

SUMMARY
Cyclin dependent kinase 14 (CDK14) and other TAIRE family kinases (CDKs 15-18) are proteins that lack functional annotation but are frequent off-targets of clinical kinase inhibitors. In this study we develop and characterize FMF-04-159-2, a tool compound that specifically targets CDK14 covalently and possesses a TAIRE kinase-biased selectivity profile. This tool compound and its reversible analog were used to characterize the cellular consequences of covalent CDK14 inhibition, including an unbiased investigation using phospho-proteomics. To reduce confounding off-target activity, washout conditions were used to deconvolute CDK14-specific effects. This investigation suggested that CDK14 plays a supporting role in cell cycle regulation, particularly mitotic progression, and identified putative CDK14 substrates. Together these results represent an important step forward in understanding the cellular consequences of inhibiting CDK14 kinase activity.
Keywords: CDK14, TAIRE kinase, covalent inhibitor, cell-cycle, mitosis, druggable genome
Graphical Abstract:
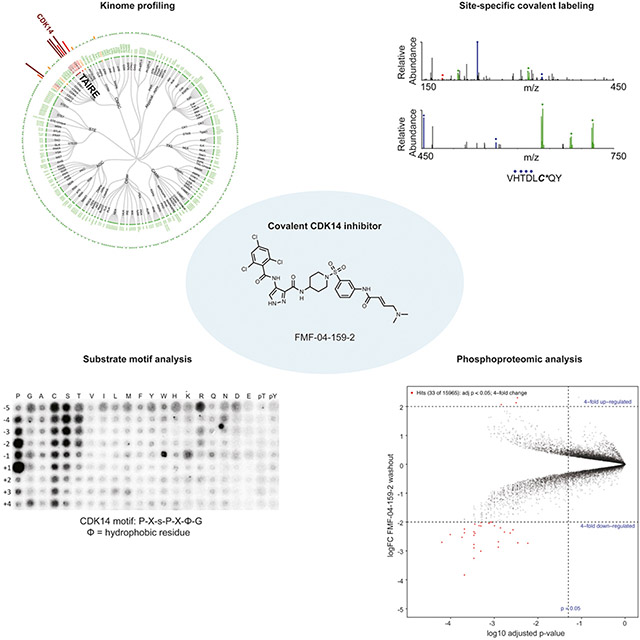
eTOC Blurb
Cyclin-Dependent Kinase 14 (CDK14) is an understudied kinase that lacks functional annotation. The authors develop and characterize a covalent CDK14 inhibitor, FMF-04-159-2, and use it to interrogate the role of CDK14 in the cell cycle and phospho-signaling.
INTRODUCTION
Cyclin-dependent kinases (CDKs) are a family of serine/threonine kinases whose activity depends on interaction with their cyclin binding partners. The CDKs have classically been categorized into three sub-families: CDKs which regulate progression through cell cycle (CDKs 1-7), CDKs which regulate various aspects of transcription (CDK7-13 and 19), and the TAIRE CDKs named for their common “TAIRE” sequence motif which is essential for binding to the G2/M cyclins, Cyclin Y and Cyclin Y-like 1 (Jiang et al., 2009),(Mikolcevic et al., 2012) (CDKs 14-18).(Malumbres, 2014) Due to their critical role in regulating important cellular processes, cell cycle and transcriptional CDKs have been well studied, and many small molecules have been developed to inhibit their kinase activity, with some obtaining FDA approval for various oncology indications.(Wu et al., 2015)’ (Ferguson and Gray, 2018)
In contrast, the TAIRE subfamily of CDKs (CDK14-18) are a sub-family of understudied CDKs without clearly described biological functions(Malumbres, 2014). Despite this, two members of the TAIRE subfamily, CDK14 and CDK16 have been implicated as potential therapeutic targets in a number of different cancers, including colorectal cancer. CDK14 (also annotated as PFTAIRE1 and PFTK) is overexpressed in human colorectal cancer patients, relative to surrounding normal colorectal lesions, and is associated with poor prognosis (Zhang et al., 2016b). CDK14 is also spontaneously amplified in a chimeric-mouse colon cancer model driven by mutant p53 and β-catenin. (Zhou et al., 2014) Knockdown of CDK16 reportedly causes mild inhibition of HCT116 colorectal cancer cell growth.(Yanagi et al., 2016)
The only reported substrate of the CDK14-Cyclin Y complex is LRP6, a transmembrane receptor protein involved in the canonical Wnt pathway. CDK14-Cyclin Y phosphorylates S1490 of LRP6 during the G2/M transition in a Wnt-independent manner. This primes LRP6 for Wnt-induced phosphorylation at PPP(S/T)P motifs by GSK3 and CK1, leading to stabilization of cytoplasmic β-catenin and activation of the canonical Wnt signaling pathway.(MacDonald and He, 2012),(Davidson et al., 2009),(Wang et al., 2016) Deregulation of Wnt signaling via loss of the Wnt pathway negative regulator APC is a hallmark of colorectal cancer.(Novellasdemunt et al., 2015) However, the functional consequences of S1490 phosphorylation and its relevance to cells in mitosis is not fully understood.(Davidson and Niehrs, 2010) Interestingly, in human HEK293 cells, ablation of LRP6 S1490 phosphorylation occurred upon RNAi-mediated knockdown of Cyclin Y and Cyclin Y-like 1. However, CDK14 knockdown showed no effect, suggesting that functional redundancy may exist among the human TAIRE kinases and providing rationale for the development of pan-TAIRE inhibitors as research tools and potential clinical leads.(Davidson et al., 2009)
In a recent study, a systemic, mass-spectrometry (MS)-based chemical proteomic profiling method (the kinobeads platform) was used to profile the targets of 243 diverse clinical kinase inhibitors in cell lysates. Although CDK14 was not detected in this kinome panel, 12% of the profiled inhibitors showed binding to either CDK16 or CDK17 (select Kd data plotted, Figure S1 A), indicating that TAIRE-kinases are frequently inhibited by clinically-relevant molecules.(Klaeger et al., 2016) However, despite their proven druggability, and association with colorectal cancer, the consequences of TAIRE kinase inhibition are poorly understood. Thus, further insight into their functions is essential to determining the underlying mechanism of action for these drugs and drug candidates. Improved understanding of the roles of TAIRE kinases in signal transduction and cellular physiology in healthy and diseased cells will also enable better rational design of drug target profiles.
To begin to address this need, we have developed a series of covalent CDK14 inhibitors with pan-TAIRE family specificity and characterized the pharmacological effects of the lead tool compound in the human colorectal cancer cell line HCT116. Additionally, we have employed the lead tool compound in combination with peptide array profiling and phospho-proteomics to characterize the acute cellular effects of pan-TAIRE kinase inhibition and identify signaling pathways involving CDK14.
RESULTS
Identification of Cys218 of CDK14 as an off-target of JNK-IN-7
In order to identify chemical starting points for the development of TAIRE-kinase family inhibitors, we implemented a combined screening and structure-guided drug design approach. The CDK family contains 20 members and the ATP-binding sites are highly conserved, which makes finding selective CDK inhibitors challenging. From our comprehensive analysis of cysteine residue locations in kinases, we noted that CDK14 possesses Cys218 in the “hinge 7” position. Based on sequence and structure alignments, a cysteine is present in this position in only 3 other kinases (JNK1, JNK2, JNK3).(Liu et al., 2013) In particular, we noticed that Cys218 is only present in CDK14 amongst all CDKs, thereby affording the opportunity to use this residue as a means to achieve selectivity for CDK14 within the CDK family. Since only a small fraction of the kinases that possess a cysteine near the ATP-binding pocket have been experimentally demonstrated to be labeled by a suitable covalent ligand, we queried our chemical proteomics database generated using the KiNativ methodology(Patricelli et al., 2011) for compounds that exhibited CDK14 binding. Our proteomic data revealed that JNK-IN-7(Zhang et al., 2012), a previously reported covalent pan-JNK inhibitor, also engages CDK14 (Figure 1).
Figure 1:

CDK14 is a covalent target of JNK-IN-7. (A) Sequence alignment of α-D loop of JNK3 and CDK14. (B) Chemical structure and biochemical potency of JNK-IN-7. (C) Kinativ profiling of JNK-IN-7 cellular selectivity at 1 μM. (D) Biotinylated JNK-IN-7 can pull down CDK14 from treated cell lysates. JNK-IN-7 shows dose dependent competition with biotin-JNK-IN-7. (E) Intact mass spectrum of recombinant CDK14 protein incubated with JNK-IN-7, resulting in mass shift compared to DMSO. (F) Biotin-JNK-IN-7 can pull down WT CDK14-flag, but not C218S CDK14-flag from HEK293 lysates, demonstrating that covalent bond formation occurs with the native protein. (G) Overlay of the crystal structure of JNK3 with the homology model of CDK14, showing spatial proximity of targeted cysteine residues. See also Figure S2.
JNK and CDK14 are categorized into different kinase sub-families (JNK and CDK respectively) and CDK14 and JNK3 have a 28.6% overall sequence homology. Therefore, we first validated this unexpected result and demonstrated that the CDK14:JNK-IN-7 interaction is specific and covalent. We used an established pull-down assay protocol(Zhang et al., 2016a) to confirm that biotinylated JNK-IN-7 can engage CDK14 in lysates derived from HEK293 cells, and that this interaction was competed out in a dose dependent manner by pre-treatment of the cells before lysis with JNK-IN-7 (Figure 1D). We next incubated recombinant CDK14 with JNK-IN-7 and observed the covalent addition of a single inhibitor molecule to the protein by LC-MS (Figure 1E). Pull-down of WT CDK14 but not C218S CDK14 from lysate, using biotin-JNK-IN-7, confirmed that Cys218 is the residue that is covalently engaged (Figure 1F). To provide molecular rationale for JNK-IN-7 covalently targeting CDK14, we examined the structural alignment of JNK3 bound to JNK-IN-7 (PDB:3V6S)(Zhang et al., 2012) and a homology model of CDK14 built using the structure of CDK12 (PDB: 5ACB)(Bosken et al., 2014). This analysis pointed to the presence of a conserved cysteine in α-D loop of both proteins, and further supported Cys218 as the site of modification in CDK14 (Figure 1G). This cysteine is not present in other CDKs, explaining this compound’s selectivity for CDK14 over the rest of the CDK family. Taken together, these results demonstrate that suitable acrylamide-modified heterocycles can efficiently label Cys218 of CDK14. However, as the phenylamino pyrimidine hinge-binding motif present in JNK-IN-7 is a generic kinase-inhibitor scaffold, we sought to identify more CDK selective ATP-site binders to help improve non-covalent potency and selectivity for the CDK-family versus other kinases.
Identification of AT7519 as a potent TAIRE-kinase inhibitor
To identify a CDK directed scaffold that could serve as a starting point for covalent inhibitor design, we screened a library of commercial and in-house kinase inhibitors using an in vitro CDK14 LanthaScreen binding assay (Invitrogen), followed by hit validation in a 33P-ATP CDK14 kinase assay (Reaction Biology). Finally, compounds that inhibited CDK14 in both biochemical assays were tested in a cellular target engagement assay. In this assay we determined whether pre-treatment of HEK293 cells with the compound of interest could block CDK14 pull-down by biotinylated JNK-IN-7 (Figure S2A, C). This screening cascade identified AT7519, a pan-CMCG kinase family inhibitor originally developed to target CDK1/2, as a remarkably potent reversible CDK14 inhibitor, with a biochemical IC50 of 19.8 ± 2.7 nM (Table 1).(Wyatt et al., 2008)
Table 1:
Biochemical IC50 data and cellular target engagement by pulldown for lead compounds discussed in this article against TAIRE kinases CDK14, CDK16, CDK17 and CDK18, and off-target CDK2. IC50s are reported as the average of triplicate (CDK14) or duplicate (CDK16-18) replicates, ± the standard error of the mean (SEM). aCellular target engagement of CDK14 by pulldown assay. Lowest concentration at which complete inhibition of CDK14 pulldown is achieved is reported. See also Figure S2 for representative western blots.
| Compoun d Name | IC50 CDK14 |
IC50 CDK14 |
IC50 CDK14 |
IC50 CDK1 4 |
IC50 CDK16 |
IC50 CDK2 |
IC50 CDK2 |
IC50 HCT11 6 |
|---|---|---|---|---|---|---|---|---|
| Lantha- screen |
Nano BRET |
Pull down |
33P kinase assay |
binding by Lantha- screen |
Z'LYTE kinase assay |
NanoB RET |
Proliferation | |
| (nM) | (nM) | (nM) | (nM) | (nM) | (nM) | (nM) | (nM) | |
| AT7519 | 19.8 ± 2.7 | 77.7 ± 5.9 | > 1000 | 11 ± 1.4 | 3.9 ± 0.7 | 28 ±3 | 969 ± 160 | 132 ± 33 |
| FMF-03-198-2 | 1.7 ± 0.7 | 143 ± 30.8 | 50 | 44 ± 4.5 | 0.9 ± 0.9 | 1.0± 0.7 | 113 ± 62.1 | 5.1 ± 1.4 |
| FMF-04-159-2 | 88.0 ± 9.6 | 39.6 ± 2.8 | 500 | 352 ± 40 | 10.1 ±1 | 8.2 ± 0.8 | 256 ± 26 | 1144 ± 190 |
| FMF-04-159-R | 139.1 ± 10.4 | 563 ± 145 | > 1000 | 367 ± 135 | 5.9 ±2 | 4.9 ± 0.5 | 493 ± 81 | 5869 ± 1191 |
To broadly assess the kinase targets of AT7519 in live cells, we performed KiNativ profiling in HCT116 cells treated with 1 μM compound for 4 h. Consistent with our previous data, we found that AT7519 potently inhibited CDK14, and also had strong activity against other TAIRE kinases, including CDK16, CDK17, and CDK18, as well as other CMCG family kinases, such as CDK9, GSK-α, and GSK-β (Figure 2F). Surprisingly, poor engagement was observed for CDK1/CDK2, and the overall selectivity profile therefore identified fewer targets than have previously been reported, potentially reflecting differences in the methodologies used, or a false negative in our data. Aside from this, the detected targets from KiNativ were largely consistent with a recent report evaluating the cellular targets of AT7519 in mixed cell lysates using the kinobeads platform.(Klaeger et al., 2016) In that study, AT7519 was found to be highly potent against CDK9, CDK16 and CDK17, and also had activity against CDK1, CDK2, CDK4, CDK12, GSKα, GSKβ and other CMCG kinases (Select Kd data plotted in Figure S1 B, Table S1).
