unfavorable but that the unfavorable membrane insertion of S4 can be counteracted by the charges in S2 and S3. Therefore, a cooperative insertion of the S2, S3 and S4 helices into the membrane is anticipated during the cellular assembly of Kv channels23, 24, which is similar to our observations during the in vitro folding of the KvAP channel.
The Cys-PEGylation assays also indicate that the pore helix in the KvAP channel is inserted into the lipid bilayer with the correct native topology prior to the formation of the tetramer. The membrane insertion of the pore helix of the Kv1.3 channel has been investigated using a PEGylation assay and the data support the membrane insertion of the pore helix in a native-like conformation in the monomeric state.43, 44 The data presently available therefore suggest that the cellular assembly of Kv channels proceeds with the insertion of channel monomers in the correct topology and mirrors our findings on the in vitro folding of the KvAP channel.
We observe that the oligomerization of the KvAP polypeptide following membrane insertion is driven, mainly, by the CTD. Deletion of the CTD affects assembly of the KvAP channel tetramers, both during in vivo biogenesis as well as during in vitro folding. Our data show that the CTD forms dimers in solution indicating that the tetramerization of the KvAP channel likely proceeds by a dimer of dimers pathway. A critical role for dimerization through the CTD playing a role in the folding process is indicated by the ability of the S6-CTD peptide to act as a inhibitor for the in vitro folding reaction. There are, in theory, two pathways to form tetramers: through the sequential addition of monomers or by the dimerization of dimers. The dimerization of dimers pathway is expected to be the efficient pathway as the sequential addition pathway has more intermediate steps en route to the tetramer, which could be deleterious as the presence of multiple intermediate steps increase the possibility of protein mis-folding during assembly.56
Tetramerization during the cellular assembly of Shaker family Kv channels is driven by the N-terminal T1 domain.8, 25 A study of the cellular assembly Kv1.3 channel has been proposed to take place through a dimer of dimers pathway even though the T1 domain that drives tetramerization is tetrameric in solution (in the isolated state).57 In the case of the majority of members of the voltage gated ion channel superfamily, tetramerization is driven by a coiled coil like domain at the C-terminus.46–49 In the case of the Kv7.1 channel, the C-terminal coiled coil domain has a tetrameric structure but studies on assembly using the isolated C-terminal domains have suggested the domain to be a dimer that associates into a tetramer in a concentration dependent manner.9 These examples suggest that tetramer formation during the cellular assembly of Kv channels may also take place through a dimer of dimers pathway.
For the KvAP channel, it is not certain as to how the KvAP dimers form tetramers. One possibility is that the CTD forms higher order assemblies at higher concentrations. Alternately, the association of the KvAP dimers may take place through the membrane embedded domains. The tetramerization of KvAP can take place, although inefficiently, without the CTD, indicating that signals for oligomerization must exist within the membrane domains. The isolated VSD has been shown to dimerize in solution depending on the detergent indicating a region in the VSD that can potentially drive subunit association.58 Further experiments will be necessary to elucidate the steps involved in this final step of the KvAP folding process.
In this study, we use the temperature dependence to control the in vitro folding of the KvAP channel. The origin of the temperature dependence can be due to changes in the conformation of the KvAP polypeptide or due to changes in the properties of the lipid bilayer with an increase in temperature. While our studies indicate insertion of the KvAP polypeptide into the lipid bilayer during folding takes place with a native-like topology, it is feasible that there are temperature dependent conformational changes following insertion that are required for tetramerization. Approaches with higher resolution than the Cys modification assay used in this study will be necessary to determine if a such a mechanism is in play. Alternately, the temperature dependence of KvAP folding could arise from changes in the properties of the lipid bilayer. KvAP is derived from an hyperthermophilic archaeon27 but the in vitro folding studies were carried out using non-archaeal lipids. It is therefore possible that the temperature dependence of KvAP folding arises due to the use of non-native lipids. We cannot test Aeropyrum lipids in our in vitro folding assay as they are not commercially available. However, in a previous study we tested folding of KvAP in 1,2-diphytanoylglycero-3-phosphocholine (DPhPC), which is an “archaeal-like’ lipid.30 For DPhPC also we observed an enhancement in the folding of KvAP with temperature, similar to the effect observed with POPC or Aso vesicles, thereby indicating the origin of temperature dependence of folding is not simply related to the use of non-archaeal lipids. Properties of the lipid bilayer such as fluidity, thickness and the surface area of the lipid bilayer are expected to change with temperature59 and the origin of the temperature dependence could be linked to changes in these bilayer properties. The mechanism of the temperature dependence of the in vitro folding of KvAP will be explored in future studies.
