

Abstract
Following damage to a peripheral nerve, injury signaling pathways converge in the cell body to generate transcriptional changes that support axon regeneration. Here, we demonstrate that dual leucine zipper kinase (DLK), a central regulator of injury responses including axon regeneration and neuronal apoptosis, is required for the induction of the pro-regenerative transcriptional program in response to peripheral nerve injury. Using a sensory neuron-conditional DLK knockout mouse model, we show a time course for the dependency of gene expression changes on the DLK pathway after sciatic nerve injury. Gene ontology analysis reveals that DLK-dependent gene sets are enriched for specific functional annotations such as ion transport and immune response. A series of comparative analyses shows that the DLK-dependent transcriptional program is distinct from that promoted by the importin-dependent retrograde signaling pathway, while it is partially shared between PNS and CNS injury responses. We suggest that DLK-dependency might provide a selective filter for regeneration-associated genes among the injury-responsive transcriptome.
Keywords: Dual leucine zipper kinase, Conditioning injury, Axon regeneration, Neuroinflammation, Neurodegeneration, Pain
Introduction
Axonal injury can result from various insults including trauma, toxins, ischemia, and progression of neurological disorders (Gerdts et al., 2016; Hill et al., 2016; Wang et al., 2012). The consequence of axonal injury ranges from death of the damaged neurons to axonal regeneration and functional recovery (Bradke et al., 2012; Liu et al., 2011). Chronic loss of presynaptic input causes devastating conditions including neural dysfunction and dystrophy of the target tissue, highlighting the need for methods to improve axon regeneration and neural repair (Gordon et al., 2011).
The ability to regenerate injured axons varies by neuronal age and identity. Axon regeneration in adult neurons is not as efficient as in younger neurons (Geoffroy et al., 2016; Kang and Lichtman, 2013; Pestronk et al., 1980; Verdu et al., 1995), and molecular pathways regulating age-dependent axon regeneration are being identified (Byrne et al., 2014; Cai et al., 2001). Moreover, injured axons in the central nervous system (CNS) have little capacity to regrow (Curcio and Bradke, 2018); instead, axonal damage often induces neuronal apoptosis (Huebner and Strittmatter, 2009; Quigley et al., 1995). In contrast, axonal regeneration in the peripheral nervous system (PNS) is robust and can reestablish functional neural connections (Magill et al., 2007; Valero-Cabré et al., 2004). As a consequence of this difference in regenerative capacity, CNS injury often leads to irreversible deficits such as cognitive impairment and paraplegia, while sensorimotor deficits resulting from PNS axon injury often improve.
The regenerative capacity of a neuron is not static. For example, a preceding lesion to a peripheral nerve potentiates axonal growth of sensory neurons in both the CNS and PNS, demonstrating that axonal damage can activate an axon regeneration program (Neumann and Woolf, 1999; Pan et al., 2003; Richardson and Issa, 1984). A pioneering study by Smith and Skene demonstrated that this “conditioning injury effect” requires gene expression, as transcriptional inhibition abolished the induction of neurite outgrowth following a pre-lesion (Smith and Skene, 1997). Moreover, recent studies have demonstrated that a nerve lesion promotes retrograde transport of injury signals and subsequent activation of transcription factors in the cell body (Abe and Cavalli, 2008; Michaelevski et al., 2010; Rishal and Fainzilber,2014). Indeed, transcriptomic analyses using microarray and RNA-sequencing (RNA-seq) techniques reveal that gene expression is markedly altered after peripheral nerve injury (Gong et al., 2016; Li et al., 2015). Importantly, bioinformatic analysis of such datasets can identify important effectors promoting axon regeneration (Ma and Willis, 2015).
The dual leucine zipper kinase (DLK) is a mitogen-activated protein kinase kinase kinase (MAP3K) that can activate the downstream cJun N-terminal kinase (JNK) pathway (Fan et al., 1996; Hirai et al., 2006; Holzman et al., 1994). DLK is an important neuronal stress response kinase with an evolutionarily conserved role in regulating the neuronal response to injury (Asghari Adib et al., 2018; Ghosh et al., 2011; Hammarlund et al., 2009; Itoh et al., 2014; Miller et al., 2009; Le Pichon et al., 2017; Watkins et al., 2013; Welsbie et al., 2013; Wlaschin et al., 2018; Xiong et al., 2010; Yan et al., 2009). We demonstrated that DLK promotes retrograde injury signaling and axon regeneration after PNS nerve injury in the mammals (Shin et al., 2012). We showed that DLK is required for injury-dependent activation of the transcription factors cJun and STAT3, and so hypothesized that DLK is also necessary for the transcriptional responses to peripheral nerve injury. Because the genetic deletion of DLK completely abolishes the effect of conditioning injury in mouse dorsal root ganglia (DRG) neurons (Shin et al., 2012), we suggest that DLK-dependent transcriptional responses to nerve injury likely include key regulators of axon regeneration. In the current study, we identify the DLK-dependent transcriptome using the sciatic nerve injury paradigm. The differentially expressed gene (DEG) analysis reveals that DLK regulates injury-responsive genes in both basal and injured conditions. By performing gene ontology analysis, we identify genes with shared functional annotations and suggest that these may serve as regulatory components of the axonal regeneration program. Finally, we perform a comparative analysis with other nerve injury datasets and find that DLK is required for a distinctive retrograde signaling pathway that regulates a regeneration program partially shared between PNS and CNS injury models.
Results
1. Sequencing of mouse DRG RNA reveals differential gene expression after sciatic nerve injury
We set out to identify DLK-dependent transcriptional changes induced by peripheral nerve injury and to investigate distinct features of the DLK-dependent transcriptome. We have previously shown that DLK is required in DRG sensory neurons for transducing retrograde signals following axonal injury and promoting regeneration of the injured axons (Shin et al., 2012). A DRG is a discrete structure containing sensory neuronal cell bodies and therefore is suitable for dissection and subsequent bioinformatic analysis. In order to define the neuronal role of the DLK pathway in transcriptional regulation, we ablated DLK expression by crossing floxed DLK mice (Miller et al., 2009) to the sensory neuron-specific advillin-cre driver (Hasegawa et al., 2007). The resulting sensory neuronal DLK knockout (KO) mice and littermate controls were injured with unilateral sciatic nerve transection, and the L4-5 DRGs from these animals were subsequently subjected to RNA isolation, ribosomal RNA depletion and transcriptome sequencing (Figure 1A). Because DRGs include non-neuronal cells such as satellite glial cells, microglia and macrophages infiltrating after injury, non-cell-autonomous responses to axon injury as well as neuronal responses will be identified in the analysis.
Figure 1.

RNA sequencing (RNA-seq) reveals DLK-dependent transcriptomic changes induced by nerve injury.
(A) Schematic diagram of the transcriptome study. Wild type (WT) and sensory neuron-specific DLK knockout (KO) mice were subjected to unilateral sciatic nerve transection and followed by collection of the L4-5 DRGs for RNA isolation. After transcriptome sequencing, differentially expressed genes (DEGs) were identified and the gene ontology analysis was performed.
(B and C) Volcano plots visualize regulation of gene expression by nerve injury either in WT (B) or in DLK KO (C). Each gene is shown as -log10(p-value) plotted against log2(fold change). Fold change was calculated by dividing the mean FPKM value at the given post-injury time point by that of the uninjured control (u). Red dots represent genes with significant expression changes (p < 0.01). The percentage and parenthesized number in blue indicate the percentage and the number of significant DEGs, up-regulated or down-regulated by injury.
(D) Gene expression changes by DLK depletion are visualized in volcano plots where each gene was shown as -log10(p-value) plotted against log2(fold change). Fold change was calculated by dividing the mean FPKM value in DLK KO by that in WT. Red dots represent genes with significant expression changes (p < 0.01). The percentage and parenthesized number in blue indicate the percentage and the number of significant DEGs, up-regulated or down-regulated in DLK KO.
To define the progression of transcriptional changes following nerve injury, we collected the DRGs at 12, 24 and 72 hours (h) after nerve transection (Figure 1A). The rationales for selecting the time points were as follows. Phosphorylation of cJun, a major downstream transcription factor of the DLK MAP3K pathway, is first observed in DRG neuronal cell bodies around 9 h after nerve injury and peaks at 12-24 h (data not shown) (Zhuang et al., 2006). Hence, the transcriptome at 12 h after injury represents early transcriptional responses to the injury signals including the DLK-JNK MAPK pathway. At 24 h after injury, extension of injured axons is observed in mouse sciatic nerve indicating an active phase of axon regeneration (Shin et al., 2014). The regeneration rate is accelerated during the following days (Pan et al., 2003), suggesting enhancement of the regeneration program in the late phase (e. g., 72 h) via transcriptional, translational and post-translational mechanisms. Each sample included pooled RNA from two animals, and three samples were subjected to transcriptome sequencing for each condition.
The sequencing resulted in an average of 30.0 million mapped reads per sample and uniquely mapped reads accounted for 27.4 million reads per sample on average. Summary of the initial alignment results are shown in Supplementary Table 1. Gene expression levels were quantified in fragments per kilobase per million mapped reads (FPKM) for each gene, which is a normalized value for the length of the gene and the sequencing depth, and compared between conditions of interest. We first examined the expression data to determine DEGs at each post-lesion time point in wild type (WT). By comparing to the gene expression profile in uninjured DRG (u), we identified increasing numbers of injury-regulated DEGs over time as presented in volcano plots in Figure 1B. At 12, 24 and 72 h after injury, 240, 830 and 1295 genes were up-regulated compared to the uninjured control, respectively, at p < 0.01 significance level. Meanwhile, 92, 456 and 1109 genes were significantly down-regulated by nerve injury at each time point (p < 0.01), demonstrating that while fewer genes are down-regulated than up-regulated in the DRG following nerve injury, both types of changes are common. The DEG analysis in WT detected many well-known regeneration-associated genes as injury-induced DEGs (e.g., Atf3, Gadd45a, Jun, Sprr1a and Npy), validating the reliability of the sequencing and analysis processes.
