

Abstract
In vivo optical imaging has enabled detailed studies of cellular dynamics in the brain of rodents in both healthy and diseased states. Such studies were made possible by three advances: surgical preparations that give optical access to the brain; strategies for in vivo labeling of cells with structural and functional fluorescent indicators; and optical imaging techniques that are relatively insensitive to light scattering by tissue. In vivo imaging in the rodent spinal cord has lagged behind than that in the brain, largely due to the anatomy around the spinal cord that complicates the surgical preparation, and to the strong optical scattering of the dorsal white matter that limits the ability to image deep into the spinal cord. Here, we review recent advances in surgical methods, labeling strategies, and optical tools that have enabled in vivo, high-resolution imaging of the dynamic behaviors of cells in the spinal cord in mice. Surgical preparations that enable long-term optical access and robust stabilization of the spinal cord are now available. Labeling strategies that have been used in the spinal cord tend to follow those that have been used in the brain, and some recent advances in genetically-encoded labeling strategies remain to be capitalized on. The optical imaging methods used to date, including two photon excited fluorescence microscopy, are largely limited to imaging the superficial layers of the spinal cord by the optical scattering of the white matter. Finally, we show preliminary data that points to the use of higher-order nonlinear optical processes, such as three photon excited fluorescence, as a means to image deeper into the mouse spinal cord.
Keywords: intravital microscopy, two-photon excited fluorescence, animal models, spinal cord, chronic imaging
Introduction
Recent developments in in vivo optical imaging technology have opened the door to visualizing the dynamic behavior of cells in different organ systems of rodents and other species in both healthy and diseased states. In the central nervous system (CNS), the use of structural and functional (e.g. calcium-sensitive fluorescent molecules) indicators along with surgical preparations that provide optical access to CNS structures, has enabled diverse cellular processes, such as neural firing and immune cell invasion, to be captured in intact neural tissue with high temporal and spatial resolution. This technology is especially beneficial for imaging CNS diseases where cell-level pathological events occur across a wide range of temporal scales, including, for example, synaptic plasticity and circuit rewiring occurring within minutes to hours after spinal cord injury (SCI) (Williams et al., 2014) and inflammatory pain (Matsumura et al., 2015; Williams et al., 2014), or the invasion and phagocytic activity of inflammatory monocytes happening over months in animal models of multiple sclerosis (MS) and amyotrophic lateral sclerosis (ALS) (Caravagna et al., 2018; Davalos & Akassoglou, 2012; Dibaj et al., 2011). Due to the fact that optical imaging is minimally-invasive and allows direct observation of the same structures over time, it enables visualization of the causative interactions among cells that drive pathological events, in contrast to histological approaches that provide only one-time “snapshots,” from which such interactions must be deduced (Misgeld et al., 2006).
In studies of the rodent brain, there have been dramatic advances in the optical approaches used and in the resulting scientific discoveries that have been enabled. For studies of how neural circuits control behavior, it is now possible to measure firing activity from hundreds to thousands of individual neurons with cell type specificity (Dombeck et al., 2010; Harvey et al., 2012), or even to visualize the spatio-temporal changes in neurotransmitter or protein kinase levels (Ma et al., 2018; Patriarchi et al., 2018), all in awake animals performing specific behavioral tasks, and with the capability for concomitant optogenetic manipulation of neural activity (Rickgauer et al., 2014). In disease models, in vivo imaging has revealed the cellular interactions that drive specific disease symptoms. In mouse models of Alzheimer’s disease, for example, such imaging has revealed that microglia swarm around and phagocytose neurons after Cx3CR1-dependent signaling (Fuhrmann et al., 2010), and that the adhesion of neutrophils in brain capillaries causes substantial cerebral blood flow reductions that contribute to memory impairment (Cruz Hernández et al., 2019).
Such in vivo imaging in the rodent brain was made possible by a combination of three technologies — First, the development of a chronic implant that allows repeated optical access to the rodent brain with good stability even when animals are awake, head-fixed, and moving under the microscope; Second, the use of exogenous and genetically encoded fluorescent indicators, such as GFP and GCaMP, to label the structure or follow function in targeted cell types with high signal to noise and low cellular toxicity in living animals; Third, the use of two-photon excited fluorescence (2PEF) imaging and other nonlinear microscopy techniques to overcome the optical scattering properties of intact tissue to enable cell structure and function to be resolved hundreds of micrometers deep into the intact brain. Although this suite of tools has been extensively adopted for a wide variety of studies in different brain areas and various neurological disease models, the use of in vivo optical imaging in the spinal cord has been more slow to emerge.
Optical imaging in the rodent spinal cord has proven to be more challenging than the brain primarily due to the more complex gross anatomy surrounding the spinal cord and the presence of highly-scattering white matter on the dorsal surface of the cord. Unlike the brain, where a sterile, sealed imaging chamber can be created by simply gluing a glass coverslip to the skull over a craniotomy (Holtmaat et al., 2009) or by thinning the skull to increase transparency (Drew et al., 2010), the spinal cord is located below thick layers of back muscles, inside the heterogenous structure of the spine, and follows the curvature of the back along the rostra-caudal axis, making surgical access more complicated. In addition, thoracic spinal segments are located above the heart and lungs, making stabilization to avoid motion artifact due to heartbeat and breathing challenging. Moreover, the white matter tracts on the dorsal surface of the spinal cord are highly optically scattering and significantly limit the ability of even scattering-insensitive microscopy approaches, such as 2PEF, to penetrate deep into the spinal cord (Farrar et al., 2012; Johannssen et al., 2013). As a result, the pioneering imaging studies of the spinal cord in living rodents, including those utilizing 2PEF, have focused on events in superficial layers of the spinal cord, such as axon dieback dynamics after injury (Lorenzana et al., 2015; Tang et al., 2015; Z. Yang et al., 2017), the activity of sensory neurons after peripheral stimulus (Johannssen et al., 2010; Ran et al., 2016), the accumulation of spinal cord-resident and invading inflammatory cells in models of neuroimmune disease or SCI (Davalos, Ryu, et al., 2012; Drew et al., 2010), and blood flow in superficial veins on the dorsal spinal cord surface (Farrar et al., 2015), for example.
In this article, we review recent strategies that enable in vivo optical studies in the mouse spinal cord. This includes methodologies for (a) gaining optical access to the spinal cord, (b) labeling cells with structural and functional fluorescent indicators, and (c) optically imaging cellular dynamics and tissue structure. We conclude with a prospectus on the potential of recent developments, such as three-photon excited fluorescence (3PEF) microscopy and implanted miniature microscopes, to enable imaging studies of deeper layers of the spinal cord than have been achieved to date.
Surgical preparations to gain optical access to the mouse spinal cord
Acute preparations
Initial efforts towards in vivo cell-resolved imaging in the spinal cord relied on acute surgical preparations, where one-time optical access to the spinal cord was obtained and animals were sacrificed after the imaging session. These surgical approaches largely paralleled those that had been used for in vivo electrophysiological studies of the rodent spinal cord and relied on similar surgical tools and restraint devices (Furue et al., 1999; Sonohata et al., 2004). Briefly, mice are anesthetized and the soft tissue and muscle over the spine are dissected away and retracted. The bone and paravertebral muscles are heavily vascularized, so bleeding must be controlled (for example with a cauterizer) during this dissection to avoid blood accumulation that could interfere with imaging. Some groups routinely intubate mice, enabling better physiological control over long-duration imaging experiments (Misgeld et al., 2007; Ran et al., 2016). The vertebrae are then stabilized around the spinal level to be imaged using pins (Light et al., 1999), custom-fabricated steel bars (Farrar et al., 2012), or commercial stabilization devices (Davalos et al., 2008). Once the vertebrae are stabilized, the spinal cord tissue is exposed by performing a dorsal laminectomy using either fine scissors or a fine surgical drill. The underlying dura may be left intact or be (partially) removed. Removing the dura modestly increases imaging depth by eliminating optical scattering from the dura and facilitates surface bulk loading of fluorescent indicators (Johannssen & Helmchen, 2010; Ran et al., 2016). Leaving the dura intact retains more physiological spinal cord integrity, reduces the risk of surgical trauma, and may reduce acute inflammation. The exposed spinal cord is then typically covered with a sterilized small glass coverslip, with sterilized artificial cerebrospinal fluid underneath. When imaging using an immersion lens a well must be built, for example from agar, around the window to enable a stable bead of immersion fluid to be maintained.
To facilitate optical access to regions of the spinal cord other than the dorsal surface, preparations that dissect through the lateral aspect of the vertebral column to expose ventrolateral areas of the cord have been developed (Cartarozzi et al., 2018), or the mouse can be rotated around the longitudinal axis by about 30–40 degrees with a more standard dorsal laminectomy to bring the less myelinated dorsal horn region of the cord to the top, enabling optical access to superficial spinal cord grey matter (Johannssen & Helmchen, 2010; Ran et al., 2016).
Acute imaging preparations typically provide high quality optical imaging, as tissue fibrosis will not have time to occur and increase optical scattering. On the other hand, acute preparations provide only a limited time window of a few hours to follow cellular behaviors and inevitably the study would be in the context of the trauma, however mild, from the surgical procedure.
Chronic preparations
The first studies that used in vivo imaging to follow cellular events over time scales longer than a few hours used repeated surgeries to gain optical access to the spinal cord. In these studies, the soft tissue was closed over the laminectomy site after each imaging session. Several approaches to protect and minimize irritation of the spinal cord tissue have been described, including placement of a synthetic matrix membrane (Di Maio et al., 2011) or saline-soaked gelfoam (Evans et al., 2014) over the laminectomy, which was then removed before each imaging session. Using such an approach, Di Maio, et al. followed the dieback trajectory of individual axons for up to four months after a dorsal root crush injury (Di Maio et al., 2011). Similarly, Dray, et al. used six successive surgical and imaging sessions to correlate vascular and axonal regenerative responses after a pin-prick injury to the dorsal spinal cord (Dray et al., 2009).