The key finding of this study is the remarkable similarity in the in vitro folding of the KvAP channel with the cellular assembly of Kv channels. There are many questions of interest in the assembly of Kv channels with one of the prominent questions being the mechanism for the domain-swap that is observed between channel subunits in certain families of Kv channels. The in vitro folding of the KvAP channel provides a simplified experimental system to explore this process.
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported by grant R01GM087546 from the NIH to FIV.
REFERENCES
- [1].Hille B (2001) Ion Channels of Excitable Membranes, Sinauer; Sunderland, MA. [Google Scholar]
- [2].Kuo MM, Haynes WJ, Loukin SH, Kung C, and Saimi Y (2005) Prokaryotic K(+) channels: from crystal structures to diversity, FEMS Microbiol Rev 29, 961–985. [DOI] [PubMed] [Google Scholar]
- [3].Long SB, Tao X, Campbell EB, and MacKinnon R (2007) Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment, Nature 450, 376–382. [DOI] [PubMed] [Google Scholar]
- [4].Kim DM, and Nimigean CM (2016) Voltage-Gated Potassium Channels: A Structural Examination of Selectivity and Gating, Cold Spring Harb Perspect Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pongs O, and Schwarz JR (2010) Ancillary subunits associated with voltage-dependent K+ channels, Physiol Rev 90, 755–796. [DOI] [PubMed] [Google Scholar]
- [6].Haitin Y, and Attali B (2008) The C-terminus of Kv7 channels: a multifunctional module, J Physiol 586, 1803–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kamnesky G, Hirschhorn O, Shaked H, Chen J, Yao L, and Chill JH (2014) Molecular determinants of tetramerization in the KcsA cytoplasmic domain, Protein Sci 23, 1403–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shen NV, Chen X, Boyer MM, and Pfaffinger PJ (1993) Deletion analysis of K+ channel assembly, Neuron 11, 67–76. [DOI] [PubMed] [Google Scholar]
- [9].Wiener R, Haitin Y, Shamgar L, Fernandez-Alonso MC, Martos A, Chomsky-Hecht O, Rivas G, Attali B, and Hirsch JA (2008) The KCNQ1 (Kv7.1) COOH terminus, a multitiered scaffold for subunit assembly and protein interaction, J Biol Chem 283, 5815–5830. [DOI] [PubMed] [Google Scholar]
- [10].Zerangue N, Jan YN, and Jan LY (2000) An artificial tetramerization domain restores efficient assembly of functional Shaker channels lacking T1, Proc Natl Acad Sci U S A 97, 3591–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dworakowska B, and Dolowy K (2000) Ion channels-related diseases, Acta Biochim Pol 47, 685–703. [PubMed] [Google Scholar]
- [12].Anderson CL, Kuzmicki CE, Childs RR, Hintz CJ, Delisle BP, and January CT (2014) Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome, Nat Commun 5, 5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Curran J, and Mohler PJ (2015) Alternative paradigms for ion channelopathies: disorders of ion channel membrane trafficking and posttranslational modification, Annu Rev Physiol 77, 505–524. [DOI] [PubMed] [Google Scholar]
- [14].Delisle BP, Anson BD, Rajamani S, and January CT (2004) Biology of cardiac arrhythmias: ion channel protein trafficking, Circ Res 94, 1418–1428. [DOI] [PubMed] [Google Scholar]
- [15].Smith JL, Anderson CL, Burgess DE, Elayi CS, January CT, and Delisle BP (2016) Molecular pathogenesis of long QT syndrome type 2, J Arrhythm 32, 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Smith JL, Reloj AR, Nataraj PS, Bartos DC, Schroder EA, Moss AJ, Ohno S, Horie M, Anderson CL, January CT, and Delisle BP (2013) Pharmacological correction of long QT-linked mutations in KCNH2 (hERG) increases the trafficking of Kv11.1 channels stored in the transitional endoplasmic reticulum, Am J Physiol Cell Physiol 305, C919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Young JC (2014) The role of the cytosolic HSP70 chaperone system in diseases caused by misfolding and aberrant trafficking of ion channels, Dis Model Mech 7, 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Papazian DM (1999) Potassium channels: some assembly required, Neuron 23, 7–10. [DOI] [PubMed] [Google Scholar]
- [19].Popot JL, Gerchman SE, and Engelman DM (1987) Refolding of bacteriorhodopsin in lipid bilayers. A thermodynamically controlled two-stage process, J Mol Biol 198, 655–676. [DOI] [PubMed] [Google Scholar]
