

Combined experiment and simulation reveals a structural mechanism of surface-catalyzed nucleation in Aβ amyloid formation.
Abstract
Understanding the structural mechanism by which proteins and peptides aggregate is crucial, given the role of fibrillar aggregates in debilitating amyloid diseases and bioinspired materials. Yet, this is a major challenge as the assembly involves multiple heterogeneous and transient intermediates. Here, we analyze the co-aggregation of Aβ40 and Aβ16–22, two widely studied peptide fragments of Aβ42 implicated in Alzheimer’s disease. We demonstrate that Aβ16–22 increases the aggregation rate of Aβ40 through a surface-catalyzed secondary nucleation mechanism. Discontinuous molecular dynamics simulations allowed aggregation to be tracked from the initial random coil monomer to the catalysis of nucleation on the fibril surface. Together, the results provide insight into how dynamic interactions between Aβ40 monomers/oligomers on the surface of preformed Aβ16–22 fibrils nucleate Aβ40 amyloid assembly. This new understanding may facilitate development of surfaces designed to enhance or suppress secondary nucleation and hence to control the rates and products of fibril assembly.
INTRODUCTION
Understanding the molecular mechanisms of peptide self-assembly into amyloid fibrils is of key importance in understanding pathological disease states (1) and in designing new functional materials (2). Aberrant self-assembly of monomeric peptides or proteins into amyloid fibrils is associated with a number of degenerative conditions, notably, Alzheimer’s and Parkinson’s disease (1, 3), in which considerable evidence now implicates soluble oligomers as the primary cause of cellular damage (4, 5). Identifying and characterizing the structural changes that occur during peptide assembly into amyloid fibrils is essential in the quest to develop strategies to combat disease and manufacture bespoke materials (1, 6).
Peptide assembly into amyloid fibrils occurs via a complex nucleation-dependent mechanism, in which subtle changes in lowly populated states can have marked effects on the rates and products of assembly (7). Elegant work has resulted in kinetic models that are able to dissect the different contributing steps in assembly, including primary nucleation, elongation, fragmentation, and secondary nucleation (8–11). Secondary nucleation is the process whereby transient binding to a fibril surface accelerates aggregation by promoting the formation of nuclei on the fibril surface. The activation energy barrier for this phase of aggregation for Aβ42 has been shown to be enthalpic (11) and distinct from that of other kinetic phases of assembly. Secondary nucleation is thought to be a specific process in which the effectiveness of nucleation can depend on the sequence and morphology of both the fibril and the assembling monomers, although the “rules” defining this specificity have yet to be elucidated. However, elucidating structural insights into these different steps in assembly, including the nature of early oligomeric species, is challenging, as circular dichroism (CD), infrared, and other spectroscopic techniques generally only observe population-average data for a whole system. Single-molecule Förster resonance energy transfer (FRET) and solid-state nuclear magnetic resonance (NMR), which have uncovered clues as to the structure of toxic versus nontoxic oligomeric species (12, 13), provide information on the average properties of the different species at different times. Native ion mobility spectrometry–mass spectrometry (IMS-MS) separates ions based on shape as well as mass and charge (14) and has been used to provide insights into the population, conformation, and ligand-binding capability of individual peptide monomers and oligomers (15). By using photo-induced cross-linking (PIC), fleeting interpeptide/intrapeptide interactions may be trapped through covalent bond formation (to encode supramolecular connectivity) (16). Molecular dynamics (MD) simulations focusing on multipeptide systems at short time scales (<1 ms) (17) can help fill the gaps between population-average data and individual structures. Such simulations can provide insights into self-assembly events in molecular detail, allowing the earliest stages of aggregation to be visualized and the course of aggregation to be tracked in all-atom detail (18–20).
The amyloid-β peptide (Aβ) is a major component of the extracellular plaques observed in Alzheimer’s disease (5, 21). Aggregation of Aβ40/42 (Fig. 1A) into amyloid fibrils has been widely studied both in vitro and in vivo (22), although numerous questions remain about its structure and role in Alzheimer’s disease progression (1, 22). Kinetic analysis of the sigmoid growth curves of Aβ40/42 aggregation has enabled their assembly mechanisms to be deconvoluted into a number of microscopic steps (7, 10). Assembly begins with a lag phase, during which time monomers and small amounts of oligomers persist (7). Monomers then undergo a rearrangement step to form a nucleus (primary nucleation) from which fibrils can grow. Further aggregate growth occurs through pathways that include elongation (whereby a monomeric peptide adds onto the end of a growing fibril), fragmentation (fibrils break into two smaller aggregates, exponentially increasing growth-competent fibril ends), and surface-catalyzed secondary nucleation (whereby nucleation is catalyzed on the fibril surface) (23). Using MD simulations, the energy landscape of Aβ40 oligomer formation has also been modeled, demonstrating the different kinetic pathways that underlie the formation of prefibrillar and nonfibrillar oligomers (17). For Aβ40, primary nucleation has been shown to be a slower process than secondary pathways such that surface-catalyzed secondary nucleation events dominate the growth rate of fibrils (10). Under quiescent conditions, the contribution of fibril fragmentation to the growth of fibrils has been shown to be negligible (9). Co-aggregation processes (i.e., where two different peptide sequences interact during aggregation but need not co-assemble) can result in more complex kinetics due to the possibility of the sequences interacting with each other to modulate aggregation (24, 25). Such a situation may occur in vivo wherein multiple sequences of different lengths of Aβ are formed (26).
Fig. 1. Co-aggregation of Aβ16–22 and Aβ40 results in accelerated aggregation kinetics for Aβ40.
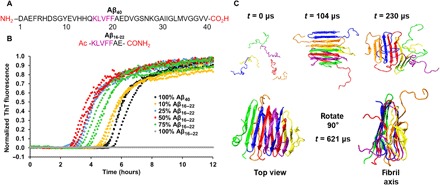
(A) Primary sequence of Aβ16–22 and Aβ40, including the groups at each terminus. The central recognition motif KLVFF is highlighted in purple. (B) ThT fluorescence assays showing that the aggregation rate of Aβ40 increases as the ratio of Aβ16–22 to Aβ40 is increased (with the total peptide concentration held constant at 40 μM). (C) Simulation snapshots of the aggregation of six Aβ40 monomers into a β-sheet–rich hexamer at an Aβ40 concentration of 5 mM. At the start of the simulation (0 μs), all the peptides are in random coils, but as the simulation progresses, they aggregate into antiparallel, in-register β sheets (104 μs). This oligomer then unfolds, losing some of its β-sheet structure (230 μs) before a rearrangement in which the β sheets rearrange, forming a stable fibril, with each Aβ40 peptide containing three β strands (621 μs) engaged in parallel intermolecular hydrogen bonding.
