

Abstract
Hereditary demyelinating neuropathies linked to peripheral myelin protein 22 (PMP22) involve the disruption of normal protein trafficking and are therefore relevant targets for chaperone therapy. Using a small molecule HSP90 inhibitor, EC137, in cell culture models, we previously validated the chaperone pathway as a viable target for therapy development. Here, we tested five commercially available inhibitors of HSP90 and identified BIIB021 and AUY922 to support Schwann cell viability and enhance chaperone expression. AUY922 showed higher efficacy, compared to BIIB021, in enhancing myelin synthesis in dorsal root ganglion explant cultures from neuropathic mice. For in vivo testing, we randomly assigned 2–3 month old C22 and 6 week old Trembler J (TrJ) mice to receive two weekly injections of either vehicle or AUY922 (2 mg/kg). By the intraperitoneal (i.p.) route, the drug was well-tolerated by all mice over the 5 month long study, without influence on body weight or general grooming behavior. AUY922 improved the maintenance of myelinated nerves of both neuropathic models and attenuated the decline in rotarod performance and peak muscle force production in C22 mice. These studies highlight the significance of proteostasis in neuromuscular function and further validate the HSP90 pathway as a therapeutic target for hereditary neuropathies.
Keywords: Neuropathy, Charcot-Marie-Tooth disease, chaperones, neuromuscular disease, myelin, C22 mice
Introduction
The heat shock (HS) pathway represents a cellular stress response, which results in elevated expression of cytoprotective chaperones or heat shock proteins (HSPs). Activation of chaperones has been shown to reduce the aggregation of misfolded proteins and alleviate disease phenotypes in various neurodegenerative disease models.1−3 It has been proposed that an increase in the availability of functional HSPs aids in the folding and the disaggregation or enhanced degradation of misfolded proteins.4−6 The activation of the HS pathway can be achieved through inhibition of HSP90, which disrupts its interaction with Heat Shock Factor-1 (HSF-1) leading to transcriptional activation of the HS response.7 Although HSP90 inhibitors have been investigated primarily for their anticancer properties, when used within a defined concentration range, they can be beneficial in the treatment of protein misfolding disorders.8
Charcot-Marie-Tooth (CMT) diseases comprise a heterogeneous group of progressive hereditary peripheral neuropathies, most often associated with overproduction of peripheral myelin protein 22 (PMP22), an aggregation-prone Schwann cell protein.9,10 Transgenic C22 mice express additional copies of the wild type (Wt) human PMP22 and reproduce the phenotypic traits of the neuropathies, including demyelination of peripheral nerves, impaired locomotor performance, and age-associated disease progression.11−13 Mislocalization and aggregation of mutant PMP22 is a culprit in early onset, severe neuropathies, modeled by the Trembler J (TrJ) mouse carrying a spontaneous mutation in the Pmp22 gene.14 In a previous study, we showed that activation of the HS pathway using EC137, a synthetic HSP90 inhibitor, reduced the aggregation of PMP22 and improved myelination in neuron-glia explant cultures from C22 mice.15 In an in vivo study of neuropathic TrJ mice, an increase in chaperone expression through intermittent fasting supported maintenance of nerve myelin and locomotor performance.16 In accordance, enhancement of the stress response by life-long calorie restriction was beneficial for peripheral nerve integrity in aged rats.15,17 Recent in vitro work further supports the importance of heat shock protein 70 (HSP70) in preventing the aggregation of misfolded PMP22 and aiding in its degradation.18 In related studies, pharmacological activation of HSP70 was shown to reverse sensory deficits in diabetic mice19 and ameliorate nerve demyelination and motor deficits in an inducible neuropathic mouse model.20 Thus, a number of experimental scenarios indicate that chaperones are critical for myelin maintenance and peripheral nerve function.
In this study, we screened five commercially available HSP90 inhibitors and identified NVP-AUY922 (referred to as AUY922 or AUY from here on) as the most effective compound in improving myelination in explant cultures from neuropathic C22 mice. This positive response correlated with the robust induction of chaperones in Schwann cells, in a dose- and time-dependent manner. In vivo administration of AUY922 preserved myelinated peripheral nerves in both C22 and TrJ models and attenuated the decline in neuromuscular performance in neuropathic C22 mice.
Results
AUY922 and BIIB021 Are Nontoxic Inducers of the Chaperone Pathway in Schwann Cells
We tested five commercially available HSP90 inhibitors, including AT13387, AUY922, BIIB021, SNX5422, and STA9090, on the viability of rat Schwann cells using the MTS assay. After 24 h of exposure, geldanamycin (GA, 50 nM), a well-known inhibitor of HSP90, significantly decreased cellular viability compared to DMSO (Figure 1A), which is in agreement with previous studies.21 Among these five tested compounds, lower dosages (50 nM) of AT13387, BIIB021, and STA9090 were well-tolerated by Schwann cells, while the higher dosages (500 nM) significantly decreased cellular viability, compared to DMSO. Surprisingly, neither concentration of AUY922 affected cell viability, while SNX5422 was toxic at both concentrations and therefore was excluded from subsequent studies.
Figure 1.

Effects of HSP90 inhibitors on Schwann cells. (A) Cell viability after treatment (24 h) with DMSO, GA (50 nM), or the indicated five HSP90 inhibitors (50 and 500 nM) was calculated and graphed, as a percentage of DMSO (vehicle). (B) HSP70 mRNA levels were quantified after 24 h of treatment with the indicated compounds (100 nM). GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as an internal control. (A, B) GA (50 nM) served as a positive control. A.U.: arbitrary units. Graphs are plotted as means ± SEM; ***P < 0.001; **P < 0.01; *P < 0.05; n.s., nonsignificant; two-tailed unpaired Student’s t-test.
Next, we determined the efficiency of these compounds in inducing the chaperone pathway, by measuring HSP70 expression in nonmyelinating Schwann cells (Figure 1B).22 After 24 h of incubation, AUY922 and BIIB021, each at 100 nM, elicited similar HSP70 transcript levels as GA (50 nM), the positive control. In comparison, incubation of the cells with AT13387 and STA9090 lacked positive effects (Figure 1B). Because AUY922 and BIIB021 elicited minimal Schwann cell toxicity and effectively induced the HS response, we chose these two compounds for further testing.
To characterize the influence of the two selected compounds on chaperone levels, we performed dosage and time course studies (Figure 2). First, nonmyelinating Schwann cells were treated with 25, 50, or 100 nM of either BIIB021 or AUY922 for 24 h and then analyzed for levels of HSP70 and HSP27 (Figure 2A). Both compounds increased HSP70 levels in a dose-dependent fashion, showing peak expression at 100 nM. Although the levels of HSP27 did not change prominently with different doses of the test compounds, cells treated with even the lowest dose showed higher HSP27 expression, as compared to the DMSO controls. However, AUY922 was more effective in increasing the levels of HSP70 and HSP27, even at lower doses, as compared to BIIB021. This finding corresponds with the higher levels of HSP70 mRNA observed upon AUY922 treatment, as compared to BIIB021 (Figure 1B). Next, time course experiments were performed over 4–48 h incubation periods (Figure 2B). As shown, 100 nM BIIB021 or AUY922 increased HSP70 levels as early as 4 h, with the expression peaking between 16 and 24 h. To study the sustainability of the induction, Schwann cells were treated with either 100 nM AUY922 or BIIB021 for 4 h (treat), followed by wash out and media replacement without drugs (chase) (Figure 2C). Cells exposed to BIIB021 or AUY922 for 4 h maintained elevated chaperone expression for at least 48 h, compared to the DMSO-treated controls. These results indicate that the exposure of Schwann cells to low concentrations of AUY922 or BIIB021 elicits robust and sustained chaperone induction, without significant cellular toxicity.
Figure 2.

Treatment with BIIB021 and AUY922 increases chaperone expression in a dose- and time-dependent manner. (A) Steady-state levels of HSP70 and HSP27 in whole Schwann cell lysates (15 μg/lane) were analyzed after 24 h of treatment with DMSO, BIIB021, or AUY922, at the specified doses. GA (50 nM) served as a positive control. (B) HSP70 and HSP27 levels were observed after treatment with 100 nM BIIB021 or AUY922 for the indicated times. (C) Chaperone pathway activation by BIIB021 or AUY922 (100 nM) was studied after 2 or 4 h (treatment), followed by 4, 24, 32, and 48 h chase time points. (A, B) GAPDH and (C) tubulin served as protein loading controls. Molecular mass on left, in kDa. Data shown are representative of n = 3 independent experiments.