While using repeat surgeries has enabled optical imaging studies that extend for weeks to months, the number of time points is ultimately limited by the complexity and animal welfare concerns of repeat surgeries. This approach also runs the risk of trauma or tissue inflammation from the repeated surgeries confounding the results. A critical development in the last ten years has been the invention of implanted chambers and associated surgical protocols that provide long-term optical access to the mouse spinal cord. These imaging chambers all solve two critical problems, each of which is more challenging for spinal cord than for brain imaging. First, the chambers create a tight seal over the exposed spinal cord to protect the tissue, prevent infection, and to contain an elastomer that occupies the empty space created by the laminectomy to reduce scar tissue growth and mechanically stabilize the spinal cord within the chamber. For brain imaging, this sealed chamber is created simply by gluing a glass coverslip to the skull (and typically does not require the elastomer), but creating such a chamber is more intricate in the spinal cord due to the more complex anatomy. Second, the chambers provide a means to mechanically stabilize the spine for imaging. For brain imaging, this mechanical stabilization is achieved using ear bars or a secondary post glued to the skull, but this mechanical stabilization tends to be a more integrated aspect of the implanted chamber for spinal cord imaging.
So far, three research groups have independently developed chronic imaging chambers for the mouse spinal cord that allow long-term optical imaging without repeat surgeries (Farrar et al., 2012; Fenrich et al., 2012; Figley et al., 2013). The initial surgical preparation, including skin incision, muscle retraction, tendon removal, and dorsal laminectomy is similar to that used in acute preparations, described above. The three approaches differ in the strategy for creating a sealed environment over the spinal cord and for providing mechanical stabilization of the spine.
In the approach described by Farrar, et al., a machined chamber was used to create both the sealed environment and to provide mechanical stabilization (Fig. 1A) (Farrar et al., 2012; Farrar et al., 2014). Briefly, a pair of small metal bars was clamped across three vertebrae and a laminectomy was performed using fine scissors, leaving the dura intact. A metal top plate with a 5-mm diameter opening was then attached to the bars with screws and a silicone elastomer was infused over the spinal cord. Before the elastomer was fully cured, a sterilized glass coverslip was fit into the hole in the top plate. The glass coverslip was gently pressed down on the still-curing silicone gel so that the underlying spinal cord tissue was firmly held by the cured silicone, but the pressure was not high enough to compress the tissue (which was sensitively indicated by blood being pushed out of the dorsal spinal vein). With optimal pressure applied during the curing of the silicone, motion artifact was smaller and the potential space for scar tissue to grow and obscure imaging was reduced. Finally, the chamber was sealed by gluing the surrounding skin to the base of the chamber with cyanoacrylate glue and dental acrylic. The top plate included threaded holes that could be used to stabilize the chamber and the mouse underneath a microscope for imaging. A slightly modified version of this imaging chamber is now commercially available from Neurotar, Inc. (Finland), and has been shown to enable mice to be awake and spine fixed while navigating in Neurotar’s Mobile Home Cage apparatus (Neurotar). This imaging chamber has been implanted in both the thoracic region (T11 – L1) as well as in the lumbar/sacral region (L2 – S2), with only minimal modification of the surgical approach (DeNotta, 2018).
Fig.1. Chronic spinal cord imaging chambers.

(A) The system developed by Farrar, et al. uses machined parts, including two small metal bars and a top plate, to create a sealed chamber that is fixed to the spine. Reproduced from (Farrar et al., 2012). (B) The approach described by Fenrich, et al. relies on small bent pieces of metal and glue to create a sealed chamber that is secured to the spine. Reproduced from (Fenrich et al., 2012; Fenrich et al., 2013a). (C) Figley, et al. sealed the laminectomy separately and then secured a mounting frame to the surrounding muscle and soft tissue over the spine. Scale bar = 10 mm. Reproduced from (Figley et al., 2013).
Fenrich, et al. used small pieces of bendable metal and dental acrylic to create an ad hoc sealed chamber and stabilization system (Fig. 1B). Briefly, metal staples were shaped so they could be placed along and underneath the pedicles of the vertebrae. Three vertebrae were surrounded and slightly lifted by two oppositely-oriented shaped staples. A bent paperclip was also placed underneath the edges of the two shaped staples so that a loop of the metal stuck out laterally from the spine. The shaped staples and bent paperclip were then anchored in place with a ring of dental acrylic. A laminectomy was performed using a drill, a thin layer of silicon elastomer was infused over the spinal cord, and the chamber was sealed by gluing a custom-cut glass coverslip in place. For imaging, the animal is stabilized under the microscope by fixing the protruding loop of the paperclip (Fenrich et al., 2013a). This group has demonstrated implantation of this chamber in the lower thoracic/upper lumbar region (T12 – L2).
In a third spinal cord imaging chamber, described by Figley, et al., a laminectomy was performed in the upper lumbar region (L2 – L3) and the exposed spinal cord covered and sealed with a pre-made, thin piece of silicone elastomer (Fig. 1C). A frame with four protruding arms was then secured to the muscle and skin surrounding the spine using tight sutures and was fitted with a glass coverslip. The arms were used to secure the animal under the imaging apparatus. The authors demonstrated the use of both metal as well as a polycarbonate material for the frame, which was more compatible with photoacoustic imaging approaches (Figley et al., 2013).
In comparing the performance of these imaging chambers, there are several factors to consider. First, it is important to assess the motion artifact, distinguishing between motion of the spine and motion of the spinal cord within the vertebrae. Because the chambers described by Farrar, et al. and by Fenrich, et al. both have metal parts that are firmly attached to the spine, the motion of the spine can be nearly completely eliminated. The system described by Figley, et al., in contrast, does not fix the vertebrae and instead holds the animal by the surrounding soft tissue, increasing the likelihood of spine motion due to respiration and heartbeat. Indeed, Figley, et al. note the presence of artifacts in optical coherence tomography images due to axial motion caused by respiration. Motion of the spinal cord within the spine can be reduced by careful application of the silicone elastomer that fills the space between the spinal cord and glass coverslip in the designs by Farrar, et al. and by Fenrich, et al.. Too little elastomer leaves excess room for the spinal cord to move, while too much elastomer leads to compression of the spinal cord, which can cause ischemia and tissue damage. Neither Farrar, et al. or Fenrich, et al. report a quantitative assessment of motion artifact, but both show image stacks that resolve individual axons without apparent blurring due to motion.
A second consideration is the duration that the window continues to provide high-quality optical access to the spinal cord, which is primarily limited by the growth of fibrous scar tissue over the spinal cord after the surgery, and the formation of air bubbles in the silicone, which can be avoided by mixing the two components of the silicone gel very gently. The chamber designs described by Farrar, et al. and Fenrich, et al. take several steps to minimize the growth of this tissue. First, both groups apply cyanoacrylate glue to the edges of bone exposed by the laminectomy, which, in our hands, seems to dramatically increase how long the chamber remains optically clear. Second, the space between the spinal cord and the glass window is filled with an inert silicone elastomer, which simply reduces the space in which scar tissue can form. These groups have demonstrated that such chambers can remain clear enough for high quality imaging for several months to a year (Farrar et al., 2012; Fenrich et al., 2012).
Finally, it is critical to assess what tissue damage results from the surgical procedure and whether there are any behavioral impacts from the chamber implantation. Using histological approaches, Farrar, et al. found an increase in density of microglia in the dorsal half of the spinal cord under the window, suggesting some tissue inflammation. This increase in microglia density was similar to that seen in cortical tissue after cranial window implantation (Xu et al., 2007; Yang et al., 2010). Similarly, Fenrich, et al. used in vivo imaging of transgenic mice to show that the window implantation led to a transient increase in the density of several classes of inflammatory cells in the dorsal spinal cord that largely resolved within a couple of weeks. Each group reported no signs of overt damage to the spinal cord. Farrar, et al. also found essentially no differences in locomotor capabilities or behavior a few days after chamber implantation.
The development of these imaging chambers has opened the door to repeat optical imaging of cellular structure and dynamics in the spinal cord of mice over timescales of months and with the number of imaging sessions limited by factors other than the surgical preparation (e.g. frequency of repeat anesthesia). Because animals can recover from the surgery before future imaging sessions, these chambers also open the door to imaging studies in awake, spine-fixed mice, paralleling developments in awake brain imaging that have proven useful in correlating neural activity patterns with behavior (Dombeck et al., 2010; Dombeck et al., 2007; Harvey et al., 2012).
Fluorescent labeling strategies
In vivo labeling strategies for structural imaging and studies of cellular dynamics in the spinal cord are largely similar to those used in the brain. We briefly review labeling strategies using exogenous dyes and genetically-encoded fluorescent indicators, emphasizing combinatorial approaches employed in specific experiments to explore cellular interactions that underlie normal and disease state processes. We review the use of endogenous optical signals in the section on imaging techniques.
Exogenous dyes
There are several strategies for local or systemic delivery of exogenous fluorescent labels to visualize the morphology of key cellular components of the spinal cord. In nearly all in vivo imaging experiments, labeling the vasculature can be a critical first step that enables navigation to the same structures in repeated imaging and provides a routine check on image quality. Blood vessels can be labeled by intravenous injection of large fluorescent markers, such as 70-kDa fluorophore-conjugated dextrans (C. Chen et al., 2017; Davalos et al., 2008; Farrar et al., 2012) or fluorescent quantum dots (Fenrich et al., 2012), which are retained in the vessel lumen for several hours. In addition, such dyes label only the blood plasma and not blood cells, so these cells appeared as dark patches, whose motion can be followed to measure blood flow speed (Farrar et al., 2015; Miyazaki et al., 2012).