Here, we combine fluorescence assays, electrospray ionization (ESI)–IMS–MS, and PIC experiments to study the structural mechanism of co-assembly of the peptide fragment Aβ16–22 (Fig. 1A), which contains the “core recognition motif” KLVFF (27) of Aβ40, with the parent Aβ40 sequence. Aβ16–22 has been shown to form fibrils with an in-register, antiparallel orientation at neutral pH (28) and has been proposed to assemble via an intermediate with out-of-register β-sheet alignment before reaching the final in-register fibril structure (29). The rate of Aβ16–22 aggregation is dependent on peptide concentration and ionic strength (29–31). Discontinuous MD (DMD) has also shown that the nucleation-dependent aggregation process of Aβ16–22 proceeds from a random coil configuration to form multilayer β-sheet fibrils, with an in-register antiparallel β-sheet orientation, in accordance with the experimental data (32). Here we show, using fluorescence quenching assays, that Aβ16–22 aggregates more rapidly than Aβ40 and that Aβ16–22 fibril formation then increases the aggregation rate of Aβ40 through a surface-catalyzed secondary nucleation mechanism, mirroring the behavior observed in kinetic analyses of Aβ40/42 aggregation (9, 10) and their co-aggregation (24). Using DMD simulations, we also show that the preformed Aβ16–22 fibrils increase the early-stage aggregation rate of Aβ40 but that the monomeric Aβ16–22 peptides do not, supporting secondary nucleation as the mechanism of enhanced Aβ40 aggregation by Aβ16–22. These experimentally validated simulations portray the structural mechanism of surface-catalyzed nucleation. This new understanding may pave the way to the generation of surfaces able to enhance or suppress assembly and may inform effective design of ligands that modulate therapeutically important amyloid assembly.
RESULTS
Aβ16–22 increases the aggregation rate of Aβ40
To determine whether the presence of Aβ16–22 affects the aggregation rate of Aβ40, we synthesized or recombinantly expressed the peptides (see Materials and Methods, Supplementary Materials, and figs. S1 and S2), purified them, and mixed them in different ratios at a constant total peptide concentration of 40 μM. The rate of aggregation was then measured using the fluorescence of thioflavin-T (ThT; Fig. 1B, Materials and Methods, and fig. S3). Initial experiments showed the expected sigmoid increase in ThT fluorescence for Aβ40 (7, 10, 33), indicating the assembly of this peptide into amyloid fibrils (Fig. 1B). While Aβ16–22 formed fibrils under the conditions used based on transmission electron microscopy (TEM) images (Fig. 2F and fig. S4), as expected (16), ThT fluorescence did not increase over 12 hours (Fig. 1B), indicating that the fibrils formed either are unable to bind ThT or do not enhance its fluorescence when bound; rotational immobilization of ThT is required for its fluorescent enhancement when bound to amyloid fibrils (34). Other amyloid dyes (NIAD-4, Congo Red, and ANS) were screened against Aβ16–22 fibrils; however, none produced a signal with which to perform kinetic assays. The increase in ThT signal in the peptide mixture thus reports on the aggregation rate of Aβ40 and how this is affected by the presence of Aβ16–22. The experiments in Fig. 1B show that, at a constant peptide concentration of 40 μM, as the molar ratio of Aβ16–22 to Aβ40 is increased, the apparent aggregation rate of Aβ40 also increases. Competition between the increased rate of Aβ40 aggregation as Aβ16–22 concentration increases and the decreased rate of aggregation of Aβ40 as its concentration correspondingly decreases results in maximal apparent rate enhancement at a 1:1 molar ratio of the two peptides (Fig. 1B). We accounted for this effect by measuring, in parallel, the t1/2 (the time at which the growth curve reaches 50% amplitude) value of aggregation of Aβ40 alone at each concentration and comparing the t1/2 values with and without Aβ16–22 added (see fig. S3). These data show that the effect saturates as would be expected for secondary nucleation events involving binding to the fibril surface.
Fig. 2. Aggregation kinetics of Aβ16–22 are unaffected by the presence of Aβ40.
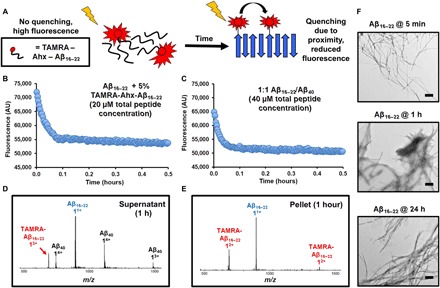
(A) Schematic showing the principle behind the fluorescence quenching assay used to determine the aggregation rate of Aβ16–22. (B) As self-assembly occurs, the TAMRA-labeled peptides [40 μM total peptide containing 5% (w/w) TAMRA-Ahx-Aβ16–22] are sequestered into the fibril structure. This brings the fluorophores into proximity, resulting in fluorescence quenching. (C) Aggregation of Aβ16–22 [containing 5% (w/w) TAMRA-Ahx-Aβ16–22] and Aβ40 at a 1:1 mol/mol ratio (40 μM total peptide concentration). A single transient, which is the median of three replicates measured, is shown. (D and E) Sedimentation and separation of the pellet and supernatant of the 1:1 mixed system and analysis of the fractions using ESI-MS after 1 hour indicate that Aβ40 is present in the (D) supernatant and in only very small amounts within the (E) pellet. (F) Under these conditions, fibrils of Aβ16–22 are present after 5 min of incubation. Scale bars, 500 nm.
To characterize the extent to which Aβ40 aggregation is accelerated by the presence of Aβ16–22, we calculated the half-time (t1/2) for each peptide mixture and normalized to the half time for the equivalent concentration of Aβ40 alone (fig. S3). The results revealed a marked, and titratable, effect of the presence of Aβ16–22 on the aggregation rate of Aβ40, demonstrating an interaction between the two peptides that accelerates the rate of assembly.
Aβ16–22 aggregates more rapidly than Aβ40 and is unaffected by the presence of Aβ40
As the assembly kinetics of Aβ16–22 could not be measured using any of the amyloid dyes surveyed at the concentrations used here, a fluorescence quenching assay was developed to determine whether Aβ16–22 aggregates more or less rapidly than Aβ40 (Fig. 2A). Similar assays have been previously used to monitor the aggregation rates of Aβ40 and Aβ42 (35), with fluorescence quenching reporting on labeled monomers coming into mutual proximity as oligomers (or fibrils) form. For these assays, Aβ16–22 N-terminally labeled with tetramethylrhodamine (TAMRA) was synthesized, including a 6-aminohexanoic acid linker (Ahx) to limit disruption to the native fibril structure that might arise due to the bulky fluorophore (TAMRA-Ahx-Aβ16–22; Supplementary Materials and figs. S1 and S4). When incubated in isolation, a 5% (w/w) TAMRA-Ahx-Aβ16–22:95% Aβ16–22 mixture (20 μM) resulted in a rapid decrease in fluorescence intensity followed by a slower phase that plateaued after 1 hour (Fig. 2B). In the presence of Aβ40 [1:1 (mol/mol) ratio, 40 μM total peptide concentration, and 2% (v/v) dimethyl sulfoxide (DMSO)], no difference in the rate of fluorescence decrease was observed, indicating that the presence of Aβ40 has no effect on Aβ16–22 aggregation (Fig. 2C). Analysis of these samples by negative-stain TEM showed the presence of fibrils after only 5 min (Fig. 2F). Sedimentation of the mixed system by centrifugation after 1 hour demonstrated that Aβ40 was present mainly in the supernatant (Fig. 2, D and E). These results demonstrate that Aβ16–22 aggregates rapidly to form amyloid-like fibrils, while Aβ40 remains soluble as monomers/oligomers. Thus, although the rate of Aβ40 aggregation is increased by the presence of Aβ16–22, limited or no co-assembly between the two peptides into fibrils was observed. By contrast, Aβ16–22 aggregation is unaffected by the presence of Aβ40. Aβ40 fibrils have been shown to adopt a parallel in-register structure involving most of the polypeptide backbone (21, 36), while Aβ16–22 has been shown to form an antiparallel β-stranded amyloid structure (28, 29). This structural incompatibility could account for the absence of co-assembly because such a structure would be less stable compared with homomeric assemblies. Furthermore, the more rapid fibril assembly of Aβ16–22 in comparison to Aβ40 disfavors co-assembly on kinetic grounds.