2. Neuronal knockout of DLK impairs transcriptional responses to nerve injury
In order to determine the involvement of DLK in gene expression changes induced by peripheral nerve injury, we first identified genes that are differentially expressed after injury from the DLK KO dataset. At 12 h after injury, the total number of significant DEGs in DLK KO was not notably different from that in WT. However, at 24 and 72 h after injury, the number of up-regulated and down-regulated DEGs at each time point was remarkably lower in DLK KO than in WT (p < 0.01) (Figure 1B-C); the DEG numbers in KO were approximately a half of those in WT at 72 h. These data demonstrate that deleting DLK expression impairs injury-induced changes in the gene expression pattern, which is essential for activation of the axonal regeneration program.
Next, we investigated the effect of DLK deficiency on the expression levels of individual genes, by comparing the expression levels in WT and the DLK mutant DRGs at each injury time point. The analysis showed that increasing numbers of DEGs were identified across the time-course, with the up-regulated and down-regulated DEGs in DLK KO reaching 442 and 563, respectively, at 72 h post-injury time point (p < 0.01) (Figure 1D). These data indicate that the requirement of DLK for transcriptome regulation becomes crucial in the later phases of injury response when gene expression changes promote the regeneration program. Notably, loss of DLK results in many up-regulated DEGs as well as down-regulated DEGs (Figure 1D), demonstrating that DLK is necessary for repression of gene expression as well as induction of gene expression by nerve injury. While the volcano plots were made with p-values to provide an overview of the differential gene expression, the DEG sets used in subsequent analyses (Figure 2 – 7) were defined by q-values, which are adjusted p-values to reduce false positives resulting from multiple testing. The DEG numbers based on q-values are listed in Supplementary Table 2 (∣Log2FC∣ > 0.35) and the complete expression data of the significant DEGs (q < 0.05) are provided in Supplementary Table 3.
Figure 2.

Quantitative reverse transcription PCR (qRT-PCR) validates the DEGs identified by RNA-seq.
(A) The RNA-seq results are shown for selected DLK-dependent DEGs. WT and DLK KO: Expression levels at 72 h after injury are shown as fold change compared to the expression levels in uninjured condition (u). DLK KO / WT: Expression levels in DLK KO are compared to those in WT, at 72 h after injury.
(B) Graphical presentation of the qRT-PCR results for the same DEGs shown in (A). Expression levels at 72 h post-injury time point were compared to those of uninjured DRGs. n = 3; *p < 0.05, **p < 0.01 and ***p < 0.001 by t test.
Figure 7.

Comparative analysis reveals the distinctive features of the DLK-dependent transcriptional response to nerve injury.
(A) The heatmap shows 16 DEGs whose expression levels are significantly affected by both KO models, DLK deletion and importinβ1 deficiency. Genes whose expression levels are significantly altered by DLK deficiency at 12 h after injury are selected and tested for their regulation by the importin pathway. The DEG data using an importinβ1 KO model were publicly available (Perry et al., 2012). Log2(fold change) that compares expression levels in KO to those in WT is indicated following the color code. The corresponding post-injury time point where the given DEG was found significant in the importinβ1 KO dataset is shown in the rightmost column.
(B) Genes whose expression levels are significantly affected by the importinβ1 deficiency at 12 h after injury are tested for their regulation by DLK. Comparison of the expression levels in KO to those in WT is represented as indicated by the color key. The post-injury time point where the given DEG appears significant in the DLK dataset is shown in the rightmost column.
(C) The heatmap show the DEGs whose expression levels are significantly altered by DLK deficiency in both DRG and retinal ganglion cell (RGC) paradigms. The DEG data in the retina were available from the study by Watkins et al. (2013), where they performed a microarray analysis on retinal transcriptome 72 h after optic nerve injury. DRG expression data mostly appeared significant at 72 h after sciatic nerve injury, except for three genes labelled by asterisk, for which 24 h expression data were used instead. Log2(fold change) is generated to compare expression levels in DLK KO to those in WT in each paradigm. Functional terms associated to the given DEGs were adopted from those annotated by Watkins et al. (2013). Genes associated with “regeneration” are highlighted in blue and genes associated to "neurite extension" are highlighted in yellow.
(D) A scatter plot showing a positive correlation between the DLK-dependent expression changes in DRG and RGC. Pearson correlation coefficient r = 0.83.
As an independent test of DEGs identified by the transcriptome analysis, we performed quantitative reverse transcription PCR (qRT-PCR) for a few genes defined as significant DEGs. From the RNA-seq analysis, for example, Sema6a and Igfbp3 were identified as DEGs up-regulated by nerve injury in a DLK-dependent manner, whereas Agtr1b was down-regulated by injury in a DLK-dependent manner at 72 h after injury (Figure 2A). Expression levels of these three genes were sufficient for qRT-PCR detection in both uninjured and injured (72 h) conditions. We confirmed the injury-induced expression changes of these genes and their DLK-dependency in a qRT-PCR experiment using RNA isolated from the DRGs of WT and DLK mice injured for 72 h (Figure 2B). Hence, two independent methods for quantifying RNA levels give very similar results for the selected DEGs.
3. Deleting DLK expression in neurons alters baseline transcript levels in DRG
We next examined whether deleting DLK in neurons affects basal transcription in uninjured DRG. Comparing FPKM values between WT and DLK KO revealed 53 genes whose expression levels are significantly up-regulated by the DLK deficiency (q < 0.01). In addition, 54 genes were significantly down-regulated in the absence of DLK (q < 0.01) (Supplementary Table 2). Hence, DLK regulates basal transcription in neurons. Interestingly, of those 107 baseline DEGs, up-regulated or down-regulated, the majority (60 genes; 56.1 %) were also detected as genes differentially regulated in WT mice after nerve injury (Figure 3). This is dramatically more overlap than would be expected by chance alone (7.5 fold over-enrichment compared to expectation, p-value = 1.31E-39; hypergeometric test). These results demonstrate that DLK is required for regulation of baseline transcription, particularly that of injury-responsive genes. Expression changes by DLK depletion and expression changes by nerve injury showed a modest positive linear relationship (Pearson correlation coefficient r = 0.49) (Figure 3). As an example of such a gene, DLK depletion results in an elevation of the Sprr1a transcript levels at baseline (Log2FC = 3.46, q = 0.003). Sprr1a is a well-known regeneration-associated gene (Bonilla et al., 2002) and is highly up-regulated by injury in WT (Log2FC = 9.49, q = 0.003 at 72 h after injury). Notably, the observed positive correlation contrasts with the notion that DLK regulates gene expression in a same direction in uninjured and injured neurons. Instead, this result suggests either 1) that the role of DLK differs in intact and injured neurons or 2) that the injury-induced regulation of the baseline DEGs is DLK-independent. To explore these possibilities, we examined the requirement of DLK for expression of these genes in injured neurons. Indeed, the injury response in 30 genes among the 60 listed genes in Figure 3 showed a strong negative correlation with the expression fold changes in injured DLK KO neurons (Pearson correlation coefficient r = −0.61) (Supplementary Table 4), indicating that the injury responses in this group of baseline DEGs is DLK-dependent. On the other hand, the expression levels of the other 30 genes from the baseline DEG list were not significantly altered by loss of DLK in injured DRG, indicating that the DLK pathway is not required for determining their response to injury (Supplementary Table 4). Collectively, these data suggest that chronic loss of DLK signaling results in a baseline alteration of the injury-associated transcription profile that is distinct from that in damaged neurons.
Figure 3.

DLK deficiency causes changes in baseline transcription levels of injury-responsive genes.
Without injury, DEG analysis between WT and DLK KO identifies 107 significant DEGs (q < 0.01). Among them, 60 genes whose expression levels are significantly altered by nerve injury in WT mice (q < 0.01) are shown in the heatmap. The heatmap presents log2(fold change) as indicated by the color key. In the left column, fold change was calculated by dividing the mean FPKM value in uninjured DLK KO mice by that in uninjured WT mice. In the right column, fold change was obtained by dividing the mean FPKM value in injured WT mice by that in uninjured WT mice. When a DEG appeared significant from multiple post-injury time points, a time point with the largest expression change was selected and the corresponding time point was indicated (#, expression is regulated in the opposite direction in unpresented time points). The fold change and FPKM data are provided in Supplementary Table 4.
4. DLK deficiency results in a partial loss of injury-induced transcriptional changes
We showed that loss of DLK altered the numbers of DEGs caused by nerve injury (Figure 1B-C, Supplementary Table 2). There are two potential explanations for this phenotype: 1) in DLK KO, gene expression changes observed in WT fail to occur and/or 2) in DLK KO, a new set of DEGs are regulated by injury that are distinct from that found in WT. To explore these two possibilities, we examined the inclusion relationship between the DEG sets from WT and DLK KO (Figure 4, list of genes presented in Supplementary Table 5). As presented in Venn diagrams, we found that a large subset of the up-regulated DEG group in WT was excluded from the DEG group in DLK KO, at both 24 h and 72 h time points (q < 0.05, injured vs. uninjured) (Figure 4A). Appearance of new DEGs in the mutant animals was less prominent (53 DEGs at 24 h and 101 DEGs at 72 h). These results demonstrate that DLK is necessary for the up-regulation of gene expression induced by nerve injury. At 12 h after injury, the inclusion relationship between the up-regulated DEG groups in WT and DLK KO was not apparent. Meanwhile, down-regulated DEGs were also largely dependent on DLK at 72 h after injury. 442 genes in the WT DEG group were not shared in the down-regulated DEG set of DLK KO (q < 0.05, injured vs. uninjured) (Figure 4B). Interestingly, there are a number of up-regulated or down-regulated DEGs that are shared between WT and DLK KO, indicating that regulation of these genes are not solely dependent on DLK. In addition, the identification of genes that exhibit injury-responsiveness only in DLK KO suggests that DLK normally represses these genes in injured WT neurons. These DLK KO-specific DEGs may reflect an inhibitory program that is suppressed by DLK after injury. Taken together, these data demonstrate that the injury-responsive transcriptome is regulated by DLK.
Figure 4.

DLK is required for injury-induced transcriptional regulation.