Locally delivered dyes can label a variety of structural components of the spinal cord for in vivo imaging with reasonable specificity. For example, topically applied or microinjected sulforhodamine 101 (SR101) robustly labels astrocytes (Johannssen & Helmchen, 2010; Nimmerjahn et al., 2004), but was recently shown to also label oligodendrocytes in the cortex (Hill et al., 2014). In the spinal cord, SR101 labeling was used together with Mitotracker Green (to label all mitochondria) and tetramethylrhodamine methyl ester (TMRM; labeling intensity reflects membrane potential) to quantify the depolarization of astrocytic mitochondria in the experimental auto-immune encephalitis (EAE) mouse model of MS (Sadeghian et al., 2016). When topically applying such dyes it is necessary to remove or make a small nick in the dura to increase penetration into the spinal cord, or the dye can be microinjected into the spinal cord tissue. In addition, intramuscular injection of classic tract tracing dyes, such as FluoroRuby, has been shown to robustly label ventral horn motoneurons that project to the injected muscle for in vivo spinal cord imaging (Cartarozzi et al., 2018). To monitor the firing pattern of neurons, calcium sensitive fluorescent indicators, such as Oregon Green 488 BAPTA-1 (OGB), can be bulk loaded into spinal cord neurons cells via microinjection (Johannssen & Helmchen, 2010; Ran et al., 2016), similar to the approaches used in the cortex (Johannssen & Helmchen, 2010; Ran et al., 2016; Stosiek et al., 2003). Calcium transients in spinal cord astrocytes can be distinguished from those in neurons by labeling with both OGB and SR101 (Cirillo et al., 2012).
Genetically-encoded fluorescent labels
A broad spectrum of different color fluorescent proteins as well as genetically-encoded fluorescent reporters of calcium concentration and membrane potential have been engineered (T. W. Chen et al., 2013; Grienberger et al., 2012; St-Pierre et al., 2015). These structural and functional indicators are genetically delivered to animals through the generation of transgenic mouse lines, viral transduction, and in utero electroporation.
Transgenic animals
A variety of transgenic mice have been created that express various color fluorescent proteins (XFPs) under the control of different cell-type specific promoters. For example, Thy1 is expressed by neurons as well as some other cell types, including thymocytes (Gordon et al., 1987; Morris, 1985), but can be made into a very specific neuronal promoter by removal of some enhancer elements in the gene (Vidal et al., 1990). Thy1-YFP mice have been used to follow the dieback of axons after SCI (Evans et al., 2014; Farrar et al., 2012; Lorenzana et al., 2015; Williams et al., 2014; Z. Yang et al., 2017), while Thy1-CFP mice were used to visualize the disintegration of axons secondary to EAE (Davalos, Ryu, et al., 2012). Other transgenic neuron labeling lines have also been used for in vivo spinal cord imaging, such as mice that express TdTomato in somatostatin expressing neurons (Sekiguchi et al., 2016).
Efforts to image inflammatory cell dynamics in the spinal cord have relied on mice that used targeted gene replacement techniques to specifically label different classes of inflammatory cells. In this approach, one copy (or sometimes both, if knockout of the gene is desired) of a gene specific to a cell type of interest is replaced with the sequence for a fluorescent protein. For example, mice with one copy of Cx3Cr1 replaced with GFP label spinal cord resident microglia and patrolling monocytes, which contribute to wound healing, while mice with one copy of CCR2 replaced with RFP label inflammatory monocytes, which have phagocytic and inflammatory functions (Benakis et al., 2014). Using Cx3Cr1-GFP mice, Dibaj, et al. showed that microglia in the spinal cord in a mouse model of ALS were more reactive to injury early in disease progression and then became less reactive when clinical signs appeared, as compared to control mice (Dibaj et al., 2011). Genetic labeling alone, however, may not always be sufficient to distinguish phenotypically different cells. For example, Cx3Cr1-GFP mice label both spinal-cord resident microglia as well as blood-borne patrolling monocytes. To distinguish these cell types, Evans, et al. used bone marrow transplants between Cx3CR1-GFP and wild type mice to create chimeric animals and used these mice to show that patrolling monocytes that invade the spinal cord from the blood, but not spinal cord resident microglia, contributed to secondary axon dieback following a dorsal column crush SCI (Evans et al., 2014).
Genetically encoded calcium indicators (GECI) enable the activity patterns of an ensemble of individual neurons to be imaged, and have come into common use in animal studies of the CNS. GECIs come into two categories: single-fluorophore GECIs, such as GCaMP, where the fluorescence intensity changes with calcium binding, and fluorescence resonance energy transfer-based (FRET-based) GECIs, such as YCnano, where calcium binding changes the efficiency of the energy transfer between two fluorescent molecules that are bound to each other. Advantages and disadvantages of different classes and specific variants of GECIs have recently been reviewed (Grienberger & Konnerth, 2012; Storace et al., 2016). Using mice expressing GCaMP under the control of the Thy1 promoter, Tang et al., showed, for example, that calcium levels in single axons in the dorsal spinal column increased after a hemisection injury to the spinal cord (Tang et al., 2015). Williams et al., used transgenic mice expressing a FRET-based GECI in spinal cord axons to show that, after a contusion injury, axons that failed to regain homeostatic control of calcium levels were more likely to degenerate (Williams et al., 2014).
Viral transfection and in utero electroporation
In addition to creating transgenic mice, both viral transfection and in utero electroporation can be used to drive expression of genetically-encoded indicators. Viral vectors can be microinjected to control the spatial distribution of the labeling. Both the cellular tropism of the virus as well as the promoter sequences used give control over the cell type in which the reporter will be expressed (Haenraets et al., 2018). Because robust expression of the reporter gene typically takes a few weeks, it may be desirable to inject the viral vector into the spinal cord in a separate procedure before the imaging window is implanted (which can be done through an intervertebral disk without the need for a laminectomy) (Kohro et al., 2015). Vectors created from adeno-associated virus (AAV) and driving expression of a fluorescent protein using a ubiquitous promoter were injected into dorsal root ganglion neurons to anterogradely label their ascending axons in the dorsal spinal column (Williams et al., 2014). Viruses also offer the opportunity to label neurons based on their anatomical projections, for example by delivering Cre-expressing viral vectors to the brain that are trans-synaptically transported, followed by intra-spinal injection of AAV vectors encoding Cre-dependent fluorescent reporters (F. Wang et al., 2016). GECIs have been virally delivered to both astrocytes (Yoshihara et al., 2018) and neurons (T. Chen et al., 2018) in the dorsal horn of the spinal cord to study astrocytic calcium responses and neural activity in animal models of acute and chronic pain, respectively. Exceptionally, AAV delivered GCaMP6 was used to measure the activity patterns of both sensory neurons and astrocytes in the dorsal horn of awake, behaving animals in response to peripheral sensory stimulation and during normal behavioral states, such as grooming (Sekiguchi et al., 2016).
Delivery of genetic reporters to spinal cord cells has also been demonstrated using embryonic electroporation, where a DNA plasmid is microinjected into the spinal canal of the embryo (at around E12.5) and electric pulses are delivered to the embryo (Saba et al., 2003). Using this approach, Nishida, et al. labeled neurons with the FRET-based GECI YCnano and evaluated the multimodality of the sensory response of individual dorsal horn neurons (Nishida et al., 2014). This same gene delivery approach was also used to structurally label dorsal spinal cord neurons with EGFP and yielded expression levels high enough to visualize the dendritic spines of individual spinal cord neurons (Matsumura et al., 2015).
High-resolution optical imaging techniques applied to the mouse spinal cord
Optical imaging technology has opened the door to visualizing a variety of biological phenomena at cellular resolution in multiple organ systems, across different species. Optical tools are well suited to studies of cell-resolved mechanisms, because the spatial and temporal resolution matches well with size of single cells and speed of even some of the fastest biological changes, such as neural firing (W. Yang et al., 2017). The success of a particular optical imaging modality in studying cell dynamics in a particular tissue depends on how sensitive the imaging tool is to the inhomogeneities and other optical properties of the tissue. The spinal cord is particularly challenging, as the densely myelinated white matter sits on the dorsal surface where optical access is achieved surgically (except for one described acute surgical preparation (Cartarozzi et al., 2018)). Because of the alternating layers of ensheathing myelin and internal cytoplasm of individual axons, and the large refractive index difference between these layers, the optical scattering length of the dorsal white matter is quite short, as compared to cortical tissue. This short scattering length leaves linear optical methods primarily probing the very surface of the spinal cord and limits even nonlinear optical imaging methods, such as 2PEF microscopy, to the superficial layers, typically within the top 100–200 µm (Farrar et al., 2012). In this section, we review a variety of different optical imaging methods that have been applied to the rodent spinal cord, briefly describe some key findings enabled by such optical imaging, and discuss the capabilities and limitations of different imaging approaches.
Linear microscopy
Wide-field reflectance and fluorescence microscopy is beneficial for meso-scale imaging of the spinal cord, and has been used, for example, to track the changes in the growth response of sparsely-labeled, severed axons in response to administered drugs (Fig. 2A) (Ruschel et al., 2015). Confocal fluorescence microscopy reduces the impact of out-of-focus background fluorescence and enables higher resolution and higher contrast imaging, allowing the visualization of the interactions of multiple cell types with dorsal axons in mouse models of neuroimmune disease, for example (Nikić et al., 2011). Spectral confocal reflectance microscopy (SCoRe) capitalizes on the wavelength-dependent reflection from myelinated fibers to visualize individual axons in the mouse spinal cord in vivo, without any labeling (Fig. 2B) (Schain et al., 2014). A miniaturized one-photon epifluorescence microscope using a graded-index (GRIN) lens as an objective that can be attached to a spine-mounted imaging chamber in a mouse has been developed, and it enables wide-field fluorescence imaging in a freely-behaving animal (Fig. 2C) (Sekiguchi et al., 2016). Using this device, Sekiguchi, et al. imaged GCaMP-expressing sensory neurons in the dorsal horn and showed that different groups of neurons were recruited in response to different cutaneous stimuli (Fig. 2D) (Sekiguchi et al., 2016). Ghosh, et al. provide a detailed schematic of the mini microscope and describe its use for recording calcium transients in vivo, while Meng, et al. outline the use of GRIN lenses for in vivo imaging of deep brain regions (Ghosh et al., 2011; Meng et al., 2019). Optical coherence tomography (OCT) relies on interference-based gating of light reflected from different depths in the sample and enables deep imaging of tissue structure in the mouse spinal cord with a resolution of a few micrometers (Fig. 2E) (Cadotte et al., 2012). Some variants of OCT imaging provide optical contrast that depends on the speed of motion of optical scatterers in the sample, enabling visualization of vascular structures and quantification of blood flow speeds (Fig. 2F) (Cadotte et al., 2012). Much of the innovation in OCT technology occurs in the context of ophthalmology applications, and is well reviewed in a paper by Fujimoto (Fujimoto, 2003). Recent applications of OCT for mouse cortical imaging are reported in Chong, et al. (Chong et al., 2015).