Monomeric Aβ16–22 can interact with monomeric and oligomeric Aβ40 through the self-recognition motif KLVFF
To determine whether Aβ16–22 and Aβ40 interact transiently in the early stages of assembly, we performed native ESI linked to IMS-MS (see Materials and Methods). This soft ionization technique has been used to identify and structurally characterize amyloid oligomers that formed from several different proteins and peptides (14, 15). Under the conditions used here, ESI-IMS-MS immediately following mixing revealed that Aβ40 copopulates a number of oligomers, ranging from monomers to pentamers (Fig. 3B, white, and table S1), consistent with previous results (33). When incubated with Aβ16–22, heteromolecular oligomers were observed (Fig. 3B, light blue), along with homomolecular oligomers of Aβ40 (Fig. 3A, white). Notably, Aβ16–22 homomolecular oligomers were not observed. The heteromolecular oligomers correspond to multiple Aβ16–22 monomers that bound to either an Aβ40 monomer or dimer (table S1). Collision cross-section (CCS) estimations from the ESI-IMS-MS analysis of the Aβ40 species in the presence or absence of Aβ16–22 indicate no discernible difference in the gas-phase cross-section of Aβ40, implying that a conformational change in monomer or oligomer structure is unlikely to be the provenance for the Aβ16–22-driven increase in the Aβ40 aggregation rate (fig. S5). Despite attempts to capture the interaction experimentally by PIC using a diazirine-labeled Aβ16–22 (Aβ*16–22; see the Supplementary Materials for synthesis, scheme S1, and fig. S1), the site of interaction could not be verified (fig. S6 and table S2) likely due to the low percentage of any heterodimers present (as assessed by total ion count, 1.0 ± 0.5%) and the lower solution concentration of Aβ16–22 arising as a consequence of its rapid aggregation.
Fig. 3. Aβ16–22 can interact with Aβ40 monomers and dimers.
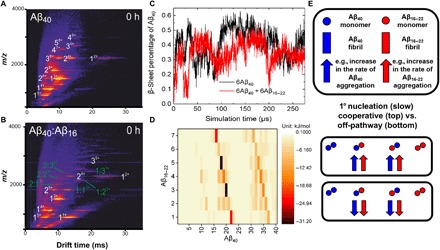
(A) Native ESI-IMS-MS drift-scope images of Aβ40 indicate the presence of multiple oligomeric species of Aβ40 (white numbers). (B) When mixed at a 1:1 mol/mol ratio with Aβ16–22 (yellow numbers), a number of heteromeric species are observed (light blue numbers) immediately following mixing. The oligomer size is given (1, 2, 3, etc.), with the charge state in superscript. (C) DMD simulation showing the percent β sheet formed by Aβ40 during aggregation in the absence (black) or presence (red) of Aβ16–22. (D) Energy contact map between one monomer of Aβ16–22 and one of Aβ40 scaled by energy (bar shown alongside), showing that residues 17 to 20 (LVFF) and 31 to 34 (IIGL) form the strongest interactions. (E) Co-aggregation can have differing effects on the primary nucleation of each peptide, depending on whether the mixed oligomers formed can progress to form mixed fibrils or are off-pathway and take no further part in the aggregation reaction. Circles represent monomers and blocks represent fibrils, with Aβ16–22 and Aβ40 in red and blue, respectively. Adapted from (24).
To further assess the nature of the interactions between Aβ16–22 and Aβ40, we performed DMD simulations (see Materials and Methods). To evaluate the role of interactions between Aβ40 and Aβ16–22 monomers (covered in this section), it was first necessary to perform DMD simulations on the aggregation of Aβ40 alone (Fig. 1C) and then a 1:1 mixture of the Aβ16–22 and Aβ40 peptide sequences at Cpeptide = 5 mM (Fig. 3C). These co-aggregation simulations starting from monomeric peptides are further discussed in the course of our analyses to rule out co-assembly (see below), and then we describe DMD analyses on the effect of Aβ16–22 fibrils on Aβ40 aggregation (see below). Simulations performed on six monomers of Aβ40 (Fig. 1C) showed that the initially unstructured peptides assemble and adopt a metastable oligomer structure by 104 μs (Fig. 1C); this structure comprises antiparallel intramolecular β strands linked by disordered regions assembled into antiparallel intermolecular sheets, with β strands stacked perpendicular to the long axis. During this oligomerization stage, the peptide conformation is similar to that observed by Zheng et al. (17). As the simulation proceeds, this oligomer loses some β-sheet content (t = 230 μs; Fig. 1C). By the end of the simulation (621 μs), peptides in oligomers undergo structural rearrangement from antiparallel β-strand conformations to the parallel β-sheet conformation observed for Aβ40 fibrils (Fig. 1C) (37). Simulations of the peptide mixtures did not show an accelerating effect of Aβ16–22 monomers on the aggregation rate of Aβ40 (see Fig. 3C and later). However, interactions between the two peptides were observed, consistent with the ESI-MS results in Fig. 3. From the DMD data, an energy contact map between the monomeric Aβ16–22 and Aβ40 peptides was calculated (Fig. 3D). The contact map indicated that Aβ16–22, specifically residues 18 to 20 (VFF), interacts strongly with residues 19 to 21 and 32 to 35 of Aβ40 (FFA and IGLM, respectively), consistent with experimental data previously reported, which indicate that KLVFF is a “self-recognition element” (27). Such an interaction between Aβ16–22 and Aβ40 oligomers, however, does not result in an acceleration of aggregation (Fig. 3C), implying that these mixed and low-abundance oligomers represent transient species that do not affect the rate of assembly (Fig. 3E).
Aβ16–22 fibrils have a larger effect on the aggregation rate of Aβ40 than Aβ16–22 monomer
To determine whether rapidly formed Aβ16–22 fibrils are the causative agents of the enhanced rate of Aβ40 aggregation in the mixed samples (Fig. 1B), we assessed the effect of preformed Aβ16–22 fibrils on Aβ40 aggregation. These experiments (Fig. 4A) showed that the presence of Aβ16–22 fibrils increases the rate of aggregation of Aβ40 in a fibril concentration–dependent manner (Fig. 4A), and addition of Aβ16–22 fibrils had a larger effect on the aggregation rate compared with the addition of monomeric (i.e., taken straight from a DMSO stock) Aβ16–22 (Fig. 4B). This suggests that aggregation is enhanced either by cross-seeding (i.e., by adding Aβ40 directly to the ends of Aβ16–22 fibrils) or by secondary nucleation of Aβ40 on the Aβ16–22 fibril surface (Fig. 4E). Sonication of fibrils fragments them, leading to a higher concentration of fibril ends. Hence, should elongation dominate the rate of fibril formation, sonication should markedly increase the rate of fibril growth. Comparison of the effects of unsonicated fibrils (fewer ends) with the same fibrils fragmented by sonication (Fig. 4C and see fig. S4 for TEM analyses) indicated that elongation was not dominant (Fig. 4C), because the average t1/2 for sonicated fibrils (6.2 ± 1.0 hour) is similar to that of its unfragmented counterpart (7.2 ± 0.7 hours). Together, the results demonstrate that the presence of rapidly formed Aβ16–22 fibrils enhances aggregation of Aβ40 in peptide mixtures by secondary nucleation, despite the presence of small amounts of mixed oligomers (as demonstrated by the ESI-IMS-MS experiments).
