

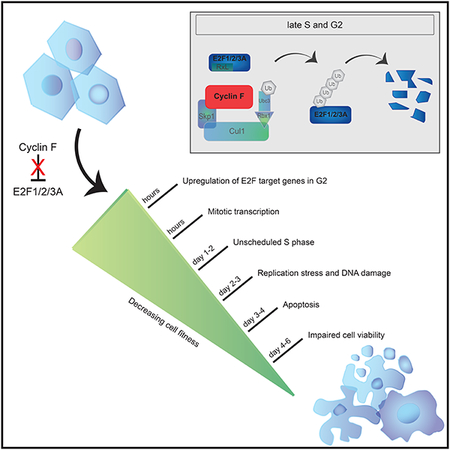
Summary
E2F1, E2F2 and E2F3A, the three activators of the E2F family of transcription factors, are key regulators of the G1/S transition, promoting transcription of hundreds of genes critical for cell cycle progression. We found that, during late S and in G2, the degradation of all three activator E2Fs is controlled by cyclin F, the substrate receptor of one of 69 human SCF ubiquitin ligase complexes. E2F1, E2F2 and E2F3A interact with the cyclin-box of cyclin F via their conserved N-terminal cyclin-binding motifs. In the short term, E2F mutants unable to bind cyclin F remain stable throughout the cell cycle, induce unscheduled transcription in G2 and mitosis, and promote faster entry into the next S-phase. However, in the long term, they impair cell fitness. We propose that, by restricting E2F activity to the S-phase, cyclin F controls one of the main and most critical transcriptional engines of the cell cycle.
Graphical Abstract
eTOC blurb
Clijsters et al. elucidate how Cyclin F controls a major transcriptional cell cycle engine. The authors show that activating E2Fs are targeted for proteasomal degradation in late S and G2, resetting the transcriptional program and preventing unscheduled entry into the next S-phase. Long-term stabilization of activator E2Fs impairs cell fitness.
Introduction
The E2F family of transcription factors regulates progression through the cell division cycle by controlling the expression of hundreds of target genes. In mammals, eight E2F genes encode three canonical activators (E2F1, E2F2 and E2F3A), four canonical repressors (E2F3B, E2F4, E2F5 and E2F6) and two atypical repressors (E2F7 and E2F8) (Bertoli et al., 2013). All E2F proteins have DNA binding domains that bind the sequence 5’-TTT[CG][CG]CGC-3’ in promoters of genes involved in cell cycle progression, DNA replication, DNA repair, apoptosis, and differentiation (Ren et al., 2002; Takahashi et al., 2000). Canonical E2Fs contain a hetero-dimerization domain for binding to either DP1 or DP2, two proteins that enhance DNA-binding, and a C-terminal transactivation domain (TA) that includes the binding domain for pocket-proteins (Bandara et al., 1993; Flemington et al., 1993; Helin et al., 1993). The TA is necessary to activate transcription after release of the pocket-proteins, RB1 (Retinoblastoma protein 1), p107 (also named Retinoblastoma-like protein 1 or RBL1), and p130 (RBL2) (Bagchi et al., 1991; Hiebert et al., 1992). The RB1-E2F pathway is often found deregulated in cancers (Dick and Rubin, 2013), emphasizing the importance of its strict regulation.