Improved Myelin Production upon Chaperone Induction in Explant Cultures from Neuropathic Mice
The effects of AUY922 and BIIB021 on the myelination capacity of peripheral glia were assessed in dorsal root ganglion (DRG) explant cultures from wild type (Wt) and neuropathic C22 mice.15 DRG explant cultures were treated with either vehicle (Veh, DMSO), AUY922 (A, 100 nM), or BIIB021 (B, 100 nM) for 2 weeks, followed by analyses for chaperone expression and myelin formation (Figure 3). As shown (Figure 3A), the levels of HSP70 are elevated in HSP90 inhibitor-treated (A and B) Wt and C22 cultures, as compared to vehicle (Veh) controls. Within the same protein lysates, myelin production was evaluated through the expression levels of myelin protein zero (P0), which constitutes the majority of peripheral myelin proteins (Figure 3A). Both AUY922- and BIIB021-treated cultures from neuropathic mice show elevated levels of P0 compared to the vehicle control, and this effect was consistent across independent culture preparations. To assess the potential contribution of DRG neurons to the increase in chaperones, we depleted Wt explants of Schwann cells by antimitotic FUdR treatment15 (SC-depleted, Figure 3B). The chaperone response of explant cultures to AUY922 (the more potent HSP90 inhibitor) without Schwann cells is muted, indicating that the increase in HSP70 expression is predominantly from the glial cells.
Figure 3.

Improved myelin production in DRG explant cultures from C22 mice after treatment with AUY922. (A) Steady-state levels of HSP70 and P0 were analyzed in vehicle (Veh)-, AUY922-(A), or BIIB021 (B)-treated explant lysates (35 μg/lane). (B) Wt DRG cultures, with (Neuron + Schwann cell) and without (depleted) Schwann cells, were treated with 100 nM AUY922 and analyzed for the indicated chaperones. (A, B) Tubulin served as a protein loading control. Molecular mass on left, in kDa. MBP-positive myelin internode lengths in explant cultures from (C) Wt and (D) C22 mice treated with vehicle, AUY922, or BIIB021 were measured (n = 100–120 segments per group) and graphed as whisker plots with median (center line), quartiles (box), and extremes (whiskers); ***P < 0.001; *P < 0.05; n.s., nonsignificant; two-tailed unpaired Student’s t-test. (E) Cultures from Wt (top panel) and C22 (lower panel) mice, treated with the indicated compounds were stained for MBP (green). Nuclei were visualized with Hoechst dye (blue). The scale bar is as shown. Data shown are representative of n = 3–4 independent experiments.
We complemented the biochemical studies on myelin production with the direct evaluation of myelin basic protein (MBP)-positive internode segments.15 Measurement and quantification of MBP-positive myelin segments in the explant cultures revealed significant increases in internode lengths in AUY922-treated Wt and C22 cultures, while the influence of BIIB021 did not reach significance in cultures from neuropathic mice (Figure 3C,D). Representative micrographs from each treatment paradigm are shown and support the positive impact of the two tested compounds on myelin formation (Figure 3E), with AUY922 being more efficacious.
AUY922 Supports Neuromuscular Performance in C22 Mice
To test the effects of AUY922 on peripheral myelin and the motor performance of neuropathic mice, Wt and C22 littermates were randomly segregated at 7 weeks of age into vehicle and AUY922 treatment cohorts. Animals were injected via the peritoneum with 2 mg/kg AUY922 twice a week, for 20 weeks. Note that this chosen dosing regimen is significantly distinct from the short-term, daily, 50 mg/kg treatment paradigm used for tumor reduction in athymic mice.23 As shown (Figure 4A), the body weight gain of the animals treated with the drug is similar to that of those injected with vehicle over the period of the study, implying no adverse effects of the drug on the overall health of the mice. Effects of AUY922 treatment on the motor performance of Wt and C22 mice were assessed on the accelerating rotarod at the beginning of the study and monthly thereafter. Since biological sex does not affect rotarod performance,24,25 values for male and female mice were combined. At baseline (7 weeks of age), there is a significant difference in the ability of Wt and C22 mice to stay on the rotating rod, and this difference becomes more pronounced at the end of the study, when the vehicle-treated groups are compared (Figure 4B,C). This is in agreement with the progressive nature of this disease in the C22 model.13,26 At baseline, the vehicle and AUY922 treatment groups of C22 mice do not differ in their latencies to fall (Figure 4B); however, at the end of the study the AUY922-treated C22 mice perform significantly better than the vehicle-treated group (Figure 4C). An ANOVA test on latency for the rotating rod was conducted for baseline and for 2, 6, 10, 14, and 20 weeks of treatment (Figure 4D). Genotype differences were observed for 2, 6, and 10 weeks of treatment. There was a tendency (P = 0.072) for a genotype difference at week 14, and the main effects of genotype [F(1, 21) = 9.74, P, 0.01] and treatment [F(1, 21) = 8.49, P, 0.01] were observed for week 20 (Figure 4D). Post hoc tests indicate that the performance of C22 AUY922 mice was not different from that of Wt vehicle-treated mice for weeks 14 and 20 (Figure 4C,D). Furthermore, post hoc tests examining treatment effects in each genotype indicated that the effect was restricted to C22 mice. Finally, a repeated measures ANOVA test between baseline and week 20 within each genotype and treatment group indicated that the C22 vehicle-treated mice showed a decreased performance [F(1, 5) = 7.39, P, 0.05] (Figure 4D).
Figure 4.
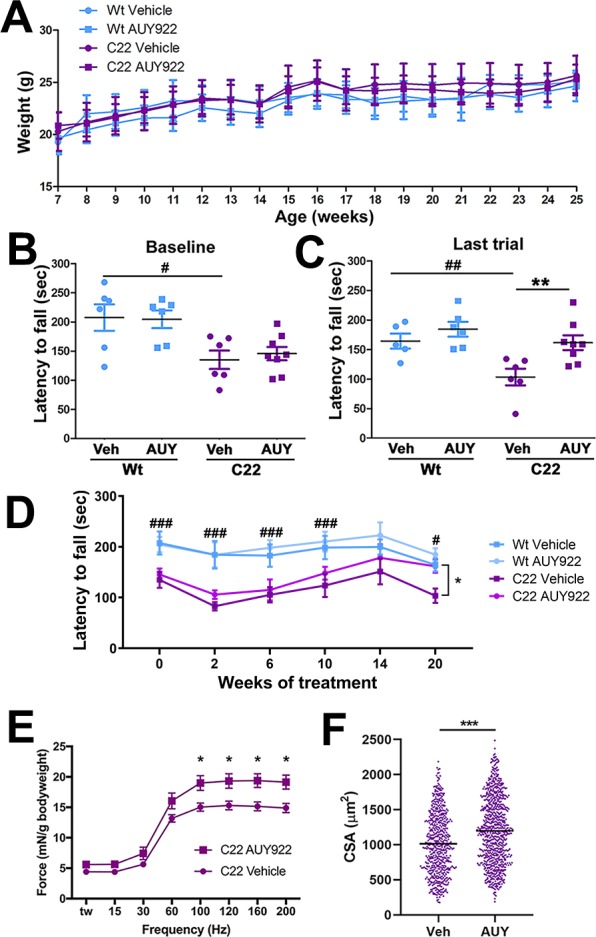
Treatment with AUY922 improves neuromuscular performance of C22 mice. (A) Mean ± SEM of the body weight of Wt and C22 mice (n = 6–8 mice per group) was plotted over the treatment period. Performances of individual animals on the accelerating rotarod (B) at baseline (7 weeks age) and (C) at the end of the treatment (25 weeks age) are shown. The bars represent the mean for each group. (D) Mean ± SEM of the rotarod performance of all groups, plotted over the treatment period. (E) Muscle force, analyzed using an in situ technique, was recorded and normalized to the animal’s body weight (mN/g = millinewtons/grams). Each point represents the mean ± SEM force. (F) Distribution of individual measures of the cross-sectional area of TA muscle from C22 mice treated with vehicle or AUY (n = 3–5 mice per group). The bars represent the mean for each group. For all graphs, # indicates a significant (#P < 0.05, ##P < 0.01, ###P < 0.001) genotype difference. *P < 0.05, **P < 0.01, and ***P < 0.001 indicate a significant treatment effect for C22 mice.
Next, to examine the effects of AUY922 on skeletal muscle strength, we performed in situ force-frequency contractile analysis on the tibialis anterior (TA) muscle.27−29 The absolute maximal tetanic force generated by the TA after sequential single stimulations of the common peroneal nerve was measured and normalized to the body weight of the animal (Figure 4E). We found a significant (∼28%) increase in force generation in C22 animals treated with AUY922, as compared to the vehicle group. In addition, an assessment of myofiber cross-sectional area within the same set of TA muscles revealed a significant improvement, or maintenance, of tissue integrity when compared to the vehicle group (Figure 4F).
For the analysis of the bioavailability of AUY922, blood and liver tissues were collected at the end of the study. While all samples were acquired within a 4 h time window of the final injection, AUY922 concentration varied in sera and liver: from 2.78 to 226 ng/mL in the sera and 101 to 304 ng/g in the liver. Overall, these results indicate that the AUY922 treatment paradigm is well-tolerated by neuropathic C22 mice and results in the attenuation of declining neuromuscular performance and myofiber atrophy.