Figure 2:

AT7519 analog FMF-03-198-2 is a pan-CMCG kinase inhibitor that targets CDK14 C218 covalently. (A) Chemical structures of AT7519 and FMF-03-198-2. (B) Intact MS analysis of recombinant CDK14 protein incubated with DMSO (top panel) or FMF-03-198-2 (bottom panel) demonstrating a mass shift corresponding to covalent addition of FMF-03-198-2 with 1:1 stoichiometry. (C) Biotin-FMF-03-198-2 can pull down WT CDK14-flag, but not C218S CDK14-flag from HEK293 lysates, demonstrating that covalent bond formation with the native protein occurs. (D) Covalent docking model of FMF-03-198-2 into a homology model of CDK14. The (1,4)-(1,3) regiochemistry is modeled to be uniquely permissive of covalent bond formation. CDK14 protein carbons shown grey, FMF-03-198-2 carbons shown green, hydrogen bonds depicted as black dotted lines. (E) Effects of AT7519 and FMF-03-198-2 on cell viability, using Cell Titer Glo. Data points are plotted as the average of three replicates ± standard error of the mean (SEM). (F) Live cell Kinativ profiling upon 4 h treatment with 1 μM AT7519 or (G) FMF-03-198-2. See also Figure S1, Figure S2 and Table S1.
Developing covalent AT7519 analogs with CDK14 activity
Developing selective inhibitors of CMCG kinases is notoriously challenging, with the majority of reported CDK inhibitors possessing extensive poly-pharmacology.(Klaeger et al., 2016) As targeting non-conserved cysteines within or near the ATP-binding pocket with a covalent warhead has been a useful strategy for improving selectivity against otherwise homologous kinases(Chaikuad et al., 2018), we focused our initial efforts on hybridizing the acrylamide warhead of JNK-IN-7 with the AT7519 scaffold.
Initially, we sought to generate a cell-penetrant, covalent CDK14 inhibitor based on the AT7519 scaffold as a proof-of principle compound, before optimizing for other properties. Through an extensive analoging campaign in which the piperidine of AT7519 was replaced with acrylamide bearing groups, varying in composition and regiochemistry, to be described in detail elsewhere, we identified the potent multi-targeted inhibitor FMF-03-198-2 (Figure 2A). FMF-03-198-2 incorporates the hinge-binding scaffold of AT7519 and the acrylamide orienting 1,3-aniline of JNK-IN-7. Covalent bond formation to CDK14 was confirmed by incubating FMF-03-198-2 with purified recombinant CDK14 protein followed by mass spectrometry analysis, which demonstrated an increase in mass corresponding to the molecular weight of FMF-03-198-2 (Figure 2B). To verify engagement of CDK14 in a cellular context, a biotinylated analog of FMF-03-198-2 (Figure S2B) was synthesized, and lysates from HEK293 cells expressing flag-tagged wildtype or C218S-CDK14 were subjected to a pull-down assay. As expected, CDK14-flag was successfully pulled down, but C218S-CDK14 was not, indicating that biotin-FMF-03-198-2 covalently labeled CDK14 at C218 in cell lysates, and that C218 was essential for compound binding activity (Figure 2C). Molecular modeling demonstrated that the 1,4 amino-piperidine, 1,3 aniline regiochemistry is exclusively permissive of covalent bond formation between CDK14 C218 and the acrylamide warhead (Figure 2D).
To test the effects of FMF-03-198-2 on cell proliferation, we treated HCT116 cells with a dose range of the compound and evaluated cell growth using Cell Titer Glo, which showed that FMF-03-198-2 was 26-fold more potent than AT7519 (IC50 = 5.1 ± 1.4 nM compared to 132 ± 33 nM) (Figure 2E). RNAi knockdown studies of CDK14 – CDK18 have demonstrated that these kinases are not essential for the 2D proliferation of colorectal cancer cell line cultures, including HCT116 cells (Table S2), unlike the other CMCG kinase targets of AT7519; for example, CDK1 depletion results in a profound effect on proliferation (Table S2).(McDonald et al., 2017) Therefore, the increased toxicity of FMF-03-198-2 against HCT116 cells was unlikely attributable to selective CDK14 inhibition, suggesting that this molecule potentially inhibited additional targets. Indeed, KiNativ profiling in HCT116 cells revealed that FMF-03-198-2 is a pan-CDK kinase inhibitor that also has potent activity against the CMCG kinases ERK5, GSK3α and GSK3β (Figure 2G). Accordingly, biotin-FMF-03-198-2 pulled down a number of CMCG kinases (including CDK2, CDK5, CDK9, CDK16 and GSK3α) from cell lysates, and when cells were pre-treated with FMF-03-198-2, dose-dependent competition was observed (Figure S2E).
While FMF-03-198-2 was not suitable as a selective probe for CDK14 and other TAIRE kinases, it confirmed that hybridization of the acrylamide warhead of JNK-IN-7 with AT7519 is a viable strategy for developing covalent CDK14 inhibitors targeting C218. Furthermore, its increased potency for TAIRE kinases (compared to AT7519), as well as its additional activity against a broad range of CMCG family kinases, enabled the use of biotin-FMF-03-198-2 as a tool for monitoring cellular target engagement for CDK14 and off-target kinases simultaneously. This ability to rapidly assess the cellular engagement of poorly characterized kinases, largely without reported downstream substrates, greatly aided our efforts to generate of more selective inhibitors. With the requisite tools in hand, we continued to optimize our pan-CMCG inhibitor to improve its selectivity.
Development and characterization of FMF-04-159-2 as a covalent CDK14 inhibitor with pan-TAIRE activity
To improve the selectivity profile of FMF-03-198-2, we subjected this compound to rounds of iterative medicinal chemistry, which resulted in development of FMF-04-159-2 (Figure 3A). Here, we built upon published SAR studies of AT7519 and related compounds which demonstrate that incorporation of a substituent at the 4-position of the benzamide ring reduces CDK2 and GSK-3β inhibition in biochemical assays, and reduces anti-proliferative activity against human cell lines.(Wyatt et al., 2008)’(Urich et al., 2014) We demonstrated that FMF-04-159-2 could covalently label CDK14 by mass spectrometry analysis, and confirmed that C218 was the site of covalent attachment by chymotryptic digestion followed by nano-LC/MS analysis of the resulting peptides (Figure 3B, C). As a chemical control to distinguish between covalent and reversible interactions, we synthesized the reversible dimethylbutanamide analog FMF-04-159-R (Figure 3A). The biochemical binding assay for CDK14 showed that FMF-04-159-2 and FMF-04-159-R have similar biochemical potencies (IC50 = 86 nM and 149 nM respectively, Figure 3D, Table 1), indicating that reversible binding activity is the primary driver of potency measured in these assays. The 33P kinase assay further supported this and also highlighted the effects of covalent inhibition, as FMF-04-159-2 was ~10-fold more potent than FMF-04-159-R (Table 1).
Figure 3:

Characterization of FMF-04-159-2 as a covalent CDK14 inhibitor with TAIRE kinase selectivity. (A) Chemical structure of FMF-04-159-2 and reversible control compound FMF-04-159-R. (B) Intact MS analysis demonstrates stoichiometric labeling of recombinant CDK14 by FMF-04-159-2. (C) Chymotryptic digestion of FMF-04-159-2 labeled CDK14 demonstrates exclusive labeling of C218 by MS/MS. (D) Biochemical CDK14 binding curves generated using a LanthaScreen assay. Data points are plotted as the average of three replicates, ± the standard error of the mean (SEM). Curves fit using nonlinear regression curve fit in GraphPad Prism 7. (E) HCT116 cytotoxicity of compounds after 72 h treatment, assessed using Cell-Titer Glo. Data points are plotted as the average of three replicates, ± the standard error of the mean (SEM). Curves fit using nonlinear regression curve fit in GraphPad Prism 7. (F) In-cell target engagement of CDK14 using the NanoBRET assay (Promega), with and without compound washout for 2 h following compound treatment for 4 h. Data points are plotted as the average of three biological replicates, ± the standard error of the mean (SEM). IC50 determined using nonlinear regression curve fit in GraphPad Prism 7. (G) Cellular kinome profiling of FMF-04-159-2 in HCT116 cells using the KiNativ platform. (H) Comparison of kinome-wide selectivity of AT7519, FMF-03-198-2 and FMF-04-159-2, assessed by KiNativ (1 μM 4 h treatment in HCT116 cells). See also Supporting Figure S2, Table S2, Table S3, Supporting Figure S3, Table S4 and Table S1
Proliferation assays in HCT116 cells revealed that FMF-04-159-2 and FMF-04-159-R were less potent than AT7519 and FMF-03-198-2 (Figure 3E, Table 1). Additionally, we observed a 5-fold difference in HCT116 cell IC50 between the reversible and covalent inhibitor pair, which suggested that a mild anti-proliferative effect is due to covalent binding (Figure 3E). This suggested that FMF-04-159-2 has a much narrower target range when compared to the parental FMF-03-198-2, given that previous studies had reported only mild impairment of cell proliferation upon siRNA-mediated knockdown of CDK14.(McDonald et al., 2017)
To quantitatively assess cellular engagement of CDK14, we used the Promega NanoBRET live cell target engagement assay (Figure 3F, Table S3).(Perez-Riverol et al., 2019) Here, we found that CDK14 was potently engaged by FMF-04-159-2 (IC50 = 39.6 ± 2.8 nM), and this engagement was sustained after a 2 h compound washout (1C50 = 56.3 ± 6.0 nM), indicative of irreversible binding. In contrast, inhibition of CDK14 by FMF-04-159-R was 5-fold less potent (IC50 = 563 ± 145) and was reduced by a further 7-fold after washout (IC50 = 3417 ± 1154), suggesting that covalent bond formation enhances cellular inhibition of CDK14, a feature not detected in biochemical assays.
To assess the cellular kinome-wide selectivity, we performed KiNativ profiling of HCT116 cells treated with 1 μM FMF-04-159-2 for 4 h, and found that it had an improved selectivity profile compared to FMF-03-198-2 (Figure 3G, H). FMF-04-159-2 potently inhibited the TAIRE kinases CDK14, CDK16, CDK17 and CDK18. Given reports of functional redundancy among the TAIRE kinases, this pan-TAIRE inhibition profile was deemed acceptable. Some residual off-target activity was detected against CDK2, and to a lesser extent CDK10.
To verify cellular engagement of the other TAIRE kinases and assess the extent of CDK2 activity, we turned to the NanoBRET assay in HCT116 cells (Perez-Riverol et al., 2019). CDK2 was inhibited by FMF-04-159-2 (IC50 = 256 ± 26 nM), and this activity was similar to that of FMF-04-159-R (IC50 = 493 ± 81 nM), consistent with the reversible nature of the interaction, as CDK2 has no equivalent cysteines in the ATP-binding site. At the 2 h timepoint (Figure S3, Table S4), FMF-04-159-2 shows nearly 20-fold weaker potency for CDK14 (IC50 = 803 ± 111) than at 6 h (Figure 3F), suggesting the time dependence of covalent bond formation and increased potency for CDK14 with time. FMF-04-159-2 and its reversible counterpart also engaged the other TAIRE kinases (CDK15, CDK16, CDK17 and CDK18) with comparable potency to each other, indicative of reversible binding (Figure S3, Table S4).