Fig. 2. Linear optical imaging in the mouse spinal cord.

(A) Sprouting of the dystrophic tip of severed GFP-labeled axons after systemic treatment with epothilone B, visualized using wide-field fluorescence microscopy. From left to right, images taken at 6 h, 1 d, and 4 d after spinal cord dorsal hemisection. The green arrows indicate regenerating axons, while the yellow and red arrows point to a forming retraction bulb and a dying axon, respectively. Images about 700-µm wide. Reproduced from (Ruschel et al., 2015). (B) Multicolor SCoRe microscopy images showing individual myelinated axons in mouse spinal cord, imaged in vivo. Scale bar = 25 µm. Reproduced from (Schain et al., 2014). (C) Schematic of a miniaturized spine-mounted wide-field fluorescence microscope used to image spinal cord neural activity in behaving mice in response to different mechanical stimuli. (D) Time-integrated fluorescence from GCaMP6-labeled dorsal horn neurons in response to an air puff on the tail (left and right; cyan) or a pinch with forceps (right; yellow). In the image on the right, neuron (i) responds only to the air puff, neuron (ii) only to the tail pinch, while neuron (iii) responds to both. Scale bar = 100 µm. C and D reproduced from (Sekiguchi et al., 2016). (E) Coronal view of the mouse spinal cord imaged in vivo with OCT, showing white and grey matter as well as the large dorsal spinal vein on the surface. (F) Contrast in speckle variance OCT depends on the motion of optical scatterers, and so highlights microvascular structures in the spinal cord. E and F reproduced from (Cadotte et al., 2012).
Nonlinear microscopy
Nonlinear microscopy has become the technique of choice for imaging with cellular resolution deep into optically scattering samples, as is nearly always encountered with in vivo imaging. Using tightly-focused, short laser pulses for excitation, nonlinear optical interactions can be confined to a micrometer-sized focal volume. This focal volume is then scanned in three dimensions, enabling an image to be formed with a variety of different nonlinear optical interactions producing the image contrast. Because excitation light scattered on the way to the focus does not contribute to the signal and because the detected signal can be attributed to the location of the laser focus, irrespective of how scattered it was, nonlinear imaging techniques are significantly more insensitive to optical scattering than linear microscopy methods. Here we review the use of 2PEF as well as some endogenous nonlinear optical interactions for imaging in the mouse spinal cord. Adur, et al., provides an accessible summary of the mechanisms of image contrast formation for some of the nonlinear optical techniques we review in this manuscript (Adur et al., 2014). Our review focuses on what these imaging approaches enable and what contrast they produce, but not on the underlying physics of the image contrast generation. While enabling the top 100–200 µm of the spinal cord to be interrogated with cellular-resolution, imaging deeper into the grey matter of the spinal cord remains beyond the current capability of these tools.
Two photon excited fluorescence microscopy
In the spinal cord, 2EPF imaging readily gives access to the dorsal white matter and to sensory neurons in the less-myelinated dorsal horn region of the spinal cord. This has enabled studies of the cellular interactions that facilitate or reduce axon degeneration in mouse models of injury or neurological disease, as well as of the neural encoding of sensory stimuli and pain. An earlier review by Helmchen and Denk provides an introduction to the general physics and in vivo application of 2PEF in neural tissue (Helmchen et al., 2005). Fenrich, et al. imaged triple transgenic mice that expressed different color fluorescent proteins in spinal cord axons as well as in different classes of immune and inflammatory cell types (Fig. 3A). After a SCI, they found that inflammatory cells invaded the spinal cord from the blood early after the injury and interacted strongly with degenerating axons, while the activity of tissue-resident inflammatory cells peaked later and contributed to the cleanup of axon debris (Fenrich et al., 2012). Similarly, Davalos, et al. used multicolor 2PEF imaging to demonstrate the importance of fibrinogen leakage into the spinal cord as a driving factor in the activation of microglia and their subsequent contribution to axon degeneration in the EAE mouse model of MS (Fig. 3B) (Davalos, Ryu, et al., 2012). Axon dieback dynamics and other morphological changes can be followed after substantial spinal cord injuries (Fig. 3C; unilateral, dorsal pin prick) or following very subtle lesions (Fig. 3D; targeted femtosecond laser cut to superficial axons) (Farrar et al., 2012). Detailed morphological changes in dendritic spines has also been followed in the mouse spinal cord over the first few hours after the initiation of peripheral inflammatory pain (Matsumara et al 2015). Using the ventrolateral surgical preparation they developed, Cartarozzi, et al. examined the response of microglia and astrocytes to inflammatory stimuli and were able to directly visualize motoneuron morphology, all in the ventral horn of the mouse spinal cord (Cartarozzi et al., 2018). In addition to imaging cell structure, 2PEF can also provide functional readouts of spinal cord physiology. For example, fluorescent labeling of blood plasma enables not just visualization of vascular structure (Fig. 3E; inset shows clotting induced by topical FeCl3 application), but also measurement of blood flow speed by tracking the motion of unlabeled red blood cells through line scans along the vessel axis (Fig. 3F) (Farrar et al., 2015). 2PEF imaging of neural activity with exogenous as well as genetically-encoded calcium indicators has also been demonstrated. Using OGB, Ran, et al. found that both heating and cooling of the hind paw evoked calcium responses in dorsal horn sensory neurons, with heat-responsive neurons responding to the absolute skin temperature without adaptation, while cold-responsive neurons, in contrast, encoded temperature change and adapted to different absolute temperatures (Fig. 3G and 3H) (Ran et al., 2016).
Fig. 3. In vivo two-photon excited fluorescence imaging of cellular structure and function in the mouse spinal cord.

(A) 2PEF imaging of four different fluorescent labels and SHG in mouse spinal cord. (top) Transgenic Thy1-CFP labels dorsal column axons (cyan), transgenic LysM-GFP labels myelo-monocytic cells such as granulocytes and macrophages (green), transgenic CD11c-EYFP labels a subset of dendritic cells, microglia, and macrophages (magenta), intravenously injected fluorescent quantum dots (QDot 655) labels the blood plasma (red), and SHG comes from endogenous collagen in the dura matter (purple). (bottom) Time series showing the division of a CD11c-expressing cell 3 d after a spinal cord injury just rostral to the imaging site. Scale bar = 100 µm and 20 µm in the top and bottom images, respectively. Reproduced from (Fenrich et al., 2013b). (B) Imaging of axons (cyan; transgenic Thy1-CFP), microglia/macrophages (green; transgenic Cx3Cr1-GFP), and fluorescently-tagged fibrinogen (red: intravenously-injected Alexa 594-fibrinogen) in the EAE mouse model of MS. The white arrowhead indicates a degenerating axon. Scale bar = 10 µm. Reproduced from (Davalos, Ryu, et al., 2012). (C) Low-magnification 2PEF imaging of dorsal axons (green; transgenic Thy1-YFP) and blood vessels (red; intravenous Texas Red-dextran) at one day after a pin prick injury to the spinal cord at the location indicated by the white asterisk (unpublished data). Scale bar = 80 µm. (D) Long-term observation of axons (transgenic Thy1-YFP) after a focal, laser injury to the dorsal surface of the spinal cord. Yellow arrow: axon degenerated beyond imaging field; red arrow: axon was stable over course of imaging with axon tip shown in insets; blue arrow: axon slowly died back from lesion; magenta circles: landmarks identified across imaging sessions. Scale bar = 100 µm. Reproduced from (Farrar et al., 2012). (E) Low-magnification 2PEF image of fluorescently-labeled blood vessels, showing the prominence of the dorsal spinal vein. The inset shows the outlined region from the main figure after topical application of FeCl3 to induce clotting. (F) Top image shows a maximum projection of a 2PEF image stack of a venule with fluorescently-labeled blood plasma. Below is a space-time image formed by taking a repeated line scan along the center of the vessel, at the position indicated with the red line in the top image. Moving red blood cells, which do not take up the intravenously injected dye, form dark streaks whose slope is proportion to the inverse of the blood flow speed. E and F reproduced from (Farrar et al., 2015). (G) In vivo 2PEF imaging of OGB-labeled sensory neurons in the spinal cord of an anesthetized mouse. Green overlays indicate neurons that responded to the application of a cooling stimulus to the hind paw. Scale bar = 50 µm. (H) Example traces of the normalized change in fluorescence from OGB-labeled neurons responding to a cooling hind paw stimulus. Scale bar represents 10% ΔF/F and 10 s. G and H reproduced from (Ran et al., 2016).