AUY922 Treatment Maintains Myelinated Axons in C22 Neuropathic Mice
Histopathological defects in peripheral nerves of C22 mice include repeated demyelination and remyelination of medium to large axons with onion bulbs and signs of acute myelin breakdown, with macrophage infiltration11.30 Cross-sectional analyses of sciatic nerves from the C22 vehicle group revealed these characteristics, when compared to the Wt vehicle group (Figure 5A). However, in our cohort of mice, the occurrences of these disease-associated pathological features are notably reduced compared with the original publication by the Huxley lab.11 Since the severity of the C22 phenotype depends on the copy number of the transgene,26 we compared archived tissue from ∼2007 to tissue from mice used in this study. We found no change in transgene copy number by qPCR. We further analyzed the genetic background and determined that historically the mice were on a mixed C57BL/6J and C57BL/6N background, but because of our maintenance breeding scheme, they are now on a predominantly CBA background. This shift in genetic background likely accounts for changes in severity from previous reports. Since all the studies described here use contemporary littermate controls, this should not affect the interpretation of the results. Indeed, we found that the nerves from AUY922-treated C22 mice contained distinct, well-myelinated axonal profiles, with a rare occurrence of degenerating fibers and onion bulbs, compared with the vehicle group (Figure 5A). We found no apparent differences in the histology of nerves from Wt mice treated with either vehicle or AUY922.
Figure 5.

AUY922 administration supports the maintenance of myelinated axons in sciatic nerves of C22 mice. (A) Cross-sectional views of nerve sections from Wt (top panels) and C22 (lower panels) male mice. Micron bar, 45 μm. (B) The cross-sectional area occupied by nerve fibers in a 40 μm × 40 μm square (n = 20–25 fibers per animal; n = 6–8 mice per group) was measured and graphed as shown. Graph plotted as means ± SEM; ***P < 0.001, across the treatment groups; #P < 0.05, across the genotypes; two-tailed unpaired Student’s t-test. Correlative analyses between axon and fiber diameter measurements were obtained from sciatic nerve cross-sectional areas from (C) Wt and (D) C22 groups. (E) Comparison of trendlines between the cohorts in parts C and D. Scatter plots comparing the g-ratios (axon diameter/fiber diameter) of individual fibers plotted as a function of axon diameters in nerves of (F) Wt and (G) C22 animals. (C–H) n = 950–1100 fibers per group. (H) Trendline comparisons of graphs in parts F and G.
We corroborated the microscopic observations with morphometric analyses of randomly selected cross-sectional nerve areas from independent mice (Figure 5B–H). The total area occupied by nerve fibers within a fixed size square is significantly increased in AUY922-treated C22 animals as compared to the vehicle group (Figure 5B). Upon comparison of axon and fiber diameters in Wt groups (Figure 5C–E), we found no deviation in the overall values (coefficient of correlation, r2 = 0.96, in both vehicle and AUY922 groups). However, in nerves from the C22 neuropathic animals, we found a delineation between the vehicle (r2 = 0.94) and AUY922 (r2 = 0.96) groups. A similar pattern is obtained when the g-ratios (axon/fiber diameter) were analyzed as a function of axon diameter (Figure 6F–H) in nerves of C22 animals (r2 = 0.03 in vehicle versus r2 = 0.23 in AUY922). This is in contrast to the trend observed in vehicle- (r2 = 0.29) and AUY922-treated (r2 = 0.28) Wt samples. A correlative analysis suggests that an increase in fiber diameter in vehicle-injected C22 animals is not accompanied by a proportional increase in axon diameter, and this is reflected in the altered g-ratio values. However, this signature is rectified with AUY922 treatment where the patterns are comparable to the Wt cohorts. Together, these results indicate that biweekly injection of AUY922 supports the maintenance of myelinated axons in C22 neuropathic mice.
Figure 6.

Improved processing of PMP22 in AUY922-treated C22 mice. (A) Sciatic nerve lysates (5 μg/lane) were treated with either EndoH (column H) or PNGaseF (column N) and probed with antihuman PMP22 antibodies. No enzyme samples served as controls (column C). EndoH-resistant (arrows) and EndoH-sensitive (arrowheads) PMP22 fractions are marked. (B) Quantification of EndoH-resistant PMP22 fractions in sciatic nerves. (C) PMP22-positive aggregates per microscopic field (0.1 mm2) were counted in longitudinal sections of sciatic nerves. (D) Representative images of anti-PMP22 antibody stained (red) nerve sections from Wt (insets) and C22 mice are shown. Arrows mark PMP22-positive aggregates. Hoechst dye (blue) was used to visualize the nuclei. The scale bars are as shown. (E) Steady-state levels of HSP70 and HSP27 in vehicle (Veh)- and AUY922 (AUY)-treated nerve lysates (30 μg/lane) were quantified from (F, G) independent Western blots. (H) Whole liver lysates (30 μg/lane) were processed for (I, J) HSP70 and HSP27 quantification. (E–J) GAPDH or tubulin served as a loading control. Molecular mass on left in kDa. (B, C, F, G, I, J) n = 3–8 mice per group and plotted as means ± SEM; ***P < 0.001; ***P < 0.01; *P < 0.05; n.s., nonsignificant; two-tailed unpaired Student’s t-test.
Subcellular Processing of PMP22 Is Improved in AUY922-Treated Neuropathic Mice
Previously, we showed impaired trafficking of the ectopic human PMP22, as indicated by a reduction in the endoglycosidase H (EndoH)-resistant protein fraction, in sciatic nerves of C22 mice.13 To investigate whether the AUY922-associated improvements in nerve morphology and neuromuscular performance are linked with improved trafficking of PMP22, we subjected sciatic nerves to biochemical analyses (Figure 6A,B). In nerves from Wt mice, the EndoH-resistant fraction of PMP22 does not change upon AUY922 treatment (83.7 ± 5.7 vs 80.0 ± 7.3). On the other hand, in samples from neuropathic mice, we observed a significant AUY922-dependent increase (∼11%) in the EndoH-resistant, membrane-associated PMP22 (54.3 ± 1.5 vs 60.2 ± 1.9), indicating improved subcellular processing.
Mistrafficking of PMP22 within Schwann cells leads to cytosolic protein aggregation, a cellular phenotype previously observed in nerves from C22 animals.13,31 To assess the effects of AUY922 administration on intracellular PMP22 aggregation, we stained longitudinal nerve sections with anti-PMP22 antibodies.13 Quantification of PMP22-positive protein aggregates within a fixed field (0.1 mm2) identified an approximately 5-fold increase in samples from C22 vehicle-treated mice, as compared to the Wt group (Figure 6C). The frequency of protein aggregates is reduced by ∼1.7-fold (5.0 ± 0.7 vs 2.9 ± 0.9) upon AUY922 treatment and is associated with improved myelin-like localization of PMP22 (Figure 6C,D). Note that, with AUY922 administration, the PMP22-like immunostaining appears uniform and is similar to nerves from Wt animals (insets in upper right corners). These results suggest that the improvements in nerve morphology are linked, in part, with enhanced processing of the overproduced PMP22.
Next, we confirmed the bioactivity of AUY922 by evaluating chaperone expression in the sciatic nerve (target tissue) and liver, where AUY922 is metabolized.23 We measured the levels of HSP70 and HSP27, as they are both regulated by HSF-1 (Figure 6E–J). In the sciatic nerves from drug-treated mice, the expressions of HSP70 and HSP27 are increased, confirming the induction of the chaperone pathway in the target tissue (Figure 6E–G). Note that the baseline levels of HSP70 are higher in neuropathic samples as compared to the Wt group, a finding that is consistent with previous publications.13,32 Similar to the nerve, quantification from independent Western blots identifies significant increases in HSP70 and HSP27 in the liver of drug-injected mice, compared to the vehicle groups (Figure 6H–J). Therefore, as suggested by our previous studies with EC137, and in cells from HSP70-deficient mice,15,18 HSP70 likely has a critical role in improving the processing of PMP22 and nerve morphology in samples from C22 mice (Figures 5 and 6). Together, these results confirm the bioavailability and bioactivity of AUY922 in the sciatic nerve.
Improved Nerve Morphology in AUY922-Treated TrJ Neuropathic Mice
Enhancements of chaperones by intermittent fasting or curcumin administration have shown benefits in TrJ neuropathic mice.16,33 Further, functional HSP70 is critical in the delivery of TrJ-PMP22 (L16P mutation) to the lysosomes for degradation.18 Therefore, we evaluated AUY922 in cohorts of male and female TrJ mice, starting the drug administration at 6 weeks of age. Nerves from vehicle-treated neuropathic mice show severe demyelination and axonal atrophy (Figure 7A, left), as described previously.34 In comparison, samples from the AUY922-treated group contain discernible myelinated axonal profiles and an improved overall nerve structure with larger caliber axons (Figure 7A, right).
Figure 7.

AUY922 promotes the maintenance of myelinated axons in TrJ mice. (A) Cross-sectional views of nerve sections from vehicle- (left) and AUY-treated (right) TrJ mice. Micron bar, 20 μm. (B–E) Morphometric analysis of nerves from vehicle- and AUY-treated TrJ mice (n = 4 mice, 320 fibers, 30–40 areas per group). (B) Percent area occupied by fibers, (C) fiber diameter, (D) myelin sheath thickness, and (E) g-ratios were graphed. (F) Rotarod performance at the baseline and at the end of treatment is shown for vehicle- or AUY-treated TrJ mice. (G) Myofiber cross-sectional area of TA muscle from the indicated groups (n = 5 mice, 695 fibers per group). (H) Sciatic nerve (30 μg/lane) and whole liver lysates (30 μg/lane), with (I, J) quantification, from vehicle (Veh)- and AUY922 (AUY)-treated TrJ mice, were assessed for HSP70. (H–J) n = 6–14 mice per group, with GAPDH serving as a loading control. Molecular mass on left, in kDa. Graphs plotted as means (C, G) ± SEM (B, F, I, J) or as whisker plots with median (center line), quartiles (box), and extremes (whiskers) (D, E); ***P < 0.001; **P < 0.01; *P < 0.05; two-tailed unpaired Student’s t-test.