To verify the concentration dependence of binding to the different CDK family kinases, we used the multi-targeted biotin-FMF-03-198-2 probe as bait in a competition pull-down assay (Figure S2B, C). Cells were treated with escalating concentrations of FMF-04-159-2 or FMF-04-159-R, followed by cell lysis and pull-down (Figure 4A). Complete engagement of CDK14 was observed at 500 nM FMF-04-159-2 and above, whereas FMF-04-159-R failed to block pull-down at concentrations up to 1 μM, consistent with the NanoBRET assay. Engagement of CDK10 was observed at concentrations above 750 nM of FMF-04-159-2 and FMF-04-159-R, while CDK16 and CDK17 were inhibited at concentrations above 750 nM of FMF-04-159-2, but not by FMF-04-159-R, despite the reversible nature of their interactions with FMF-04-159-2. Only slight inhibition of CDK2 by FMF-04-159-2 and FMF-04-159-R was observed at concentrations above 500 nM in this assay. The pull-down assay was also utilized to investigate the effects of compound washout (Figure 4B). CDK14 engagement was sustained for up to 2 h after washout by 1 μM FMF-04-159-2, whereas CDK2, CDK10 and CDK17 were no longer inhibited at this time point after washout and CDK16 target engagement was almost fully removed.
Figure 4:

Characterization of the target engagement and cellular effects of FMF-04-159-2 and FMF-04-159-R in HCT116 cells. (A) Target engagement of FMF-04-159-2 and FMF-04-159-R with 4 h treatment at the indicated concentration, assessed using biotin-FMF-03-198-2 pull down, followed by immunoblotting. (B) Target engagement of FMF-04-159-2 and FMF-04-159-R, treated for 4 h at 1 μM before washout for the indicated time, assessed using biotin-FMF-03-198-2 pull down followed by immunoblotting. (C) Immunoblot for known phospho-substrates of targets of FMF-04-159-2 identified by KiNativ. HCT116 cells were treated for either 6h or 4 h followed by 2 h compound washout with the indicated inhibitors. (D) Double thymidine-synchronized HCT116 CDK14 knockout cellsexpressing WT or C218S CDK14 were released and allowed to progress through the cell cycle, subject to treatment with 1 μM FMF-04-159-2 or FMF-04-159-R for 4 h windows, followed by compound washout. Cells were collected at indicated times after release. (E) FACS PI cell cycle analysis of 24 h treatment of HCT116 CDK14 knockout cells expressing WT or C218S CDK14. Reported as mean ± standard error for percentage of cells in each cell cycle phase from n=3 biological replicates. See also Figure S4.
Cumulatively these results suggest that FMF-04-159-2 is a potent, covalent inhibitor of CDK14 in cells, but will also engage non-covalently with CDK2, CDK10 and the TAIRE family kinases at micromolar concentrations, consistent with the reversible CDK binding conferred by the pyrazole amide tridentate hinge binding motif of AT7519. FMF-04-159-2 should only be used as a pharmacological probe of CDK14 when controlled for by compound washout experiments and by use of reversible control compound FMF-04-159-R. Our structured set of preliminary experiments in the HCT116 cell line established experimental conditions relevant to mapping cellular consequences of CDK14 inhibition, namely 1 μM treatment with FMF-04-159-R or FMF-04-159-2 with a 2 h washout control.
Mapping cellular consequences of CDK14 versus pan-TAIRE inhibition
LRP6 is basally phosphorylated by the Drosophila homolog of CDK14, L63, during mitosis specifically in a Wnt-independent manner, and is otherwise a substrate of GSK kinases.(Davidson et al., 2009) Initially, we assessed LRP6 phosphorylation in unsynchronized HCT116 cells, where we did not observe substantial reduction of phosphorylated LRP6 S1490 levels, the only known substrate of CDK14 (Figure 4C). Given the cell cycle dependence of this CDK14-mediated phosphorylation, large changes in CDK14-dependent LRP6 phosphorylation are expected to be challenging to detect in unsynchronized cells, and this was confirmed by our initial observations (Figure 4C).(Davidson et al., 2009)
As we had found that FMF-04-159-2 had some activity against CDK2, we examined the phosphorylation status of reported CDK2 substrates in parallel.(Lundberg and Weinberg, 1998) Interestingly, we did not observe major reductions in the levels of phosphorylated RB S807/811 after treatment with 1 μM FMF-04-159-2 and FMF-04-159-R, while a partial reduction was seen upon treatment with AT7519 at 1 μM, which was rescued by compound washout (Figure 4C). Inhibition of Nucleophosmin (NPM) T199 phosphorylation was observed at levels comparable to AT7519 upon treatment with either FMF-04-159-2 or FMF-04-159-R but was fully rescued upon compound washout for FMF-04-159-2, but not FMF-04-159-R (Figure 4C). This data corroborated that the experimental conditions identified by the competition cellular target engagement studies are suitable for examining the downstream effects of CDK14 inhibition.
CDK14-Cyclin Y expression peaks in mitosis, and this is the phase of the cell cycle in which CDK14 is reported to phosphorylate LRP6 at S1490.(Davidson and Niehrs, 2010; Mikolcevic et al., 2012; Wang et al., 2016) Thus, we examined LRP6 phosphorylation in the context of double thymidine-synchronized cells, treated with FMF-04-159-2 or FMF-04-159-R in 4 h windows, harvested either with or without 2 h drug washout. In this setting, a partial reduction of pLRP6 (22 – 35 %) was seen during mitosis upon FMF-04-159-2 treatment, which was reached around 8 to 10 h after synchronization release, as reflected by peak expression of Cyclin B1 and pNPM T199, followed by increased expression of pH3 S10 (Figure 4D). This was only partially rescued when the reversible inhibitor FMF-04-159-R was used, consistent with the hypothesis that the multiple TAIRE kinases inhibited reversibly by FMF-04-159-R can also phosphorylate LRP6 in human cells.(Davidson et al., 2009)
CDK14-Cyclin Y has also been reported to play a role in cell cycle progression in promoting the G1/S phase transition, although CDK14 expression peaks in mitosis.(Shu et al., 2007)’(Yang et al., 2015)’(Pang et al., 2007) To assess cell cycle-related consequences of TAIRE kinase inhibition, CDK14 knockout HCT116 cells expressing either WT or C218S CDK14 were analyzed by FACS after treatment with FMF-04-159-2 or FMF-04-159-R (Figure 4E). Significant effects on cell cycle were observed, with increased numbers of cells in G2/M upon FMF-04-159-2 treatment (two-way ANOVA padj = .0001). Treatment with the reversible inhibitor FMF-04-159-R resulted in a more modest effect, resulting in increases in cells in G1 and G2/M and a reduction in S-phase cells. Similar effects were observed in both the WT and C218S CDK14-expressing cells, indicating that these effects were not solely due to covalent CDK14 inhibition.
Cell cycle data from HCT116 CDK14 CRISPR KO corroborates the observation that CDK14 covalent inhibition alone is not solely responsible for the observed cell cycle effects, as CDK14 KO cells did not show significant cell cycle differences at baseline compared to CDK14 WT or parental HCT116 cells (Figure S4A-C). Treatment with FMF-04-159-2 in the HCT116 CDK14 KO cells results in a more modest G2/M accumulation, or partial rescue, compared to the WT cells (Figure S4D), suggesting that the non-CDK14 targets of FMF-04-159-2 are also contributing the observed cell cycle effects. Taken together, these data suggest that under the above-mentioned experimental conditions, CDK14 kinase activity does not play a driving role in regulating cell cycle progression in the HCT116 cell line. The reversible binding effects of FMF-04-159-2/-R, namely pan-TAIRE kinase inhibition and/or CDK2 inhibition, may be responsible for some of the observed G2/M arrest, suggesting possible functional redundancy in within the TAIRE family.
The effects on cell cycle upon FMF-04-159-2 treatment were also observed in additional cell lines of differing tissues of origin: PATU-8988T (pancreatic cancer), MDA-MD-231 (breast cancer) and HepG2 (liver cancer). The magnitude of G2/M accumulation varied between different cell types, though the effect overall was consistent with what was observed in the HCT116 cells (Figure S4E-G). We also investigated the phenotypic effects of treatment with FMF-04-159-2 and did not observe major effects on actin cytoskeleton in HCT116 cells (Figure S4H).
FMF-04-159-2 as a tool for determining CDK14 substrates and phenotypes
Having established conditions in which CDK14 inhibition is maximized while CDK2 inhibition is minimized (Figure 4), we sought to further characterize the immediate consequences of CDK14 inhibition versus pan-TAIRE kinase inhibition, as well as potentially identify substrates of CDK14 using FMF-04-159-2 as a chemical tool through dual proteomics and phospho-proteomics experiments.
For the dual proteomics and phospho-proteomics experiment, HCT116 cells were treated with FMF-04-159-2, with and without a 2 h washout step, before subjecting either total peptides or enriched phospho-peptides to tandem mass tag labeling (TMT) and quantitative LC-MS. Few significant changes in total protein levels were observed proteome-wide upon compound treatment (Figure S5A-B), concurrent with the absence of reported direct transcriptional roles for the TAIRE kinases in the literature. Phospho-proteomics was conducted, and though direct treatment with FMF-04-159-2 resembled AT7519 treatment (Figure 5A, Figure S5C-D), compound washout resulted in fewer significantly affected phospho-peptides (435 versus 1305 2-fold up- or down-regulated sites) (Figure 5A-D).
Figure 5:

Cellular consequence of TAIRE kinase inhibition. (A) Volcano plot showing phospho-proteomics results for FMF-04-159-2 4hr treatment, analyzed using Bioconductor in R to assess fold change relative to DMSO. (B) Volcano plot showing phospho-proteomics results for FMF-04-159-2 4hr treatment followed by 2 h compound washout, analyzed using Bioconductor in R to assess fold change relative to DMSO. (C) Relative normalized TMT channel intensities for hits from FMF-04-159-2 direct treatment, reported as mean ± standard error. (D) Relative normalized TMT channel intensities for hits from FMF-04-159-2 washout condition with strong scores for CDK14 phosphorylation motif consensus, reported as mean ± standard error. (E) In vitro kinase assay results for phosphorylation of peptide library array by CDK14/Cyclin Y. (F) Top ten GO enrichment terms (Ashburner et al., 2000) for significantly downregulated phospho-peptides identified as hits from compound washout with strong scores for CDK14 phosphorylation motif consensus. FDR reported from Fisher’s exact test. See also Figure S5 and Figure S6.
To further triage the results of phosphoproteomic analysis, we conducted in vitro phospho-motif characterization of CDK14 using a peptide-scanning substrate array (Figure 5E). We identified P-X-s-P-X-ϕ-G (ϕ=hydrophobic) as the CDK14 phosphorylation motif. Reassuringly, this motif is in general agreement with the known substrate LRP6 S1490 (PP-s-PATE). We characterized another TAIRE kinase, CDK16, using the same peptide-scanning substrate array methodology, and established that this kinase has an identical consensus motif, with higher in vitro activity than CDK14 (Figure S6A). We observed the same consensus motif with CDK18, though with lower in vitro activity (Figure S6B). The identified TAIRE phospho-motif was distinct from the CDK2 motif, X-X-X-X-X-s-P-X-K-X, which was also shared by CDK3 (Figure S6C, D). This motif was used to score putative substrates based on phospho-peptide sequence matching to the predicted motif. Our peptide-scanning substrate array data provide not only a sequence motif to score putative substrates and bootstrap our phosphoproteomic analysis, but also further evidence for strong functional redundancy of the TAIRE kinases, given that CDK14, CDK16 and CDK18 likely regulate an overlapping pool of substrates based on the identical consensus phosphorylation motif.
All phospho-sites detected in the experiment were scored against the CDK14 phospho-motif, which identified 41 likely substrates from the set of 2-fold significantly down-regulated phospho-sites in the washout condition. Unfortunately, none of these high scoring substrates had available phospho-antibodies, antibodies or recombinant proteins commercially available to validate the identified putative substrates in a cellular context. Therefore, we synthesized four 15-mer peptides based on the protein sequences surrounding high scoring phospho-sites identified in washout proteomics. We also identified the sites on recombinant Rb protein that are phosphorylated by CDK14, Rb S608 and Rb S780 (Figure S6E), and synthesized two 15-mer control peptides based on the sequences surrounding these residues to be used as positive controls. We incubated recombinant CDK14 protein, ATP, and the target peptide, and assessed serine/threonine phosphorylation by mass spectrometry. We found that both positive controls, as well as 3 of the 4 putative substrate peptides could be phosphorylated at the expected sites by CDK14/Cyclin Y in vitro (Figure S6F-J), with MS/MS spectra passing a 1% false discovery rate. In all five cases, phosphorylation was dramatically reduced by pre-treatment with FMF-04-159-2 (Figure S6). These data demonstrate that the putative substrate peptides identified by the phospho-proteomics experiment can indeed be phosphorylated by CDK14 in vitro. Further work is required to fully validate their parental proteins as bona fide CDK14 substrates in a cellular context.
The distribution of the 41 sites prioritized from the washout condition among reported cellular processes was calculated using gene ontology (GO) enrichment.(Ashburner et al., 2000) This analysis showed strong enrichment for terms related to mitosis, cell cycle and cell division (Figure 5F). Though LRP6 itself was not detected, this further suggests that CDK14 plays a supporting, but not driving, role in mitosis, and that the covalent action of FMF-04-159-2 impairs mitotic cell cycle progression. Taken together, these results demonstrate the utility of FMF-04-159-2 as a tool compound to investigate CDK14 kinase biology, and point to a role for CDK14 in mitosis and cell division. The weak anti-proliferative effects of FMF-04-159-2 indicate potential functional redundancy amongst the CDKs in driving mitosis and cell cycle progression.