Fig. 4. Aβ16–22 fibrils increase the aggregation rate of Aβ40 to a greater extent than Aβ16–22 monomers.
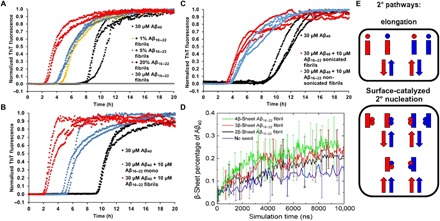
(A) Increased concentrations (% w/w) of Aβ16–22 fibrils were added to Aβ40 monomers (as shown in the key), and the aggregation rate was measured by ThT fluorescence. (B) Direct comparison of the effect of Aβ16–22 monomers (i.e., taken straight from a DMSO stock) and Aβ16–22 fibrils on Aβ40 aggregation. (C) Effect of sonicating the Aβ16–22 fibrils on the Aβ40 aggregation rate shows little effect compared with the data shown in (A) (see text for details). (D) Plots of the percent β sheet formed by Aβ40 in the absence (blue) or presence of preformed two (black), three (red), or four (green) β-sheet Aβ16–22, determined using DMD, showing that an increased Aβ16–22 fibril size increases the rate of Aβ40 aggregation. (E) During co-aggregation experiments, both elongation and surface-catalyzed mechanisms can occur; each has a different effect on the rate of assembly of each peptide (the same notation is used as in Fig. 3E, with circles representing monomers, blocks representing fibrils, and Aβ16–22 and Aβ40 in red and blue, respectively). Adapted from (24).
DMD simulations of the aggregation of six Aβ40 peptides were also performed in the presence of preformed Aβ16–22 fibrils of different sizes (two, three, and four β sheets) at an Aβ40 concentration of 1 mM to model the dynamic process of the secondary nucleation event. The results (Fig. 4D) showed that the largest Aβ16–22 fibril (i.e., four β sheets; green trace in Fig. 4D) led to the largest increase in the rate of β-sheet formation by Aβ40. Given that the presence of Aβ16–22 monomers had no observable effect on Aβ40 assembly (Fig. 3C), these simulations are thus qualitatively concordant with the experimental findings that the fibrillar structure of Aβ16–22 is the dominant influence on the aggregation rate of Aβ40. Such behavior is consistent with that observed for Aβ40/42 co-aggregation for which a kinetic model has been established (24).
Aβ40 and Aβ16–22 form distinct homomolecular fibrils
The peptide composition of the final fibril structure(s) represents a further means to discern the difference between surface-catalyzed secondary nucleation and co-assembly exploiting fibril ends. A surface-catalyzed mechanism would most likely produce homomolecular fibrils of Aβ40, as once they have formed on the Aβ16–22 fibril surface, the Aβ40 nuclei would dissociate and form pure Aβ40 fibrils. In contrast, co-assembly involving fibril ends should result in mixed fibrils, in which Aβ16–22 seeds are segmentally separated from fibril regions containing Aβ40 monomers.
Negative-stain TEM images taken at the end of the aggregation reaction showed Aβ40 fibrils with similar gross morphology when incubated in isolation or co-aggregated with Aβ16–22 (Fig. 5, A and B). Similarly, quantitation of ThT fluorescence at the end-point of aggregation in mixed samples and quantitation of the same concentration of Aβ40 incubated alone were indistinguishable (fig. S3), supporting the hypothesis that homomolecular Aβ40 fibrils are formed at the end of the assembly reaction. Last, PIC was used to explore whether homo- or heteromolecular fibrils had formed (Fig. 5C and Materials and Methods). To perform PIC experiments, a diazirine label was placed on F20 of Aβ16–22 (Aβ*16–22) (16). Control experiments demonstrated that Aβ*16–22 has a similar effect on the rate of Aβ40 aggregation as its unmodified counterpart (fig. S3). PIC experiments performed 5 min and 24 hours after initiating assembly failed to detect cross-links between Aβ*16–22 and Aβ40 (Fig. 5C, fig. S6, and table S2). Instead, all identifiable cross-links were consistent with intermolecular/intramolecular Aβ*16–22 or solvent adducts, as previously identified in Aβ*16–22-containing fibrils by Preston et al. (16), indicating that co-assembly into fibrils either is very rare and cannot be detected despite the sensitivity of ESI-MS or does not occur.
Fig. 5. Aβ16–22and Aβ40do not co-assemble during co-aggregation.
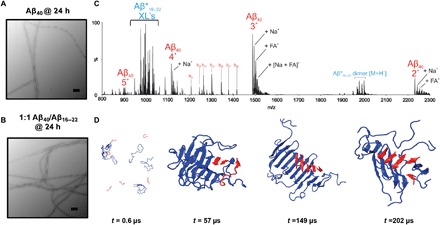
Negative-stain TEM analysis of Aβ40 incubated for 24 hours in the (A) absence or (B) presence of Aβ16–22. Scale bars, 200 nm. (C) PIC of mixtures of diazirine-labeled Aβ16–22 (Aβ*16–22) and Aβ40 incubated for 24 hours and then irradiated for 30 s. Only homomolecular Aβ16–22 cross-links are observed, indicating that the fibrils are not copolymerized at the end of the reaction (the inset depicts the mechanism of PIC of the diazirine group. (D) DMD simulation snapshots of co-aggregation of Aβ40 (blue) and Aβ16–22 (red) indicate that separate homomolecular oligomers are formed at t = 202 μs.
To provide a molecular image of co-assembly, we further analyzed the DMD simulations in which six Aβ40 and six Aβ16–22 monomers were mixed and their aggregation behavior was monitored versus time at Cpeptide = 5 mM (Fig. 5D). The simulations showed that in the early stages of assembly (t = 0.6 μs), a mixture of monomeric and oligomeric Aβ40 was present. As the simulation progressed (t = 57 μs), all Aβ40 peptides coalesced into one β-sheet–rich oligomer, with Aβ16–22 intercalated within the structure. Throughout the simulation, monomeric Aβ16–22 was observed to bind transiently to other monomeric Aβ16–22 peptides or the KLVFF motif of Aβ40, in accordance with the data presented above. Last, at the end of the simulation (t = 202 μs), the peptides form distinct oligomeric domains, with Aβ40 and Aβ16–22 forming separate sheets.