- [20].Popot JL, and Engelman DM (2016) Membranes Do Not Tell Proteins How To Fold, Biochemistry 55, 5–18. [DOI] [PubMed] [Google Scholar]
- [21].Popot JL, and Engelman DM (1990) Membrane protein folding and oligomerization: the two-stage model, Biochemistry 29, 4031–4037. [DOI] [PubMed] [Google Scholar]
- [22].Cymer F, von Heijne G, and White SH (2015) Mechanisms of integral membrane protein insertion and folding, J Mol Biol 427, 999–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang L, Sato Y, Hessa T, von Heijne G, Lee JK, Kodama I, Sakaguchi M, and Uozumi N (2007) Contribution of hydrophobic and electrostatic interactions to the membrane integration of the Shaker K+ channel voltage sensor domain, Proc Natl Acad Sci U S A 104, 8263–8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tu L, Wang J, Helm A, Skach WR, and Deutsch C (2000) Transmembrane biogenesis of Kv1.3, Biochemistry 39, 824–836. [DOI] [PubMed] [Google Scholar]
- [25].Li M, Jan YN, and Jan LY (1992) Specification of subunit assembly by the hydrophilic amino-terminal domain of the Shaker potassium channel, Science 257, 1225–1230. [DOI] [PubMed] [Google Scholar]
- [26].Rosenberg RL, and East JE (1992) Cell-free expression of functional Shaker potassium channels, Nature 360, 166–169. [DOI] [PubMed] [Google Scholar]
- [27].Ruta V, Jiang Y, Lee A, Chen J, and MacKinnon R (2003) Functional analysis of an archaebacterial voltage-dependent K+ channel, Nature 422, 180–185. [DOI] [PubMed] [Google Scholar]
- [28].Schmidt D, Cross SR, and MacKinnon R (2009) A gating model for the archeal voltage-dependent K(+) channel KvAP in DPhPC and POPE:POPG decane lipid bilayers, J Mol Biol 390, 902–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, and MacKinnon R (2003) X-ray structure of a voltage-dependent K+ channel, Nature 423, 33–41. [DOI] [PubMed] [Google Scholar]
- [30].Devaraneni PK, Devereaux JJ, and Valiyaveetil FI (2011) In vitro folding of KvAP, a voltage-gated K+ channel, Biochemistry 50, 10442–10450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lee SY, Lee A, Chen J, and MacKinnon R (2005) Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane, Proc Natl Acad Sci U S A 102, 15441–15446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sambrook J, Fritsch EF, and Maniatis T (1989) Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- [33].Komarov AG, Linn KM, Devereaux JJ, and Valiyaveetil FI (2009) Modular strategy for the semisynthesis of a K+ channel: investigating interactions of the pore helix, ACS Chem Biol 4, 1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Harbury PB, Zhang T, Kim PS, and Alber T (1993) A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants, Science 262, 1401–1407. [DOI] [PubMed] [Google Scholar]
- [35].Sivashanmugam A, Murray V, Cui C, Zhang Y, Wang J, and Li Q (2009) Practical protocols for production of very high yields of recombinant proteins using Escherichia coli, Protein Sci 18, 936–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lu J, and Deutsch C (2001) Pegylation: a method for assessing topological accessibilities in Kv1.3, Biochemistry 40, 13288–13301. [DOI] [PubMed] [Google Scholar]
- [37].Devaraneni PK, Conti B, Matsumura Y, Yang Z, Johnson AE, and Skach WR (2011) Stepwise insertion and inversion of a type II signal anchor sequence in the ribosome-Sec61 translocon complex, Cell 146, 134–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hildebrandt ER, Davis DM, Deaton J, Krishnankutty RK, Lilla E, and Schmidt WK (2013) Topology of the yeast Ras converting enzyme as inferred from cysteine accessibility studies, Biochemistry 52, 6601–6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Howe V, and Brown AJ (2017) Determining the Topology of Membrane-Bound Proteins Using PEGylation, Methods Mol Biol 1583, 201–210. [DOI] [PubMed] [Google Scholar]