(A) For the DEG group up-regulated after nerve injury (q < 0.05, injured vs. uninjured), DEGs in WT and DEGs in DLK KO are compared at each post-injury time point. Venn diagrams show the number of DEGs that are common between the two genotypes and the number of DEGs that are unique to each genotype. At 24 h and 72 h after injury, the DLK deficiency causes a failure in injury-induced up-regulation of many DEGs, while a gain of novel DEGs is less prominent.
(B) For the DEG group down-regulated after nerve injury (q < 0.05, injured vs uninjured), DEGs in WT and DEGs in DLK KO are compared for each post-injury time point. Venn diagrams show the number of common and unique DEGs. At 72 h after injury, a large number of DEGs are failed to be down-regulated in DLK KO while a gain of novel DEGs is less prominent.
5. DLK-dependent injury-induced DEGs include known regeneration-associated genes
Since DLK is required for injury-induced transcriptional changes and axon regeneration (Shin et al., 2012), we hypothesized that DLK regulates expression of genes that are involved in axon regeneration. To investigate the roles of the DLK-dependent DEGs for axon regeneration, we first examined whether regeneration-associated genes previously reported by other groups are regulated by injury in a DLK-dependent manner. Among 23 regeneration-associated genes whose DRG expression levels significantly change following sciatic nerve injury (Finelli et al., 2013; Li et al., 2015; Ma and Willis, 2015), 18 of them showed sufficiently high expression levels for statistical testing in our RNA-sequencing analysis (Figure 5). The expression analysis demonstrated that 16 genes, with the exception of Cebpa and Klf7, appeared as significant DEGs in our WT dataset. Cebpa and Klf7 also showed upregulation after injury (Log2FC = 0.33 and Log2FC = 0.73, respectively, at 72 h after injury) consistent with previous reports, but the expression changes were not statistically significant perhaps due to the small number of samples in our sequencing analysis (n = 3). Interestingly, the vast majority of the 16 significant DEGs displayed various degrees of DLK-dependency (q < 0.05) (Figure 5). For instance, although Atf3 and Sox11 are significantly up-regulated DEGs in both genotypes, WT and DLK KO, their expression levels are significantly lower in DLK KO than in WT at all time points tested, demonstrating that DLK is required for the robust up-regulation of these regeneration-associated genes. Sprr1a, Adcyap1, and Jun are also significantly up-regulated in both genotypes but showed DLK-dependency only at certain time points. Ten genes, including Npy, Itga7, Gal, Atf4, Cdkn1a, Gap43, Crem, Basp1, Fos, and Hoxd1, exhibited different temporal patterns of regulation between WT and DLK. Overall, DLK-dependent regulation appeared more prominent at 72 h after injury, based on the expression fold changes between the genotypes. Meanwhile, Onecut1 was the only significant DEG which did not show marked differences between the genotypes in the injury-induced regulation (Figure 5). Collectively, regulation of regeneration-associated genes exhibit strong DLK-dependency, suggesting that DLK regulates the transcriptional program accompanying axon regeneration.
Figure 5.
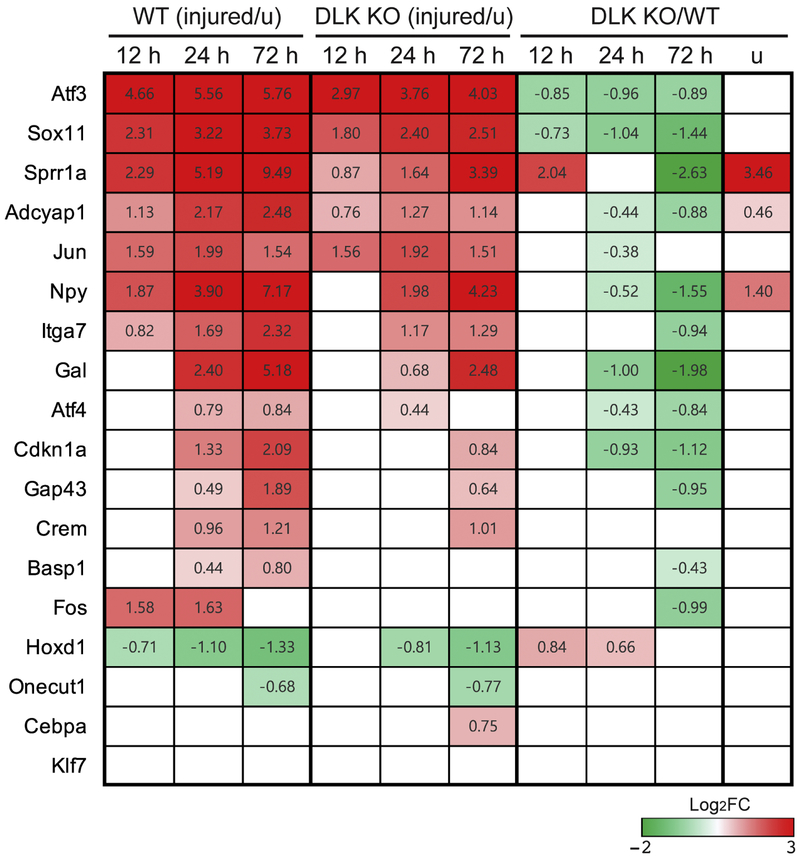
The majority of known regeneration-associated genes exhibits DLK-dependency for injury-induced transcriptional regulation.
The heatmap shows expression data for 18 previously reported regeneration-associated genes. Significant expression changes (q < 0.05) are quantified by log2(fold change) and indicated by the color key. Injury-induced changes in WT are presented in the leftmost three columns with a time-course, injury-induced regulation in DLK KO is displayed in the next three columns and comparison between DLK KO and WT at each time point is shown in the rightmost four columns. White color indicates no statistical significance.
6. DLK-dependent injury-induced genes display distinct functional enrichment
Our transcriptome analysis resulted in a total of 898 genes that are differentially expressed between WT and DLK KO (q < 0.05 at one or more post-injury time points). An effective bioinformatic method to evaluate a gene set and its molecular functions is to examine the enrichment of the annotated biological terms within the gene group. Using the Database for Annotation, Visualization and Integrated Discovery (DAVID) analysis (Huang et al., 2009a, 2009b), we sorted biological pathways highly enriched in the entire DEG set that are differentially regulated in DLK KO. Interestingly, the analysis showed a particular enrichment of functional annotations relating to ion transport, immune response, and response to pain and mechanical stimulus (FDR < 0.05), all of which are biological events associated with nerve injury and axon regeneration (Figure 6A) (Enes et al., 2010; Niemi et al., 2013; Tedeschi et al., 2016; Udina et al., 2008; von Hehn et al., 2012).
Figure 6.

Gene ontology analysis reveals functional annotation terms enriched in the DLK-dependent DEG groups.
(A) Functional enrichment test using the Database for Annotation, Visualization and Integrated Discovery (DAVID) analysis identifies biological pathway terms that are significantly enriched in the DLK-dependent DEG group. The DEGs whose expression levels are significantly different between WT and DLK KO at one or more post-injury time points (q < 0.05) were subjected to the analysis. Count represents the number of DEGs associated with the biological pathway term. FDR is adjusted p-values for multiple comparisons. Fold enrichment is defined as the ratio of the proportion of the associated DEGs to the proportion of associated genes in the reference genome and quantifies the degree of overrepresentation of a biological term in the DEG set. Significantly enriched terms are grouped according to their biological function. FDR < 0.05.
(B) Gene ontology study analyzing individual DLK-dependent DEG groups identified from the 72 h post-injury time point (q < 0.05). Red, functional terms related to ion transport; green, functional terms related to immune response; blue, functional terms related to pain response. FDR < 0.05. Results from other post-injury time points are listed in Supplementary Table 6.
(C) k-means clustering of the DEGs whose expression levels are significantly different between WT and DLK KO at one or more post-injury time point (q < 0.05). Graphical presentation of the expression data shows the temporal pattern of log2(fold change) in cluster 1, 3 and 4. The fold change is defined as the mean expression level in DLK KO divided by that in WT. Individual DEGs are shown in gray lines while an average log2(fold change) level in the cluster is shown in blue (mean ± SD). The expression data of cluster 2, 5 and 6, which did not yield any significantly enriched terms, are shown in Supplementary Table 6.
(D) Functional enrichment test to identify biological pathway terms significantly enriched in each DEG cluster. Cluster 1 was particularly enriched for ion transport-related functional terms while cluster 4 was enriched for immune response-associated terms (FDR < 0.05). A pain sensation-related term was identified from cluster 3.
In order to test whether these biological functions correlate with specific gene expression patterns, we next performed the gene ontology analysis in individual DEG subgroups identified from each injury time point (i.e. 12 h, 24 h, 72 h and uninjured) (q < 0.05, WT vs. DLK KO). Notably, we found that ion transport genes, such as the Alpha2delta2 subunit of voltage-gated calcium channels, Cacna2d2, are particularly enriched in the DEG group from 72 h post-injury time point (Figure 6B, labeled by red font color, Supplemenatary Table 6). This result shows that the DLK-dependent regulation of the ion transport genes is prominent in the late phase of injury response, consistent with the notion that ion channels including Cacna2d2 are down-regulated after nerve injury and such expression changes support the transition of the injured adult neurons back to the growing phase (Tedeschi et al., 2016). To further investigate temporal regulation of the enriched functional annotations, we clustered the DEGs based on their expression profile over time and then performed the gene ontology analysis on each cluster. By using the k-Means clustering method, the 898 DEGs were grouped into six clusters having different temporal profiles of the ratio of DLK KO to WT expression values (FPKM in DLK KO divided by FPKM in WT, named “KO-to-WT ratio” hereafter) (Figure 6C and Supplementary Table 6). For example, cluster 1 showed an increase in the KO-to-WT ratio over time, indicating that the gene expression is normally repressed by the DLK pathway in later time points. At 72 h, the gene expression is either reduced in WT but sustained in the absence of DLK, or induced in WT and up-regulated even higher in DLK KO. We found that ranking functional annotation terms in the individual clusters generated highly enriched terms with statistical significance (Figure 6D). The functional annotations relating to ion transport were significantly enriched in the cluster 1. Indeed, the cluster 1 genes included the calcium channel subunit Cacna2d2 (Supplementary Table 6). This finding indicates that the DLK pathway is responsible for down-regulation of ion transport genes in the late phase of injury response and suggest that DLK might promote axonal regeneration by repressing the electrophysiological activity of injured neurons. The full DAVID analysis results for significant functional annotations are provided in Supplementary Table 6.