DISCUSSION
Kinases, a large family of enzymes that catalyze the transfer of a phosphate from ATP to a specific substrate (Avendaño and Menéndez, 2008), are involved in almost every known signal transduction pathway in the cell. Understanding the kinases that regulate these pathways has led to the successful development of kinase inhibitors for diseases such as cancer, where cell growth is deregulated. (Ferguson and Gray, 2018) To date, there are 38 FDA approved kinase inhibitors, making kinases one of the most productively targeted human gene families. (Wu et al., 2015) It is well-recognized that the majority of approved kinase inhibitors have more than one kinase target, and this polypharmacology is postulated to confer an advantage over highly selective compounds in the context of heterogeneous and highly adaptive cancers .(Klaeger et al., 2017), (Davis et al., 2011) However, the poly-pharmacology of clinical kinase inhibitors is rarely rationally designed, and in some cases is the root of undesirable toxicity. (Gujral et al., 2014)
Research in academia and industry is heavily biased towards kinases with well-defined roles in cellular signaling. (Fedorov et al., 2010) Consequently, the biological function of ~ 25% of kinases remains completely unknown, and ~50% of kinases are largely uncharacterized. (Knapp et al., 2013) However, cancer driver mutations and the potential drug targets identified by genetic knockdown are distributed throughout the kinome, establishing a case for expanding the scope of current research efforts. (McDonald et al., 2017), (Fedorov et al., 2010), (Tsherniak et al., 2017) An example of such a subfamily of kinases is the TAIRE CDKs, which include CDK14-18. Although genetic studies have implicated CDK14 and CDK16 as therapeutic targets especially in the context of colorectal cancers (Zhang et al., 2016b), (Zhu et al., 2016), (Zhou et al., 2014), not much is known about their biological roles and substrate preferences.
The need for annotating the understudied branches of the kinome is broadly recognized, and is even a key component of the NIH “Illuminating the Druggable Genome” initiative.(Fleuren et al., 2016),(Fedorov et al., 2010),(Rodgers et al., 2018) However, the absence of foundational data on the biochemical and biological functions of kinases of interest presents a huge challenge to small molecule chemical probe development. In the absence of characterized cellular inhibition phenotypes or substrates, innovative solutions must be found for establishing cellular target engagement. For example, in the course of this study, we developed a biotinylated probe, biotin-FMF-03-198-2, which acts to pull down CMCG family kinases from cell lysates. This assay using biotin-FMF-03-198-2 is broadly applicable to other kinase inhibitor projects and enables analysis of cellular target engagement by ATP-competitive small molecules for many kinases, including those that are understudied. We were able to use this tool to monitor the selectivity of synthesized compounds for CMGC kinases under various experimental conditions, eventually resulting in the development of FMF-04-159-2, that covalently targets CDK14 at C218 and possesses pan-TAIRE kinase biased selectivity profile.
Characterization of FMF-04-159-2 through pull-down assays and live cell target engagement using the NanoBRET platform verified sustained, covalent inhibition of CDK14, and reversible binding to the remaining TAIRE kinases CDKs 15-18. Reversible off-target activity against CDK2 was also measured. In the course of the development of FMF-04-159-2, compounds with increased selectivity for CDK14 and the TAIRE kinases showed reduced cytotoxic effects; FMF-04-159-2 was more than 200-fold less potent than FMF-03-198-2 in HCT116 cell proliferation assays. The mild cytotoxicity of FMF-04-159-2 may be due to the residual off-target reversible CDK2 activity. This is in agreement with RNAi data in the publicly available PROJECT DRIVE(McDonald et al., 2017) database. Taken together, these results suggest that selective CDK14 or pan-TAIRE inhibition is not a promising method for inhibiting colorectal cancer cell growth.
Analysis of known downstream substrates of the CDKs targeted by FMF-04-159-2 showed the expected effects of kinase inhibition. We were able to pharmacologically validate that LRP6 is a target of the human TAIRE-kinases in mitosis through compound treatment in synchronized cells, as evidenced by a maximal reduction of LRP6 pS1490 of 35%, as LRP6 is known to be phosphorylated by other kinases.(Davidson and Niehrs, 2010) We did not observe a meaningful reduction in LRP6 S1490 phosphorylation in unsynchronized cells upon compound treatment, consistent with reported results.
Few direct substrates other than LRP6 have been identified and validated for CDK14. Development of FMF-04-159-2 as a covalent CDK14 inhibitor provided the opportunity to identify potential substrates, and better characterize the immediate cellular consequences of CDK14 kinase inhibition. The GO analysis terms derived from phospho-proteomics experiments performed on cells treated with FMF-04-159-2 were very similar to those produced by cells treated with AT7519. This could be due to the dominant effects of CDK2 inhibition but does not rule out the involvement of the pan-TAIRE kinase inhibition activity shared by both molecules impairing cell cycle progression, particularly in mitosis. Integrating kinase motif studies with significantly down-regulated phospho-sites in the compound washout condition allowed for identification of 41 additional putative CDK14 substrates. Pathway analysis of these putative substrates suggested that CDK14 plays a role in mitotic cell cycle regulation and further supported CDK14’s role in mitotic progression, in agreement with cell cycle analysis data. These phenotypes warrant further investigation.
Finally, the availability of a narrow-spectrum TAIRE family kinase tool compound enables study of this class of kinases upon acute inhibition, complementing previous genetic studies. As there is evidence that the TAIRE kinases have some redundancies, the ability to rapidly inhibit these kinases in concert may also aid in initial substrate and functional annotation.
STAR METHODS TEXT
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nathanael S. Gray (Nathanael Gray@dfci.harvard.edu). Requested compounds will be provided following completion of an MTA.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HCT116 cells (adult male) were grown in McCoy’s 5A media (Life Technologies) and HEK293 cells (fetal), MDA-MB-231 (adult female), HepG2 (adolescent male) and PATU-8988T (adult female) were grown in Dulbecco's Modified Eagle's Medium (Invitrogen). All base media was supplemented with 10% FBS (Sigma) and 100U/mL Penicillin-Streptomycin (Gibco). All cells were cultured at 37Ό in a humidified chamber in the presence of 5% CO2. Cell lines were purchased from ATCC, with the exception of PATU-8988T cells, which were purchased from DSMZ. All cell lines were tested for mycoplasma on a monthly basis.
METHOD DETAILS
Pull Down / Cellular Target Engagement Protocol
HEK293 cells overexpressing a CDK14-flag fusion protein were treated with candidate compounds for 4 hours. Cells were washed with PBS, harvested and lysed in Pierce IP buffer with protease and phosphatase inhibitors (Roche). Lysates were clarified by centrifugation, then incubated with 1 uM biotin-FMF-03-198 (or 1uM biotin-ATP mimetic) overnight at 4 °C. To enhance pulldown, lysates were incubated at room temperature for an additional 2 hours. Lysates were then incubated with streptavidin agarose (Thermo Fisher #20349) for 2 hours at 4 °C. Agarose beads were was hed 3 times with Pierce IP buffer, then boiled in 2x LDS + 10% β–mercaptoethanol at 95 °C for 5 minutes. Proteins of interest engaged by the biotinylated compound were then assessed using western blotting.
For washout pull downs, medium containing inhibitors was removed after 4-hour compound treatment to effectively 'wash out' the compound. Cells were washed 1x with PBS, 1x with media. Fresh media was replaced, and cells were allowed to grow in the absence of inhibitor. Cells were then harvested at indicated times after washout (usually 2 hours unless otherwise noted) for lysis and subsequent pull down, as described above.
Antiproliferation Assay Protocol
HCT116 cells were plated in 384-well plates at 750 cells/well in 50ul fresh media and were treated the next day with 0.1 μl compounds in four-fold dilution series using the Janus pinner. Cells were incubated with compounds for 72 hours in 37 °C 5% CO2. Anti-proliferative effects of these compounds were then assessed using Cell Titer Glo (Promega # G7571) according to manufacturer protocol by measuring luminescence using an Envision plate-reader. All proliferation assays were performed in biological quadruplicate. IC50 values were determined using a non-linear regression curve fit in GraphPad Prism 6. N=4 biological replicates were used for each treatment condition.
In vitro Kinase Assays
LanthaScreen Eu kinase binding assays were conducted for CDK14/CycY as performed in the commercial assay service by Life Technologies, but included a 30 minute pre-incubation step of the kinase and Eu-labelled anti-GST antibody with candidate compounds to facilitate covalent bond formation. CDK14/CycY protein was purchased from Life Technologies.
For some compounds, LanthaScreen Eu kinase binding assays were conducted for CDK14/CycY at Life Technologies. LanthaScreen Eu kinase binding assays were conducted for CDK16/CycY at Life Technologies. Z'LYTE kinase assays were conducted for CDK2/CycA at Life Technologies using Km ATP concentrations. CDK14 33P kinase assays were performed by Reaction Biology Corp.
For RB phosphorylation, assayed using phospho-specific antibodies, CDK14/CycY (Life Technologies # PV6382) was used to phosphorylate truncated RB protein (Signal Chem # R05-55G-50). Reaction was allowed to proceed with 500uM ATP at 30 °C for 15 minutes. The reaction was stopped by addition of 2X LDS + 10% β–mercaptoethanol followed by incubation at 95 °C for 5 minutes. Western blotting was performed as described above, with the indicated phospho-specific RB antibodies (Cell Signaling) used to assess phosphorylation.
Intact Mass Spectrometry Analysis
Un-tagged CDK14 (amino acids 124-169) was expressed and purified using baculovirus. This recombinant CDK14 protein was incubated with DMSO or a 10-fold molar excess of FMF-03-198-2 or FMF-04-159-2 at room temperature for 2 hours in buffer (50 mM HEPES, 150 mM NaCl, 10% Glycerol, 2 mM TCEP, pH 7.5). Reacted protein (5 μg) was desalted on a self packed column (500 μm I.D. packed with 4 cm POROS 50R2, Applied Biosystems, Framingham, MA), eluted with a ballistic HPLC gradient (0-100% B in 1 minute; A=0.1 M acetic acid in water; B=0.1 M acetic acid in acetonitrile), and introduced to an LTQ ion trap mass spectrometer via electrospray ionization. The mass spectrometer collected profile data over m/z 300-2000. Mass spectra were deconvoluted using MagTran 1.03b2 (Zhang and Marshall, 1998).
Nano-LC/MS analysis of labeled CDK14
Protein labeled as described above was denatured with rapigest (0.1% final concentration), reduced (10 mM DTT, 56 °C, 30 minutes), alkylated (22.5 mM IAA, RT, dark), and digested with chymotrypsin overnight at 37 °C. Digests were acidified with 10% TFA, incubated at 37 °C for 30 minutes, and centrifuged at 13,000 rpm for 10 minutes at 4 °C to remove rapigest byproducts. Supernatant was then desalted by C18 and peptides dried by vacuum centrifugation. Dried peptides were resuspended in 50% acetonitrile with 1% formic acid, 100 mM ammonium acetate and analyzed by CE-MS using a ZipChip CE system and autosampler (908 Devices, Boston, MA) interfaced to a QExactive HF mass spectrometer (ThermoFisher Scientific). Peptides were loaded for 30 seconds and CE separation performed at 500 V/cm on an HR chip for 10 minutes with a background electrolyte consisting of 1% formic acid in 50% acetonitrile. Pressure assist was utilized and started at 1 minute. The mass spectrometer was operated in data dependent mode and subjected the 5 most abundant ions in each MS scan (60k resolution, 1E6 target, lock mass enabled) to MS/MS (15k resolution, 2E5 target, 100 ms max inject time). Dynamic exclusion was enabled with a repeat count of 1 and an exclusion time of 6 seconds. MS/MS data was extracted to .mgf using mulitplierz (Askenazi et al., 2009; Perez-Riverol et al., 2019) and searched against a custom database containing the sequence of CDK14 using Mascot version 2.2. Search parameters specified fixed carbamidomethylation of cysteine, and variable oxidation (methionine) and variable FMF-03-198-2 or FMF-04-159-2 modification (cysteine). Precursor mass tolerance was set to 10 ppm and product ion tolerance was 25 mmu. Peptide sequence confirmation was performed using mzStudio (Ficarro et al., 2017).
Cell Line Generation
HEK293 cells were transfected with pCMV-CDK14-myc/DDK (Origene # RC224426) using the Neon electroporation system according to manufacturer’s protocol (Life Technologies). The C218S CDK14 was generated using QuikChange (Agilent) according to manufacturer’s protocol, using the following primers:
fwd: GTGCACACTGATTTATCTCAGTACATGGAC
rev: GTCCATGTACTGAGATAAATCAGTGTGCAC
Sequence verification was performed using the following primer:
GAAGAAGAAGGGACAC
Cloning was performed using NEB 10-beta cells (NEB # C3019I).
HCT116 CDK14 knockout cell lines were generated by cloning CDK14-targeting sgRNAs into the Bbsl site of pX458 (Addgene # 48138). The genomic sequence complementary to the CDK14-directed guide RNA that was cloned into pX458 and used in the genome editing experiments is: TCATGACATCATCCATACCA.