- [40].Howe V, Chua NK, Stevenson J, and Brown AJ (2015) The Regulatory Domain of Squalene Monooxygenase Contains a Re-entrant Loop and Senses Cholesterol via a Conformational Change, J Biol Chem 290, 27533–27544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pathania A, Gupta AK, Dubey S, Gopal B, and Sardesai AA (2016) The Topology of the l-Arginine Exporter ArgO Conforms to an Nin-Cout Configuration in Escherichia coli: Requirement for the Cytoplasmic N-Terminal Domain, Functional Helical Interactions, and an Aspartate Pair for ArgO Function, J Bacteriol 198, 3186–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang Y, Toei M, and Forgac M (2008) Analysis of the membrane topology of transmembrane segments in the C-terminal hydrophobic domain of the yeast vacuolar ATPase subunit a (Vph1p) by chemical modification, J Biol Chem 283, 20696–20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gajewski C, Dagcan A, Roux B, and Deutsch C (2011) Biogenesis of the pore architecture of a voltage-gated potassium channel, Proc Natl Acad Sci U S A 108, 3240–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Delaney E, Khanna P, Tu L, Robinson JM, and Deutsch C (2014) Determinants of pore folding in potassium channel biogenesis, Proc Natl Acad Sci U S A 111, 4620–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Saito F, Noda H, and Bode JW (2015) Critical evaluation and rate constants of chemoselective ligation reactions for stoichiometric conjugations in water, ACS Chem Biol 10, 1026–1033. [DOI] [PubMed] [Google Scholar]
- [46].Howard RJ, Clark KA, Holton JM, and Minor DL Jr. (2007) Structural insight into KCNQ (Kv7) channel assembly and channelopathy, Neuron 53, 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ludwig J, Owen D, and Pongs O (1997) Carboxy-terminal domain mediates assembly of the voltage-gated rat ether-a-go-go potassium channel, EMBO J 16, 6337–6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Powl AM, O’Reilly AO, Miles AJ, and Wallace BA (2010) Synchrotron radiation circular dichroism spectroscopy-defined structure of the C-terminal domain of NaChBac and its role in channel assembly, Proc Natl Acad Sci U S A 107, 14064–14069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tsuruda PR, Julius D, and Minor DL Jr. (2006) Coiled coils direct assembly of a cold-activated TRP channel, Neuron 51, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990) Basic local alignment search tool, J Mol Biol 215, 403–410. [DOI] [PubMed] [Google Scholar]
- [51].Butt TR, Edavettal SC, Hall JP, and Mattern MR (2005) SUMO fusion technology for difficult-to-express proteins, Protein Expr Purif 43, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jiang Y, Ruta V, Chen J, Lee A, and MacKinnon R (2003) The principle of gating charge movement in a voltage-dependent K+ channel, Nature 423, 42–48. [DOI] [PubMed] [Google Scholar]
- [53].Sato Y, Sakaguchi M, Goshima S, Nakamura T, and Uozumi N (2003) Molecular dissection of the contribution of negatively and positively charged residues in S2, S3, and S4 to the final membrane topology of the voltage sensor in the K+ channel, KAT1, J Biol Chem 278, 13227–13234. [DOI] [PubMed] [Google Scholar]
- [54].Mishima E, Sato Y, Nanatani K, Hoshi N, Lee JK, Schiller N, von Heijne G, Sakaguchi M, and Uozumi N (2016) The topogenic function of S4 promotes membrane insertion of the voltage-sensor domain in the KvAP channel, Biochem J 473, 4361–4372. [DOI] [PubMed] [Google Scholar]
- [55].Hessa T, White SH, and von Heijne G (2005) Membrane insertion of a potassium-channel voltage sensor, Science 307, 1427. [DOI] [PubMed] [Google Scholar]
- [56].Powers ET, and Powers DL (2003) A perspective on mechanisms of protein tetramer formation, Biophys J 85, 3587–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tu L, and Deutsch C (1999) Evidence for dimerization of dimers in K+ channel assembly, Biophys J 76, 2004–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chakrapani S, Cuello LG, Cortes DM, and Perozo E (2008) Structural dynamics of an isolated voltage-sensor domain in a lipid bilayer, Structure 16, 398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kucerka N, Nieh MP, and Katsaras J (2011) Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature, Biochim Biophys Acta 1808, 2761–2771. [DOI] [PubMed] [Google Scholar]
- [60].Humphrey W, Dalke A, and Schulten K (1996) VMD: visual molecular dynamics, J Mol Graph 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.