We found that genes annotated with sensory response to pain are enriched in the DEG group from 72 h post-injury time point (q < 0.05, WT vs. DLK KO) (Figure 6B, labeled in blue). This result is in line with the development of chronic neuropathic pain after nerve injury (Basbaum et al., 2009; Ji and Strichartz, 2004) and support the involvement of DLK in the regulation of pain hypersensitivity. Consistent with our finding, Wlaschin and colleagues have recently reported that DLK is required for injury-induced regulation of pain-associated genes, spinal microgliosis, and mechanical allodynia following a spared-nerve injury to branches of the sciatic nerve (Wlaschin et al., 2018). As microgliosis contributes to the development of neuropathic pain (von Hehn et al., 2012), the authors highlight that Csf1, a neuronally-expressed cytokine that promotes microgliosis after nerve injury (Guan et al., 2016), is induced in a DLK-dependent manner in both the spared-nerve injury and sciatic nerve transection, the same injury used in our study. Indeed, in our gene ontology analysis results, we also found Csf1 in the immune-associated gene set that is enriched in the 72 h post-injury DEG group (q < 0.05, WT vs. KO). Furthermore, immune-related functional annotations, including cellular response to interferon-beta and innate immune response, are enriched in the baseline DEGs identified from uninjured WT and DLK KO DRGs as well as in the 72 h late post-injury DEG group (Figure 6B, labelled in green, and Supplementary Table 6). The DAVID analysis results based on the gene clusters show that immune response-related biological pathways are enriched highly in cluster 4, where the mean KO-to WT ratio is low in uninjured and late post-injury conditions (Figure 6C and 6D). This result suggests an important role of DLK in the regulation of immune reaction in DRG at basal level and in response to nerve injury. As the DRG samples include immune cells, this result can be partially attributed to a non-cell-autonomous effect, such as impaired recruitment and/or proliferation of immune cells in DLK KO animals. Collectively, by utilizing DLK-dependent DEG groups sorted by their temporal expression patterns, the functional annotation analysis demonstrates the requirement of DLK in the regulation of genes involved in ion transport pathway, pain sensation, and defense responses, and further implicates these biological functions in the regulation of axonal regeneration.
7. The DLK-dependent transcriptome is distinct from Importinβ1-dependent transcriptome in the injury signaling pathway
In nerve injury responses, axonal importins regulate retrograde transport of injury signals via binding of cargoes containing nuclear localization signals. Hanz et al. (2003) and Perry et al. (2012) previously reported that importinβ1 is required for the transport of injury signals from the damaged nerve to the cell body where these injury signals regulate transcriptional responses (Hanz et al., 2003; Perry et al., 2012). DLK is also required for the retrograde transport of injury signals necessary for the accumulation of activated cJun and STAT3 in the nuclei of injured DRG neurons (Shin et al., 2012). As two major pathways controlling retrograde injury signals, we asked whether the DLK and importinβ1 pathways regulate overlapping or distinct downstream signals. To correlate the DLK-dependent transcriptome to the importinβ1-dependent transcriptome, we compared our DEG data to those reported in a microarray study by Perry et al. (2012), where axonal importinβ1 KO mice were used for expression analysis at 6, 12 and 18 h post-injury time points. Because 12 h after injury was commonly examined in the two datasets, we focused on the 12 h data points. Then, to compensate for possible differences in the time-courses generated by the two independent studies, we used the importinβ1 DEG data from all three post-injury time points. We first checked consistency between the injury responses in WT mice from each dataset. A positive correlation between the expression fold changes in the two datasets indicates that the transcriptional injury response appears consistent between the independent studies (Pearson correlation coefficient r = 0.48). Next, we determined whether the DLK-dependent significant DEGs at 12 h (WT vs. DLK KO, q < 0.05) were identified in the importinβ1-dependent DEG set. Sixteen of the 119 DLK-dependent DEGs (13.4 %) were found to be significantly different in the importinβ1 expression data (WT vs. importinβ1 KO, p < 0.05) (Figure 7A). Conversely, we checked whether the importinβ1-dependent significant DEGs at 12 h were differentially regulated in our DLK dataset at any time point. We found that only 46 genes among the 816 importinβ1-dependent DEGs (5.6 %) were significantly dependent on DLK (Figure 7B). The low concordance rates indicate that DLK-dependent DEGs and importinβ1-dependent DEGs do not highly overlap (not significantly overenriched in Figure 7A; 1.3 fold over-enrichment, p-value = 0.04 in Figure 7B; hypergeometric test). Moreover, comparison of the KO-to-WT ratios further demonstrates that even the overlapping DEGs do not show positive correlation between the expression changes resulting from loss of DLK and loss of importinβ1 (Pearson correlation coefficient r = − 0.05 for Figure 7A, r = − 0.29 for Figure 7B). Collectively, these results suggest that DLK regulates retrograde injury signals that are distinct from those promoted by the importinβ1 pathway.
8. DLK-dependent transcriptional changes after nerve injury share similar signatures between DRG neurons and retinal ganglion cells (RGCs)
RGC axons are often used as a model of CNS axon injury due to the stereotyped architecture of the retina and the optic nerve. After injury to the optic nerve, cell death becomes prominent in RGCs and the injured axons fail to regenerate, unlike the robust regenerative response of DRG axons after PNS nerve injury. This difference in regenerative potential of RGCs and DRGs suggests that differences in the injury-dependent transcriptome might underlie the contrasting outcomes in the CNS versus PNS. Previous studies (Watkins et al., 2013; Welsbie et al., 2013) demonstrated that DLK is required for RGC cell death after injury. Moreover, Watkins et al. characterized the transcriptional responses to optic nerve injury, comparing retinal microarray data from WT and a DLK KO line after optic nerve crush (Watkins et al., 2013). Utilizing their published data, we examined how the role of DLK differs in the CNS versus PNS by comparing the DLK-dependent DEGs in the RGC model (q < 0.05) to the DLK-dependent DEG group identified in our DRG neuronal study (q < 0.05). The retinal microarray experiment was performed at a single post-injury time point, 72 h. We searched for the RGC DEGs in our list of significant DEG groups at any analyzed time point (i.e. 12, 24 and 72 h). Among the 201 DLK-dependent DEGs in RGCs, we found that 41 genes (20.4 %) were also identified as DLK-dependent DEGs in the DRG data (Figure 7C). Those shared DEGs mostly showed significance at 72 h time point in our dataset, except for three genes that were significant only at 24 h (indicated by asterisk; Oprl1, Slc2a6 and Ddit3). Remarkably, we found that the expression fold changes (the KO-to-WT ratios) of individual genes show strong positive correlation between the two datasets (Pearson correlation coefficient r = 0.83) (Figure 7D); the directionality of gene regulation by the DLK deficiency, i.e. up- or down-regulation, was mostly consistent between the DRG and RGC datasets. Interestingly, most of the RGC DEGs annotated with "regeneration" in the original report were also found as significant DEGs in DRG neurons (8 out of 9 total, gene symbol colored in light blue in Figure 7C). All three of "neurite extension" genes defined in the RGC DEG set were also shared by the DRG DEG group (gene symbol highlighted in yellow in Figure 7C). These pro-regenerative genes contrast with apoptotic genes, where only 4 out of 12 apoptosis-related genes in the original paper were found in the significant DEG list from our database (p < 0.01, Fisher exact test). Therefore, while damaged axons fail to regenerate in RGCs, the regulation of the signature regeneration-associated genes was comparable between RGC and DRG injured neurons. These data suggest that the DLK-dependent regeneration program may not be a major limiting factor of CNS regeneration. Instead, differences in other injury-responsive pathways, such as apoptotic pathways, may be critical in determining response to nerve injury.
Taken together, our data demonstrate that DLK is required for transcriptional changes after nerve injury, including for genes functionally associated with axon regeneration, and that DLK also regulates basal transcription of injury-induced genes. DLK-dependent DEG clusters highlight several functional annotations for potential involvement in axonal regeneration or other injury-dependent processes. Finally, our data support the hypothesis that DLK regulates a distinct retrograde signaling pathway that promotes a transcriptional regeneration program shared in the CNS and PNS.
Discussion
Retrograde injury signals play pivotal roles in inducing the axonal regeneration program in damaged peripheral neurons (Rishal and Fainzilber, 2014). DLK regulates the axon regeneration pathway as loss-of-function mutants in DLK orthologs from C. elegans, Drosophila and mice each impair injury signaling and axonal regrowth (Asghari Adib et al., 2018; Hammarlund et al., 2009; Shin et al., 2012; Tedeschi and Bradke, 2013; Xiong et al., 2010; Yan et al., 2009). In mouse peripheral neurons, DLK is required for retrograde transport of injury signals such as JIP3 and phospho-STAT3 and the subsequent activation of transcription factors including cJun (Shin et al., 2012). Since such transcriptional regulation is necessary for the conditioning injury effect (Smith and Skene, 1997), these previous studies have demonstrated that DLK is an essential regulator of the transcriptional regeneration program in the injured PNS. In support of this idea, DLK is also required for normal transcriptional regulation in a Neuro-2a cell line (Blondeau et al., 2016) as well as in the mouse retina after optic nerve crush (Le Pichon et al., 2017; Watkins et al., 2013). In the current study, we identified the DLK-dependent transcriptome in injured DRG neurons wherein regenerative responses are robust after sciatic nerve lesion. These data are the first to reveal the requirement of DLK in the regenerative transcriptional response after sciatic nerve injury for unbiased target genes by using the next generation sequencing method.