Different regulatory mechanisms converge to allow E2F transcription activity to peak at the G1/S transition. In activator E2Fs, binding to pocket-proteins masks the TA, resulting in target gene silencing. When cells commit to enter the cell cycle in response to mitogens and nutrient availability, cyclin-dependent-kinases (CDKs) phosphorylate pocket-proteins causing their release from activator E2Fs and, consequently, promoting a transcriptional program that contributes to progression through the S-phase (Bertoli et al., 2013; Guardavaccaro and Pagano, 2006; Van Den Heuvel and Dyson, 2008). At the end of the S-phase, E2F-mediated transcription is turned off through negative feedback loops and the subsequent activation of atypical repressors (Li et al., 2008a; Westendorp et al., 2012). Two major components of the negative feedback, cyclin A2 and CDK2 (the products of two E2F target genes), form an active kinase complex that phosphorylates E2F1 and inhibits its transcription activity (Dynlacht et al., 1994; Krek et al., 1994; Mudryj et al., 1991; Pagano et al., 1992). Moreover, the F-box protein SKP2 has been suggested to promote the degradation of E2F1 in S and G2 (Marti et al., 1999). E2F1 and E2F3A have also been reported to be degraded via the Anaphase Promoting Complex/Cyclosome (APC/C) ubiquitin ligase in either mitosis (Peart et al., 2010) or during quiescence and differentiation (Ping et al., 2012; Singh and Dagnino, 2017). How E2F2 is negatively regulated has remained elusive. Altogether, tight regulation results in the oscillating and alternating activity of activator and repressor E2Fs. However, the complex molecular mechanisms that control inactivation of the E2F-mediated transcription are not well understood.
Cyclin F (encoded by CCNF) is the founding member of the family of F-box proteins, the substrate receptors of the SCF (SKP1, CUL1, F-box protein) ubiquitin ligase complexes (Bai et al., 1994). In contrast to most cyclins, cyclin F does not bind CDKs. Besides a handful of cyclin F’s interactors, such as BMYB (Klein et al., 2015), p27 (Sharma et al., 2012), AKT (Choudhury et al., 2017), and CDC6 (Walter et al., 2016), various cyclin F’s substrates were identified in the last decade: the centrosomal protein CP110 and the spindle-associated protein NuSAP are degraded in G2 to control centrosome homeostasis and mitotic fidelity (D’Angiolella et al., 2010; Emanuele et al., 2011). RRM2 is degraded in G2 to control the availability of dNTPs during DNA replication and repair (D’Angiolella et al., 2012). The exonuclease Exo1 is degraded in response to replication stress (Elia et al., 2015). Lastly, the stem-loop binding protein SLBP is degraded in G2 to control histone H2A.X levels after genotoxic stress (Dankert et al., 2016). Our current investigation elucidates a cyclin F-dependent mechanism limiting E2F-mediated gene transcription to the S-phase and provides a model explaining how the oscillation of E2F activity is controlled to ensure cell fitness.
Results
Cyclin F interacts with and promotes the ubiquitylation of E2F1, E2F2, and E2F3A
As a first step to understand how the transcriptional activity of E2F is turned off after it peaks in S-phase, we examined the protein levels of activating E2Fs as cells progress through the cell cycle. Therefore, we immunoblotted lysates from synchronized RPE1-hTERT cells that were collected at different times after a thymidine block. Levels of E2F1, E2F2 and E2F3A (from now on, E2F1/2/3A, unless specified) decreased ~4–6 hours after release from the thymidine block, when most cells were in S and G2, as judged by the phosphorylation of Histone H3 and flow cytometry analysis (Fig. 1A and Fig. S1A–B). To determine whether E2F1/2/3A protein levels are controlled by degradation through the ubiquitin-proteasome-system, RPE1-hTERT cells were synchronized in G2 and treated with either the proteasome inhibitor MG-132 or the neddylation inhibitor MLN-4924. Compared to untreated control cells, levels of E2F1/2/3A increased in both cases (Fig. 1B). In contrast, the levels of the E2F dimerization partner DP1 remained unchanged. These experiments show that all three activator E2Fs are degraded in late S and during G2 in a proteasome- and neddylation-dependent manner.
Figure 1. E2F1, E2F2, and E2F3A bind to and are ubiquitylated by cyclin F.

A. RPE-1 cells were synchronized in G1/S by a thymidine block and then released in fresh medium for the indicated hours. Cells were collected and processed for FACS analysis (Fig. S1A), and Western blot. The graph on the right summarizes the results of the FACS analysis. TR, thymidine block and release.
B. RPE-1 cells were synchronized in G2 by a thymidine block followed by a release in fresh medium for 8 hours, and treated with either MG-132 or MLN-4924 for the indicated hours. Cells were then collected, lysed and immunoblotted.