Aβ40 oligomer dynamics on the surface of Aβ16–22 fibrils
To obtain a molecular image of the process of secondary nucleation, we performed DMD simulations, in which six Aβ40 monomers were mixed with preformed fibrils of Aβ16–22 at CAβ40 = 5 mM (Fig. 6A). At the early stage of the simulation (t = 0.29 μs), three Aβ40 peptides were present in an oligomer: One Aβ40 peptide was associated at the end of the fibril, and the remaining two Aβ40 peptides were elongated across the fibril surface. At this stage (t = 0.29 μs), the Aβ40 peptides in the oligomer and on the surface were observed to adopt a predominantly random coil conformation with small amounts of β-strand structure [note that an elongated monomeric structure was also observed in simulations performed by Barz and Strodel (19) in exploring the secondary nucleation of Aβ42 on the surface of Aβ11–42]. The β sheets were next observed to act as templates for peptides present in a random coil conformation (1.93 μs) and to pull them more fully to the fibril surface. Thus, as the simulation progressed, the Aβ40 peptides remaining in solution were recruited by those on the fibril surface. Once the oligomer became fully associated with the fibril surface, the amount of β-sheet structure in the surface-associated oligomer increased (t = 7.7 μs); antiparallel β strands formed via inter- and intramolecular hydrogen bonding, leading to sheet formation consistent with the early stages observed in the simulations performed for Aβ40 alone (t = 104 μs; Fig. 1C). Last, the surface-associated Aβ40 peptides were joined in an ordered oligomer (t = 29.0 and 77.7 μs). Related “bind and reorganize” processes for secondary nucleation were observed in simulations performed by Schwierz et al. (38) using Aβ9–40 as a model. As noted above, Aβ40 peptides attached both to the lateral surface and to the end of the Aβ16–22 fibril during the simulation, with the Aβ40 C-terminal region attaching more frequently to the lateral surface of the fibril than to the fibril ends at C = 5 mM (fig. S7). To assess the consistency of the results, we repeated the simulation three times; two of the three independent runs gave results similar to those described above, while for the final run, a greater number of associations to the fibril end were observed. Collectively, these results provide molecular images of surface-catalyzed nucleation, in which a random coil peptide is catalytically converted into a β-sheet fibrillar structure on a fibril surface.
Fig. 6. Aβ16–22fibrils catalyze Aβ40assembly through secondary surface nucleation.
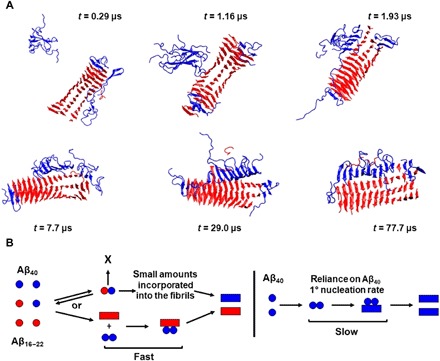
(A) Simulation snapshots of the process by which Aβ16–22 fibrils (red) increase the aggregation rate of Aβ40 (blue) through a surface-catalyzed secondary nucleation. (B) A schematic description of the mechanism is also included, with Aβ40 in blue and Aβ16–22 in red.
DISCUSSION
In this work, we used ESI-MS, PIC, and DMD to study the co-assembly mechanism of Aβ16–22 and Aβ40 into amyloid, demonstrating the power of using integrated approaches to study structural determinants of molecular assembly processes. We show that mixed Aβ16–22/Aβ40 heteromeric oligomers form but that these are transient and lowly populated (~1%) and do not significantly affect the rate of aggregation. In contrast, Aβ16–22 has a high propensity to self-associate into homomolecular fibrils, and these fibrils accelerate Aβ40 assembly by monomer/oligomer interactions through secondary nucleation at the fibril surface. Recent modeling of amyloid assembly kinetics has revealed the importance of primary nucleation, secondary nucleation, and fibril elongation in fibril growth mechanisms (7, 10, 23). Notably, a kinetic model has been described for the co-aggregation of Aβ40/42 (24). The experimental data presented here for co-aggregation of Aβ16–22 and Aβ40 qualitatively agree with this model, whereas our DMD simulations illustrate that while all primary/secondary nucleation and elongation processes occur simultaneously, secondary nucleation is the dominant process in Aβ40 fibril formation kinetics during co-assembly with Aβ16–22, which is consistent with the findings for the self-assembly mechanism of Aβ40 observed previously (9, 10, 24). Moreover, Aβ40 assembly intermediates on the surface of Aβ16–22 fibrils resemble those formed spontaneously in solution for Aβ40 alone, implying that the fibril surface catalyzes the assembly reaction without modifying the molecular mechanism, at least for the simulations performed here. Whether this holds for other sequences and co-assembly reactions will require further exploration (notably, which features of a fibril and the assembling monomer determine compatibility with secondary nucleation from a fibril surface).
Overall, the current study thus serves to emphasize the marked differences in aggregation behavior that are observed during co-aggregation compared to homomolecular self-assembly and underscores the need to use multiple methods to understand aggregation mechanisms in molecular detail. Significant current interest centers on characterizing distinct molecular steps leading to amyloid fibril formation, with secondary nucleation considered as playing a key role in causing toxicity (11, 39). Recently, kinetic analyses have been augmented by mapping the free-energy landscapes defining different microscopic phases in the aggregation pathway (11), providing insight to facilitate development of strategies that modulate the thermodynamically distinct surface-monomer interactions characteristic of secondary nucleation. However, to design therapeutically useful modulators of amyloid aggregation requires that this understanding is complemented with structural insights of the molecular recognition between fibrils and monomers, set within the context of other interactions occurring during aggregation (e.g., monomer-nuclei interactions). We have shown here that Aβ40 monomers and oligomers dock onto the fibril surface, which catalyzes the assembly of antiparallel strand formation in close situ to the parent Aβ16–22 fiber. Whether this is the end-point product or further reorganization is required to generate the final amyloid structure requires further study (longer simulation time). In this context, metastable amyloid structures have been observed for the Iowa mutant of Aβ40 using solid-state NMR, in which antiparallel fibrils were observed as trapped intermediates in the assembly process to the final all-parallel fibril structure (40).
Together, the results demonstrate that kinetic analyses and theory together with MD provide a powerful arsenal and capability to visualize secondary nucleation in structural and kinetic detail. Such approaches may allow informed targeting of this process to either prevent or accelerate secondary nucleation for therapeutic purposes and peptide material assembly. Co-aggregation adds an additional layer of complexity in understanding molecular assembly yet represents an opportunity to manipulate these supramolecular assembly processes, as demonstrated here for the model system involving Aβ16–22 and Aβ40. Evidently, Aβ40 shows a propensity to aggregate via secondary nucleation from its own fibril surface or that of other peptide sequences, as shown here for fibrils of Aβ16–22. Hence, this work begins to address the molecular recognition events required for secondary nucleation to occur on a fibril surface and may inform strategies to modulate the aggregation of Aβ40 under conditions in which secondary nucleation dominates fibril growth.
MATERIALS AND METHODS
Synthesis of N-Fmoc TFMD-Phe and Aβ peptides
N-Fmoc TFMD-Phe was synthesized using the method described by Smith et al. (41) and further minor changes in protecting group (scheme S1). Aβ16–22, TAMRA-Ahx-Aβ16–22, and Aβ*16–22 were synthesized via both automated and manual solid-phase peptide synthesis and dissolved into DMSO stock solutions before use (fig. S1). Aβ40 was synthesized recombinantly using the method of Walsh et al. (42) and modifications by Stewart et al. (43). To ensure that Aβ40 was monomeric before use, the peptide was purified by size exclusion chromatography, lyophilized, and stored at −4°C (fig. S2).