Expression studies in peripheral nerve injury paradigms have provided a useful platform for identifying important regulators of axonal regeneration because genes whose expression levels change dramatically after injury are likely to be involved in the injury response. However, these transcriptional responses are not limited to functionally important genes, and so the expression data are often insufficient to pinpoint the genes regulating the regeneration pathway. To overcome this limitation, additional paradigms can be added to the bioinformatic analysis for effective filtering. For example, Chandran et al. (2016) have compared the injury-induced expression changes between the PNS and CNS using a meta-analysis, highlighting the functional importance of PNS-specific regulation (Chandran et al., 2016). Likewise, Tedeschi et al. (2016) have successfully identified a group of candidate suppressors of axon growth by combining three criteria: up-regulated as mouse embryos mature, down-regulated as cultured DRG neurons are shifted to the elongation stage, and down-regulated after conditioning injury to the sciatic nerve (Tedeschi et al., 2016). In order to strengthen our strategy to identify regeneration-associated genes, we utilized a genetic filter, the loss of DLK, because DLK is an essential and evolutionarily conserved regulator of the regenerative response.
As an upstream kinase of a MAPK pathway, DLK can regulate the activity of the AP-1 transcription factor (Collins et al., 2006; Fan et al., 1996; Ghosh et al., 2011; Itoh et al., 2009). Therefore, we first examined whether DLK is required for the basal transcription in uninjured DRGs. We found that a specific group of genes were differently expressed in the DLK-deficient mice. Interestingly, the majority of these basally-regulated DLK-dependent genes overlap with the genes that are injury-regulated in wild type neurons. This result shows that the DLK pathway regulates these injury-associated genes even at the basal state in adults, consistent with the recent finding that DLK regulates basal MAPK signaling in DRG neurons (Summers et al., 2018). Hence, while DLK is best understood as a stress kinase (Asghari Adib et al., 2018; Farley and Watkins, 2018; Tedeschi and Bradke, 2013), it also plays an important role in uninjured neurons. Interestingly, these baseline DEGs show a modest positive correlation between the expression changes in uninjured DLK KO and injured WT animals, indicating that the DLK pathway might play different regulatory roles in intact and injured neurons. The DLK pathway may have a distinct molecular function in uninjured neurons, or, the baseline transcriptional changes could result from the chronic loss of DLK activity. As DLK is required for neural development, loss of DLK during development may activate a compensatory pathway that basally turns on a surveillance program that involves the injury-responsive genes. Alternatively, the change in baseline DEGs may reflect dysregulation of a DLK-dependent negative feedback mechanism that normally blunts expression of injury-dependent transcripts in uninjured neurons.
Axonal regeneration in the DLK KO mice is significantly delayed (Shin et al., 2012). Consistent with the physiological phenotype, we found that global transcriptional responses that unfold after nerve injury are markedly reduced by the loss of DLK. Interestingly, both the injury-dependent upregulation and downregulation of gene expression is largely dependent on DLK. The role of the DLK-JNK pathway for transcriptional activation is well known (Asghari Adib et al., 2018), however our results also indicate that DLK promotes transcriptional repression. Consistent with this notion, cJun can inhibit Smad3 transcriptional activity in non-neuronal cells (Dennler et al., 2000). In addition, we found that DLK-dependent DEGs in the early injury response (12 h) include genes that are negative regulators of gene expression such as Sox11, which is required for axonal regrowth (Perry et al., 2018). Hence there may be multiple mechanisms by which DLK promotes transcriptional repression after injury.
Using functional annotation analysis, we showed that the DLK-dependent DEG group is enriched for genes that cluster in a number of functional groups. Specifically, genes related to ion transmembrane transport (e.g. ion channels) are enriched in the cluster where the KO-to-WT ratio increases over time. Consistently, the functional terms related to ion transport were most represented in the DEG list from the 72 h post-injury time point. Therefore, the DLK pathway normally represses ion transport genes after injury, while expression of those genes is derepressed in DLK KO. Indeed, previous studies have described synaptic decline phenotypes in many different injury models. Synaptic input declines after neuronal damage (Navarro et al., 2007; Purves, 1975), and dendritic spines are reduced in the PS2APP mouse model of Alzheimer’s disease in a DLK-dependent manner (Le Pichon et al., 2017). Furthermore, Li et al. have recently demonstrated that Wallenda, a DLK orthologue in Drosophila, is responsible for limiting the levels of synaptic proteins during embryogenesis and, later at the larval stage, in response to "stress signals" elicited by an axon transport defects (Li et al., 2017). In line with these findings, our expression data in mice support the model that the DLK pathway represses synaptic transmission genes in response to axonal injury. Interestingly, the repression of neuronal activity has recently been implicated in the shift of injured neurons from transmitting to growing mode, which consequently promotes axonal regeneration. For example, Tedeschi and colleagues have shown that expression of a calcium channel subunit Cacna2d2 is induced as neurons mature but repressed once they encounter peripheral injury. Exogenously expressing Cacna2d2 suppresses axon regeneration in the PNS whereas pharmacologically inhibiting the channel improves regeneration of ascending axons after spinal cord injury, pinpointing this channel gene as a negative regulator of axon growth and regeneration. (Tedeschi et al., 2016).
In our study, Cacna2d2 was identified as a DEG that is significantly down-regulated by injury in a DLK-dependent manner, highlighting the role of DLK in the repression of synaptic genes in damaged neurons. Therefore, we suggest that the DLK pathway might promote effective regrowth of peripheral axons in part by down-regulating ion channel/synaptic transmission genes, which would then drive the mature neurons to revert to a growth state.
Other biological functions enriched in the DLK-dependent DEG group include immune response and pain sensation. Immune response-related functional terms were highly represented within the DEG sets identified from the uninjured DRGs and the late post-injury time point. In accordance with this finding, the DLK pathway has recently been implicated in spinal cord microgliosis in mouse models of amyotrophic lateral sclerosis and mechanical allodynia (Le Pichon et al., 2017; Wlaschin et al., 2018) as well as neuroinflammation in a fly model of TDP-43-induced neurotoxicity (Zhan et al., 2015). The JNK MAPK, a major kinase downstream of DLK, is also responsible for inflammation in DRGs following sciatic nerve injury (Wang et al., 2018). Importantly, our study utilized a sensory neuron-specific DLK KO line, and therefore support an instructive role of the neuronal DLK pathway in recruiting immune cells to the damaged neural circuit via the regulation of cytokine genes (e.g. Csf1). Indeed, neuron-macrophage interactions promote the conditioning injury effect and axon regeneration of DRG neurons (Kwon et al., 2015; Lindborg et al., 2018; Niemi et al., 2013). Therefore, DLK may activate the pro-regenerative program in part via a non-cell-autonomous mechanism that promotes gliosis and inflammation. Experimental validation of the role of DLK pathway in the recruitment of immune cells to DRG is an important future direction. In addition, the enrichment of the pain response terms among DLK-dependent DEGs is consistent with the recent finding that DLK regulates pain sensitivity following peripheral nerve injury (Wlaschin et al., 2018). Additionally, the DLK-dependent activation of the pain response-pathway might be another route for regulating the axon regeneration program, as stimulating a nociceptive TRPV1 channel with an agonist, capsaicin, can induce the regenerative potential of DRG neurons (Frey et al., 2018).
Our analysis of the DLK- and injury-dependent transcriptomes in DRG neurons allows us to compare our findings with prior work in the field. For example, DLK is required for the retrograde transport of injury signals, as are importins, which couple local translation and axon transport toward the nucleus after injury (Hanz et al., 2003; Yudin et al., 2008). Here we explored whether DLK and importins cooperate to regulate the same retrograde signaling system or, instead, promote distinct injury signals that independently contribute to the axon regeneration program. Our comparative analysis between the target genes of the two pathways revealed that the transcriptional changes regulated by DLK and importinβ1 show little overlap. Comparison between the DLK-dependent DEG group and the importinβ1-dependent DEG group yielded only a short list of common DEGs, partially due to the limitation resulting from utilizing data generated by the two independently controlled experiments (Perry et al., 2012). Furthermore, the directionality of expression changes of the common DEGs are inconsistent between the importinβ1 KO and the DLK KO models. These data strongly support the model that the DLK and importin pathways function independently. The DLK pathway is stimulated by MAP4Ks and cytoskeletal disruption while activation of the importin pathway is associated with local translation of importinβ1 and RANBP (Hanz et al., 2003; Larhammar et al., 2017; Valakh et al., 2015; Yudin et al., 2008), and so the two pathways might be effectors for distinct molecular triggers of injury signals.
Our goal was to investigate the transcriptional program that supports robust axon regeneration after nerve injury, and therefore we performed our analysis using peripheral neurons, which regenerate robustly. Moreover, this gave us the opportunity to assess how this PNS transcriptional response differs from that in the CNS following a similar nerve injury. We compared the DLK-dependent DEGs previously reported in mouse retina (Watkins et al., 2013) to the DLK-dependent DEGs identified in DRGs and found that the DEG subgroup shared between the two datasets displayed remarkably similar expression change patterns. These data demonstrate that the DLK-dependent transcriptome shows some commonality between the PNS and the CNS. Nonetheless, DEGs that are unique to the PNS or CNS could differentiate the regeneration outcomes after traumatic injury. Indeed, Venkatesh et al. (2018) recently demonstrated that target genes of the cJun and STAT3 transcription factors, both regulated by the DLK pathway, exhibit low chromatin accessibility in cortical neurons, suggesting that the DLK-dependent transcriptional regulation may not be fully active in the CNS (Venkatesh et al., 2018). Therefore, the DLK pathway may signal a common neuronal stress response in both the PNS and CNS, but cell type specific epigenetic mechanisms may shape how this common pathway induces either PNS pro-regenerative or CNS pro-apoptotic transcriptional responses (Mahar and Cavalli, 2018; Shin and Cho, 2017).
This study demonstrates that DLK, a central regulator of diverse neural injury responses, regulates a large portion of the transcriptional response to peripheral nerve injury. Gene ontology analysis of the DEG results support over-enrichment of particular biological functions in the DLK-dependent transcriptome. With the expression data provided, interesting future research directions include functional validation of DLK target genes and formation of new hypothesis about the involvement of specific biological processes in axon regeneration and neural injury responses. In addition, future studies of DLK-dependent transcriptional regulation at the single cell level (Hu et al., 2016; Lisi et al., 2017) will enhance our understanding of the role of DLK for both the resident DRG cell population and the immune cells recruited following injury.