We quantified these morphological measurements from vehicle- and drug-treated TrJ mice and found statistically significant improvements in the nerve tissue area occupied by fibers, which correlates with an increase in nerve fiber diameter (Figure 7B,C). The thickness of myelin around individual axons is also improved, leading to an overall improvement in nerve myelination as reflected by a reduction in g-ratio (Figure 7D,E). However, we did not observe any obvious improvements in the motor behavior or locomotion of AUY922-treated TrJ mice. The rotarod data shown in Figure 7F are representative of several independent cohorts of mice and indicate no treatment effect. In agreement, we failed to identify changes in TA myofiber cross-sectional area upon drug treatment (Figure 7G).
For each mouse, we tested the bioactivity of the drug in target tissues by analyzing the levels of HSP70 in the nerve and liver, as above (Figure 6E–J). The levels of HSP70, which are significantly elevated in TrJ neuropathic nerves as compared with Wt,16 did not increase further upon treatment with AUY922 (Figure 7H,I). In comparison, the liver of injected mice showed a statistically significant increase in HSP70 upon AUY922 treatment (Figure 7H,J). Note that all tissues were collected within 4–6 h of the last drug injection to facilitate the detection of bioactivity. Overall, the results from TrJ neuropathic mice indicate significant improvements in nerve morphology, without detectable benefits in neuromuscular performance or TA myofiber size.
Discussion
In this study, we show enhanced neuromuscular function and improved peripheral nerve morphology in AUY922-treated C22 neuropathic mice. These benefits are accompanied by correction in the subcellular processing of the overexpressed, disease-causing PMP22 protein. In comparison, in the more severe, PMP22 mutant TrJ neuropathic mice, the improvements in nerve myelination failed to affect neuromuscular performance. Overall, the findings presented here agree with other reports on the benefits of chaperone-inducing compounds in various neurodegenerative conditions.35,36 Further optimization of compound delivery, including dosage, timing, and route of administration, may aid in improving drug efficacy in hereditary peripheral neuropathies.
The benefits of HS pathway activation have been documented in various protein misfolding disorders of the CNS;1−3,37,38 however, there have been fewer studies in the PNS. Induction of the HS stress pathway, including increased expression of chaperones by a 5 month long intermittent fasting regimen in TrJ mice, improved peripheral nerve morphology and myelination.16 Significantly, the improvements in nerve morphology were paralleled with maintenance of motor performance, despite disease progression in ad libitum fed neuropathic mice. A more direct correlation between the subcellular processing of PMP22 and chaperones is evident from in vitro studies, where EC137, a synthetic HSP90 inhibitor, successfully activated the HS pathway and improved myelination by Schwann cells from neuropathic C22 mice.15 Due to the lack of availability of EC137, here we screened a set of commercially available compounds, with similar pharmacological properties. Due to their antiproliferative properties, the potential effects of HSP90 inhibitors on cellular toxicity are of major concern,39 particularly when chronic, long-term administration is needed. However, at lower dosages, these drugs can robustly activate the stress pathways without cell death, which is beneficial for protein misfolding diseases.8
Identification of the ideal therapeutic compound for hereditary peripheral neuropathies poses specific challenges, as within a neuropathic nerve there is a heterogeneous population of Schwann cells, with regards to the differentiation state.40 Initially, we selected low, nanomolar drug concentration ranges in the MTS assay, which did not affect the viability of nonmyelinating, mitotic Schwann cells. In the context of myelination, which requires the differentiation of Schwann cells, we identified AUY922 (100 nM) as the most effective drug in increasing myelin synthesis, with BIIB021 closely behind (Figure 3). We tested both of these compounds in cohorts of mice and found that oral administration of BIIB021 in neuropathic animals proved to be a challenge, while intraperitoneal (i.p.) injections with this compound gave an inconsistent read out on bioactivity in target nerve tissue. The mechanism for AUY922-mediated nerve improvement likely involved HSP70, as this specific chaperone has been shown to alleviate protein aggregation in several neurodegenerative diseases, including PMP22-associated neuropathies.4,18,37,38,41 Indeed, HSP70 was robustly induced in the liver and nerve of drug-treated C22 mice. A critical role for HSP70 in our treatment paradigm is further supported by the study where the crossing of HSP70-knockout with TrJ mice exacerbated the neuropathic phenotype.42 Nonetheless, while AUY922 is considered to be a “classical HSP90 inhibitor” that upregulates HSP70 and HSP27, HSP90α knockdown studies in cultured cells indicate multiple targets.43 Therefore, in addition to the inhibition of HSP90 and upregulation of HSP70 and HSP27, additional mechanisms may have contributed to the beneficial effects of AUY922 treatment on neuropathic mice.
As originally described, C22 mice used in this study had a strong phenotype within weeks of birth, including an unsteady gait and sudden reaction to loud noises.11 Subsequently, the mice developed distinct motor disabilities, with nerve demyelination and muscle atrophy by about 6 months of age.31,44,45 Our study was initiated at 7 weeks of age, as only at this age we detected significant impairment of the C22 mice on the rotarod, as compared with age-matched Wt littermates (Figure 5B). In our laboratory, we have been breeding the C22 mice for nearly 15 years, and over time the animals became less phenotypic and are now similar to what has been described for the C3 mice.26 However, by genotyping we detect the human PMP22 transgene by PCR; the copy number is unchanged from tissue banked from mice studied several years ago, and the human protein is highly expressed.13 Still, these mildly affected C22 mice benefitted from the AUY922 therapy, which we distinguished by improvements in both nerve and muscle morphology. In comparison, the more severely affected TrJ mice only showed improvements in nerve morphology without behavioral or skeletal muscle benefits. The difference in the response of the C22 and TrJ mice to AUY922 therapy could be the results of underlying developmental deficits in the neuromuscular system in TrJ that are refractory to HSP therapy.46 The heightened inflammation in nerves of TrJ mice, as compared to the C22 model,13,47 could be an additional factor in impacting the response. Furthermore, at the baseline, nerves from both TrJ and C22 mice have elevated levels of HSP70;32 however, only in the C22 samples did we detect a significant increase upon AUY922 treatment. The variance in drug effectiveness could be the result of differences in drug metabolism, as well as the genetic defects, and emphasizes the need for optimization of drug therapies for the various forms of PMP22-linked neuropathies, in mice and humans.
Albeit the availability of animal models and advancements in the understanding of CMT1A pathobiology, the therapeutic options for affected patients are limited. One of the promising therapeutic candidates, ascorbic acid, elicited prominent improvement of the neuropathic phenotype in C22 mice;12 however, it has failed in independently conducted clinical trials.48,49 Progesterone antagonists are another class of therapeutic drugs which promoted improvements in motor performance in rats that overexpress PMP22.50 The high toxicity and potential side effects of available progesterone antagonists, however, impeded further testing in human clinical trials.51 Another small molecule, rapamycin, an activator of autophagy and an immunosuppressant, improved the myelin structure of TrJ sciatic nerves without significant benefits in motor performance.47 The rapamycin study emphasizes the potential distinct response of nerve and muscle tissue to drugs, when using systemic administration. Besides natural and engineered drugs, dietary supplements have been explored to alleviate neuropathic symptoms. For example, curcumin, and more recently pyruvate supplementation in conjunction with NT-3 gene therapy, has shown benefits in improving nerve morphology and motor performance in TrJ mice.33,52 Curcumin, which is known to work through the HSP70 pathway,42 could be readily tested as a food supplement in CMT1A patients. Together, these preclinical studies in rodents emphasize the need for stringent evaluation of potential drug candidates, preferably in more than one independent animal model.
As of today, there have been two human clinical trials for CMT1A, both using orally available molecules. The first trial tested ascorbic acid (vitamin C) to correct the expression of the overproduced PMP22;12,53 however, this multicenter trial with nearly 500 CMT1A patients proved unsuccessful in providing benefits.49 A recent international Phase 3 clinical trial for CMT1A used pleiotropic drug therapy, including a low dose combination of baclofen, naltrexone, and D-sorbitol.54 Formal publication on the results from this clinical trial has not been made public. In a recent study, PMP22 antisense oligonucleotides (ASOs) were utilized to treat C22 mice and CMT1A rats, and a 50% reduction was reported in the PMP22 mRNA, with significant improvements in myelinated axons.55 Therefore, an additional approach could involve the combined use of small molecules, gene therapy, dietary supplements such as curcumin, phospholipids, and/or exercise. While the underlying subcellular pathogenesis of hereditary demyelinating neuropathies is complex and might be significantly distinct across the genes involved, the neuromuscular system has robust plasticity,56 which aids repair. While optimization of HSP90 inhibitor drug dosing and the route of administration needs improvement for efficacy, our results suggest that activation of the chaperone pathway alone, or in combination with another therapeutic approach, may provide benefits in ameliorating the neuropathic phenotype in affected individuals.