ThT fluorescence assays
Samples were prepared in a 96-well nonbinding plate (Corning Costar 3881, Corning Life Sciences, Amsterdam, the Netherlands) sealed with clear sealing film (BMG Labtech, Aylesbury, Bucks, UK) and were incubated in a FLUOstar OPTIMA plate reader (BMG Labtech, Aylesbury, Bucks, UK) for 20 hours at 37°C without agitation. Samples had a volume of 95 μl containing 10 μM ThT in 100 mM ammonium bicarbonate (pH 7.4) and a final concentration of 1% (v/v) DMSO. For seeding experiments, Aβ16–22 was incubated at 50 μM for at least 24 hours in the same buffer as described above, with the presence of fibrils confirmed by TEM (described below). Before the assay, the fibrils were probe-sonicated for 5 s at 22% amplitude to generate “seeds.” The ThT experiments used excitation and emission filters of 430 and 485 nm. Each ThT experiment shown was repeated in independent assays on three different occasions, with the traces shown in this work being representative of all repeats.
Transmission electron microscopy
TEM images were taken at the end of each experiment by removing 5 μl from the necessary well and incubating this sample on carbon-formvar grids for 30 s before staining with 2% (w/v) uranyl acetate solution for an additional 30 s, as described by Preston et al. (27). Images were taken on a JEM-1400 (JEOL Ltd., Tokyo, Japan) or a Tecnai F12 TEM. Images were taken on a JEM-1400 (JEOL Ltd., Tokyo, Japan) or a Tecnai T12 (FEI, Hillsboro, OR, USA) TEM. Images were taken using either an ATM charge-coupled device (CCD) camera or a Gatan UltraScan 1000 XP (994) CCD camera (JEM-1400) or an UltraScan 100XP (994) CCD camera (Tecnai F12). Once taken, images were processed using ImageJ [National Institutes of Health (NIH)].
General sedimentation protocol
Samples were taken at the desired time point and centrifuged (20 min, 14,000g, 4°C). Each sample was then separated into pellet and supernatant fractions, lyophilized overnight, and disaggregated in hexafluoroisopropanol (HFIP) for at least 2 hours. HFIP was removed under a stream of N2, and the peptides were taken up in DMSO before analysis by high-resolution MS (Bruker HCT ion-trap MS).
Fluorescence quenching assays
Wild-type Aβ16–22 was spiked with 5% (w/w) TAMRA-Ahx-Aβ16–22 and incubated either in isolation or at a 1:1 ratio with Aβ40 (total peptide concentration, 40 μM) in 100 mM ammonium bicarbonate buffer (pH 7.4) with a final concentration of 2% (v/v) DMSO. Samples were placed in quartz cuvettes and analyzed using a temperature-controlled fluorimeter at 37°C. Time points were taken every 30 s for the duration of the experiment, and TEM images (as described above) were taken at the end of each experiment to ensure the presence of fibrils. The TAMRA fluorophore was excited at 520 nm, and emission was recorded at 600 nm to reduce the inner filter effect.
ESI-IMS-MS analysis
All samples were prepared as described above and left to incubate at 37°C without agitation for 5 min. A SYNAPT HDMS quadrupole time-of-flight MS (Micromass UK Ltd., Waters Corp., Manchester, UK), equipped with a TriVersa NanoMate (Advion Biosciences, Ithaca, NY, USA) automated nano-ESI interface, was used in this study. The instrument has a traveling-wave IMS device situated in between the quadrupole and the time-of-flight analyzers, as described in detail elsewhere. Samples were analyzed by positive ionization nano-ESI, with a capillary voltage of 1.4 kV and a nitrogen-nebulizing gas pressure of 0.8 psi. The following instrumental parameters were set: cone voltage, 60 V; source temperature, 60°C; backing pressure, 4.7 mbar; ramped traveling speed, 7 to 20 V; traveling wave speed, 400 m s−1; IMS nitrogen gas flow, 20 ml min−1; IMS cell pressure, 0.55 mbar. The mass/charge ratio (m/z) scale was calibrated using aq. CsI cluster ions. CCS measurements were estimated using a calibration obtained by analysis of denatured proteins (cytochrome c, ubiquitin, and alcohol dehydrogenase) and peptides (tryptic digests of alcohol dehydrogenase and cytochrome c), with known CCSs obtained elsewhere from drift tube ion mobility measurements (15, 33). Data were processed using MassLynx v4.1 and Driftscope software supplied with a mass spectrometer.
Photo-induced covalent cross-linking
A 1:1 ratio of Aβ16–22/Aβ*16–22 or Aβ*16–22/Aβ40 (40 μM total peptide concentration) in 100 mM ammonium bicarbonate buffer (pH 7.4) with a final concentration of 1% (v/v) DMSO was incubated in Eppendorf tubes for either 5 min or 24 hours. Samples were then irradiated for 30 s using a homebuilt light-emitting diode lamp at 365 nm, then removed, lyophilized overnight, taken up in HFIP for at least 2 hours, and vortexed to ensure that any aggregates were disrupted. HFIP was then removed under a stream of N2, and the sample was resuspended in 50:50 (v/v) MeCN/H2O + 0.05% formic acid to a final concentration of ~40 μM. Any cross-links were then analyzed using the method previously described and the ESI-IMS-MS system as described above (16).
DMD and PRIME20 force field
The simulation approach applied in this work is DMD, a fast alternative to traditional MD, in combination with the PRIME20 force field, a four-bead-per-residue coarse-grained protein model developed by the Hall group (44). In the PRIME20 model, each of the 20 different amino acids contains three backbone spheres (NH, CαH, and CO) and one side-chain sphere (R) with a distinct hard sphere diameter (effective van der Waals radius) and distinct side chain–to–backbone distances (R-CαH, R-NH, and R-CO). The backbone hydrogen bonding interaction is modeled as a directional square well potential. In the original PRIME20 force field, the potential function between any two side-chain beads on the 20 different amino acids (except glycine) is modeled as a single-well potential, containing 210 different square well widths and 19 different square well depths using the 5.5 Å heavy atom criteria. In this work, we follow Cheon’s approach to apply a double-square well potential instead of the single-square well for side chain–side chain interaction (45). All the other nonbonded interactions are modeled as hard sphere interactions. A detailed description of the derivation of the geometric and energetic parameters of the PRIME20 model is given in our earlier work (46).
Simulation procedure
DMD/PRIME20 simulations were performed on the following systems: (i) six Aβ40 monomeric peptides; (ii) six monomeric Aβ40 peptides with six monomeric Aβ16–22 peptides; and (iii) six Aβ40 monomeric peptides in the presence of preformed two, three, and four β-sheet Aβ16–22 protofilaments. The two, three, and four β-sheet Aβ16–22 protofilaments contain 21, 42, and 71 peptides, respectively. Each simulation was performed at two different total peptide concentrations (1 and 5 mM). Similar seeding simulations were performed in a previous work (45). The simulations were performed in the canonical ensemble (fixed number of particles, volume, and temperature). The reduced temperature was defined to be T* = kBT/εHB, where the hydrogen bonding energy εHB = 12.47 kJ/mol. The reduced temperature is related to real temperature by using the equation T/K = 2288.46T* − 115.79. The reduced temperature T* is chosen to be 0.20, which corresponds to a real temperature of 342 K. The system was maintained at a constant temperature by applying the Andersen thermostat. We performed 3 to 10 independent runs for each system.