Materials and methods
Mice and surgical procedure
DLK floxed allele (DLK F) (Map3k12tm1.1Adia), DLK deletion allele (DLK D) (Map3k12tm1.2Adia) (Miller et al., 2009) and advillin-Cre driver line (Hasegawa et al., 2007) were described previously. DLK KO animals (DLK F/D; advillin-Cre) and the littermate control (DLK F/WT) mice were generated by crossing DLK F/F female with DLK D/WT; advillin-Cre male. Three-months or older mice were used for sciatic nerve transection surgery. Briefly, mice were anesthetized with isoflurane and the sciatic nerve was unilaterally exposed through a small incision made to the skin and muscles at mid-thigh level. Then, the sciatic nerve was transected by surgical scissors and the incision was closed by nylon suture. The animals were then subjected to post-operation care until euthanized for analysis. Mouse husbandry, surgical procedure and post-operation care were performed under the supervision of Division of Comparative Medicine at Washington University.
RNA preparation and sequencing
At 12, 24 or 72 h after nerve lesion, L4-5 DRG tissues were dissected for RNA isolation and the tail tissues were collected for confirmative genotyping experiment. From the mice in the 12 h group, the contralateral DRGs were also collected as uninjured (u) control. Total RNA was isolated from the DRG tissues with RNAqueous-Micro Total RNA Isolation Kit (ThermoFisher, AM1931). Then, RNA from two individual mice were pooled per sample and subjected to DNase I reaction by using RNA Clean & Concentrator kit (Zymo research). RNA integrity was determined using an Agilent Bioanalyzer and ribosomal RNA was removed by a hybridization method using Ribo-ZERO kits (Illumina). mRNA was then fragmented in buffer containing 40mM Tris Acetate pH 8.2, 100mM Potassium Acetate and 30mM Magnesium Acetate and heating to 94 degrees for 150 seconds. mRNA was reverse transcribed to yield cDNA using Superscript III RT enzyme (Life Technologies, per manufacturer’s instructions) and random hexamers. A second strand reaction was performed to yield ds-cDNA. cDNA was blunt ended, had an A base added to the 3’ ends, and then had Illumina sequencing adapters ligated to the ends. Ligated fragments were then amplified for 14 cycles using primers incorporating unique index tags. Fragments were sequenced on an Illumina HiSeq-2500 using single reads extending 50 bases targeting 30 million reads per sample.
RNA-seq data analysis
Deep sequencing data corresponding to the sense strand of the input RNA were considered and dealt with throughout the present study. Before mapping, the adaptor sequences of raw reads were trimmed by means of Cutadapt (Martin, 2011). In adaptor-trimming step, the read lengths over 15 bp and with Phred quality score (>=30) were filtered for further analysis. The filtered reads were aligned to reference mouse genome (mm10) via STAR software (Dobin et al., 2013).
Two samples whose total mapping rates were noticeably low, one from WT 12 h after injury and the other from DLK KO 12 h after injury, were excluded from the following analyses. Only uniquely mapped reads were used for subsequent analyses. The correlations between independent biological replicates of each experiment were determined by means of the NumPy library in Python. The mapped reads from RNA sequencing was quantified in FPKM via Cufflinks version 2.2.1 (Trapnell et al., 2010), based on mouse GENCODE VM15. DEGs were found by mean of the Cuffdiff program, a part of the Cufflinks software package, which filters out genes with expression levels insufficient for statistical testing. The DEGs with zero FPKM values were excluded from subsequent analyses. Plots were generated by Prism (GraphPad) and heatmaps for comparative analysis were created by Excel (Microsoft). To cluster DEGs based on expression levels, k-Means Clustering/Hierarchical Clustering tools in MeV version 4.8 were used (Howe et al., 2011). Enrichment of biological pathway terms was tested with the DAVID Bioinformatics Resources, version 6.8 (Huang et al., 2009a, 2009b).
qRT-PCR
DRGs were collected from three WT and three DLK KO mice for triplication of the experiment, at 72 h after nerve injury. RNA samples were prepared from the DRG tissues as described above. Then, purified RNA was used for first-strand cDNA synthesis with qScript cDNA SuperMix (Qaunta Biosciences, 95048). qRT-PCR was performed by using PerfeCTa SYBR Green FastMix (Quanta Biosciences, 95073). The relative mRNA levels were calculated by the ΔΔCt method. Primers used: Sema6a - ACAGCCTGCCCCCTAAAGT (forward), AGCTCCTCTTATATTCGAGCCC (reverse); Igfbp3 - CCAGGAAACATCAGTGAGTCC (forward), GGATGGAACTTGGAATCGGTCA (reverse); Agtr1b – TGGCTTGGCTAGTTTGCCG (forward), ACCCAGTCCAATGGGGAGT (reverse). Gapdh levels were used as an internal control.
Supplementary Material
Supplementary Table 1. Summary of the transcriptome sequencing and initial mapping results. Highlighted in pink indicates the two lowest mapping rates. We have excluded the sample pair for DEG analysis.
Supplementary Table 2. Number of DEGs (q < 0.01 or q < 0.05). All DEGs showed ∣Log2FC∣ > 0.35.
Supplementary Table 3. Expression data of significant DEGs (q < 0.05)
Supplementary Table 4. Analysis of baseline DEGs, related to Figure 3. Baseline DEGs (DLK KO vs. WT, uninjured DRG) whose expression levels are significantly altered by nerve injury in WT mice are listed based on two q-value cutoff levels (q < 0.05 and q < 0.01). The baseline DEG group at q < 0.05 was significantly enriched with injury-responsive genes (5.2 fold over-enrichment compared to expectation, p-value = 1.97E-53; hypergeometric test) as confirmed for the q < 0.01 DEG group (see Results). For these DEG lists, baseline expression differences between two genotypes, maximal expression fold changes by injury in WT, and expression differences between two genotypes at time points corresponding to the WT injury response are presented as log2(fold change) in heatmaps. The relationship between the fold changes are test by Pearson correlation test. FPKM values for each condition are shown.
Supplementary Table 5. Shared and specific DEGs in WT and DLK KO at different post-injury time points, related to Figure 4. Genes included in the Venn diagrams in Figure 4 are listed.
Supplementary Table 6. DAVID analysis results for significant functional annotations, related to Figure 6. (A) Functional enrichment test results using DAVID analysis identifies biological pathway terms that are significantly enriched in the DLK-dependent DEG group. The DEGs whose expression levels are significantly different between WT and DLK KO at one or more post-injury time points (q < 0.05) were subjected to the analysis. Count represents the number of DEGs associated with the biological pathway term. FDR is adjusted p-values for multiple comparisons. Fold enrichment is defined as the ratio of the proportion of the associated DEGs to the proportion of associated genes in the reference genome and quantifies the degree of overrepresentation of a biological term in the DEG set. Significantly enriched terms are grouped according to their biological function. FDR < 0.05.
(B) Gene ontology study analyzing individual DLK-dependent DEG groups identified from different post-injury time points (i.e. uninjured, 12 h, 24 h and 72 h after injury; q < 0.05). The DEG group from the 12 h experiment did not result in significantly enriched biological pathway terms. FDR < 0.05.
(C) Functional enrichment test to identify biological pathway terms significantly enriched in each DEG cluster. Cluster 1 was particularly enriched for ion transport-related functional terms while cluster 4 was enriched for immune response-associated terms (FDR < 0.05). A pain sensation-related term was identified from cluster 3. Clusters 2, 5 or 6 did not generate significantly enriched biological pathway terms.
(C) k-means clustering of the DEGs whose expression levels are significantly different between WT and DLK KO at one or more post-injury time point (q < 0.05). Graphical presentation of the expression data shows the temporal pattern of log2(fold change) in clusters 2, 5 and 6. The fold change is defined as the mean expression level in DLK KO divided by that in WT. Individual DEGs are shown in gray lines while an average log2(fold change) level in the cluster is shown in blue (mean ± SD).
Highlights.
DLK is required for injury-induced transcriptional regulation in the PNS.
DLK regulates baseline transcription in dorsal root ganglion neurons.
DLK-dependent injury-responsive genes are enriched for particular functions.
DLK regulates a distinct pro-regenerative retrograde signaling pathway.
Acknowledgments
We thank the Genome Technology Access Center at Washington University for library generation and RNA sequencing, Bo Zhang and Erin Frey for helpful comments on the manuscript, Aldrin Yim for preliminary sequence analysis, and members of the DiAntonio laboratory for helpful discussions.
Funding:
This work was supported by the Korea Research Fellowship (NRF-2015H1D3A1066313) to J.E.S., the National Research Foundation of Korea (NRF) grants to J.E.S. (NRF-2017R1C1B2008356) and Y.C. (NRF-2016R1C1B2006675), Health Technology R&D Project to Y.C. (HI17C1459), and the National Institutes of Health grant (NS065053) and Children’s Discovery Institute support to A.D. H.H. was in part supported by the Basic Science Research Program funded by Korean Ministry of Education (NRF-2016R1A6A3A11933750). The funding sources had no involvement in study design and data analysis.
Footnotes
Declarations of interest: none
Conflict of interest statement
The authors have no conflicts of interest to declare.