Methods
Mouse Colonies and Genotyping
A founder pair of C22 mice (MGI: 2183770) obtained from Dr. Clair Huxley11 were bred on C57Bl/6 or on CBA/CaJ background for multiple generations. Heterozygous Trembler J (TrJ, MGI: 1856217) mice on a C57BL/6J background were bred to wild type C57BL6/J mice, obtained from Jackson laboratories. All animals were maintained under SPF conditions within the University of Florida animal care facilities and strictly in compliance with procedures approved by the Institutional Animal Care and Use Committee (IACUC). For genotyping C22 mice, DNA was obtained from tail biopsies of less than 8 day old pups and analyzed by PCR using the following primer sets: C22- 5′ TTCTGCTGCCTGTGAGGAC 3′ and 5′ GGGTGAAGAGTTGGCAGAAG 3′ which yield a 209 bp product. The endogenous mouse PMP22 was identified using the following primers: 5′ GGTTGCCAAACTGGAGTGAT 3′ and 5′ CGGCTCTGTCAAGATTAGCC 3′ yielding a 458 bp product. TrJ mice were genotyped as described.34 At weaning age, littermates were segregated by genotype and sex and randomly assigned to vehicle and AUY922 treatment groups. All efforts were made to reduce the number of animals used and to minimize their discomfort.
Analysis of Transgene Copy Number and Genetic Background
To investigate the milder phenotype observed in our present colony of C22 mice, we analyzed the PMP22 transgene copy number and the genetic background of the mice used in this study, in comparison to archived material from 10 years ago, when the phenotype more closely matched previous reports. The liver was used as the source of genomic DNA, and QPCR was performed for the human PMP22 transgene using mouse Pmp22 as an internal standard, as described.26 Four archived samples, and four samples from mice used in the present study, were analyzed. No change in transgene copy number was detected between the two sets of tissue samples. As an alternative explanation for the change in phenotype, we also examined the genetic background of the mice using a panel of 48 SNP markers spanning the autosomes and X chromosome. These markers are used routinely for genetic quality control at The Jackson Laboratory. This analysis revealed that the archived samples were a mix of C57BL/6J and C57BL/6N genetic backgrounds, whereas the mice used in the current study were predominantly CBA/Ca but still carried some heterozygous C57BL/6 alleles on a subset of chromosomes. On the basis of these analyses, we conclude the change in phenotype is due to the change in genetic background. However, as all studies described here used contemporaneous littermate controls and did not rely on historical data for comparison, this does not influence the interpretation of our results.
Cell Culture Models
Nonmyelinating Schwann cell cultures were established from the sciatic nerves of postnatal day 2 rats, as described.10 The cells were maintained in DMEM (Gibco, Thermo Fisher, Waltham, MA) and supplemented with 10% FCS (HyClone, Thermo Fisher), 100 μg/mL bovine pituitary extract (Biomedical Technologies Inc., Stoughton, MA), and 5 μM forskolin (Calbiochem, Millipore, Burlington, MA). Dorsal-root ganglion (DRG) explants were established from embryonic day 12–13 Wt and C22 embryos.15 Briefly, DRGs were dissociated in 0.25% trypsin (Gibco) and plated onto collagen-coated cell culture wells. DNA isolated from each embryo was used for genotyping, as described above. All explants were maintained in MEM (Gibco), 10% FCS (Hyclone), 0.3% glucose (Sigma-Aldrich, St. Louis, MO), 10 mM HEPES (Gibco), and 100 ng/mL nerve growth factor (Harlan Bioproducts for Science, Indianapolis, IN) for 7 days. The cultures were then supplemented with 50 μg/mL ascorbate for an additional 7 days to promote myelin formation. For Schwann cell-depleted neuronal cultures, explants were subjected to alternate-day treatment with 5-fluoro-2′-deoxyuridine (FUdR) for 10 days and then continued on the same paradigm described above.15
In Vitro Pharmacologic Treatment Paradigms
HSP90 inhibitor compounds, including AT13387 (S1163), AUY922 (S1069), BIIB021 (S1175), SNX5422 (S2656), and STA9090 (S1159), were purchased from Selleckchem (Houston, TX) and stored at a stock concentration of 1 mM in DMSO. Primary Schwann cells were treated with HSP90 inhibitors at the indicated concentrations in complete media (see above), 24 h after seeding. DMSO served as the vehicle control while geldanamycin (GA) was used as a positive control for heat shock pathway activation. The DRG explant cultures were maintained for 7 days in ascorbate-containing media prior to treatment with either DMSO, AUY922 (100 nM), or BIIB021 (100 nM), every third day (72 h apart). Cultures were procured, 24 h after the third treatment, for either biochemical or immunochemical analyses.15
Cell Viability Assay
Schwann cells were plated at a seeding density of 104 cells/well in a 96-well plate (Nunc, Thermo Fisher), coated with poly-l-lysine (Sigma), and treated with either DMSO or an HSP90 inhibitor at the desired concentrations for 24 h.15 At the end of the treatment, cells were incubated in a mixture of MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (333 μg/mL) and phenazine methosulfate (25 μM) for 2 h at 37 °C, producing the soluble formazan product (Promega, Madison, WI). The formazan product was measured spectrophotometrically at 490 nm and graphed as a percent of DMSO-treated controls using GraphPad Prism v5.0 software.
Quantitative RT-PCR
Rat Schwann cells, treated with either DMSO or the selected HSP90 inhibitor compounds (100 nM), were harvested in TRIzol (Invitrogen, Carlsbad, CA), and RNA was isolated as per the manufacturer’s instructions. A 1 μg portion of total RNA was used to synthesize cDNA using the SuperScript III first strand synthesis kit (Invitrogen). The same volume of undiluted cDNA from each sample was used for real time (RT)-PCR analysis, using the SYBR GreenER qPCR kit (Invitrogen) and QuantiTect Primer for HSP70 (QT00370489) or GAPDH (QT00199633). The normalized transcript levels of HSP70 relative to geldanamycin (GA) were determined using the 2–ΔΔCT method.57 Values obtained were analyzed and graphed with the help of GraphPad Prism v5.0 software.
AUY922 Administration, Rotarod Testing, and Serum and Liver Analyses
During the course of the study, the body weight of each mouse was recorded twice per week. Baseline rotarod measurements were obtained before the start of the compound treatment at 12 weeks of age for C22 and at 6 weeks of age for TrJ mice. The mice were trained the first 2 days at 5 rpm for 60 s: three trials/day, with 30 min breaks.16 On the third day, mice were tested on the rotarod, accelerating from 16 to 36 rpm in steps of 4 rpm increase/min.42 The control groups were injected intraperitoneally (i.p.) twice/week with the vehicle consisting of 10% DMSO, 5% Tween-20, and 85% saline.23 The treatment groups received 2 mg/kg AUY922, using the same route and vehicle for administration. Dosage for AUY922 was determined on the basis of the half-life of the compound in plasma23 and results of the in vitro experiments (see Figures 2 and 3). Rotarod testing was done on all groups monthly, where all mice underwent the same 3 day testing. The time on the rotarod before falling was recorded for each mouse and graphed. The study was terminated after 20 weeks of drug treatment. At termination, the mice were sacrificed within 4 h after the final injection, and blood and tissue samples were collected. Blood was collected into tubes with clot activator gel (BD 365967, Franklin Lakes, NJ) and centrifuged at 10 000 rpm for 5 min to isolate serum. Serum and liver samples were sent to Charles River Laboratories (Wilmington, MA) for processing and analysis of AUY922 concentration via protein precipitation, followed by LC-MS/MS using glafenine and carbamazepine as internal standards.
In Situ Isometric Twitch Torque Analyses
The isometric twitch torque analysis was performed on the tibialis anterior (TA) muscle and anterior tibial tendon. Under anesthesia, the skin and fascia surrounding the distal hindlimb were surgically removed exposing the TA. A braided (4–0) silk surgical suture (Teleflex Medical, Wayne, PA) was secured around the anterior tibial tendon before all tendons to the foot were detached. Mice were positioned in dorsal recumbence on a preheated physiology table to maintain body temperature at 37 °C. A clamp was used to secure the hindlimb at 90° at the knee, and the paw was positioned to the physiology table using transpore surgical tape (3M). The anterior tibial tendon was secured to a 300C-LR-FP muscle lever (Aurora Scientific, Aurora, ON, Canada). Cathode and anode electrodes were inserted distal to the fibular to stimulate the peroneal nerve. Under control of the Dynamic Muscle Control (DMC) and Analysis (DMA) Software suite (Aurora Scientific), optimal electrode placement was determined by repositioning of the electrodes and stimulating the nerve at 1 Hz until the maximum twitch amplitude was recorded for a given position. Optimal length-tension was determined by performing the isometric twitch stimulation at an increasing range of amplitudes and tensions until the maximum twitch amplitude was observed. Three successive tetanic stimulations (200 Hz, 100 pulses per train, 60 s between independent stimuli) were performed, and the muscle was allowed to rest for 5 min. Single stimulations at 15, 30, 60, 100, 120, 160, and 200 Hz were then performed with 30 s between each successive frequency, and the resulting torque was recorded and analyzed using DMC and DMA (Aurora Scientific).