Imaging with endogenous nonlinear optical signals
In addition to nonlinear excitation of fluorescent indicators, there are several nonlinear optical processes that produce strong signals from endogenous structures and molecules in the spinal cord and can provide useful image contrast. Because these signals depend on a nonlinear optical interaction, they naturally provide high resolution, 3D imaging capabilities. As with 2PEF, the penetration depth of these imaging modalities in the spinal cord is strongly limited by the optical scattering of the dorsal white matter. Coherent anti-Stokes Raman scattering (CARS) microscopy enables imaging with contrast based on the concentration of molecular bonds with a particular vibrational spectrum. A detailed explanation of the mechanisms of image formation in CARS microscopy, with an overview of in vivo and ex vivo applications can be found in Min, et al. (Min et al., 2011). In vivo, CARS has primarily been used to visualize carbon-hydrogen bonds and because of the large number of such bonds that are present in lipids, CARS images mainly show the myelin in the spinal cord. Combined 2PEF and CARS imaging shows the tight wrapping of myelin around individual axons (Fig. 4A) and the intercalation of microglia in myelinated fiber tracts (Fig. 4B) (Bélanger et al., 2012). Second harmonic generation (SHG) and third harmonic generation (THG) are nonlinear optical processes that rely on coherent conversion of two or three incident laser photons into a single photon. The underlying physics of how signal generation for SHG and THG depends on sample properties is reviewed in Weigelin, et al. (Weigelin et al., 2016). In in vivo imaging, SHG is primarily seen from non-centrosymmetric molecules, such as collagen (Zipfel et al., 2003). In the spinal cord, the dominant source of SHG is thus from the collagen-rich dural membrane (Fig. 3A). For tightly-focused laser beams, the THG produced on either side of the laser focus is out of phase and cancels in the far field, yielding no signal from isotropic, uniform samples. At an optical interface, however, the THG does not perfectly cancel, leading to a detectible signal (Squier et al., 1998). THG imaging thus tends to visualize bold optical interfaces in a sample, where there are substantial changes in refractive index or nonlinear susceptibility. The alternating layers of lipid-rich myelin and watery cytoplasm found in the dorsal white matter produce such bold optical interfaces, so THG imaging highlights myelinated fiber tracts (Fig. 4C) (Farrar et al., 2011), and enables, for example, visualization of myelin disruption following a local ischemic injury (Fig. 4D). For 1,320-nm excitation light, the THG signal is resonantly enhanced by Soret band transitions in hemoglobin, leading to bright THG from red blood cells, enabling label-free imaging of vascular structure and quantification of blood flow speeds (Dietzel et al., 2014; Koizumi et al., 2018) (Fig. 4E). THG requires higher per pulse energies at the laser focus to produce a robust signal as compared to SHG or 2PEF imaging, necessitating the use of lower repetition rate lasers (typically few MHz) to avoid thermal damage to the sample.
Fig. 4. Imaging of endogenous nonlinear optical signals in the mouse spinal cord.

(A) 2PEF imaging of YFP labeled axons (left), CARS imaging of myelin (middle), and an overlay of both modalities (right). Scale bar = 15 µm. (B) 2PEF of GFP labeled microglia (red) and CARS imaging of myelin (green). Scale bar = 15 µm. A and B reproduced from (Bélanger et al., 2012). (C) 2PEF imaging of YFP labeled axons (green) and THG (grey), which highlights myelin. The magnified image to the right (at location of blue box) shows the THG signal from myelin (arrowheads) wrapping tightly around a YFP labeled axon. Imaging done using 1,040-nm excitation light. Reproduced from (Farrar et al., 2011). (D) THG images taken using 1,320-nm excitation light at the surface of the spinal cord before (left) and 30 min after (right) photothrombotic occlusion of a surface venule. THG contrast is produced both by the myelin and by red blood cells inside the vessel (visible as streaks in the image on the left due to their motion). After the occlusion, only stationary red blood cells (which show a characteristic donut shape) are visible and the nearby myelin has begun to degenerate (unpublished data). (E) (top) 3PEF imaging of a spinal cord capillary labeled with an intravenous injection of FITC-dextran. (bottom) Space-time images from repetitive line scans along the axis of the capillary with THG and 3PEF signals detected simultaneously, showing the mutual exclusivity of these two signals in blood, with red blood cells producing THG while excluding the fluorescent dye that labels the blood plasma (unpublished data).
Conclusions and future outlook
The surgical preparations, labeling strategies, and optical techniques applied to spinal cord imaging to date have enabled long-term studies of cellular structure and activity in the more superficial layers of the mouse spinal cord. Modest tweaks to the surgical approaches, adoption of recent developments in genetically-encoded labeling strategies, and utilization of higher-order nonlinear optical processes for imaging all hold promise for increasing the reach of in vivo imaging for studies of the murine spinal cord.
Surgical preparations
The chronic imaging chambers developed so far have primarily been implanted in the lower thoracic and upper lumbar regions of the spinal cord. Imaging in more rostral regions of the spinal cord could open the door to studies of sensory response and motor control of the forelimbs, which mice use for more dexterous tasks, such as reaching and grasping. The lower thoracic region sits near the top of the arch of the mouse’s back, where the spine is relatively straight, and the chamber and surgical approach will likely need to be modestly modified to work well in the upper thoracic and cervical regions. Recent work by Haghayegh Jahromi, et. al showed that acute optical imaging of the cervical spinal cord (C2–C5) was feasible (Haghayegh Jahromi et al., 2017). These authors found differences in the architecture of the vasculature in the cervical vs. lumbar spinal cord and directly visualized the invasion of circulating T-cells into the cervical spinal cord in the EAE mouse model (Haghayegh Jahromi et al., 2017). For studies of the neural correlates of behavior, chronic spinal cord chambers offer the opportunity to image neural activity in awake mice. This has already been demonstrated by fixing the spine (using the imaging chamber) and the head of mice under a microscope, as well as by using a miniaturized microscopes that was mounted to the imaging chamber (Sekiguchi et al., 2016). For spine-fixed studies in awake animals in spinal cord regions outside the lower thoracic region, the natural angle the spine makes relative to the horizontal will need to be accommodated by tipping the microscope.
Labeling strategies
In studies of neural structure and dynamics in the brain, the use of mice expressing Cre recombinase in specific neural sub-classes (defined by the expression of embryonic transcription factors) bred to mice that will express a reporter gene only when Cre is present has dramatically increased the degree to which the expression of genetically-encoded reporters can be carefully controlled, leading to efficient cell-type specific labeling in mice (Wang et al., 2016). In the spinal cord, such strategies have been used for cell type identification in post-mortem analysis (Sapir et al., 2004), for optical imaging in explanted spinal cord preparations (Bellardita et al., 2017), to control expression of channelrhodopsin to specific classes of neurons (Christensen et al., 2016), and in studies of the stimulus-evoked activity in dorsal root ganglion neurons (Chisholm et al., 2018; Emery et al., 2016). To date, these more modern genetic targeting strategies have seen only occasional use for in vivo imaging in the spinal cord (Sekiguchi et al., 2016). Newly-developed reporter lines offer strong and stable expression of fluorescent proteins or GECIs that is restricted to Cre-expressing cells and can be temporally controlled with doxycycline treatment (Daigle et al., 2018; Madisen et al., 2015). New developments in viral gene delivery may also offer new labeling capabilities. Chan, et al., developed modified AAV variants that efficiently cross the blood brain barrier, opening the door to systemic administration of viral vectors for spinal cord labeling. Additionally, these researchers have shown that AAV capsids can be altered to produce vectors that target neurons or glial cells with high specificity (Challis et al., 2019; Chan et al., 2017). These new transgenic and viral genetic labeling strategies could be of great utility for future in vivo spinal cord imaging studies.
Imaging tools
Optical imaging in the spinal cord is complicated by motion artifact, optical aberrations, and strong scattering from myelinated axons. For imaging of micrometer-scale features or for resolving calcium transients, even a few micrometers of motion artifact can be problematic. This much motion artifact can occur due to movement of the spinal cord relative to the vertebrae, which cannot be remedied with improved fixation. In plane motion can corrected with post-processing techniques, provided the imaging frame rate is high enough to avoid aliasing (Bélanger et al., 2012). With 2PEF or other optically-sectioning imaging modalities, motion along the z-axis must be corrected in real time. Laffray, et al. used a piezoelectric device to move the microscope objective and tracked reflected light from the spinal cord surface to follow the motion and were able to adaptively compensate for axial motion while imaging the mouse spinal cord (Laffray et al., 2011). Motion artifact is more pronounced in awake animals, so compensation techniques will be especially important for studies of the neural control of limb motion, for example. The cylindrical shape of the spinal cord likely contributes to optical aberrations that degrade signal strength and resolution with increased imaging depth. The average refractive index of the dorsal white matter is higher than the underlying grey matter due to the high lipid content and it is shaped like a cylindrical meniscus lens, leading to astigmatism. The use of adaptive optics to compensate for these aberrations could improve imaging performance (Ji et al., 2012; Park et al., 2017).
The white matter on the dorsal surface of the spinal cord has a very short scattering length, which limits the penetration depth of even 2PEF imaging to the top 100–200 µm. In contrast, 2PEF is able to penetrate over 800 µm into the mouse cortex. The limited imaging depth in the spinal cord restricts the kinds of scientific questions that can be addressed. The neurons that form the central pattern generator circuits that coordinate limb motion, for example, are located at depths of 400 µm and greater in adult mice. In the brain, GRIN lenses or prisms implanted into the cortex have been used to reach structures that are deeper than can be imaged from the cortical surface, but at the cost of damage to overlying tissue (Flusberg et al., 2005; Jung et al., 2004; Levene et al., 2004). Such approaches have not yet been tried in the spinal cord. The use of higher order nonlinear optical processes for fluorescence excitation also offers a path to greater penetration into the spinal cord. The maximum depth that can be achieved with 2PEF microscopy is ultimately limited by excitation of fluorescence from the sample surface. To image deeper into the sample, the laser energy must be increased to compensate for excitation light that is lost to optical scattering. At some depth, the amount of fluorescence excited at the sample surface (where the laser energy is high, but it is spread over a large area) approaches the amount of fluorescence excited at the laser focus (where the laser is focused to a micrometer-sized spot, but the remaining energy in the pulse is lower). This leads to an apparent increase in the background fluorescence as a function of depth (Theer et al., 2003). The shorter the scattering length of the sample, the shallower this limit is reached. With three photon excited fluorescence (3PEF) imaging, this signal to background limit to the imaging depth is much harder to reach. In brain, 3PEF has enabled cell structure and calcium transients to be imaged in the hippocampus, through the intact cortex and white matter (Horton et al., 2013; Ouzounov et al., 2017), as well as imaging up to 500 µm into the cortex through the intact skull (T. Wang et al., 2018). 3PEF imaging, similar to THG, requires different laser parameters than 2PEF or SHG imaging to be successful. First, longer excitation wavelengths are needed to three photon pump fluorescent species, with 1.3 µm and 1.7 µm light providing efficient three-photon excitation of many green and red emitting fluorescent species, respectively. Wavelengths between 1.3 and 1.6 µm are too strongly absorbed by water for in vivo imaging. Second, in order to efficiently drive three-photon excitation (or THG), higher per pulse energies must be delivered to the laser focus, typically 1–2 nJ per pulse. When imaging deep in a sample, much higher pulse energies must be applied to the sample to overcome the losses due to optical scattering and deliver this energy to the laser focus (as high as 120–150 nJ for maximum depth spinal cord imaging, in our hands). To avoid average power mediated sample damage with these energies, lower repetition rate lasers are needed, in the ~1–2 MHz range. In preliminary data, we compared the imaging depth that could be achieved with 2PEF vs. 3PEF imaging of FITC-dextran labeled blood vessels in the spinal cord of a live, anesthetized mouse. As expected, the background increased markedly below depths of about 100 µm with 2PEF imaging (Fig. 5, left). 3PEF imaging, however, enabled blood vessels to be reliably resolved to a depth of 500 µm into the spinal cord (Fig. 5, right).