Accession code
The next-generation sequencing data will be deposited in the Sequence Read Archive of the National Center for Biotechnology Information upon acceptance of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe N, and Cavalli V (2008). Nerve injury signaling. Curr. Opin. Neurobiol. 18, 276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asghari Adib E, Smithson LJ, and Collins CA (2018). An axonal stress response pathway: degenerative and regenerative signaling by DLK. Curr. Opin. Neurobiol. 53, 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, and Julius D (2009). Cellular and Molecular Mechanisms of Pain. Cell 139, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondeau A, Lucier J-F, Matteau D, Dumont L, Rodrigue S, Jacques P-É, and Blouin R (2016). Dual leucine zipper kinase regulates expression of axon guidance genes in mouse neuronal cells. Neural Dev. 11, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla IE, Tanabe K, and Strittmatter SM (2002). Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. J. Neurosci. 22, 1303–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradke F, Fawcett JW, and Spira ME (2012). Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat. Rev. Neurosci. 13, 183–193. [DOI] [PubMed] [Google Scholar]
- Byrne AB, Walradt T, Gardner KE, Hubbert A, Reinke V, and Hammarlund M (2014). Insulin/IGF1 Signaling Inhibits Age-Dependent Axon Regeneration. Neuron 81, 561–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, and Filbin MT (2001). Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J. Neurosci. 21, 4731–4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran V, Coppola G, Nawabi H, Omura T, Versano R, Huebner EA, Zhang A, Costigan M, Yekkirala A, Barrett L, et al. (2016). A Systems-Level Analysis of the Peripheral Nerve Intrinsic Axonal Growth Program. Neuron 89, 956–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C. a, Wairkar YP, Johnson SL, and DiAntonio A (2006). Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron 51, 57–69. [DOI] [PubMed] [Google Scholar]
- Curcio M, and Bradke F (2018). Axon Regeneration in the Central Nervous System: Facing the Challenges from the Inside. Annu. Rev. Cell Dev. Biol. 34, annurev-cellbio-100617-062508. [DOI] [PubMed] [Google Scholar]
- Dennler S, Prunier C, Ferrand N, Gauthier JM, and Atfi A (2000). c-Jun inhibits transforming growth factor beta-mediated transcription by repressing Smad3 transcriptional activity. J. Biol. Chem. 275, 28858–28865. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enes J, Langwieser N, Ruschel J, Carballosa-Gonzalez MM, Klug A, Traut MH, Ylera B, Tahirovic S, Hofmann F, Stein V, et al. (2010). Electrical Activity Suppresses Axon Growth through Cav1.2 Channels in Adult Primary Sensory Neurons. Curr. Biol. 20, 1154–1164. [DOI] [PubMed] [Google Scholar]
- Fan G, Merritt SE, Kortenjann M, Shaw PE, and Holzman LB (1996). Dual leucine zipper-bearing kinase (DLK) activates p46SAPK and p38mapk but not ERK2. J. Biol. Chem. 271, 24788–24793. [DOI] [PubMed] [Google Scholar]
- Farley MM, and Watkins TA (2018). Intrinsic Neuronal Stress Response Pathways in Injury and Disease. Annu. Rev. Pathol. Mech. Dis. 13, 93–116. [DOI] [PubMed] [Google Scholar]
- Finelli MJ, Wong JK, and Zou H (2013). Epigenetic Regulation of Sensory Axon Regeneration after Spinal Cord Injury. J. Neurosci. 33, 19664–19676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey E, Karney-Grobe S, Krolak T, Milbrandt J, and DiAntonio A (2018). TRPV1 Agonist, Capsaicin, Induces Axon Outgrowth after Injury via Ca2+/PKA Signaling. Eneuro 5, ENEURO.0095-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy CG, Hilton BJ, Tetzlaff W, and Zheng B (2016). Evidence for an Age-Dependent Decline in Axon Regeneration in the Adult Mammalian Central Nervous System. Cell Rep. 15, 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Milbrandt J, and DiAntonio A (2016). Axon Self-Destruction: New Links among SARM1, MAPKs, and NAD+ Metabolism. Neuron 89, 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AS, Wang B, Pozniak CD, Chen M, Watts RJ, and Lewcock JW (2011). DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J. Cell Biol. 194, 751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L, Wu J, Zhou S, Wang Y, Qin J, Yu B, Gu X, and Yao C (2016). Global analysis of transcriptome in dorsal root ganglia following peripheral nerve injury in rats. Biochem. Biophys. Res. Commun. 478, 206–212. [DOI] [PubMed] [Google Scholar]
- Gordon T, Tyreman N, and Raji M a (2011). The basis for diminished functional recovery after delayed peripheral nerve repair. J. Neurosci. 31, 5325–5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Guan AK, Evans-Reinsch Z, Braz J, Devor M, et al. (2016). Injured sensory neuron–derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat. Neurosci. 19, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Nix P, Hauth L, Jorgensen EM, and Bastiani M (2009). Axon regeneration requires a conserved MAP kinase pathway. Science 323, 802–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanz S, Perlson E, Willis D, Zheng J-Q, Massarwa R, Huerta JJ, Koltzenburg M, Kohler M, van-Minnen J, Twiss JL, et al. (2003). Axoplasmic Importins Enable Retrograde Injury Signaling in Lesioned Nerve. Neuron 40, 1095–1104. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Abbott S, Han B-X, Qi Y, and Wang F (2007). Analyzing somatosensory axon projections with the sensory neuron-specific Advillin gene. J. Neurosci. 27, 14404–14414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CS, Coleman MP, and Menon DK (2016). Traumatic Axonal Injury: Mechanisms and Translational Opportunities. Trends Neurosci. 39, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai S, Cui DF, Miyata T, Ogawa M, Kiyonari H, Suda Y, Aizawa S, Banba Y, and Ohno S (2006). The c-Jun N-terminal kinase activator dual leucine zipper kinase regulates axon growth and neuronal migration in the developing cerebral cortex. J. Neurosci. 26, 11992–12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzman LB, Merritt SE, and Fan G (1994). Identification, Molecular Cloning, and Characterization of Dual Leucine Zipper Bearing Kinase. J. Biol. Chem. 269, 30808–30817. [PubMed] [Google Scholar]
- Howe EA, Sinha R, Schlauch D, and Quackenbush J (2011). RNA-Seq analysis in MeV. Bioinformatics 27, 3209–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Huang K, Hu Y, Du G, Xue Z, Zhu X, and Fan G (2016). Single-cell RNA-seq reveals distinct injury responses in different types of DRG sensory neurons. Sci. Rep. 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huebner EA, and Strittmatter SM (2009). Axon Regeneration in the Peripheral and Central Nervous Systems In Cell Biology of the Axon. Results and Problems in Cell Differentiation, (Springer, Berlin, Heidelberg: ), pp. 305–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh A, Horiuchi M, Bannerman P, Pleasure D, and Itoh T (2009). Impaired regenerative response of primary sensory neurons in ZPK/DLK gene-trap mice. Biochem. Biophys. Res. Commun. 383, 258–262. [DOI] [PubMed] [Google Scholar]
- Itoh T, Horiuchi M, Ikeda RH, Xu J, Bannerman P, Pleasure D, Penninger JM, Tournier C, and Itoh A (2014). ZPK/DLK and MKK4 form the critical gateway to axotomy-induced motoneuron death in neonates. J. Neurosci. 34, 10729–10742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji R-R, and Strichartz G (2004). Cell signaling and the genesis of neuropathic pain. Sci. STKE 2004, reE14. [DOI] [PubMed] [Google Scholar]
- Kang H, and Lichtman JW (2013). Motor axon regeneration and muscle reinnervation in young adult and aged animals. J. Neurosci. 33, 19480–19491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon MJ, Shin HY, Cui Y, Kim H, Le Thi AH, Choi JY, Kim EY, Hwang DH, and Kim BG (2015). CCL2 Mediates Neuron-Macrophage Interactions to Drive Proregenerative Macrophage Activation Following Preconditioning Injury. J. Neurosci. 35, 15934–15947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larhammar M, Huntwork-Rodriguez S, Rudhard Y, Sengupta-Ghosh A, and Lewcock JW (2017). The Ste20 Family Kinases MAP4K4, MINK1, and TNIK Converge to Regulate Stress-Induced JNK Signaling in Neurons. J. Neurosci. 37, 11074–11084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang YV, Asghari Adib E, Stanchev DT, Xiong X, Klinedinst S, Soppina P, Jahn TR, Hume RI, Rasse TM, et al. (2017). Restraint of presynaptic protein levels by Wnd/DLK signaling mediates synaptic defects associated with the kinesin-3 motor Unc-104. Elife 6, e24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Xue C, Yuan Y, Zhang R, Wang Y, Wang Y, Yu B, Liu J, Ding F, Yang Y, et al. (2015). The transcriptional landscape of dorsal root ganglia after sciatic nerve transection. Sci. Rep. 5, 16888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisi V, Singh B, Giroux M, Guzman E, Painter MW, Cheng Y-C, Huebner E, Coppola G, Costigan M, Woolf CJ, et al. (2017). Enhanced Neuronal Regeneration in the CAST/Ei Mouse Strain Is Linked to Expression of Differentiation Markers after Injury. Cell Rep. 20, 1136–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindborg JA, Niemi JP, Howarth MA, Liu KW, Moore CZ, Mahajan D, and Zigmond RE (2018). Molecular and cellular identification of the immune response in peripheral ganglia following nerve injury. J. Neuroinflammation 15, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Tedeschi A, Park KK, and He Z (2011). Neuronal intrinsic mechanisms of axon regeneration. Annu. Rev. Neurosci. 34, 131–152. [DOI] [PubMed] [Google Scholar]
- Ma TC, and Willis DE (2015). What makes a RAG regeneration associated? Front. Mol. Neurosci. 8, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magill CK, Tong A, Kawamura D, Hayashi A, Hunter D. a, Parsadanian A, Mackinnon SE, and Myckatyn TM (2007). Reinnervation of the tibialis anterior following sciatic nerve crush injury: a confocal microscopic study in transgenic mice. Exp. Neurol. 207, 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahar M, and Cavalli V (2018). Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 19, 323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet.Journal 17, 10–12. [Google Scholar]
- Michaelevski I, Segal-ruder Y, Shalem O, Medzihradszky KF, Rozenbaum M, Coppola G, Geschwind DH, Pilpel Y, Burlingame AL, Fainzilber M, et al. (2010). Signaling to transcription networks in the neuronal retrograde injury response. Sci. Signal. 3, ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, and Diantonio A (2009). A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat. Neurosci. 12, 387–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro X, Vivó M, and Valero-Cabré A (2007). Neural plasticity after peripheral nerve injury and regeneration. Prog. Neurobiol. 82, 163–201. [DOI] [PubMed] [Google Scholar]