Western Blot Analyses
Cell harvesting and tissue homogenization were done in sodium dodecyl sulfate (SDS) gel sample buffer (62.5 mM Tris pH 6.8, 10% glycerol, 3% SDS), supplemented with protease and phosphatase inhibitors.15 Protein concentrations were measured using BCA assay (Pierce, Thermo Fisher). Digestions using endoglycosidase H (EndoH) or N-glycosidase F (PNGaseF) enzymes (New England Biolabs) were performed to assess the subcellular processing of PMP22, as described.58 Equal amounts of proteins for each experiment were separated on denaturing SDS gels and transferred to nitrocellulose membrane (0.45 or 0.22 μm pore size) (Bio-Rad, Hercules, CA). Membranes were blocked in 5% milk (in Tris-buffered saline with 0.05% Tween-20) and incubated with the indicated primary antibodies (Table 1) overnight at 4 °C. Bound antibodies were detected with antirabbit, antigoat, antimouse IgG, or antichicken IgY HRP-linked secondary antibodies (Sigma) and visualized with the chemiluminescence detection method (PerkinElmer Life Sciences, Waltham, MA). Films were digitally imaged using a GS-800 densitometer (Bio-Rad) and were formatted for printing, using Adobe Photoshop.
Table 1. Primary Antibodies Used in This Studya.
| dilution |
||||
|---|---|---|---|---|
| species | antigen | source and catalog no. | WB | IS |
| rabbit | HSP70 | Stressgen; SPA-812 | 1:3000 | n/a |
| rabbit | HSP70 | Abcam; ab137680 | 1:2000 | n/a |
| goat | HSP27 | Santa Cruz Biotechnology, Inc.; sc-1049 | 1:1000 | n/a |
| mouse | GAPDH | Encor Biotechnology, Inc.; MCA-1D4 | 1:10 000 | n/a |
| rabbit | GAPDH | Encor Biotechnology, Inc.; RPCA-GAPDH | 1:8000 | n/a |
| mouse | tubulin | Sigma, St Louis, MO, USA; T6199 | 1:2000 | n/a |
| mouse | tubulin | Encor Biotechnology, Inc.; MCA-1B12 | 1:10 000 | n/a |
| chicken | P0 | Encor Biotechnology, Inc. | 1:500 | n/a |
| rat | MBP | Chemicon; MAB386 | n/a | 1:500 |
| rabbit | PMP22 | Chittoor et al., 2013 | 1:1000 | 1:250 |
| rabbit | laminin | Sigma; L9393 | n/a | 1:300 |
WB: Western blotting. IS: immunostaining. n/a: not applicable.
Immunostaining
Explant cultures were fixed in 4% paraformaldehyde (EMS, Hatfield, PA) and permeabilized in 100% ice-cold methanol (Fisher Scientific, Hampton, NH). After blocking with 5% normal goat serum, samples were incubated with anti-MBP antibodies, overnight at 4 °C. Bound antibodies were detected with Alexa Fluor 488 goat antirat IgG (Molecular Probes, Eugene, OR). Coverslips were mounted using the Prolong Antifade kit (Molecular Probes). Proximal regions of sciatic nerves were sectioned (5 μm thickness) and processed for immunostaining with anti-PMP22 antibodies, as described.13 AlexaFluor 594-conjugated goat antirabbit antibodies were used to detect the bound primary antibodies. Samples which were processed in parallel without incubation with primary antibodies served as the negative controls. Images were obtained using a SPOT digital camera (Diagnostic Instrumentals, Sterling Heights, MI), with a Nikon Eclipse E800 or an Olympus DSU spinning disc confocal (Tokyo, Japan) microscope, using identical exposure settings. Images were processed using Photoshop (Adobe Systems).
Myelin Internode Length Measurement
DRG cultures were stained for MBP as described above, to label myelin internode segments. The MBP-positive internodes were measured using ImageJ software (NIH). Measurements from three independent experiments, per treatment per genotype, were graphed using GraphPad Prism software.
Morphometric Analyses of the Sciatic Nerve
Proximal ends of sciatic nerves from vehicle- and AUY922-treated groups were fixed by immersion in ice-cold 2% paraformaldehyde and 2% glutaraldehyde, in 0.1 M sodium cacodylate buffer at 4 °C.59 Plastic sections, stained with toluidine blue, were prepared by the Robert P. Apkarian Integrated Electron Microscopy Core at Emory University and imaged with a light microscope (Zeiss Axioscop 2 plus). Axon diameter, fiber diameter, myelin sheath thickness (n = 320–1100 fibers per group), and total area occupied by nerve fibers (n = 20–40 areas per group) were measured using ImageJ software (NIH).16 The g-ratio was calculated as the axon diameter/fiber diameter, using the respective values. The myelin sheath thickness was calculated as [(fiber perimeter – axon perimeter)/2π].60
Cross-Sectional Area Analyses of TA Muscles
Fresh-frozen TA muscles from C22 and TrJ mice, treated with vehicle or AUY922, were sectioned at 10 μm thickness and immunostained with rabbit antilaminin antibody to outline the individual myofibers. AlexaFluor 488-conjugated goat antirabbit secondary antibody was used to detect the bound primary antibody, and Hoechst was used to label nuclei. Images were captured using a Nikon DS digital camera fitted on a Nikon Eclipse E800 microscope. Myofiber cross-sectional areas of individual fibers were measured using ImageJ software (NIH) and exported to GraphPad Prism v8.0.1 for analysis.
Data Analyses
For all comparisons, mean ± SEM was calculated, and statistical differences were determined using unpaired the two-tailed Student’s t-test. P-values <0.05 (*), <0.01 (**), and <0.001 (***) were considered to be significant. For in situ torque analysis, significance was determined using two-way ANOVA with Sidak’s multiple comparison between individual groups and frequencies. For longitudinal rotarod analysis, significance was determined using two-way ANOVA with Fisher’s LSD post hoc test.
Acknowledgments
We thank members of the Notterpek lab for assistance with various technical aspects of this project over a 3 year period, including genotyping and weighing the mice, behavioral testing, morphological studies, and sample collection. We acknowledge the Robert P. Apkarian Integrated Electron Microscopy Core at Emory University for semithin sectioning and toluidine blue processing of sciatic nerves and Greg Ballard for help with the SNP genome scan. This project was supported by NIH-NINDS Grants NS091435 (L.N.) and NS098523 (R.W.B.).
Glossary
Abbreviations
- PMP22
peripheral myelin protein 22
- CMT1A
Charcot–Marie–Tooth disease type 1A
- HSP
heat shock protein
- Wt
wild type
Author Contributions
Research design: V.G.C.-V., and L.N. Experimental work: V.G.C.-V., H.B., A.G.T., D.F., K.H.M., R.W.B., and L.N. Data analyses and interpretation: V.G.C.-V., H.B., A.G.T., D.F., K.H.M., R.W.B., T.C.F., and L.N. Writing, review, and revision of the manuscript: V.G.C.-V., H.B., A.G.T., D.F., R.W.B., T.C.F., and L.N.
The authors declare no competing financial interest.
Notes
Chemical compounds: NVP-AUY922 (PubChem CID: 10096043); BIIB021 (PubChem CID: 16736529); AT13387 (PubChem CID: 11955716); SNX5422 (PubChem CID: 44195571); STA9090 (PubChem CID: 66577018).