C. HEK-293T cells were transfected with an empty vector (EV) or the indicated STREP-tagged F-box protein constructs (FBPs). Whole cell extracts (WCEs) were subjected to affinity-purification (AP) with Streptavidin resin and immunoblotted.
D. HEK-293T cells were transfected with an EV or constructs expressing FLAG-tagged members of the E2F transcription factor family and their hetero-dimerization partners. WCEs were subjected to immune-purification (IP) with an anti-FLAG resin and immunoblotted.
E. The experiment was performed as in (C), except that HEK-293T cells were transfected with the indicated constructs.
F. HEK-293T cells were co-transfected with FLAG-tagged activator E2Fs, STREP-tagged cyclin F, and either HA-tagged Ubiquitin or Ubiquitin(K48R) mutant. WCEs were subjected to IP with an anti-FLAG resin under denaturing conditions and immunoblotted.
Next, we aimed at identifying the ubiquitin ligase(s) that targets E2F1/2/3A for proteasomal degradation. The accumulation of E2F1/2/3A upon treatment with MLN-4924 suggested that the three proteins are degraded via a cullin-ring ubiquitin ligase (CRL) complex, possibly one of 69 SCF complexes, since SCFs (also known as CRL1s) are often involved in cell cycle control (Skaar et al., 2013; Zheng et al., 2016). To identify the CRL’s substrate receptor that interacts with E2F3A, we screened a panel of human F-box proteins. Nine F-box proteins were expressed in HEKC293T cells (with the neddylation inhibitor MLN-4924 added for 2 hours prior to harvesting) and immunoprecipitated to evaluate their interaction with E2F3A. We found that the only F-box protein able to coimmunoprecipitate endogenous E2F3A was cyclin F (Fig. 1C). We then asked which E2F proteins interact with endogenous cyclin F. All nine E2F family members and their dimerization partners were expressed in HEK-293T cells and immunoprecipitated to evaluate their interaction with cyclin F. We found that all three activating E2Fs, but not the other family members, DP1, and DP2, were able to coimmunoprecipitate endogenous cyclin F, although to a different extent (Fig. 1D). Next, activator E2Fs and E2F3B were expressed in HEK-293T cells and immunoprecipitated to evaluate their interaction with various CRL’s substrate receptors. We found that E2F1/2/3A, but not E2F3B coimmunoprecipitated endogenous cyclin F, but not other F-box proteins (Fig. 1E). In contrast to a previous report indicating binding of SKP2 with E2F1 (Marti et al., 1999), but in agreement with the observation that deletion of the SKP2 binding site in E2F1 does not stabilize this protein (Koseoglu et al., 2010), we did not observe any interaction between SKP2 and E2F1. Finally, in contrast to Drosophila E2f1 (the only activator E2f present in fly), which is degraded through to its binding to Pcna and Cdt2, we did not observe any interaction between E2F1/2/3A and CDT2, the substrate receptor of one of ~40 mammalian CRL4 complexes. This is not surprising since mammalian E2F1/2/3A, contrary to fly E2f1, do not contain a PCNA interacting domain. The interaction between cyclin F and E2F1/2/3A was additionally confirmed at endogenous protein levels (Fig. S1C). Finally, expression of cyclin F together with wild type (WT) Ubiquitin, but not Ubiquitin(K48R), promoted the ubiquitylation of E2F1/2/3A (Fig. 1F). The ubiquitylation is F-box dependent, as cyclin F(LP35/36AA), a mutant in which the conserved Lys and Pro of the F-box domain were mutated to Ala, was unable to promote the ubiquitylation of E2F1/2/3A (Fig. S1D).
These results show that cyclin F physically interacts with E2F1/2/3A and promotes their ubiquitylation via a K48 linkage, suggesting that SCFcyclin F targets E2F1/2/3A for proteasomal degradation. This is in agreement with the fact that cyclin F protein levels peak in G2, correlating with the timing of E2F1/2/3A degradation (Fig. 1A).