- Neumann S, and Woolf CJ (1999). Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 23, 83–91. [DOI] [PubMed] [Google Scholar]
- Niemi JP, DeFrancesco-Lisowitz A, Roldán-Hernández L, Lindborg JA, Mandell D, and Zigmond RE (2013). A critical role for macrophages near axotomized neuronal cell bodies in stimulating nerve regeneration. J. Neurosci. 33, 16236–16248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YA, Misgeld T, Lichtman JW, and Sanes JR (2003). Effects of neurotoxic and neuroprotective agents on peripheral nerve regeneration assayed by time-lapse imaging in vivo. J. Neurosci. 23, 11479–11488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RB-T, Doron-mandel E, Iavnilovitch E, Rishal I, Dagan SY, Tsoory M, Coppola G, McDonald MK, Gomes C, Geschwind DH, et al. (2012). Subcellular knockout of importin β1 perturbs axonal retrograde signaling. Neuron 75, 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RB-T, Hezroni H, Goldrich MJ, and Ulitsky I (2018). Regulation of Neuroregeneration by Long Noncoding RNAs. Mol. Cell 72, 553–567.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestronk A, Drachman DB, and Griffin JW (1980). Effects of Aging on Nerve Sprouting and Regeneration. Exp. Neurol. 70, 65–82. [DOI] [PubMed] [Google Scholar]
- Le Pichon CE, Meilandt WJ, Dominguez S, Solanoy H, Lin H, Ngu H, Gogineni A, Sengupta Ghosh A, Jiang Z, Lee S-H, et al. (2017). Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci. Transl. Med. 9, eaag0394. [DOI] [PubMed] [Google Scholar]
- Purves D (1975). Functional and structural changes in mammalian sympathetic neurones following interruption of their axons. J. Physiol. 252, 429–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, Nickells RW, Kerrigan LA, Pease ME, Thibault DJ, and Zack DJ (1995). Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Invest. Ophthalmol. Vis. Sci. 36, 774–786. [PubMed] [Google Scholar]
- Richardson PM, and Issa VM (1984). Peripheral injury enhances central regeneration of primary sensory neurones. Nature 309, 791–793. [DOI] [PubMed] [Google Scholar]
- Rishal I, and Fainzilber M (2014). Axon–Soma Communication in Neuronal Injury. Nat. Rev. Neurosci. 15, 32–42. [DOI] [PubMed] [Google Scholar]
- Shin JE, and Cho Y (2017). Epigenetic Regulation of Axon Regeneration after Neural Injury. Mol. Cells 40, 10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V, and DiAntonio A (2012). Dual Leucine Zipper Kinase Is Required for Retrograde Injury Signaling and Axonal Regeneration. Neuron 74, 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Geisler S, and DiAntonio A (2014). Dynamic regulation of SCG10 in regenerating axons after injury. Exp. Neurol. 252, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DS, and Skene JH (1997). A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J. Neurosci. 17, 646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers DW, Milbrandt J, and DiAntonio A (2018). Palmitoylation enables MAPK-dependent proteostasis of axon survival factors. Proc. Natl. Acad. Sci. U. S. A. 201806933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi A, and Bradke F (2013). The DLK signalling pathway--a double-edged sword in neural development and regeneration. EMBO Rep. 14, 605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi A, Dupraz S, Laskowski CJ, Xue J, Ulas T, Beyer M, Schultze JL, and Bradke F (2016). The Calcium Channel Subunit Alpha2delta2 Suppresses Axon Regeneration in the Adult CNS. Neuron 92, 419–434. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udina E, Furey M, Busch S, Silver J, Gordon T, and Fouad K (2008). Electrical stimulation of intact peripheral sensory axons in rats promotes outgrowth of their central projections. Exp. Neurol. 210, 238–247. [DOI] [PubMed] [Google Scholar]
- Valakh V, Frey E, Babetto E, Walker LJ, and DiAntonio A (2015). Cytoskeletal disruption activates the DLK/JNK pathway, which promotes axonal regeneration and mimics a preconditioning injury. Neurobiol. Dis. 77, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero-Cabré A, Tsironis K, Skouras E, Navarro X, and Neiss WF (2004). Peripheral and Spinal Motor Reorganization after Nerve Injury and Repair. J. Neurotrauma 21, 95–108. [DOI] [PubMed] [Google Scholar]
- Venkatesh I, Mehra V, Wang Z, Califf B, and Blackmore MG (2018). Developmental chromatin restriction of pro-growth gene networks acts as an epigenetic barrier to axon regeneration in cortical neurons. Dev. Neurobiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdu E, Buti M, and Navarro X (1995). The effect of aging on efferent nerve fibers regeneration in mice. Brain Res. 696, 76–82. [DOI] [PubMed] [Google Scholar]
- von Hehn CA, Baron R, and Woolf CJ (2012). Deconstructing the Neuropathic Pain Phenotype to Reveal Neural Mechanisms. Neuron 73, 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Medress Z. a, and Barres B. a (2012). Axon degeneration: Molecular mechanisms of a self-destruction pathway. J. Cell Biol. 196, 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang S, Liu T, Wang H, Liu K, Wang Q, and Zeng W (2018). Sarm1/Myd88-5 Regulates Neuronal Intrinsic Immune Response to Traumatic Axonal Injuries. Cell Rep. 23, 716–724 [DOI] [PubMed] [Google Scholar]
- Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, and Lewcock JW (2013). DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc. Natl. Acad. Sci. U. S. A. 110, 4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Yang Z, Ge Y, Mitchell KL, Zhou X, Martin SE, Berlinicke CA, Hackler L Jr., Fuller J, Fu J, et al. (2013). Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc. Natl. Acad. Sci. U. S. A. 110, 4045–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlaschin JJ, Gluski JM, Nguyen E, Silberberg H, Thompson JH, Chesler AT, and Le Pichon CE (2018). Dual leucine zipper kinase is required for mechanical allodynia and microgliosis after nerve injury. Elife 7, e33910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, DiAntonio A, and Collins C. a (2010). Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J. Cell Biol. 191, 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Wu Z, Chisholm AD, and Jin Y (2009). The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell 138, 1005–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudin D, Hanz S, Yoo S, Iavnilovitch E, Willis D, Gradus T, Vuppalanchi D, Segal-Ruder Y, Ben-Yaakov K, Hieda M, et al. (2008). Localized regulation of axonal RanGTPase controls retrograde injury signaling in peripheral nerve. Neuron 59, 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan L, Xie Q, and Tibbetts RS (2015). Opposing roles of p38 and JNK in a Drosophila model of TDP-43 proteinopathy reveal oxidative stress and innate immunity as pathogenic components of neurodegeneration. Hum. Mol. Genet. 24, 757–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Z-Y, Wen Y-R, Zhang D-R, Borsello T, Bonny C, Strichartz GR, Decosterd I, and Ji R-R (2006). A Peptide c-Jun N-Terminal Kinase (JNK) Inhibitor Blocks Mechanical Allodynia after Spinal Nerve Ligation: Respective Roles of JNK Activation in Primary Sensory Neurons and Spinal Astrocytes for Neuropathic Pain Development and Maintenance. J. Neurosci. 26, 3551–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Summary of the transcriptome sequencing and initial mapping results. Highlighted in pink indicates the two lowest mapping rates. We have excluded the sample pair for DEG analysis.
Supplementary Table 2. Number of DEGs (q < 0.01 or q < 0.05). All DEGs showed ∣Log2FC∣ > 0.35.
Supplementary Table 3. Expression data of significant DEGs (q < 0.05)
Supplementary Table 4. Analysis of baseline DEGs, related to Figure 3. Baseline DEGs (DLK KO vs. WT, uninjured DRG) whose expression levels are significantly altered by nerve injury in WT mice are listed based on two q-value cutoff levels (q < 0.05 and q < 0.01). The baseline DEG group at q < 0.05 was significantly enriched with injury-responsive genes (5.2 fold over-enrichment compared to expectation, p-value = 1.97E-53; hypergeometric test) as confirmed for the q < 0.01 DEG group (see Results). For these DEG lists, baseline expression differences between two genotypes, maximal expression fold changes by injury in WT, and expression differences between two genotypes at time points corresponding to the WT injury response are presented as log2(fold change) in heatmaps. The relationship between the fold changes are test by Pearson correlation test. FPKM values for each condition are shown.
Supplementary Table 5. Shared and specific DEGs in WT and DLK KO at different post-injury time points, related to Figure 4. Genes included in the Venn diagrams in Figure 4 are listed.
Supplementary Table 6. DAVID analysis results for significant functional annotations, related to Figure 6. (A) Functional enrichment test results using DAVID analysis identifies biological pathway terms that are significantly enriched in the DLK-dependent DEG group. The DEGs whose expression levels are significantly different between WT and DLK KO at one or more post-injury time points (q < 0.05) were subjected to the analysis. Count represents the number of DEGs associated with the biological pathway term. FDR is adjusted p-values for multiple comparisons. Fold enrichment is defined as the ratio of the proportion of the associated DEGs to the proportion of associated genes in the reference genome and quantifies the degree of overrepresentation of a biological term in the DEG set. Significantly enriched terms are grouped according to their biological function. FDR < 0.05.
(B) Gene ontology study analyzing individual DLK-dependent DEG groups identified from different post-injury time points (i.e. uninjured, 12 h, 24 h and 72 h after injury; q < 0.05). The DEG group from the 12 h experiment did not result in significantly enriched biological pathway terms. FDR < 0.05.
(C) Functional enrichment test to identify biological pathway terms significantly enriched in each DEG cluster. Cluster 1 was particularly enriched for ion transport-related functional terms while cluster 4 was enriched for immune response-associated terms (FDR < 0.05). A pain sensation-related term was identified from cluster 3. Clusters 2, 5 or 6 did not generate significantly enriched biological pathway terms.
(C) k-means clustering of the DEGs whose expression levels are significantly different between WT and DLK KO at one or more post-injury time point (q < 0.05). Graphical presentation of the expression data shows the temporal pattern of log2(fold change) in clusters 2, 5 and 6. The fold change is defined as the mean expression level in DLK KO divided by that in WT. Individual DEGs are shown in gray lines while an average log2(fold change) level in the cluster is shown in blue (mean ± SD).