Fig. 5. Three photon excited fluorescence microscopy enables much deeper imaging than two photon excited fluorescence in mouse spinal cord.
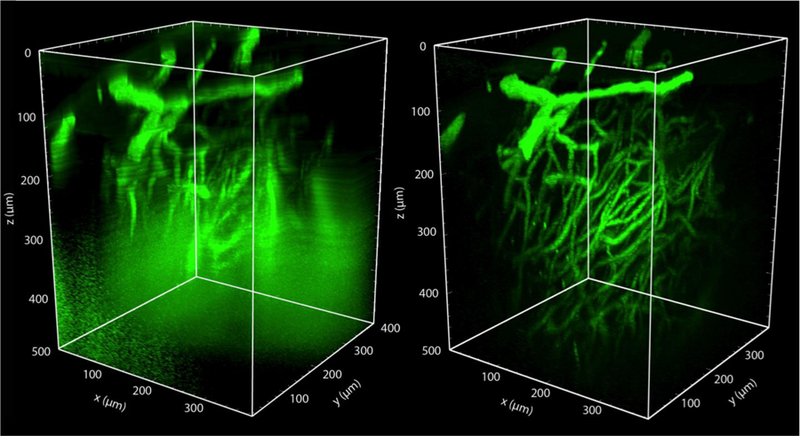
(left) Rendered 2PEF image stack of the spinal cord vasculature from a live, anesthetized mouse, labeled with an intravenous injection of 5% FITC-dextran. Image was taken with 800-nm excitation light (Chameleon, Coherent). (right) 3PEF imaging of the same region using 1,320-nm excitation light (Opera-F, Coherent). Out of plane fluorescence excitation leads to an increasing background with depth using 2PEF, ultimately limiting the imaging depth. This background is suppressed with 3PEF imaging, enabling visualization of individual capillaries as deep as 500 µm into the spinal cord (unpublished data).
Acknowledgments
This work was supported with funding from the National Science Foundation NeuroNex program (DBI-1707312 to CBS), the New York State Spinal Cord Injury Research Board (C32094GG to CBS; pre-doctoral fellowship C32630GG to YC), the National Institutes of Health (NS096669 to CBS), and the Craig H. Neilsen foundation (296332 to CBS). We thank Yu-Hsien Cheng for assistance with the drawings in Fig. 1.
List of Abbreviations:
- 2PEF
two photon excited fluorescence
- 3PEF
three photon excited fluorescence
- AAV
adeno-associated virus
- ALS
amyotrophic lateral sclerosis
- CARS
coherent anti-Stokes Raman scattering
- CNS
central nervous system
- EAE
experimental autoimmune encephalitis
- FITC
fluorescein isothiocyanate
- FRET
fluorescence resonant energy transfer
- GECI
genetically encoded calcium indicator
- GRIN
gradient index
- MS
multiple sclerosis
- OCT
optical coherence tomography
- OGB
Oregon Green Bapta
- SCI
spinal cord injury
- SCoRe
spectral confocal reflectance microscopy
- SHG
second harmonic generation
- SR101
sulforhodamine 101
- THG
third harmonic generation
- “X”FP
fluorescent protein, “X” = C for cyan, G for green, Y for yellow
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adur J, Carvalho HF, Cesar CL, & Casco VH (2014). Nonlinear optical microscopy signal processing strategies in cancer. Cancer Inform, 13, 67–76. doi: 10.4137/CIN.S12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger E, Crepeau J, Laffray S, Vallee R, De Koninck Y, & Cote D (2012). Live animal myelin histomorphometry of the spinal cord with video-rate multimodal nonlinear microendoscopy. J Biomed Opt, 17(2), 021107. doi: 10.1117/1.JBO.17.2.021107 [DOI] [PubMed] [Google Scholar]
- Bellardita C, Caggiano V, Leiras R, Caldeira V, Fuchs A, Bouvier J, … Kiehn O (2017). Spatiotemporal correlation of spinal network dynamics underlying spasms in chronic spinalized mice. Elife, 6. doi: 10.7554/eLife.23011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benakis C, Garcia-Bonilla L, Iadecola C, & Anrather J (2014). The role of microglia and myeloid immune cells in acute cerebral ischemia. Front Cell Neurosci, 8, 461. doi: 10.3389/fncel.2014.00461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadotte DW, Mariampillai A, Cadotte A, Lee KK, Kiehl TR, Wilson BC, … Yang VX (2012). Speckle variance optical coherence tomography of the rodent spinal cord: in vivo feasibility. Biomed Opt Express, 3(5), 911–919. doi: 10.1364/BOE.3.000911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caravagna C, Jaouen A, Desplat-Jego S, Fenrich KK, Bergot E, Luche H, … Debarbieux F (2018). Diversity of innate immune cell subsets across spatial and temporal scales in an EAE mouse model. Sci Rep, 8(1), 5146. doi: 10.1038/s41598-018-22872-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartarozzi LP, Rieder P, Bai X, Scheller A, Oliveira ALR, & Kirchhoff F (2018). In vivo two-photon imaging of motoneurons and adjacent glia in the ventral spinal cord. J Neurosci Methods, 299, 8–15. doi: 10.1016/j.jneumeth.2018.01.005 [DOI] [PubMed] [Google Scholar]
- Challis RC, Ravindra Kumar S, Chan KY, Challis C, Beadle K, Jang MJ, … Gradinaru V (2019). Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat Protoc, 14(2), 379–414. doi: 10.1038/s41596-018-0097-3 [DOI] [PubMed] [Google Scholar]
- Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, … Gradinaru V (2017). Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci, 20(8), 1172–1179. doi: 10.1038/nn.4593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Zhang YP, Sun Y, Xiong W, Shields LBE, Shields CB, … Xu XM (2017). An In Vivo Duo-color Method for Imaging Vascular Dynamics Following Contusive Spinal Cord Injury. J Vis Exp (130). doi: 10.3791/56565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Taniguchi W, Chen QY, Tozaki-Saitoh H, Song Q, Liu RH, … Zhuo M (2018). Top-down descending facilitation of spinal sensory excitatory transmission from the anterior cingulate cortex. Nat Commun, 9(1), 1886. doi: 10.1038/s41467-018-04309-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, … Kim DS (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature, 499(7458), 295–300. doi: 10.1038/nature12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm KI, Khovanov N, Lopes DM, La Russa F, & McMahon SB (2018). Large Scale In Vivo Recording of Sensory Neuron Activity with GCaMP6. eNeuro, 5(1). doi: 10.1523/ENEURO.0417-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong SP, Merkle CW, Cooke DF, Zhang T, Radhakrishnan H, Krubitzer L, & Srinivasan VJ (2015). Noninvasive, in vivo imaging of subcortical mouse brain regions with 1.7 mum optical coherence tomography. Opt Lett, 40(21), 4911–4914. doi: 10.1364/OL.40.004911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen AJ, Iyer SM, Francois A, Vyas S, Ramakrishnan C, Vesuna S, … Delp SL (2016). In Vivo Interrogation of Spinal Mechanosensory Circuits. Cell Rep, 17(6), 1699–1710. doi: 10.1016/j.celrep.2016.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo G, De Luca D, & Papa M (2012). Calcium imaging of living astrocytes in the mouse spinal cord following sensory stimulation. Neural Plast, 2012, 425818. doi: 10.1155/2012/425818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz Hernández JC, Bracko O, Kersbergen CJ, Muse V, Haft-Javaherian M, Berg M, … Schaffer CB (2019). Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nature Neuroscience doi: 10.1038/s41593-018-0329-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle TL, Madisen L, Hage TA, Valley MT, Knoblich U, Larsen RS, … Zeng H (2018). A Suite of Transgenic Driver and Reporter Mouse Lines with Enhanced Brain-Cell-Type Targeting and Functionality. Cell, 174(2), 465–480 e422. doi: 10.1016/j.cell.2018.06.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, & Akassoglou K (2012). In vivo imaging of the mouse spinal cord using two - photon microscopy. J Vis Exp(59), e2760. doi: 10.3791/2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Lee JK, Smith WB, Brinkman B, Ellisman MH, Zheng B, & Akassoglou K (2008). Stable in vivo imaging of densely populated glia, axons and blood vessels in the mouse spinal cord using two-photon microscopy. J Neurosci Methods, 169(1), 1–7. doi: 10.1016/j.jneumeth.2007.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, … Akassoglou K (2012). Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun, 3, 1227. doi: 10.1038/ncomms2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNotta S (2018). In-vivo Multiphoton Excited Fluoresence Microscopy Of The Spinal Cord. (Ph.D.), Cornell University. [Google Scholar]
- Di Maio A, Skuba A, Himes BT, Bhagat SL, Hyun JK, Tessler A, … Son YJ (2011). In vivo imaging of dorsal root regeneration: rapid immobilization and presynaptic differentiation at the CNS/PNS border. J Neurosci, 31(12), 4569–4582. doi: 10.1523/JNEUROSCI.4638-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibaj P, Steffens H, Zschuntzsch J, Kirchhoff F, Schomburg ED, & Neusch C (2011). In vivo imaging reveals rapid morphological reactions of astrocytes towards focal lesions in an ALS mouse model. Neurosci Lett, 497(2), 148–151. doi: 10.1016/j.neulet.2011.04.049 [DOI] [PubMed] [Google Scholar]