Cloning was performed using NEB Stable cells (NEB #C3040I). Single cells expressing GFP 48 hours after transfection using the Neon electroporation system (Life Technologies) were sorted into 96 well plates using FACS. Clones were verified both by western blotting and genomic DNA PCR of the edited region. CDK14 was cloned into the Xbal/Notl sites of the pCDH-CMV-MCS vector backbone (System Biosciences# CD510B-1). CDK14-flag wild type or C218S was re-introduced to the HCT116 CDK14 knockout cells using lentiviral infection.
KiNativ Live Cell Profiling Protocol
HCT116 cells were plated in fresh media in 15cm plates and treated for 4 hours with candidate compounds. For washout conditions, compound-containing media was removed from cells, and cells were washed 1x with PBS and 1x with media before fresh media was replaced. Cells were incubated for an additional 2 hours after washout. To harvest cells, plates were washed 1x with cold PBS, then collected by scraping and centrifugation. Cell pellets were snap-frozen in liquid nitrogen. The remainder of the KiNativ profiling experiment was performed by ActivX Biosciences (La Jolla, CA).
Fluorescence-Activated Cell Sorting Analysis (FACS) for Cell Cycle Analysis
Cells were plated and treated the next day with the indicated compounds for 24 hours. Cells were trypsinized, washed once in phosphate-buffered saline (PBS), and fixed overnight with 80% ethanol in PBS at −20°C. Cells were washed three times with PBS. Cells were stained in PBS containing 0.1% Triton X-100, 25 μg/mL propidium iodide (PI, Molecular Probes), and 0.2 mg/mL RNase A (Sigma) overnight at 4 °C. Cells were filtered through a 0.2um filter prior to data acquisition on a BD LSR II. Data were analyzed using FlowJo. N=3 biological replicates were used for each treatment condition.
Inhibitor Treatment and Western Blotting Experiments
HCT116 cells were plated and treated for 6 hours or 4 hours followed by compound washout for 2 hours with indicated compounds at 1 uM. Cells were dissociated using trypsin, washed with PBS and lysed in RIPA buffer + protease and phosphatase inhibitor (Roche) + nuclear (Pierce). Samples were normalized and prepped in 4x LDS + 10% β-mercaptoethanol and boiled for 5 minutes at 95 °C. Lysates were probed for specified proteins by western blotting using the Bolt system (Life Technologies).
NanoBRET Live Cell Target Engagement
HCT116 cells were transiently transfected with luciferase fusion-expressing CDK constructs and a Cyclin Y carrier (plasmids courtesy of Promega). Cells were treated with compound and an ATP-mimetic Tracer 20-24 hours later and assayed 4 hours after treatment using the Intracellular TE Nano-Glo kit according to manufacturer protocol (Promega #N2160). N=3 biological replicates were used for each treatment condition.
Immunohistochemistry for Phalloidin Staining
HCT116 cells with CDK14 wild type or KO (CRISPR-generated clonal lines) were plated in 6 well plates with a single coverslip in each well, and treated the next day with DMSO or FMF-04-159-2 for 24 h. Cells were washed once with PBS, fixed with 4% formaldehyde in PBS for 15 minutes at room temperature, then washed three times with PBS. Cells were incubated in the dark at room temperature for 1 hour with Phalloidin-488 (Santa Cruz # sc-363791) according to manufacturer instructions. Cells were washed three times with PBS, then coverslips were mounted on slides with ProLong Gold Antifade Reagent with DAPI (Cell Signaling # 8961) and cured in the dark at room temperature overnight before storage at 4°C. Slides were imaged using a Zeiss Axiostar microscope using a 20X objective, and images were captured using a Zeiss Axiocam and ZENIite software. N=3 biological replicates were used for each treatment condition.
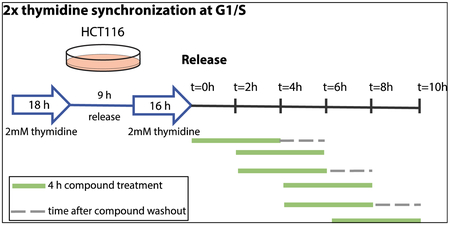
Double Thymidine Synchronization with Compound Treatment
100,000 cells per well were plated in a 6-well tissue culture-treated plate. The next day, cells were treated with 2mM thymidine final concentration (from 100mM stock) for 18 h. Cells were washed 2x with media, and fresh media was replaced. After 8 h, cells were treated again with 2mM thymidine as before for 16 h. Cells were released by washing 2x with media, and fresh media was replaced. Cells were treated with 1 uM FMF-04-159-2 for 4 h time periods, and were either harvested, or where indicated, compound was washed out by washing cells 2x with media and replacing with fresh media, and cells were incubated further for 2 h (schematic below). Cells were harvested at the indicated timepoint using trypsin, and western blot was performed as described above.
Peptide Library Assay for Phospho-motif Identification
Assay was performed as described previously (Hutti et al., 2004). The spot densities from the dot array were quantified and normalized by each row. These values were used to score the amino acid sequence surrounding the serine or threonine of each identified phospho-peptide, by summing the corresponded dots of each substrate. Two outputs were generated: Raw scores and relative rankings. In the relative rankings, CDK14/16 score was ranked in comparison to 189 other serine/threonine kinase substrate motif scores (Johnson J.L. et al., unpublished).
CDK16/Cyclin Y (cat # PV6379) was purchased from Life Technologies, CDK14/Cyclin Y (cat # P15-10G-05) and CDK18/Cyclin Y (cat # P11-10G-05) were purchased from SignalChem. CDK2/Cyclin E1 (cat # C29-18G-10) and CDK3/Cyclin E1 (cat # C30-10G-10) were also purchased from SignalChem.
Proteomics and Phospho-proteomics Cell Lysis, Protein Digest and TMT Labeling
Cells were lysed by homogenization (QIAshredder) in lysis buffer (2% SDS, 150 mM NaCl, 50 mM Tris pH 7.4). Lysates were reduced with 5 mM DTT, alkylated with 15 mM iodoacetamide for 30 minutes in the dark, alkylation reactions quenched with 50 mM freshly prepared DTT and proteins precipitated by methanol/chloroform precipitation. Digests were carried out in 200 mM EPPS pH 8.5 in presence of 2% acetonitrile (v/v) with LysC (Wako, 2mg/ml, used 1:75) for 3 hours at room temperature and after subsequent addition of trypsin (Promega #V5111, stock 1:75) over night at 37°C.
Missed cleavage rate was assayed from a small aliquot by mass spectrometry. For whole proteome analysis, digests containing approximately 60 μg of peptide material were directly labeled with TMT reagents (Thermo Fisher Scientific). Labeling efficiency and TMT ratios were assayed by mass spectrometry, while labeling reactions were stored at −80°C. After quenching of TMT labeling reactions with hydroxylamine, TMT labeling reactions were mixed, solvent evaporated to near completion and TMT labeled peptides purified and desalted by acidic reversed phase C18 chromatography. Peptides were then fractionated by alkaline reversed phase chromatography into 96 fractions and combined into 12 samples. Before mass spectrometric analysis, peptides were desalted over Stage Tips (Rappsilber et al., 2003).
Proteomics and Phospho-proteomics Phosphopeptide Enrichment
Phosphopeptides were enriched from digested material containing approximately 4 mg of peptide material per sample by Fe-NTA chromatography (Thermo Fisher Scientific #A32992). After TMT labeling, phosphopeptides were subjected to alkaline reverse fractionation by a linear acetonitrile gradient into 96 fractions and combined into 24 samples. TMT labeled, fractionated phosphopeptides were desalted over Stage Tips (Rappsilber et al., 2003) prior to mass spectrometry analysis.
Proteomics and Phospho-proteomics Mass Spectrometry Analysis
Data were collected by a MultiNotch MS3 TMT method (McAlister et al., 2014) using an Orbitrap Lumos mass spectrometer (Thermo Fisher Scientific) coupled to a Proxeon EASY-nLC 1200 liquid chromatography (LC) system (Thermo Fisher Scientific). The 100 μm inner diameter capillary column used was packed with C18 resin (SepPax Technologies Inc. 1.8 μm). Peptides of each fraction were separated over 3 h acidic acetonitrile gradients by LC prior to mass spectrometry (MS) injection. The first scan of the sequence was an MS1 spectrum (Orbitrap analysis; resolution 120,000; mass range 400–1400 Th). MS2 analysis followed collision-induced dissociation (CID, CE=35) with a maximum ion injection time of 150 ms (350 ms for phosphopeptides) and an isolation window of 0.7 Da. In order to obtain quantitative information, MS3 precursors were fragmented by high-energy collision-induced dissociation (HCD) and analyzed in the Orbitrap at a resolution of 50,000 at 200 Th.
For phosphopeptides, MS2 spectra were recorded with an isolation window of 0.5 Da and multistage activation (MSA) with a neutral loss of 97.9763 Da. In the MS3 for charge states of 2 and 3, an isolation window of 1.2 m/z and for charge states 4-6 an isolation window of 0.8 m/z was chosen. MS3 injection time for phosphopeptides was 350 ms at a resolution of 50,000. Further details on LC and MS parameters and settings used were described recently (Paulo et al., 2016a).
Peptides were searched with a SEQUEST-based in-house software against a human database with a target decoy database strategy and a false discovery rate (FDR) of 2% set for peptide-spectrum matches following filtering by linear discriminant analysis (LDA) and a final collapsed protein-level FDR of 2%. Quantitative information on peptides was derived from MS3 scans. Quant tables were generated requiring an MS2 isolation specificity of >65% for each peptide and a sum of TMT s/n of >150 over all channels for any given peptide and exported to Excel and further processed therein. To quantify the confidence assignment of phosphorylation sites, a modified version of the Ascore algorithm was used. For confident site localization, Ascore values of >13 (p <= 0.05) were required (Huttlin et al., 2010). Details of the TMT intensity quantification method and further search parameters applied were described recently (Paulo et al., 2016b).
In Vitro Phosphorylation of Synthetic Peptides followed by MS/MS
Recombinant CDK14/Cyclin Y (Life Technologies) was preincubated with or without 1 μM FMF-04-159-2 for 1 h at 37 °C. The kin ase was then reacted with synthetic peptides (100:1 substrate to enzyme ratio) in kinase buffer (50 mM Tris pH 7.5, 150 mM NaCl, 5% glycerol, 1 mM TCEP, 20 mM MgCl2, 1 mM ATP) for 18 h at 37 °C. Reactions were desalted and analyzed using a ZipChip (908 Devices, Cambridge MA) capillary electrophoresis source on a Q Exactive HF mass spectrometer using a 10 min run method as described previously (Ficarro et al., 2016). MS/MS spectra of synthetic phosphopeptides were annotated using mzStudio (Ficarro et al., 2017). Spectra were searched with Mascot using no enzyme specificity against a forward and reverse human proteome database.
Chemistry Synthetic Schemes
Scheme 1
Scheme 2
Scheme 3
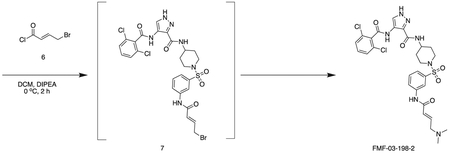
General Chemistry Methods
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. 1H NMR spectra were recorded on 500 MHz Bruker Avance Ill spectrometer, and chemical shifts are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as s (singlet), br (broad singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Mass spectra were obtained on a Waters Acquity UPLC. Preparative HPLC was performed on a Waters Sunfire C18 column (19 mm × 50 mm, 5 μM) using a gradient of 15–95% methanol in water containing 0.05% trifluoroacetic acid (TFA) over 22 min (28 min run time) at a flow rate of 20 mL/min. Assayed compounds were isolated and tested as TFA salts. Purities of assayed compounds were in all cases greater than 95%, as determined by reverse-phase HPLC analysis.
Experimental Details for Individual Compound Synthesis
tert butyl (1-((3-nitrophenyl)sulfonyl)piperidin-4-yl)carbamate
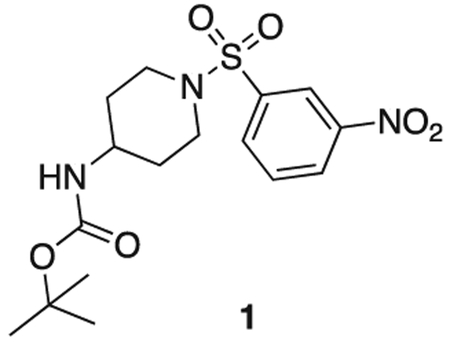
tert-butyl piperidin-4-ylcarbamate (1.0 g, 5.0 mmol), 3-nitrobenzenesulfonyl chloride (1.22 g, 5.5 mmol), triethylamine (1.1 mL, 7.5 mmol) were stirred at r.t. in DCM (20 mL) for 2 h. The reaction mixture was concentrated in vacuo, and diluted with sat. aq sodium bicarbonate (50 mL) and extacted with 100 mL DCM three times. The organics were combined, dried over sodium sulfate, filtered, and concentrated in vacuo. The product was used without further purification. Off-white solid (1.8 g, 4.7 mmol). MS (ESI) mlz 386 (M + H)+. Expected mass from chemical formula C16H23N3O6: 385.44 Da.