Supplementary Material
Acknowledgments
We thank M. Iadanza for help with taking and processing the EM images. We also thank N. Khan for excellent technical support and members of our laboratories for excellent discussions during the course of the work. Funding: This work was supported by NIH grant R01 EB006006 and, in part, by National Science Foundation (NSF) Research Triangle Materials Research Science and Engineering Centers (MRSEC) grant DMR-1121107, NSF grant CBET 581606, and EPSRC grants EP/N035267/1, EP/N013573/1, and EP/KO39292/1. S.E.R. acknowledges funding from the ERC (grant agreement number 322408) and the Wellcome Trust (204963). S.J.B. gratefully acknowledges BBSRC for PhD studentship grant BB/J014443/1. The EM was purchased with funding from the Wellcome Trust (108466/Z/15/Z and 094232/Z/10/Z). Y.W. and C.K.H. gratefully acknowledge the support of a Cheney Visiting Scholar Fellowship from University of Leeds. Author contributions: S.J.B. performed the experimental work; Y.W. performed simulations; K.L.S. performed expression and purification of Aβ40; S.E.R., C.K.H., and A.J.W. designed the research; S.J.B., Y.W., A.E.A., S.E.R., C.K.H., and A.J.W. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data available from authors upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav8216/DC1
General materials and methods for organic synthesis
Synthesis of N-Fmoc–protected TFMD-Phe
General materials and methods for Aβ16–22 solid-phase peptide synthesis
General materials and methods for HPLC purification
Analytical MS and HPLC data for synthetic peptides
General materials and methods for recombinant peptide synthesis
Additional characterization and analyses
CCS analysis of Aβ40 in the presence and absence of Aβ16–22
Scheme S1. Synthesis of TFMD-Phe.
Fig. S1. HRMS and analytical HPLC traces of Aβ16–22 and its variants.
Fig. S2. SEC trace of Aβ40 indicates that there is a single peak, and ESI-IMS-MS indicates that in the gas phase Aβ40 is largely monomeric.
Fig. S3. Supplementary ThT data.
Fig. S4. Supplementary negative-stain TEM images.
Fig. S5. Analysis of the CCS values for Aβ40 in the absence or presence of Aβ16–22 over different IMS experiments.
Fig. S6. PIC analysis of 1:1 Aβ*16–22/Aβ40 at 5 min and 24 hours.
Fig. S7. Plot of the average number of hydrogen bonding and side chain–side chain.
Table S1. The expected and observed m/z values for monomeric and oligomeric Aβ40 in isolation and in the presence of a 1:1 ratio of Aβ16–22.
Table S2. Assignments of each of the major peaks observed in fig. S6A.
Data file S1. MD snapshots as pdb files Fig. 1 t = 0.
Data file S2. MD snapshots as pdb files Fig. 1 t = 104.
Data file S3. MD snapshots as pdb files Fig. 1 t = 230.
Data file S4. MD snapshots as pdb files Fig. 1 t = 621.
Data file S5. MD snapshots as pdb files Fig. 6 t = 0.29.
Data file S6. MD snapshots as pdb files Fig. 6 t = 1.16.
Data file S7. MD snapshots as pdb files Fig. 6 t = 1.93.
Data file S8. MD snapshots as pdb files Fig. 6 t = 7.7.
Data file S9. MD snapshots as pdb files Fig. 6 t = 29.
Data file S10. MD snapshots as pdb files Fig. 6 t = 77.7.
REFERENCES AND NOTES
- 1.Knowles T. P. J., Vendruscolo M., Dobson C. M., The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Adler-Abramovich L., Gazit E., The physical properties of supramolecular peptide assemblies: From building block association to technological applications. Chem. Soc. Rev. 43, 6881–6893 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Iadanza M. G., Jackson M. P., Hewitt E. W., Ranson N. A., Ranson S., A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 19, 755–773 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Kayed R., Head E., Thompson J. L., McIntire T., Milton S. C., Cotman C. W., Glabe C. G., Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Haass C., Selkoe D. J., Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Eisenberg D., Jucker M., The amyloid state of proteins in human diseases. Cell 148, 1188–1203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen S. I. A., Vendruscolo M., Dobson C. M., Knowles T. P. J., From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 421, 160–171 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Xue W.-F., Homans S. W., Radford S. E., Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc. Natl. Acad. Sci. U.S.A. 105, 8926–8931 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen S. I. A., Linse S., Luheshi L. M., Hellstrand E., White D. A., Rajah L., Otzen D. E., Vendruscolo M., Dobson C. M., Knowles T. P. J., Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 9758–9763 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meisl G., Yang X., Hellstrand E., Frohm B., Kirkegaard J. B., Cohen S. I. A., Dobson C. M., Linse S., Knowles T. P. J., Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc. Natl. Acad. Sci. U.S.A. 111, 9384–9389 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen S. I. A., Cukalevski R., Michaels T. C. T., Šarić A., Törnquist M., Vendruscolo M., Dobson C. M., Buell A. K., Knowles T. P. J., Linse S., Distinct thermodynamic signatures of oligomer generation in the aggregation of the amyloid-β peptide. Nat. Chem. 10, 523–531 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fusco G., Chen S. W., Williamson P. T. F., Cascella R., Perni M., Jarvis J. A., Cecchi C., Vendruscolo M., Chiti F., Cremades N., Ying L., Dobson C. M., de Simone A., Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 358, 1440–1443 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Varela J. A., Rodrigues M., de S., Flagmeier P., Gandhi S., Dobson C. M., Klenerman D., Lee S. F., Optical structural analysis of individual α-synuclein oligomers. Angew. Chem. Int. Ed. 57, 4886–4890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernstein S. L., Dupuis N. F., Lazo N. D., Wyttenbach T., Condron M. M., Bitan G., Teplow D. B., Shea J. E., Ruotolo B. T., Robinson C. V., Bowers M. T., Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem. 1, 326–331 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young L. M., Tu L.-H., Raleigh D. P., Ashcroft A. E., Radford S. E., Understanding co-polymerization in amyloid formation by direct observation of mixed oligomers. Chem. Sci. 8, 5030–5040 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Preston G. W., Radford S. E., Ashcroft A. E., Wilson A. J., Covalent cross-linking within supramolecular peptide structures. Anal. Chem. 84, 6790–6797 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Zheng W., Tsai M.-Y., Chen M., Wolynes P. G., Exploring the aggregation free energy landscape of the amyloid-β protein (1-40). Proc. Natl. Acad. Sci. U.S.A. 113, 11835–11840 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gurry T., Stultz C. M., Mechanism of amyloid-β fibril elongation. Biochemistry 53, 6981–6991 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Barz B., Strodel B., Understanding amyloid-β oligomerization at the molecular level: The role of the fibril surface. Chem. A Eur. J. 22, 8768–8772 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Barz B., Liao Q., Strodel B., Pathways of amyloid-β aggregation depend on oligomer shape. J. Am. Chem. Soc. 140, 319–327 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Qiang W., Yau W.-M., Lu J.-X., Collinge J., Tycko R., Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature 541, 217–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Strooper B., Karran E., The cellular phase of Alzheimer’s disease. Cell 164, 603–615 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Törnquist M., Michaels T. C. T., Sanagavarapu K., Yang X., Meisl G., Cohen S. I. A., Knowles T. P. J., Linse S., Secondary nucleation in amyloid formation. Chem. Commun. 54, 8667–8684 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Cukalevski R., Yang X., Meisl G., Weininger U., Bernfur K., Frohm B., Knowles T. P. J., Linse S., The Aβ40 and Aβ42 peptides self-assemble into separate homomolecular fibrils in binary mixtures but cross-react during primary nucleation. Chem. Sci. 6, 4215–4233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarell C. J., Stockley P. G., Radford S. E., Assessing the causes and consequences of co-polymerization in amyloid formation. Prion 7, 359–368 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jucker M., Walker L. C., Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tjernberg L. O., Näslund J., Lindqvist F., Johansson J., Karlström A. R., Thyberg J., Terenius L., Nordstedt C., Arrest of β-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 271, 8545–8548 (1996). [DOI] [PubMed] [Google Scholar]