- Dietzel S, Pircher J, Nekolla AK, Gull M, Brandli AW, Pohl U, & Rehberg M (2014). Label-free determination of hemodynamic parameters in the microcirculaton with third harmonic generation microscopy. PLoS One, 9(6), e99615. doi: 10.1371/journal.pone.0099615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombeck DA, Harvey CD, Tian L, Looger LL, & Tank DW (2010). Functional imaging of hippocampal place cells at cellular resolution during virtual navigation. Nat Neurosci, 13(11), 1433–1440. doi: 10.1038/nn.2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombeck DA, Khabbaz AN, Collman F, Adelman TL, & Tank DW (2007). Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron, 56(1), 43–57. doi: 10.1016/j.neuron.2007.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray C, Rougon G, & Debarbieux F (2009). Quantitative analysis by in vivo imaging of the dynamics of vascular and axonal networks in injured mouse spinal cord. Proc Natl Acad Sci U S A, 106(23), 9459–9464. doi: 10.1073/pnas.0900222106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew PJ, Shih AY, Driscoll JD, Knutsen PM, Blinder P, Davalos D, … Kleinfeld D (2010). Chronic optical access through a polished and reinforced thinned skull. Nat Methods, 7(12), 981–984. doi: 10.1038/nmeth.1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery EC, Luiz AP, Sikandar S, Magnusdottir R, Dong X, & Wood JN (2016). In vivo characterization of distinct modality-specific subsets of somatosensory neurons using GCaMP. Sci Adv, 2(11), e1600990. doi: 10.1126/sciadv.1600990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans TA, Barkauskas DS, Myers JT, & Huang AY (2014). Intravital imaging of axonal interactions with microglia and macrophages in a mouse dorsal column crush injury. J Vis Exp (93), e52228. doi: 10.3791/52228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar MJ, Bernstein IM, Schlafer DH, Cleland TA, Fetcho JR, & Schaffer CB (2012). Chronic in vivo imaging in the mouse spinal cord using an implanted chamber. Nat Methods, 9(3), 297–302. doi: 10.1038/nmeth.1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar MJ, Rubin JD, Diago DM, & Schaffer CB (2015). Characterization of blood flow in the mouse dorsal spinal venous system before and after dorsal spinal vein occlusion. J Cereb Blood Flow Metab, 35(4), 667–675. doi: 10.1038/jcbfm.2014.244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar MJ, & Schaffer CB (2014). A procedure for implanting a spinal chamber for longitudinal in vivo imaging of the mouse spinal cord. J Vis Exp (94). doi: 10.3791/52196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar MJ, Wise FW, Fetcho JR, & Schaffer CB (2011). In vivo imaging of myelin in the vertebrate central nervous system using third harmonic generation microscopy. Biophys J, 100(5), 1362–1371. doi: 10.1016/j.bpj.2011.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenrich KK, Weber P, Hocine M, Zalc M, Rougon G, & Debarbieux F (2012). Long-term in vivo imaging of normal and pathological mouse spinal cord with subcellular resolution using implanted glass windows. J Physiol, 590(16), 3665–3675. doi: 10.1113/jphysiol.2012.230532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenrich KK, Weber P, Rougon G, & Debarbieux F (2013a). Implanting glass spinal cord windows in adult mice with experimental autoimmune encephalomyelitis. J Vis Exp (82), e50826. doi: 10.3791/50826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenrich KK, Weber P, Rougon G, & Debarbieux F (2013b). Long- and short-term intravital imaging reveals differential spatiotemporal recruitment and function of myelomonocytic cells after spinal cord injury. J Physiol, 591(19), 4895–4902. doi: 10.1113/jphysiol.2013.256388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figley SA, Chen Y, Maeda A, Conroy L, McMullen JD, Silver JI, … DaCosta RS (2013). A spinal cord window chamber model for in vivo longitudinal multimodal optical and acoustic imaging in a murine model. PLoS One, 8(3), e58081. doi: 10.1371/journal.pone.0058081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flusberg BA, Cocker ED, Piyawattanametha W, Jung JC, Cheung EL, & Schnitzer MJ (2005). Fiber-optic fluorescence imaging. Nat Methods, 2(12), 941–950. doi: 10.1038/nmeth820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, … Herms J (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci, 13(4), 411–413. doi: 10.1038/nn.2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto JG (2003). Optical coherence tomography for ultrahigh resolution in vivo imaging. Nat Biotechnol, 21(11), 1361–1367. doi: 10.1038/nbt892 [DOI] [PubMed] [Google Scholar]
- Furue H, Narikawa K, Kumamoto E, & Yoshimura M (1999). Responsiveness of rat substantia gelatinosa neurones to mechanical but not thermal stimuli revealed by in vivo patch-clamp recording. J Physiol, 521 Pt 2, 529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh KK, Burns LD, Cocker ED, Nimmerjahn A, Ziv Y, Gamal AE, & Schnitzer MJ (2011). Miniaturized integration of a fluorescence microscope. Nat Methods, 8(10), 871–878. doi: 10.1038/nmeth.1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JW, Chesa PG, Nishimura H, Rettig WJ, Maccari JE, Endo T, … Silver J (1987). Regulation of Thy-1 gene expression in transgenic mice. Cell, 50(3), 445–452. [DOI] [PubMed] [Google Scholar]
- Grienberger C, & Konnerth A (2012). Imaging calcium in neurons. Neuron, 73(5), 862–885. doi: 10.1016/j.neuron.2012.02.011 [DOI] [PubMed] [Google Scholar]
- Haenraets K, Albisetti GW, Foster E, & Wildner H (2018). Adeno-associated Virus-mediated Transgene Expression in Genetically Defined Neurons of the Spinal Cord. J Vis Exp (135). doi: 10.3791/57382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghayegh Jahromi N, Tardent H, Enzmann G, Deutsch U, Kawakami N, Bittner S, … Engelhardt B (2017). A Novel Cervical Spinal Cord Window Preparation Allows for Two-Photon Imaging of T-Cell Interactions with the Cervical Spinal Cord Microvasculature during Experimental Autoimmune Encephalomyelitis. Front Immunol, 8, 406. doi: 10.3389/fimmu.2017.00406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Coen P, & Tank DW (2012). Choice-specific sequences in parietal cortex during a virtual-navigation decision task. Nature, 484(7392), 62–68. doi: 10.1038/nature10918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F, & Denk W (2005). Deep tissue two-photon microscopy. Nat Methods, 2(12), 932–940. doi: 10.1038/nmeth818 [DOI] [PubMed] [Google Scholar]
- Hill RA, & Grutzendler J (2014). In vivo imaging of oligodendrocytes with sulforhodamine 101. Nat Methods, 11(11), 1081–1082. doi: 10.1038/nmeth.3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, … Wilbrecht L (2009). Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc, 4(8), 1128–1144. doi: 10.1038/nprot.2009.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton NG, Wang K, Kobat D, Clark CG, Wise FW, Schaffer CB, & Xu C (2013). In vivo three-photon microscopy of subcortical structures within an intact mouse brain. Nat Photonics, 7(3). doi: 10.1038/nphoton.2012.336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji N, Sato TR, & Betzig E (2012). Characterization and adaptive optical correction of aberrations during in vivo imaging in the mouse cortex. Proc Natl Acad Sci U S A, 109(1), 22–27. doi: 10.1073/pnas.1109202108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannssen HC, & Helmchen F (2010). In vivo Ca2+ imaging of dorsal horn neuronal populations in mouse spinal cord. J Physiol, 588(Pt 18), 3397–3402. doi: 10.1113/jphysiol.2010.191833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannssen HC, & Helmchen F (2013). Two-photon imaging of spinal cord cellular networks. Exp Neurol, 242, 18–26. doi: 10.1016/j.expneurol.2012.07.014 [DOI] [PubMed] [Google Scholar]
- Jung JC, Mehta AD, Aksay E, Stepnoski R, & Schnitzer MJ (2004). In vivo mammalian brain imaging using one- and two-photon fluorescence microendoscopy. J Neurophysiol, 92(5), 3121–3133. doi: 10.1152/jn.00234.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohro Y, Sakaguchi E, Tashima R, Tozaki-Saitoh H, Okano H, Inoue K, & Tsuda M (2015). A new minimally-invasive method for microinjection into the mouse spinal dorsal horn. Sci Rep, 5, 14306. doi: 10.1038/srep14306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi K, Hattori Y, Ahn SJ, Buendia I, Ciacciarelli A, Uekawa K, … Iadecola C (2018). Apoepsilon4 disrupts neurovascular regulation and undermines white matter integrity and cognitive function. Nat Commun, 9(1), 3816. doi: 10.1038/s41467-018-06301-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laffray S, Pages S, Dufour H, De Koninck P, De Koninck Y, & Cote D (2011). Adaptive movement compensation for in vivo imaging of fast cellular dynamics within a moving tissue. PLoS One, 6(5), e19928. doi: 10.1371/journal.pone.0019928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levene MJ, Dombeck DA, Kasischke KA, Molloy RP, & Webb WW (2004). In vivo multiphoton microscopy of deep brain tissue. J Neurophysiol, 91(4), 1908–1912. doi: 10.1152/jn.01007.2003 [DOI] [PubMed] [Google Scholar]
- Light AR, & Willcockson HH (1999). Spinal laminae I-II neurons in rat recorded in vivo in whole cell, tight seal configuration: properties and opioid responses. J Neurophysiol, 82(6), 3316–3326. doi: 10.1152/jn.1999.82.6.3316 [DOI] [PubMed] [Google Scholar]
- Lorenzana AO, Lee JK, Mui M, Chang A, & Zheng B (2015). A surviving intact branch stabilizes remaining axon architecture after injury as revealed by in vivo imaging in the mouse spinal cord. Neuron, 86(4), 947–954. doi: 10.1016/j.neuron.2015.03.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Jongbloets BC, Xiong WH, Melander JB, Qin M, Lameyer TJ, … Zhong H (2018). A Highly Sensitive A-Kinase Activity Reporter for Imaging Neuromodulatory Events in Awake Mice. Neuron, 99(4), 665–679 e665. doi: 10.1016/j.neuron.2018.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, … Zeng H (2015). Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron, 85(5), 942–958. doi: 10.1016/j.neuron.2015.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura S, Taniguchi W, Nishida K, Nakatsuka T, & Ito S (2015). In vivo two-photon imaging of structural dynamics in the spinal dorsal horn in an inflammatory pain model. Eur J Neurosci, 41(7), 989–997. doi: 10.1111/ejn.12837 [DOI] [PubMed] [Google Scholar]