1-((3-nitrophenyl)sulfonyl)piperidin-4-amine
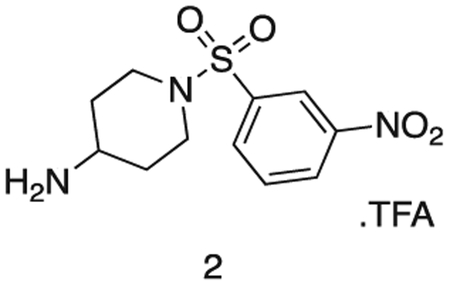
Intermediate 1 (1.8 g, 4.7 mmol) and TFA (2 mL) were stirred in DCM (20 mL) at r.t. for 16 h. The reaction mixture was concentrated in vacuo to afford the product as a white solid (1.87 g, 4.7 mmol). 1H NMR (500 MHz, DMSO-d6) δ 9.09 (s, 2H), 8.56 (dd, J = 8.2, 2.2 Hz, 1H), 8.39 (t, J = 2.0 Hz, 1H), 8.19 (dt, J = 7.8, 1.2 Hz, 1H), 7.97 (t, J = 8.0 Hz, 1H), 3.72 (d, J = 9.1 Hz, 1H), 3.61 (ddt, J = 10.5, 6.6, 3.9 Hz, 2H), 3.13 (dd, J = 7.4, 4.2 Hz, 3H), 1.98 (dd, J = 13.5, 3.8 Hz, 2H), 1.58 (qd, J = 11.9, 4.2 Hz, 2H). MS (ESI) m/z 286 (M + H)+. Expected mass from chemical formula C16H23N3O6; 285.32 Da.
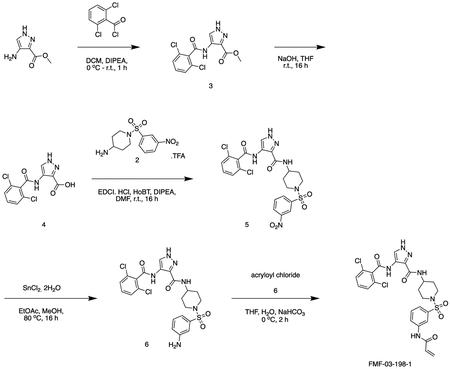
methyl 4-(2,6-dichlorobenzamido)-1H-pyrazole-3-carboxylate
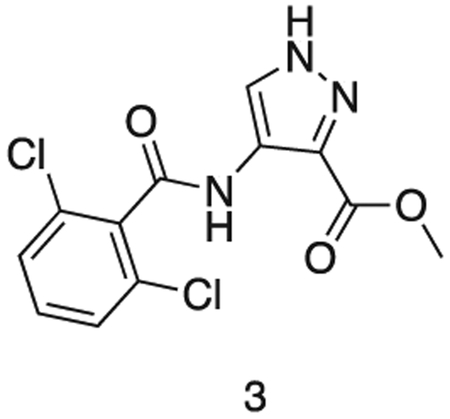
methyl 4-amino-1H-pyrazole-3-carboxylate (1.0 g, 7.09 mmol) and triethylamine (1.5 mL, 8.5 mmol) were stirred in dioxane (10 mL) at 0°C. A solution of 2,6-dichlorobenzoyl chloride (1.5 g, 7.17 mmol) in THF (5 mL) was added dropwise until the starting material was consumed. The reaction was filtered, and the resultant solid washed with dioxane (3 × 20 mL). The filtrates were combined and used directly in the next reaction.
MS (ESI) m/z 315 (M + H)+. Expected mass from chemical formula C12H9N3O3: 314.12 Da.
4-(2,6-dichlorobenzamido)-1H-pyrazole-3-carboxylic acid
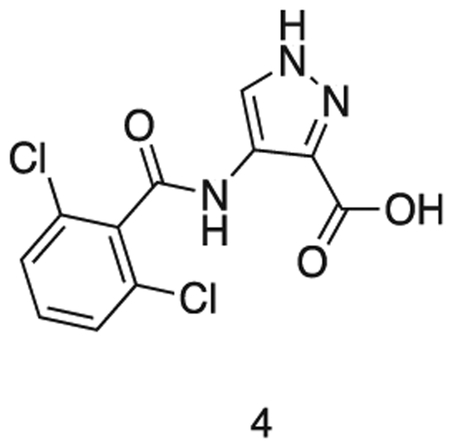
Intermediate 3 in dioxane (15 mL) was added to 2N aq. NaOH (15 mL). The solution was stirred at r.t. for 6 h. The reaction mixture was concentrated in vacuo and dissolved in water (30 mL). The solution was cooled to 0 °C and conc. HCI added dropwise to pH 1. The precipitate was filtered and washed with water (3 × 5 mL). The precipitate was azeotroped with toluene, to afford the title compound as a pale gray solid (1.58 g, 0.52 mmol). 1H NMR (500 MHz, DMSO-d6) δ 9.86 (s, 1H), 8.28 (s, 1H), 7.57 – 7.44 (m, 5H). MS (ESI) m/z 301 (M + H)+. Expected mass from chemical formula C11H7N3O3; 300.10 Da.
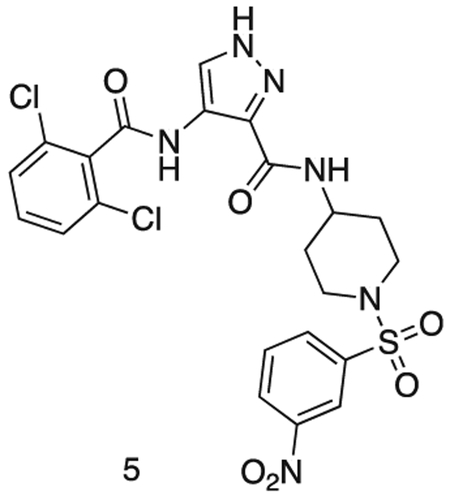
4-(2,6-dichlorobenzamido)-N-(1-((3-nitrophenyl)sulfonyl)piperidin-4-yl)-1H-pyrazole-3-carboxamide
Intermediate 2 (144 mg, 0.36 mmol), intermediate 4 (100 mg, 0.33 mmol), HoBt (60 mg, 0.44 mmol), EDCI.HCI (80 mg, 0.42 mmol) were dissolved in DMF (2 mL) and stirred at r.t. for 16 h. The reaction was diluted with sat. aq sodium bicarbonate (25 mL) and extracted with EtOAc (3 × 50 mL). The organics were combined, dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (50 – 100 % EtOAc in Hexanes) to afford the title compound as a yellow solid (150 mg, 0.26 mmol).
MS (ESI) m/z 568 (M + H)+. Expected mass from chemical formula C22H20N6O6S: 567.40 Da.
N-(1-((3-aminophenyl)sulfonyl)piperidin-4-yl)-4-(2,6-dichlorobenzamido)-1H-pyrazole-3-carboxamide
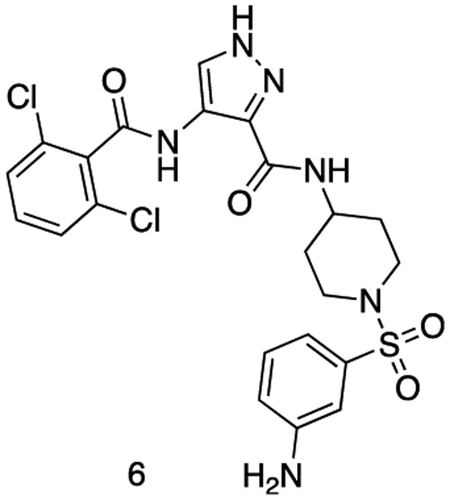
Intermediate 5 (155 mg, 0.26 mmol), SnCl2.2H2O (146 mg, 0.65 mmol) and HCI (cat.) were dissolved in EtOAc (4 mL) and MeOH (1 mL). The reaction was stirred at 80 °C for 16 h. The reaction mixture was cooled to r.t. and quenched with sat. aq sodium bicarbonate (5 mL).The reaction was diluted with sat. aq sodium bicarbonate (25 mL) and extracted with EtOAc (3 × 50 mL). The organics were combined, dried over sodium sulfate, filtered, and concentrated in vacuo to afford the title compound as a yellow powder. (122 mg, 0.22 mmol). 1H NMR (500 MHz, DMSO-d6) δ 10.13 (s, 1H), 8.43 (dd, J = 8.2, 3.7 Hz, 1H), 8.39 – 8.31 (m, 1H), 7.59 – 7.54 (m, 2H), 7.54 – 7.46 (m, 2H), 7.24 (td, J = 7.8, 3.4 Hz, 1H), 6.92 (dt, J = 9.1, 2.1 Hz, 1H), 6.85 – 6.77 (m, 2H), 5.63 (d, J = 3.1 Hz, 2H), 3.71 (dtd, J = 11.2, 7.5, 4.1 Hz, 1H), 3.66 – 3.54 (m, 2H), 2.67 – 2.55 (m, 2H), 2.31 (td, J = 12.0, 2.7 Hz, 1H), 1.80 (dd, J = 12.9, 3.5 Hz, 2H), 1.70 (pd, J = 12.4, 11.1, 3.0 Hz, 2H). MS (ESI) m/z 538 (M + H)+. Expected mass from chemical formula C22H22N6O4S: 537.42 Da.