- 28.Balbach J. J., Ishii Y., Antzutkin O. N., Leapman R. D., Rizzo N. W., Dyda F., Reed J., Tycko R., Amyloid fibril formation by Aβ16-22, a seven-residue fragment of the Alzheimer’s β-amyloid peptide, and structural characterization by solid state NMR. Biochemistry 39, 13748–13759 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Hsieh M. C., Liang C., Mehta A. K., Lynn D. G., Grover M. A., Multistep conformation selection in amyloid assembly. J. Am. Chem. Soc. 139, 17007–17010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petty S. A., Decatur S. M., Experimental evidence for the reorganization of β-strands within aggregates of the Aβ(16-22) peptide. J. Am. Chem. Soc. 127, 13488–13489 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Senguen F. T., Lee N. R., Gu X., Ryan D. M., Doran T. M., Anderson E. A., Nilsson B. L., Probing aromatic, hydrophobic, and steric effects on the self-assembly of an amyloid-β fragment peptide. Mol. BioSyst. 7, 486–496 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Cheon M., Chang I., Hall C. K., Spontaneous formation of twisted Aβ16-22 fibrils in large-scale molecular-dynamics simulations. Biophys. J. 101, 2493–2501 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young L. M., Mahood R. A., Saunders J. C., Tu L. H., Raleigh D. P., Radford S. E., Ashcroft A. E., Insights into the consequences of co-polymerisation in the early stages of IAPP and Aβ peptide assembly from mass spectrometry. Analyst 20, 6990–6999 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biancalana M., Koide S., Molecular mechanism of thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 1804, 1405–1412 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garai K., Frieden C., Quantitative analysis of the time course of Aβ oligomerization and subsequent growth steps using tetramethylrhodamine-labeled Aβ. Proc. Natl. Acad. Sci. U.S.A. 110, 3321–3326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu J.-X., Qiang W., Yau W. M., Schwieters C. D., Meredith S. C., Tycko R., Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 154, 1257–1268 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aleksis R., Oleskovs F., Jaudzems K., Pahnke J., Biverstål H., Structural studies of amyloid-β peptides: Unlocking the mechanism of aggregation and the associated toxicity. Biochimie 140, 176–192 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Schwierz N., Frost C. V., Geissler P. L., Zacharias M., From Aβ filament to fibril: Molecular mechanism of surface-activated secondary nucleation from all-atom MD simulations. J. Phys. Chem. B 121, 671–682 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Aprile F. A., Sormanni P., Perni M., Arosio P., Linse S., Knowles T. P. J., Dobson C. M., Vendruscolo M., Selective targeting of primary and secondary nucleation pathways in Aβ42 aggregation using a rational antibody scanning method. Sci. Adv. 3, e1700488 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiang W., Yau W.-M., Luo Y., Mattson M. P., Tycko R., Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 109, 4443–4448 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith D. P., Anderson J., Plante J., Ashcroft A. E., Radford S. E., Wilson A. J., Parker M. J., Trifluoromethyldiazirine: An effective photo-induced cross-linking probe for exploring amyloid formation. Chem. Commun. 2008, 5728–5730 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Walsh D. M., Thulin E., Minogue A. M., Gustavsson N., Pang E., Teplow D. B., Linse S., A facile method for expression and purification of the Alzheimer’s disease-associated amyloid β-peptide. FEBS J. 276, 1266–1281 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stewart K. L., Hughes E., Yates E. A., Middleton D. A., Radford S. E., Molecular origins of the compatibility between glycosaminoglycans and Aβ40 amyloid fibrils. J. Mol. Biol. 429, 2449–2462 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheon M., Chang I., Hall C. K., Extending the PRIME model for protein aggregation to all 20 amino acids. Proteins 78, 2950–2960 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheon M., Hall C. K., Chang I., Structural conversion of Aβ17–42 peptides from disordered oligomers to U-shape protofilaments via multiple kinetic pathways. PLOS Comput. Biol. 11, e1004258 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheon M., Chang I., Hall C. K., Influence of temperature on formation of perfect tau fragment fibrils using PRIME20/DMD simulations. Protein Sci. 21, 1514–1527 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav8216/DC1
General materials and methods for organic synthesis
Synthesis of N-Fmoc–protected TFMD-Phe
General materials and methods for Aβ16–22 solid-phase peptide synthesis
General materials and methods for HPLC purification
Analytical MS and HPLC data for synthetic peptides
General materials and methods for recombinant peptide synthesis
Additional characterization and analyses
CCS analysis of Aβ40 in the presence and absence of Aβ16–22
Scheme S1. Synthesis of TFMD-Phe.
Fig. S1. HRMS and analytical HPLC traces of Aβ16–22 and its variants.
Fig. S2. SEC trace of Aβ40 indicates that there is a single peak, and ESI-IMS-MS indicates that in the gas phase Aβ40 is largely monomeric.
Fig. S3. Supplementary ThT data.
Fig. S4. Supplementary negative-stain TEM images.
Fig. S5. Analysis of the CCS values for Aβ40 in the absence or presence of Aβ16–22 over different IMS experiments.
Fig. S6. PIC analysis of 1:1 Aβ*16–22/Aβ40 at 5 min and 24 hours.
Fig. S7. Plot of the average number of hydrogen bonding and side chain–side chain.
Table S1. The expected and observed m/z values for monomeric and oligomeric Aβ40 in isolation and in the presence of a 1:1 ratio of Aβ16–22.
Table S2. Assignments of each of the major peaks observed in fig. S6A.
Data file S1. MD snapshots as pdb files Fig. 1 t = 0.
Data file S2. MD snapshots as pdb files Fig. 1 t = 104.
Data file S3. MD snapshots as pdb files Fig. 1 t = 230.
Data file S4. MD snapshots as pdb files Fig. 1 t = 621.
Data file S5. MD snapshots as pdb files Fig. 6 t = 0.29.
Data file S6. MD snapshots as pdb files Fig. 6 t = 1.16.
Data file S7. MD snapshots as pdb files Fig. 6 t = 1.93.
Data file S8. MD snapshots as pdb files Fig. 6 t = 7.7.
Data file S9. MD snapshots as pdb files Fig. 6 t = 29.
Data file S10. MD snapshots as pdb files Fig. 6 t = 77.7.