The degradation of E2F1, E2F2, and E2F3A in late S and in G2 is mediated by cyclin F
To investigate the role of cyclin F in the degradation of E2F1/2/3A, we silenced cyclin F in U2OS cells by treating them for 24–48 hours with a previously validated siRNA oligo (D’Angiolella et al., 2010, 2012; Dankert et al., 2016). Depletion of cyclin F increased E2F1/2/3A protein levels similar to established cyclin F substrates (Fig. 2A and Fig. S2A). E2F1/2/3A mRNA levels also displayed some increase, possibly because they are themselves E2F target genes (Fig. S2B). Importantly, the increase in E2F1/2/3A protein levels was reversed by co-expressing an siRNA-insensitive version of cyclin F cDNA (Fig. S2C), demonstrating that the accumulation of E2F1/2/3A is not due to an off-target effect of cyclin F siRNA.
Figure 2. The degradation of E2F1/2/3A in late S and in G2 is mediated by cyclin F.

A. U2OS cells were transfected with either a non-targeting (N/T) si-RNA or CCNF si-RNA for the indicated hours. Cells were then collected and processed for FACS analysis (Fig. S2A), qRT-PCR (Fig. S2B) and Western blot. The asterisks indicate non-specific bands.
B. RPE-1 cells were transfected with either a N/T si-RNA or CCNF si-RNA, synchronized by a TR for the indicated hours. Cells were then collected and processed for FACS analysis (Fig. S2E) and Western blot.
C. Quantification of the densitometric intensity of four independent experiments, included that shown in (B) represented as percentage change compared to time 0 (mean ± SEM).
D. Live U2OS cells stably co-expressing Cherry-PCNA and Venus-E2F2 were imaged by fluorescence microscopy. Still images of control cells (upper panel) and cells treated with CCNF si-RNA (bottom panel) are shown at the indicated times.
E-G. Live U2OS cells stably co-expressing Cherry-PCNA and either Venus-E2F1 (E), Venus-E2F2 (F) or Venus-E2F3A (G) were imaged by fluorescence microscopy in three independent experiments. The intensity of fluorescence was plotted against the time from the disappearance of PCNA dots (time 0). The graphs show the average fluorescence (mean ± SEM) of individual cells after transfection with CCNF si-RNA (blue) compared to N/T si-RNA (red).
H. Live U2OS cells stably co-expressing Cherry-PCNA and Venus-E2F1 were imaged by fluorescence microscopy. Still images of control cells (upper panel) and cells treated with CCNF si-RNA (bottom panel) are shown at the indicated times.
I-J. Live U2OS cells stably co-expressing Cherry-PCNA and Venus-E2F1 (green), Venus-E2F2 (red) or Venus-E2F3A (blue) were imaged by fluorescence microscopy in three independent experiments. The intensity of fluorescence was plotted against the time from nuclear envelope breakdown (NEB) (time 0). The graphs show the average fluorescence (mean ± SEM) of all individual cells after transfection with N/T si-RNA (I) or CCNF si-RNA (J).
Next, to examine the timing of the cyclin F-mediated E2F1/2/3A degradation, we silenced cyclin F in RPE1-hTERT cells and U2OS cells synchronized by a thymidine block and release. Depletion of cyclin F inhibited the degradation of E2F1/2/3A in late S and in G2 (Fig. 2B–C and Fig. S2D) without affecting cell cycle progression, as indicated by the levels of phosphorylated Histone H3 and flow cytometry analysis (Fig. 2B and Fig. S2E).