References
- Jiang Y. Q.; Wang X. L.; Cao X. H.; Ye Z. Y.; Li L.; Cai W. Q. (2013) Increased heat shock transcription factor 1 in the cerebellum reverses the deficiency of Purkinje cells in Alzheimer’s disease. Brain Res. 1519, 105–11. 10.1016/j.brainres.2013.04.059. [DOI] [PubMed] [Google Scholar]
- Lin P. Y.; Simon S. M.; Koh W. K.; Folorunso O.; Umbaugh C. S.; Pierce A. (2013) Heat shock factor 1 over-expression protects against exposure of hydrophobic residues on mutant SOD1 and early mortality in a mouse model of amyotrophic lateral sclerosis. Mol. Neurodegener. 8, 43. 10.1186/1750-1326-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmar B.; Lu C. H.; Greensmith L. (2014) The role of heat shock proteins in Amyotrophic Lateral Sclerosis: The therapeutic potential of Arimoclomol. Pharmacol. Ther. 141 (1), 40–54. 10.1016/j.pharmthera.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Jinwal U. K.; Akoury E.; Abisambra J. F.; O’Leary J. C. 3rd; Thompson A. D.; Blair L. J.; Jin Y.; Bacon J.; Nordhues B. A.; Cockman M.; Zhang J.; Li P.; Zhang B.; Borysov S.; Uversky V. N.; Biernat J.; Mandelkow E.; Gestwicki J. E.; Zweckstetter M.; Dickey C. A. (2013) Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J. 27 (4), 1450–9. 10.1096/fj.12-220889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattoo R. U.; Sharma S. K.; Priya S.; Finka A.; Goloubinoff P. (2013) Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J. Biol. Chem. 288 (29), 21399–411. 10.1074/jbc.M113.479253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho M.; Herrera A. E.; Orzaez M.; Perez-Paya E. (2014) Inactivation of Apaf1 reduces the formation of mutant huntingtin-dependent aggregates and cell death. Neuroscience 262, 83–91. 10.1016/j.neuroscience.2013.12.062. [DOI] [PubMed] [Google Scholar]
- Ali A.; Bharadwaj S.; O’Carroll R.; Ovsenek N. (1998) HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol. Cell. Biol. 18 (9), 4949–60. 10.1128/MCB.18.9.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide S. D.; Morimoto R. I. (2005) Heat shock response modulators as therapeutic tools for diseases of protein conformation. J. Biol. Chem. 280 (39), 33097–100. 10.1074/jbc.R500010200. [DOI] [PubMed] [Google Scholar]
- Lupski J. R.; Garcia C. A. (1992) Molecular genetics and neuropathology of Charcot-Marie-Tooth disease type 1A. Brain Pathol. 2 (4), 337–49. 10.1111/j.1750-3639.1992.tb00710.x. [DOI] [PubMed] [Google Scholar]
- Notterpek L.; Ryan M. C.; Tobler A. R.; Shooter E. M. (1999) PMP22 accumulation in aggresomes: implications for CMT1A pathology. Neurobiol. Dis. 6 (5), 450–60. 10.1006/nbdi.1999.0274. [DOI] [PubMed] [Google Scholar]
- Huxley C.; Passage E.; Manson A.; Putzu G.; Figarella-Branger D.; Pellissier J. F.; Fontes M. (1996) Construction of a mouse model of Charcot-Marie-Tooth disease type 1A by pronuclear injection of human YAC DNA. Hum. Mol. Genet. 5 (5), 563–9. 10.1093/hmg/5.5.563. [DOI] [PubMed] [Google Scholar]
- Passage E.; Norreel J. C.; Noack-Fraissignes P.; Sanguedolce V.; Pizant J.; Thirion X.; Robaglia-Schlupp A.; Pellissier J. F.; Fontes M. (2004) Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat. Med. 10 (4), 396–401. 10.1038/nm1023. [DOI] [PubMed] [Google Scholar]
- Chittoor V. G.; Sooyeon L.; Rangaraju S.; Nicks J. R.; Schmidt J. T.; Madorsky I.; Narvaez D. C.; Notterpek L. (2013) Biochemical characterization of protein quality control mechanisms during disease progression in the C22 mouse model of CMT1A. ASN Neuro 5 (5), e00128 10.1042/AN20130024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortun J.; Dunn W. A. Jr.; Joy S.; Li J.; Notterpek L. (2003) Emerging role for autophagy in the removal of aggresomes in Schwann cells. J. Neurosci. 23 (33), 10672–80. 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju S.; Madorsky I.; Pileggi J. G.; Kamal A.; Notterpek L. (2008) Pharmacological induction of the heat shock response improves myelination in a neuropathic model. Neurobiol. Dis. 32 (1), 105–15. 10.1016/j.nbd.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madorsky I.; Opalach K.; Waber A.; Verrier J. D.; Solmo C.; Foster T.; Dunn W. A. Jr.; Notterpek L. (2009) Intermittent fasting alleviates the neuropathic phenotype in a mouse model of Charcot-Marie-Tooth disease. Neurobiol. Dis. 34 (1), 146–54. 10.1016/j.nbd.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opalach K.; Rangaraju S.; Madorsky I.; Leeuwenburgh C.; Notterpek L. (2010) Lifelong calorie restriction alleviates age-related oxidative damage in peripheral nerves. Rejuvenation Res. 13 (1), 65–74. 10.1089/rej.2009.0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittoor-Vinod V. G.; Lee S.; Judge S. M.; Notterpek L. (2015) Inducible HSP70 is critical in preventing the aggregation and enhancing the processing of PMP22. ASN Neuro 7 (1), 175909141556990. 10.1177/1759091415569909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban M. J.; Pan P.; Farmer K. L.; Zhao H.; Blagg B. S.; Dobrowsky R. T. (2012) Modulating molecular chaperones improves sensory fiber recovery and mitochondrial function in diabetic peripheral neuropathy. Exp. Neurol. 235 (1), 388–96. 10.1016/j.expneurol.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Li C.; Fowler S. C.; Zhang Z.; Blagg B. S. J.; Dobrowsky R. T. (2018) Targeting Heat Shock Protein 70 to Ameliorate c-Jun Expression and Improve Demyelinating Neuropathy. ACS Chem. Neurosci. 9 (2), 381–390. 10.1021/acschemneuro.7b00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata Y. (2005) Hsp90 inhibitor geldanamycin and its derivatives as novel cancer chemotherapeutic agents. Curr. Pharm. Des. 11 (9), 1131–8. 10.2174/1381612053507585. [DOI] [PubMed] [Google Scholar]
- Saibil H. (2013) Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 14 (10), 630–42. 10.1038/nrm3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles S. A.; Massey A.; Raynaud F. I.; Sharp S. Y.; Box G.; Valenti M.; Patterson L.; de Haven Brandon A.; Gowan S.; Boxall F.; Aherne W.; Rowlands M.; Hayes A.; Martins V.; Urban F.; Boxall K.; Prodromou C.; Pearl L.; James K.; Matthews T. P.; Cheung K. M.; Kalusa A.; Jones K.; McDonald E.; Barril X.; Brough P. A.; Cansfield J. E.; Dymock B.; Drysdale M. J.; Finch H.; Howes R.; Hubbard R. E.; Surgenor A.; Webb P.; Wood M.; Wright L.; Workman P. (2008) NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 68 (8), 2850–60. 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- Hickey M. A.; Zhu C.; Medvedeva V.; Lerner R. P.; Patassini S.; Franich N. R.; Maiti P.; Frautschy S. A.; Zeitlin S.; Levine M. S.; Chesselet M. F. (2012) Improvement of neuropathology and transcriptional deficits in CAG 140 knock-in mice supports a beneficial effect of dietary curcumin in Huntington’s disease. Mol. Neurodegener. 7, 12. 10.1186/1750-1326-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Bokov A.; Gelfond J.; Soto V.; Ikeno Y.; Hubbard G.; Diaz V.; Sloane L.; Maslin K.; Treaster S.; Rendon S.; van Remmen H.; Ward W.; Javors M.; Richardson A.; Austad S. N.; Fischer K. (2014) Rapamycin extends life and health in C57BL/6 mice. J. Gerontol., Ser. A 69 (2), 119–30. 10.1093/gerona/glt056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhamme C.; King R. H.; ten Asbroek A. L.; Muddle J. R.; Nourallah M.; Wolterman R.; Baas F.; van Schaik I. N. (2011) Myelin and axon pathology in a long-term study of PMP22-overexpressing mice. J. Neuropathol. Exp. Neurol. 70 (5), 386–98. 10.1097/NEN.0b013e318217eba0. [DOI] [PubMed] [Google Scholar]
- Huguet A.; Medja F.; Nicole A.; Vignaud A.; Guiraud-Dogan C.; Ferry A.; Decostre V.; Hogrel J. Y.; Metzger F.; Hoeflich A.; Baraibar M.; Gomes-Pereira M.; Puymirat J.; Bassez G.; Furling D.; Munnich A.; Gourdon G. (2012) Molecular, physiological, and motor performance defects in DMSXL mice carrying > 1,000 CTG repeats from the human DM1 locus. PLoS Genet. 8 (11), e1003043 10.1371/journal.pgen.1003043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk D. J.; Todd A. G.; Lee S.; Soustek M. S.; ElMallah M. K.; Fuller D. D.; Notterpek L.; Byrne B. J. (2015) Peripheral nerve and neuromuscular junction pathology in Pompe disease. Hum. Mol. Genet. 24 (3), 625–36. 10.1093/hmg/ddu476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd A. G.; McElroy J. A.; Grange R. W.; Fuller D. D.; Walter G. A.; Byrne B. J.; Falk D. J. (2015) Correcting Neuromuscular Deficits With Gene Therapy in Pompe Disease. Ann. Neurol. 78 (2), 222–34. 10.1002/ana.24433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley C.; Passage E.; Robertson A. M.; Youl B.; Huston S.; Manson A.; Saberan-Djoniedi D.; Figarella-Branger D.; Pellissier J. F.; Thomas P. K.; Fontes M. (1998) Correlation between varying levels of PMP22 expression and the degree of demyelination and reduction in nerve conduction velocity in transgenic mice. Hum. Mol. Genet. 7 (3), 449–58. 10.1093/hmg/7.3.449. [DOI] [PubMed] [Google Scholar]