FMF-03-198-2 (E)-4-(2,6-dichlorobenzamido)-N-(1-((3-(4-(dimethylamino)but-2-enamido)phenyl)sulfonyl)piperidin-4-yl)-1H-pyrazole-3-carboxamide
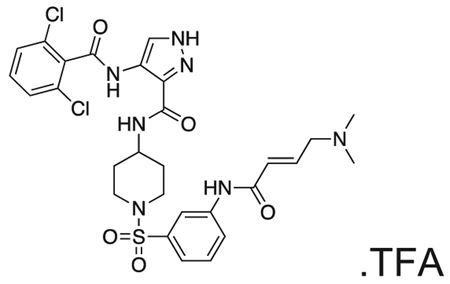
Intermediate 6 (50 mg, 0.09 mmol) and DIPEA (100 μL, 1.1 mmol) were dissolved in DCM (10 mL). The reaction was cooled to 0 °C and (E)-4-bromobut-2-enoyl chloride (26 mg, 0.14 mmol) in DCM (1 mL) was added dropwise until the starting material was consumed. The reaction mixture was concentrated in vacuo. The residue was purified by HPLC to afford the title compound (2 mg, 0.004 mmol) as a white powder. 1H NMR (500 MHz, DMSO-d6) δ 13.44 (s, 1H), 10.73 (s, 1H), 10.11 (s, 1H), 10.01 (s, 1H), 8.45 (d, J = 8.4 Hz, 1H), 8.35 (s, 1H), 8.18 (t, J = 2.0 Hz, 1H), 7.95 – 7.88 (m, 1H), 7.66 – 7.48 (m, 4H), 7.45 (dt, J = 7.8, 1.3 Hz, 1H), 6.79 (dt, J = 14.8, 7.2 Hz, 1H), 6.51 – 6.40 (m, 1H), 3.97 (d, J = 7.1 Hz, 2H), 3.77 – 3.67 (m, 1H), 3.63 (d, J = 11.5 Hz, 2H), 2.81 (s, 6H), 2.34 (d, J = 11.9 Hz, 2H), 1.80 (d, J = 12.3 Hz, 2H), 1.70 (d, J = 12.7 Hz, 2H). MS (ESI) m/z 649 (M + H)+. Expected mass from chemical formula C28H31Cl2N7O5S: 648.56
FMF-04-159-2 (E)-N-(1-((3-(4-(dimethylamino)but-2-enamido)phenyl)sulfonyl)piperidin-4-yl)-4-(2,4,6-trichlorobenzamido)-1H-pyrazole-3-carboxamide
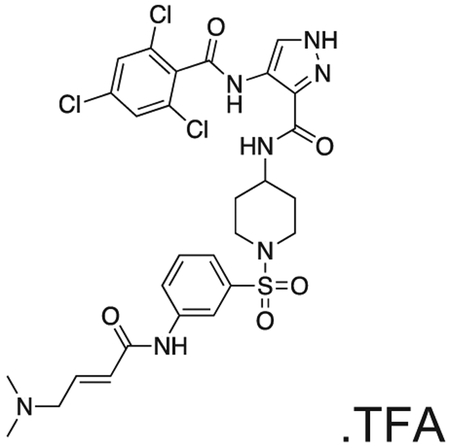
The compound was prepared according to Scheme 3 (17 mg, 0.024 mmol) as a white powder. 1H NMR (500 MHz, DMSO-d6) δ 13.42 (s, 1H), 10.70 (s, 1H), 10.18 (s, 1H), 9.84 (s, 1H), 8.42 (d, J = 8.2 Hz, 1H), 8.35 (d, J = 1.5 Hz, 1H), 8.17 (t, J = 2.0 Hz, 1H), 8.00 – 7.85 (m, 1H), 7.79 (s, 2H), 7.62 (t, J = 8.0 Hz, 1H), 7.45 (dt, J = 7.9, 1.3 Hz, 1H), 6.79 (dt, J = 14.8, 7.1 Hz, 1H), 6.50 – 6.37 (m, 1H), 3.96 (d, J = 7.2 Hz, 2H), 3.83 – 3.68 (m, OH), 3.63 (d, J = 11.6 Hz, 2H), 2.81 (s, 6H), 2.41 – 2.29 (m, 2H), 1.87 – 1.64 (m, 4H). MS (ESI) m/z 689 (M + H)+. Expected mass from chemical formula C28H30Cl3N7O5S: 683.00
FMF-04-153-1 (Biotin-FMF-03-198-2) 4-(2,6-dichlorobenzamido)-N-(1-((3-((E)-5-methyl-8,22-dioxo-26-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)-12,15,18-trioxa-5,9,21-triazahexacos-2-enamido)phenyl)sulfonyl)piperidin-4-yl)-1H-pyrazole-3-carboxamide
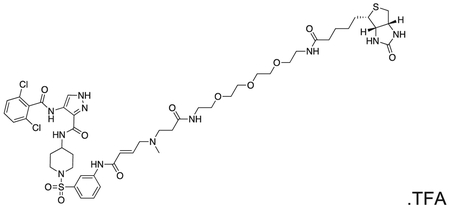
The compound was prepared according to Scheme 3 (4 mg, 0.003 mmol) as a white powder. MS (ESI) m/z 1108 (M + H)+, 554 [(M + H)+/2]. Expected mass from chemical formula C48H65Cl2N11O11S2 : 1107.13
FMF-04-159-R (FMF-05-176-1) N-(1-((3-(4-(dimethylamino)butanamido)phenyl)sulfonyl)piperidin-4-yl)-4-(2,4,6-trichlorobenzamido)-1H-pyrazole-3-carboxamide
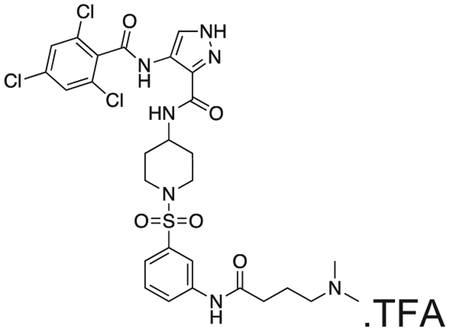
FMF-04-159-2 (25 mg) and palladium on charcoal (wet, 10%, 5 mg) were suspended in EtOH and stirred at r.t. under H2 for 16 h. The reaction was filtered over celite and the filtrate concentrated under vaccum. The residue was purified by HPLC to yield the title compound as a colourless oil (2 mg, 0.003 mmol). 1H NMR (500 MHz, DMSO-d6) δ 13.43 (s, 1H), 10.40 (s, 1H), 10.18 (s, 1H), 8.42 (d, J = 8.2 Hz, 1H), 8.35 (s, 1H), 8.12 (d, J = 2.1 Hz, 1H), 7.85 – 7.81 (m, 1H), 7.80 (s, 2H), 7.58 (t, J = 8.0 Hz, 1H), 7.40 (dt, J = 7.8, 1.2 Hz, 1H), 3.76 – 3.66 (m, 1H), 3.62 (d, J = 11.6 Hz, 2H), 3.08 (dd, J = 9.7, 6.5 Hz, 2H), 2.78 (s, 6H), 2.45 (t, J = 7.1 Hz, 2H), 2.35 (t, J = 12.1 Hz, 2H), 1.97 – 1.89 (m, 2H), 1.81 (d, J = 12.1 Hz, 2H), 1.70 (d, J = 12.5 Hz, 2H). MS (ESI) m/z 686 (M + H)+. Expected mass from chemical formula C34H35Cl2N7O5 S: 685.13
QUANTIFICATION AND STATISTICAL ANALYSIS
Antiproliferation Assay
IC50 values were determined using a non-linear regression curve fit in GraphPad Prism 6. N=4 biological replicates were used for each treatment condition. Values are reported as mean +/− SEM, as also indicated in the figure legends.
In Vitro Kinase Assays
IC50 values were determined using a non-linear regression curve fit in GraphPad Prism 6. N=3 biological replicates were used for each treatment condition. Values are reported as mean +/− SEM, as also indicated in the figure legends.
NanoBRET Assay
IC50 values were determined using a non-linear regression curve fit in GraphPad Prism 6. N=3 biological replicates were used for each treatment condition. Values are reported as mean +/− SEM, as also indicated in the figure legends.
Intact Mass and Compound Labeling
Mass spectra were deconvoluted using MagTran 1.03b2 (Zhang and Marshall, 1998). MS/MS data was extracted to .mgf using mulitplierz (Askenazi et al., 2009; Perez-Riverol et al., 2019) and searched against a custom database containing the sequence of CDK14 using Mascot version 2.2. Peptide sequence confirmation was performed using mzStudio (Ficarro et al., 2017).
FACS
Data were analyzed using FlowJo. N=3 biological replicates were used for each treatment condition. Bar height represents mean percentage of cells in each cell cycle phase, and error bars represent SEM, as also indicated in figure legends.
Immunohistochemistry
images were captured using a Zeiss Axiocam and ZENIite software. ImageJ was used to add scale bar. N=3 biological replicates were used for each treatment condition. Representative images are shown.
Proteomics and Phospho-proteomics
Reporter ion intensities were normalized and scaled using the R framework. Statistical analysis was carried out in R using the Bioconductor package Limma.(Huber et al., 2015) Each phospho-site identified was scored for relative agreement with the identified CDK14 substrate motif. A score cutoff of 0.5 was used to indicate likely CDK14 substrate phospho-sites.
DATA AND SOFTWARE AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD012692 (Perez-Riverol et al., 2019).
Supplementary Material
Table S1. Complete KiNativ profiling data for AT7519, FMF-03-198-2 and FMF-04-159-2. Related to Figure 2 and Figure 3. All experiments were performed at 1 μM for 4 h in HCT116 cells. Repeated kinase names indicate different sites of modification identified.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti PFTAIRE-1 (C-3) | Santa Cruz Biotechnology | Cat # sc-376366 |
| Mouse monoclonal anti Flag | Sigma Aldrich | Cat # F1804-50UG |
| Rabbit polyclonal anti Cyclin Y | Bethyl Laboratories | Cat # A302-376A |
| Rabbit polyclonal anti PCTAIRE-1 | Cell Signaling Technologies | Cat # 4852 |
| Rabbit polyclonal anti PCTAIRE-2 | Sigma Aldrich | Cat # SAB1306378 |
| Rabbit monoclonal anti CDK2 | Cell Signaling Technologies | Cat # 2546 |
| Rabbit monoclonal anti CDK9 | Cell Signaling Technologies | Cat # 2316 |
| Rabbit monoclonal anti CDK10 | Cell Signaling Technologies | Cat # 36106 |
| Rabbit polyclonal anti Cyclin K | Bethyl Laboratories | Cat # A301-939A |
| Mouse monoclonal anti CDK5 | Cell Signaling Technologies | Cat # 12134 |
| Rabbit monoclonal anti GSK3α | Cell Signaling Technologies | Cat # 4337 |
| Rabbit monoclonal anti Phospho-LRP6 S1490 | Cell Signaling Technologies | Cat # 2568 |
| Rabbit monoclonal anti LRP6 | Cell Signaling Technologies | Cat # 2560 |
| Rat monoclonal anti Pol II pSer2 | EMD Millipore | Cat # 04-1571 |
| Rabbit polyclonal anti Pol II (N-20) | Santa Cruz Biotechnology | Cat # sc-899 |
| Rabbit polyclonal anti Phospho-NPM T199 | Cell Signaling Technologies | Cat # 3451 |
| Rabbit polyclonal anti NPM | Cell Signaling Technologies | Cat # 3452 |
| Rabbit monoclonal anti Phospho-RB S608 | Cell Signaling Technologies | Cat # 8147 |
| Rabbit monoclonal anti Phospho-RB S780 | Cell Signaling Technologies | Cat # 8180 |
| Rabbit polyclonal anti Phospho-RB S795 | Cell Signaling Technologies | Cat # 9301 |
| Rabbit monoclonal anti Phospho-RB S807/811 | Cell Signaling Technologies | Cat # 8516 |
| Mouse monoclonal anti RB | Cell Signaling Technologies | Cat # 9309 |
| Rabbit monoclonal anti active β-catenin | Cell Signaling Technologies | Cat # 8814 |
| Rabbit monoclonal anti β-catenin | Cell Signaling Technologies | Cat # 9582 |
| Rabbit polyclonal anti Cyclin B1 | Cell Signaling Technologies | Cat # 4138 |
| Rabbit monoclonal anti Phospho-Histone H3 S10 | Cell Signaling Technologies | Cat # 53348 |
| Mouse monoclonal anti Histone H3 | Cell Signaling Technologies | Cat # 14269 |
| Mouse monoclonal anti Tubulin | Cell Signaling Technologies | Cat # 3873 |
| Mouse monoclonal anti Actin | Cell Signaling Technologies | Cat # 3700 |
| Bacterial and Virus Strains | ||
| NEB 10-beta | NEB | Cat # C3019I |
| NEB Stable | NEB | Cat # C3040I |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| CDK14 / Cyclin Y | Life Technologies | Cat # PV6382 |
| CDK14 / Cyclin Y | Signal Chem | Cat # P15-10G-05 |
| CDK16 / Cyclin Y | Life Technologies | Cat # PV6379 |
| CDK18 / Cyclin Y | Signal Chem | Cat # P11-10G-05 |
| RB | Signal Chem | Cat # R05-55G-50 |
| CDK2 / Cyclin A | Signal Chem | Cat # C29-18G-10 |
| CDK3 / Cyclin A | Signal Chem | Cat # C30-10G-10 |
| CellTiter Glo | Promega | Cat # G7571 |
| Thymidine | Sigma Aldrich | Cat # T1895-5G |
| Peptide: RbS608 TAADMYLSPVRSPKK |
Tufts University Dept. of Physiology | Custom synthesis |
| Peptide: RbS780 STRPPTLSPIPHIPR |
Tufts University Dept. of Physiology | Custom synthesis |
| Peptide: CDC20 KEAAGPAPSPMRAAN |
Tufts University Dept. of Physiology | Custom synthesis |
| Peptide: CCAR2 NQPLLKSPAPPLLH |
Tufts University Dept. of Physiology | Custom synthesis |
| Peptide: CENPF KIFTTPLTPSQYYSG |
Tufts University Dept. of Physiology | Custom synthesis |
| Peptide: ERF EAGGPLTPRRVSS |
Tufts University Dept. of Physiology | Custom synthesis |
| Critical Commercial Assays | ||
| LanthaScreen Eu (CDK14 / Cyclin Y) | Invitrogen | Assay ID 1839 |
| LanthaScreen Eu (CDK16 / Cyclin Y) | Invitrogen | Assay ID 1838 |
| Z’LYTE (CDK2 / Cyclin A) | Invitrogen | Assay ID 314 |
| CDK14 33P Kinase Assay | Reaction Biology Corp | |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| HCT116 | ATCC | CCL-247 |
| HEK293 | ATCC | CRL-1573 |
| MDA-MB-231 | ATCC | HTB-26 |
| HepG2 | ATCC | HB-8065 |
| PATU-8988T | DSMZ | Cat # ACC 162 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| CDK14 C218S QuikChange forward: GTGCACACTGATTTATCTCAGTACATGGAC | IDT | Custom synthesis |
| CDK14 C218S QuikChange reverse: GTCCATGTACTGAGATAAATCAGTGTGCAC | IDT | Custom synthesis |
| CDK14 QuikChange sequencing: GAAGAAGAAGGGACAC | IDT | Custom synthesis |
| sgCDK14 gRNA forward: caccgTCATGACATCATCCATACCA | IDT | Custom synthesis |
| sgCDK14 gRNA reverse: aaacTGGTATGGATGATGTCATGAc | IDT | Custom synthesis |
| Recombinant DNA | ||
| pCMV6-CDK14-myc/flag | Origene | Cat # RC224426 |
| pCMV6-CDK14 C218S-myc/flag | This paper | n/a |
| pX458 | Addgene | Cat # 48138 |
| pX458-sgCDK14 | This paper | n/a |
| pCDH-CMV-MCS- | System Biosciences | Cat # CD510B-1 |
| NanoBRET CDK2 | Promega | n/a |
| NanoBRET CDK14 | Promega | n/a |
| NanoBRET CDK15 | Promega | n/a |
| NanoBRET CDK16 | Promega | n/a |
| NanoBRET CDK17 | Promega | n/a |
| NanoBRET CDK18 | Promega | n/a |
| NanoBRET Cyclin Y (carrier DNA) | Promega | n/a |
| Software and Algorithms | ||
| Prism | GraphPad | |
| FlowJo | FlowJo, LLC | |
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
| ZEN lite | Carl Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen-lite.html |
| TIDE | Brinkman et al., 2014 | https://tide.deskgen.com/ |
| GO | Ashburner et al., 2000 | http://geneontology.org/page/go-enrichment-analysis |
| R - Bioconductor | Huber et al., 2015 | https://www.bioconductor.org/packages/release/bioc/html/limma.html |
| Other | ||
| Streptavidin-agarose beads | Thermo Fisher | Cat # 20349 |
| ProLong Gold DAPI Antifade | Cell Signaling Technologies | Cat # 8961 |
| Phalloidin CruzFluor-488 | Santa Cruz | Cat # sc-363791 |
SIGNIFICANCE.