- Meng G, Liang Y, Sarsfield S, Jiang WC, Lu R, Dudman JT, … Ji N (2019). High-throughput synapse-resolving two-photon fluorescence microendoscopy for deep-brain volumetric imaging in vivo. Elife, 8. doi: 10.7554/eLife.40805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min W, Freudiger CW, Lu S, & Xie XS (2011). Coherent nonlinear optical imaging: beyond fluorescence microscopy. Annu Rev Phys Chem, 62, 507–530. doi: 10.1146/annurev.physchem.012809.103512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, & Kerschensteiner M (2006). In vivo imaging of the diseased nervous system. Nat Rev Neurosci, 7(6), 449–463. doi: 10.1038/nrn1905 [DOI] [PubMed] [Google Scholar]
- Misgeld T, Nikic I, & Kerschensteiner M (2007). In vivo imaging of single axons in the mouse spinal cord. Nat Protoc, 2(2), 263–268. doi: 10.1038/nprot.2007.24 [DOI] [PubMed] [Google Scholar]
- Miyazaki K, Masamoto K, Morimoto N, Kurata T, Mimoto T, Obata T, … Abe K (2012). Early and progressive impairment of spinal blood flow-glucose metabolism coupling in motor neuron degeneration of ALS model mice. J Cereb Blood Flow Metab, 32(3), 456–467. doi: 10.1038/jcbfm.2011.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R (1985). Thy-1 in developing nervous tissue. Dev Neurosci, 7(3), 133–160. doi: 10.1159/000112283 [DOI] [PubMed] [Google Scholar]
- Neurotar. (2019). Spinal cord surgery set Retrieved from https://www.neurotar.com/product/spinal-cord-surgery-set/
- Nikić I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM, … Kerschensteiner M (2011). A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med, 17(4), 495–499. doi: 10.1038/nm.2324 [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN, & Helmchen F (2004). Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods, 1(1), 31–37. doi: 10.1038/nmeth706 [DOI] [PubMed] [Google Scholar]
- Nishida K, Matsumura S, Taniguchi W, Uta D, Furue H, & Ito S (2014). Three-dimensional distribution of sensory stimulation-evoked neuronal activity of spinal dorsal horn neurons analyzed by in vivo calcium imaging. PLoS One, 9(8), e103321. doi: 10.1371/journal.pone.0103321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouzounov DG, Wang T, Wang M, Feng DD, Horton NG, Cruz-Hernandez JC, … Xu C (2017). In vivo three-photon imaging of activity of GCaMP6-labeled neurons deep in intact mouse brain. Nat Methods, 14(4), 388–390. doi: 10.1038/nmeth.4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Kong L, Zhou Y, & Cui M (2017). Large-field-of-view imaging by multi-pupil adaptive optics. Nat Methods, 14(6), 581–583. doi: 10.1038/nmeth.4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriarchi T, Cho JR, Merten K, Howe MW, Marley A, Xiong WH, … Tian L (2018). Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science, 360(6396). doi: 10.1126/science.aat4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran C, Hoon MA, & Chen X (2016). The coding of cutaneous temperature in the spinal cord. Nat Neurosci, 19(9), 1201–1209. doi: 10.1038/nn.4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickgauer JP, Deisseroth K, & Tank DW (2014). Simultaneous cellular-resolution optical perturbation and imaging of place cell firing fields. Nat Neurosci, 17(12), 1816–1824. doi: 10.1038/nn.3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruschel J, Hellal F, Flynn KC, Dupraz S, Elliott DA, Tedeschi A, … Bradke F (2015). Axonal regeneration. Systemic administration of epothilone B promotes axon regeneration after spinal cord injury. Science, 348(6232), 347–352. doi: 10.1126/science.aaa2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba R, Nakatsuji N, & Saito T (2003). Mammalian BarH1 confers commissural neuron identity on dorsal cells in the spinal cord. J Neurosci, 23(6), 1987–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghian M, Mastrolia V, Rezaei Haddad A, Mosley A, Mullali G, Schiza D, … Smith KJ (2016). Mitochondrial dysfunction is an important cause of neurological deficits in an inflammatory model of multiple sclerosis. Sci Rep, 6, 33249. doi: 10.1038/srep33249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapir T, Geiman EJ, Wang Z, Velasquez T, Mitsui S, Yoshihara Y, … Goulding M (2004). Pax6 and engrailed 1 regulate two distinct aspects of renshaw cell development. J Neurosci, 24(5), 1255–1264. doi: 10.1523/JNEUROSCI.3187-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schain AJ, Hill RA, & Grutzendler J (2014). Label-free in vivo imaging of myelinated axons in health and disease with spectral confocal reflectance microscopy. Nat Med, 20(4), 443–449. doi: 10.1038/nm.3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi KJ, Shekhtmeyster P, Merten K, Arena A, Cook D, Hoffman E, … Nimmerjahn A (2016). Imaging large-scale cellular activity in spinal cord of freely behaving mice. Nat Commun, 7, 11450. doi: 10.1038/ncomms11450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonohata M, Furue H, Katafuchi T, Yasaka T, Doi A, Kumamoto E, & Yoshimura M (2004). Actions of noradrenaline on substantia gelatinosa neurones in the rat spinal cord revealed by in vivo patch recording. J Physiol, 555(Pt 2), 515–526. doi: 10.1113/jphysiol.2003.054932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squier J, Muller M, Brakenhoff G, & Wilson KR (1998). Third harmonic generation microscopy. Opt Express, 3(9), 315–324. [DOI] [PubMed] [Google Scholar]
- St-Pierre F, Chavarha M, & Lin MZ (2015). Designs and sensing mechanisms of genetically encoded fluorescent voltage indicators. Curr Opin Chem Biol, 27, 31–38. doi: 10.1016/j.cbpa.2015.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storace D, Sepehri Rad M, Kang B, Cohen LB, Hughes T, & Baker BJ (2016). Toward Better Genetically Encoded Sensors of Membrane Potential. Trends Neurosci, 39(5), 277–289. doi: 10.1016/j.tins.2016.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, & Konnerth A (2003). In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci U S A, 100(12), 7319–7324. doi: 10.1073/pnas.1232232100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang P, Zhang Y, Chen C, Ji X, Ju F, Liu X, … Zhang L (2015). In vivo two-photon imaging of axonal dieback, blood flow, and calcium influx with methylprednisolone therapy after spinal cord injury. Sci Rep, 5, 9691. doi: 10.1038/srep09691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theer P, Hasan MT, & Denk W (2003). Two-photon imaging to a depth of 1000 microns in living brains by use of a Ti:Al2O3 regenerative amplifier. Opt Lett, 28(12), 1022–1024. [DOI] [PubMed] [Google Scholar]
- Vidal M, Morris R, Grosveld F, & Spanopoulou E (1990). Tissue-specific control elements of the Thy-1 gene. EMBO J, 9(3), 833–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Belanger E, Paquet ME, Cote DC, & De Koninck Y (2016). Probing pain pathways with light. Neuroscience, 338, 248–271. doi: 10.1016/j.neuroscience.2016.09.035 [DOI] [PubMed] [Google Scholar]
- Wang T, Ouzounov DG, Wu C, Horton NG, Zhang B, Wu CH, … Xu C (2018). Three - photon imaging of mouse brain structure and function through the intact skull. Nat Methods, 15(10), 789–792. doi: 10.1038/s41592-018-0115-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelin B, Bakker GJ, & Friedl P (2016). Third harmonic generation microscopy of cells and tissue organization. J Cell Sci, 129(2), 245–255. doi: 10.1242/jcs.152272 [DOI] [PubMed] [Google Scholar]
- Williams PR, Marincu BN, Sorbara CD, Mahler CF, Schumacher AM, Griesbeck O, … Misgeld T (2014). A recoverable state of axon injury persists for hours after spinal cord contusion in vivo. Nat Commun, 5, 5683. doi: 10.1038/ncomms6683 [DOI] [PubMed] [Google Scholar]
- Xu HT, Pan F, Yang G, & Gan WB (2007). Choice of cranial window type for in vivo imaging affects dendritic spine turnover in the cortex. Nat Neurosci, 10(5), 549–551. doi: 10.1038/nn1883 [DOI] [PubMed] [Google Scholar]
- Yang G, Pan F, Parkhurst CN, Grutzendler J, & Gan WB (2010). Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nat Protoc, 5(2), 201–208. doi: 10.1038/nprot.2009.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, & Yuste R (2017). In vivo imaging of neural activity. Nat Methods, 14(4), 349–359. doi: 10.1038/nmeth.4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Xie W, Ju F, Khan A, & Zhang S (2017). In vivo two-photon imaging reveals a role of progesterone in reducing axonal dieback after spinal cord injury in mice. Neuropharmacology, 116, 30–37. doi: 10.1016/j.neuropharm.2016.12.007 [DOI] [PubMed] [Google Scholar]
- Yoshihara K, Matsuda T, Kohro Y, Tozaki-Saitoh H, Inoue K, & Tsuda M (2018). Astrocytic Ca(2+) responses in the spinal dorsal horn by noxious stimuli to the skin. J Pharmacol Sci, 137(1), 101–104. doi: 10.1016/j.jphs.2018.04.007 [DOI] [PubMed] [Google Scholar]
- Zipfel WR, Williams RM, Christie R, Nikitin AY, Hyman BT, & Webb WW (2003). Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proc Natl Acad Sci U S A, 100(12), 7075–7080. doi: 10.1073/pnas.0832308100 [DOI] [PMC free article] [PubMed] [Google Scholar]