- Fortun J.; Go J. C.; Li J.; Amici S. A.; Dunn W. A. Jr.; Notterpek L. (2006) Alterations in degradative pathways and protein aggregation in a neuropathy model based on PMP22 overexpression. Neurobiol. Dis. 22 (1), 153–64. 10.1016/j.nbd.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Amici S. A.; Dunn W. A. Jr.; Notterpek L. (2007) Developmental abnormalities in the nerves of peripheral myelin protein 22-deficient mice. J. Neurosci. Res. 85 (2), 238–49. 10.1002/jnr.21118. [DOI] [PubMed] [Google Scholar]
- Khajavi M.; Shiga K.; Wiszniewski W.; He F.; Shaw C. A.; Yan J.; Wensel T. G.; Snipes G. J.; Lupski J. R. (2007) Oral curcumin mitigates the clinical and neuropathologic phenotype of the Trembler-J. mouse: a potential therapy for inherited neuropathy. Am. J. Hum. Genet. 81 (3), 438–53. 10.1086/519926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notterpek L.; Shooter E. M.; Snipes G. J. (1997) Upregulation of the endosomal-lysosomal pathway in the trembler-J neuropathy. J. Neurosci. 17 (11), 4190–200. 10.1523/JNEUROSCI.17-11-04190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Ma J.; Zhao H.; Blagg B. S.; Dobrowsky R. T. (2012) Induction of heat shock protein 70 (Hsp70) prevents neuregulin-induced demyelination by enhancing the proteasomal clearance of c-Jun. ASN Neuro 4 (7), e00102 10.1042/20120047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A.; Kwon Y. T. (2017) Protein Quality Control by Molecular Chaperones in Neurodegeneration. Front. Neurosci. 11, 185. 10.3389/fnins.2017.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino T.; Murao N.; Namba T.; Takehara M.; Adachi H.; Katsuno M.; Sobue G.; Matsushima T.; Suzuki T.; Mizushima T. (2011) Suppression of Alzheimer’s disease-related phenotypes by expression of heat shock protein 70 in mice. J. Neurosci. 31 (14), 5225–34. 10.1523/JNEUROSCI.5478-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifondorwa D. J.; Jimenz-Moreno R.; Hayes C. D.; Rouhani H.; Robinson M. B.; Strupe J. L.; Caress J.; Milligan C. (2012) Administration of Recombinant Heat Shock Protein 70 Delays Peripheral Muscle Denervation in the SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Neurol Res. Int. 2012, 170426. 10.1155/2012/170426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans C. G.; Chang L.; Gestwicki J. E. (2010) Heat shock protein 70 (hsp70) as an emerging drug target. J. Med. Chem. 53 (12), 4585–602. 10.1021/jm100054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneja M.; Burns J.; Saporta M. A.; Timmerman V. (2019) Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J. Neurol., Neurosurg. Psychiatry 90 (1), 58–67. 10.1136/jnnp-2018-318834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobkova N. V.; Garbuz D. G.; Nesterova I.; Medvinskaya N.; Samokhin A.; Alexandrova I.; Yashin V.; Karpov V.; Kukharsky M. S.; Ninkina N. N.; Smirnov A. A.; Nudler E.; Evgen’ev M. (2013) Therapeutic effect of exogenous hsp70 in mouse models of Alzheimer’s disease. J. Alzheimer's Dis. 38 (2), 425–35. 10.3233/JAD-130779. [DOI] [PubMed] [Google Scholar]
- Okamoto Y.; Pehlivan D.; Wiszniewski W.; Beck C. R.; Snipes G. J.; Lupski J. R.; Khajavi M. (2013) Curcumin facilitates a transitory cellular stress response in Trembler-J mice. Hum. Mol. Genet. 22 (23), 4698–705. 10.1093/hmg/ddt318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Koay Y. C.; McAlpine S. R. (2017) Redefining the Phenotype of Heat Shock Protein 90 (Hsp90) Inhibitors. Chem. - Eur. J. 23 (9), 2010–2013. 10.1002/chem.201604807. [DOI] [PubMed] [Google Scholar]
- Norreel J. C.; Vinay L.; Fontes M.; Clarac F. (2003) Close relationship between motor impairments and loss of functional motoneurons in a Charcot-Marie-Tooth type 1A model. Neuroscience 116 (3), 695–703. 10.1016/S0306-4522(02)00741-8. [DOI] [PubMed] [Google Scholar]
- Szigeti K.; Lupski J. R. (2009) Charcot-Marie-Tooth disease. Eur. J. Hum. Genet. 17 (6), 703–10. 10.1038/ejhg.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scurry A. N.; Heredia D. J.; Feng C. Y.; Gephart G. B.; Hennig G. W.; Gould T. W. (2016) Structural and Functional Abnormalities of the Neuromuscular Junction in the Trembler-J Homozygote Mouse Model of Congenital Hypomyelinating Neuropathy. J. Neuropathol. Exp. Neurol. 75 (4), 334–46. 10.1093/jnen/nlw004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicks J.; Lee S.; Harris A.; Falk D. J.; Todd A. G.; Arredondo K.; Dunn W. A. Jr.; Notterpek L. (2014) Rapamycin improves peripheral nerve myelination while it fails to benefit neuromuscular performance in neuropathic mice. Neurobiol. Dis. 70, 224–36. 10.1016/j.nbd.2014.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareyson D.; Reilly M. M.; Schenone A.; Fabrizi G. M.; Cavallaro T.; Santoro L.; Vita G.; Quattrone A.; Padua L.; Gemignani F.; Visioli F.; Laura M.; Radice D.; Calabrese D.; Hughes R. A.; Solari A. (2011) Ascorbic acid in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL and CMT-TRAUK): a double-blind randomised trial. Lancet Neurol. 10 (4), 320–8. 10.1016/S1474-4422(11)70025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis R. A.; McDermott M. P.; Herrmann D. N.; Hoke A.; Clawson L. L.; Siskind C.; Feely S. M.; Miller L. J.; Barohn R. J.; Smith P.; Luebbe E.; Wu X.; Shy M. E. (2013) High-dosage ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A: results of a randomized, double-masked, controlled trial. JAMA Neurol 70 (8), 981–7. 10.1001/jamaneurol.2013.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sereda M. W.; Meyer zu Horste G.; Suter U.; Uzma N.; Nave K. A. (2003) Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat. Med. 9 (12), 1533–7. 10.1038/nm957. [DOI] [PubMed] [Google Scholar]
- Pareyson D.; Marchesi C. (2009) Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 8 (7), 654–67. 10.1016/S1474-4422(09)70110-3. [DOI] [PubMed] [Google Scholar]
- Sahenk Z.; Yalvac M. E.; Amornvit J.; Arnold W. D.; Chen L.; Shontz K. M.; Lewis S. (2018) Efficacy of exogenous pyruvate in Trembler(J) mouse model of Charcot-Marie-Tooth neuropathy. Brain Behav 8 (10), e01118 10.1002/brb3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareyson D.; Schenone A.; Fabrizi G. M.; Santoro L.; Padua L.; Quattrone A.; Vita G.; Gemignani F.; Visioli F.; Solari A. (2006) A multicenter, randomized, double-blind, placebo-controlled trial of long-term ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL): the study protocol [EudraCT no.: 2006–000032–27]. Pharmacol. Res. 54 (6), 436–41. 10.1016/j.phrs.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Chumakov I.; Milet A.; Cholet N.; Primas G.; Boucard A.; Pereira Y.; Graudens E.; Mandel J.; Laffaire J.; Foucquier J.; Glibert F.; Bertrand V.; Nave K. A.; Sereda M. W.; Vial E.; Guedj M.; Hajj R.; Nabirotchkin S.; Cohen D. (2014) Polytherapy with a combination of three repurposed drugs (PXT3003) down-regulates Pmp22 over-expression and improves myelination, axonal and functional parameters in models of CMT1A neuropathy. Orphanet J. Rare Dis 9, 201. 10.1186/s13023-014-0201-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H. T.; Damle S.; Ikeda-Lee K.; Kuntz S.; Li J.; Mohan A.; Kim A.; Hung G.; Scheideler M. A.; Scherer S. S.; Svaren J.; Swayze E. E.; Kordasiewicz H. B. (2018) PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models. J. Clin. Invest. 128 (1), 359–368. 10.1172/JCI96499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko C. P.; Robitaille R. (2015) Perisynaptic Schwann Cells at the Neuromuscular Synapse: Adaptable, Multitasking Glial Cells. Cold Spring Harbor Perspect. Biol. 7 (10), a020503. 10.1101/cshperspect.a020503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K. J.; Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 (4), 402–8. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Pareek S.; Notterpek L.; Snipes G. J.; Naef R.; Sossin W.; Laliberte J.; Iacampo S.; Suter U.; Shooter E. M.; Murphy R. A. (1997) Neurons promote the translocation of peripheral myelin protein 22 into myelin. J. Neurosci. 17 (20), 7754–62. 10.1523/JNEUROSCI.17-20-07754.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amici S. A.; Dunn W. A. Jr.; Murphy A. J.; Adams N. C.; Gale N. W.; Valenzuela D. M.; Yancopoulos G. D.; Notterpek L. (2006) Peripheral myelin protein 22 is in complex with alpha6beta4 integrin, and its absence alters the Schwann cell basal lamina. J. Neurosci. 26 (4), 1179–89. 10.1523/JNEUROSCI.2618-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie M. J.; Stein R. B. (1983) The relationship between axon diameter, myelin thickness and conduction velocity during atrophy of mammalian peripheral nerves. Brain Res. 259 (1), 41–56. 10.1016/0006-8993(83)91065-X. [DOI] [PubMed] [Google Scholar]