CDK14 and other TAIRE cyclin dependent kinase family members (CDKs 15-18) are frequent drug off-targets, despite their biological functions remaining poorly understood. Here we describe the development of FMF-04-159-2, a tool compound that specifically targets CDK14 covalently. FMF-04-159-2 has a TAIRE-kinase biased reversible binding selectivity profile, with substantial off-target activity only detected against CDK2. In addition to validating LRP6 as a substrate of CDK14 in mitosis, unbiased investigation into the consequences of pan-TAIRE/CDK2 and covalent CDK14 inhibition through phospho-proteomics suggested that CDK14 plays a supporting role in cell cycle regulation. We characterize the cellular consequences of covalent CDK14 kinase inhibition, and describe characteristics of putative substrates of these kinases. When used in combination with FMF-04-159-R, or a compound washout control, FMF-04-159-2 is a valuable tool for further study of CDK14 kinase function.
Highlights.
A covalent, cell permeable inhibitor of CDK14 was developed.
Methods for assessing cellular engagement of TAIRE-family kinases are described.
Kinase motif analysis and phosphoproteomics identified putative CDK14 substrates.
ACKNOWLEDGEMENTS
The authors would like to acknowledge Eric Wang and Milka Kostic for helpful comments on the manuscript. The authors would also like to acknowledge Matthew Robers and the scientists at Promega, for helpful discussion and protocol sharing related to the NanoBRET cellular target engagement assays.
This work was supported by R35CA197588, P50-GM107618, U24-DK116204 R21CA188881, R01CA219850, the DFCI Strategic Research Initiative and the Chleck Foundation.
Abbreviations Used
- DIEA
diisopropylethyamine
- XPhos
2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl
- Dba
dibenzylideneacetone
- DCM
dichloromethane
- DMF
dimethylformamide
Footnotes
DECLARATION OF INTERESTS
L.C.C. is a founder and member of the scientific advisory board (SAB) and holds equity in Agios Pharmaceuticals and Petra Pharmaceuticals. L.C.C. is also a member of the BOD of Agios and an observer on the board of directors (BOD) of Petra. Petra provides partial support for his laboratory. Both Agios and Petra are developing drugs for cancer therapies.
J.M. is a member of the SAB of Devices.
N.S.G. is a founder, SAB member and equity holder in Gatekeeper, Syros, Petra, C4, B2S, and Soltego. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Her2llc, Deerfield, and Sanofi.
N.S.G., F.M.F. and Z.M.D. are inventors on a patent application covering chemical matter in this publication owned by Dana–Farber Cancer Institute.
P. K. S. is a founder, SAB member and equity holder in Merrimack Pharmaceutical and Glencoe Software; he is on the Board of Directors of Applied Biomath and the SAB of RareCyte Inc. In the last five years the Sorger lab has received research funding from Novartis and Merck. P. K. S. declares that none of these relationships are directly or indirectly related to the content of this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askenazi M, Parikh JR, and Marto JA (2009). mzAPI: a new strategy for efficiently sharing mass spectrometry data. Nat Methods 6, 240–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avendaño C, and Menéndez JC (2008). Chapter 9 - Drugs That Inhibit Signalling Pathways for Tumor Cell Growth and Proliferation In Medicinal Chemistry of Anticancer Drugs (Amsterdam: Elsevier; ), pp. 251–305. [Google Scholar]
- Bosken CA, Farnung L, Hintermair C, Merzel Schachter M, Vogel-Bachmayr K, Blazek D, Anand K, Fisher RP, Eick D, and Geyer M (2014). The structure and substrate specificity of human Cdk12/Cyclin K. Nature communications 5, 3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikuad A, Koch P, Laufer SA, and Knapp S (2018). The Cysteinome of Protein Kinases as a Target in Drug Development. Angewandte Chemie (International ed in English) 57, 4372–4385. [DOI] [PubMed] [Google Scholar]
- Davidson G, and Niehrs C (2010). Emerging links between CDKcell cycle regulators and Wnt signaling. Trends in cell biology 20, 453–460 [DOI] [PubMed] [Google Scholar]
- Davidson G, Shen J, Huang YL, Su Y, Karaulanov E, Bartscherer K, Hassler C, Stannek P, Boutros M, and Niehrs C (2009). Cell cycle control of wnt receptor activation. Developmental cell 77, 788–799. [DOI] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, and Zarrinkar PP (2011). Comprehensive analysis of kinase inhibitor selectivity. Nature biotechnology 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Fedorov O, Muller S, and Knapp S (2010). The (un)targeted cancer kinome. Nature chemical biology 6, 166–169. [DOI] [PubMed] [Google Scholar]
- Ferguson FM, and Gray NS (2018). Kinase inhibitors: the road ahead. Nature reviews Drug discovery 77, 353–377. [DOI] [PubMed] [Google Scholar]
- Ficarro SB, Alexander WM, and Marto JA (2017). mzStudio: A Dynamic Digital Canvas for User-Driven Interrogation of Mass Spectrometry Data. Proteomes 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficarro SB, Browne CM, Card JD, Alexander WM, Zhang T, Park E, McNally R, Dhe-Paganon S, Seo HS, Lamberto I, et al. (2016). Leveraging Gas-Phase Fragmentation Pathways for Improved Identification and Selective Detection of Targets Modified by Covalent Probes. Anal Chem 88, 12248–12254. [DOI] [PubMed] [Google Scholar]
- Fleuren ED, Zhang L, Wu J, and Daly RJ (2016). The kinome 'at large' in cancer. Nature reviews Cancer 16, 83–98. [DOI] [PubMed] [Google Scholar]
- Gujral TS, Peshkin L, and Kirschner MW (2014). Exploiting polypharmacology for drug target deconvolution. Proceedings of the National Academy of Sciences of the United States of America 111, 5048–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, and Turk BE (2004). A rapid method for determining protein kinase phosphorylation specificity. Nat Methods 7, 27–29. [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Gao Y, Yang T, Zhu X, and Chen J (2009). Cyclin Y, a novel membrane-associated cyclin, interacts with PFTK1. FEBS letters 583, 2171–2178. [DOI] [PubMed] [Google Scholar]
- Klaeger S, Gohlke B, Perrin J, Gupta V, Heinzlmeir S, Helm D, Qiao H, Bergamini G, Handa H, Savitski MM, et al. (2016). Chemical Proteomics Reveals Ferrochelatase as a Common Off-target of Kinase Inhibitors. ACS chemical biology 11, 1245–1254. [DOI] [PubMed] [Google Scholar]
- Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig PA, Reinecke M, Ruprecht B, Petzoldt S, Meng C, et al. (2017). The target landscape of clinical kinase drugs. Science (New York, NY) 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp S, Arruda P, Blagg J, Burley S, Drewry DH, Edwards A, Fabbro D, Gillespie P, Gray NS, Kuster B, et al. (2013). A public-private partnership to unlock the untargeted kinome. Nature chemical biology 9, 3–6. [DOI] [PubMed] [Google Scholar]
- Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, and Gray NS (2013). Developing irreversible inhibitors of the protein kinase cysteinome. Chemistry & biology 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg AS, and Weinberg RA (1998). Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Molecular and cellular biology 18, 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, and He X (2012). Frizzled and LRP5/6 Receptors for Wnt/β-Catenin Signaling. Cold Spring Harbor Perspectives in Biology 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M (2014). Cyclin-dependent kinases. Genome biology 15, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald ER 3rd, de Week A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K, et al. (2017). Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 170, 577–592 e510. [DOI] [PubMed] [Google Scholar]
- Mikolcevic P, Rainer J, and Geley S (2012). Orphan kinases turn eccentric: a new class of cyclin Y-activated, membrane-targeted CDKs. Cell cycle (Georgetown, Tex) 11, 3758–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novellasdemunt L, Antas P, and Li VS (2015). Targeting Wnt signaling in colorectal cancer. A Review in the Theme: Cell Signaling: Proteins, Pathways and Mechanisms. American journal of physiology Cell physiology 309, C511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang EY, Bai AH, To KF, Sy SM, Wong NL, Lai PB, Squire JA, and Wong N (2007). Identification of PFTAIRE protein kinase 1, a novel cell division cycle-2 related gene, in the motile phenotype of hepatocellular carcinoma cells. Hepatology (Baltimore, Md) 46, 436–445. [DOI] [PubMed] [Google Scholar]
- Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, et al. (2011). In situ kinase profiling reveals functionally relevant properties of native kinases. Chemistry & biology 18, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, O'Connell JD, Everley RA, O'Brien J, Gygi MA, and Gygi SP (2016a). Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J Proteomics 148, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, O'Connell JD, and Gygi SP (2016b). A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J Am Soc Mass Spectrom 27, 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic acids research 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, and Mann M (2003). Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75, 663–670. [DOI] [PubMed] [Google Scholar]
- Rodgers G, Austin C, Anderson J, Pawlyk A, Colvis C, Margolis R, and Baker J (2018). Glimmers in illuminating the druggable genome. Nature reviews Drug discovery 17, 301–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu F, Lv S, Qin Y, Ma X, Wang X, Peng X, Luo Y, Xu BE, Sun X, and Wu J (2007). Functional characterization of human PFTK1 as a cyclin-dependent kinase. Proceedings of the National Academy of Sciences of the United States of America 104, 9248–9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urich R, Grimaldi R, Luksch T, Frearson JA, Brenk R, and Wyatt PG (2014). The design and synthesis of potent and selective inhibitors of Trypanosoma brucei glycogen synthase kinase 3 for the treatment of human african trypanosomiasis. Journal of medicinal chemistry 57, 7536–7549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Jia Y, Fei C, Song X, and Li L (2016). Activation/Proliferation-associated Protein 2 (Caprin-2) Positively Regulates CDK14/Cyclin Y-mediated Lipoprotein Receptor-related Protein 5 and 6 (LRP5/6) Constitutive Phosphorylation. The Journal of biological chemistry 291, 26427–26434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Nielsen TE, and Clausen MH (2015). FDA-approved small-molecule kinase inhibitors. Trends in pharmacological sciences 36, 422–439. [DOI] [PubMed] [Google Scholar]
- Wyatt PG, Woodhead AJ, Berdini V, Boulstridge JA, Carr MG, Cross DM, Davis DJ, Devine LA, Early TR, Feltell RE, et al. (2008). Identification of N-(4-piperidinyl)-4-(2,6-dichlorobenzoylamino)-1 H-pyrazole-3-carboxamide (AT7519), a novel cyclin dependent kinase inhibitor using fragment-based X-ray crystallography and structure based drug design. Journal of medicinal chemistry 51, 4986–4999. [DOI] [PubMed] [Google Scholar]
- Yanagi T, Tachikawa K, Wilkie-Grantham R, Hishiki A, Nagai K, Toyonaga E, Chivukula P, and Matsuzawa S (2016). Lipid Nanoparticle-mediated siRNA Transfer Against PCTAIRE1/PCTK1/Cdk16 Inhibits In Vivo Cancer Growth. Molecular therapy Nucleic acids 5, e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Zhu J, Huang H, Yang Q, Cai J, Wang Q, Zhu J, Shao M, Xiao J, Cao J, et al. (2015). PFTK1 Promotes Gastric Cancer Progression by Regulating Proliferation, Migration and Invasion. PloS one 10, e0140451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim J, et al. (2012). Discovery of potent and selective covalent inhibitors of JNK. Chemistry & biology 19, 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Kwiatkowski N, Olson CM, Dixon-Clarke SE, Abraham BJ, Greifenberg AK, Ficarro SB, Elkins JM, Liang Y, Hannett NM, et al. (2016a). Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nature chemical biology 12, 876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Liu R, Tang C, Xi Q, Lu S, Chen W, Zhu L, Cheng J, Chen Y, Wang W, et al. (2016b). PFTK1 regulates cell proliferation, migration and invasion in epithelial ovarian cancer. International journal of biological macromolecules 85, 405–416. [DOI] [PubMed] [Google Scholar]
- Zhang Z, and Marshall AG (1998). A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J Am Soc Mass Spectrom 9, 225–233. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Rideout WM 3rd, Bressel A, Yalavarthi S, Zi T, Potz D, Farlow S, Brodeur J, Monti A, Reddipalli S, et al. (2014). Spontaneous genomic alterations in a chimeric model of colorectal cancer enable metastasis and guide effective combinatorial therapy. PloS one 9, e105886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Liu C, Liu F, Wang Y, and Zhu M (2016). Knockdown of PFTAIRE Protein Kinase 1 (PFTK1) Inhibits Proliferation, Invasion, and EMT in Colon Cancer Cells. Oncology research 24, 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Complete KiNativ profiling data for AT7519, FMF-03-198-2 and FMF-04-159-2. Related to Figure 2 and Figure 3. All experiments were performed at 1 μM for 4 h in HCT116 cells. Repeated kinase names indicate different sites of modification identified.
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD012692 (Perez-Riverol et al., 2019).