To pinpoint when the degradation of activator E2Fs started in relation to the S and G2 phases, we imaged the levels of their fluorescent versions together with a fluorescent marker of the S-phase (i.e., Cherry-tagged PCNA). The end of the S-phase was defined as time 0, which is the moment in which discrete nuclear Cherry-PCNA foci (i.e., DNA replication factories) disappear and the PCNA staining becomes uniform. In control U2OS cells, Venus-E2F2 levels started to substantially decline before the end of S. However, in cells depleted of cyclin F, Venus-E2F2 remained stable in S and increased in G2, the latter was due to the combination of decreased degradation and sustained transcription though an exogenous promoter (Fig. 2D, quantified in 2F). Similar results were obtained when filming U2OS cells expressing either Venus-E2F1 (Fig. 2E) or Venus-E2F3A (Fig. 2G). During G2, when cyclin F is expressed, Venus-E2F1 levels were low, but increased around the start of nuclear envelope breakdown (NEB) [which corresponds to the time when cyclin F levels decrease (Mavrommati et al., 2018) (Fig. 1A)] and remained high during the next G1 (Fig. 2H–I). Upon cyclin F silencing, Venus-E2F1 fluorescent levels were already high in G2 and remained largely unchanged during mitosis and G1 (Fig. 2H and 2J). Similar results were obtained with Venus-E2F2 and Venus-E2F3A (Fig. 2I–J). These results suggest that, in cycling cells passing through G1, SCFcyclin F is the only ubiquitin ligase mediating the degradation of activating E2Fs, and that APC/C’s role in targeting E2F1 and E2F3A occurs not in G1, but in quiescent and differentiating cells, as previously suggested (Ping et al., 2012; Singh and Dagnino, 2017). Moreover, these experiments show that cyclin F’s role in promoting the degradation of activating E2F proteins is independent of their transcription since cyclin F affects also the stability of exogenous proteins expressed under a constitutive promoter.
We concluded that, in late S and G2, cyclin F targets E2F1/2/3A for proteolysis.
Conserved N-terminal CY motifs are required for E2F1/2/3A binding to cyclin F and their degradation in S and G2
We mapped the cyclin F binding motifs in E2F1/2/3A. We have previously shown that cyclin F substrates interact with cyclin F via their cyclin-binding (CY) motifs (i.e., RxL or RxI) that act as degradation motifs (degrons) (D’Angiolella et al., 2010, 2012; Dankert et al., 2016). E2F1, E2F2, and E2F3A have eight, three, and two putative CY motifs, respectively, which could function as degrons for cyclin F recognition (Fig. 3A). The E2F1(R90A/L92A), E2F2(R87A/L89A) and E2F3A(R126A/L128A) mutants, but not the other CY mutants, failed to coimmunoprecipitate endogenous cyclin F (Fig. 3B and Fig. S3A–C). Although unable to bind cyclin F, the three CY mutants (from now on, E2F-RL/AA mutants) were still able to coimmunoprecipitate the dimerization partner DP1 (Fig. S3D). We also confirmed that E2F1/2/3A bind to cyclin F in a canonical manner, which is through cyclin F’s hydrophobic patch in its cyclin-box. Indeed, cyclin F(ML309/313AA) and cyclin F(L313A), two cyclin-box mutants (D’Angiolella et al., 2010), failed to coimmunoprecipitate endogenous E2F1/2/3A (Fig. 3C). These results show that cyclin F and E2F1/2/3A interact via cyclin F’s cyclin-box and the conserved N-terminal CY motifs in activator E2Fs. Next, we validated the subcellular localization of the E2F-RL/AA mutants and found that all three localized to the nucleus like their WT counterparts (Fig. 3D). Significantly, co-expression of cyclin F promoted the in vivo ubiquitylation and degradation of WT E2F1/2/3A, but not the E2F-RL/AA mutants (Fig. 3E and Fig. S3E). The observed reduction in E2F1/2/3A protein levels upon cyclin F transfection was rescued by the addition of the proteasome inhibitor MG-132 (Fig. S3E), confirming that cyclin F promotes the proteasome-mediated proteolysis of E2F1/2/3A.
Figure 3. Conserved N-terminal CY motifs are required for the binding of E2F1/2/3A to cyclin F and their degradation in S and G2.

A. Schematic representation of activator E2Fs. Potential CY motifs are indicated. Functional CY motifs are indicated in orange.
B. The experiment was performed as in (1D), except that HEK-293T cells were transfected with the indicated constructs.
C. The experiment was performed as in (1C), except that HEK-293T cells were transfected with the indicated constructs.
D. RPE-1 cells were transduced with lentiviruses expressing FLAG-FLAG-STREP-STREP (FF-SS)-tagged WT activating E2Fs, or their respective E2F-RL/AA mutants under the control of a doxycycline-inducible promoter. Cells were treated with doxycycline and, 24 hours later, processed for immune-fluorescence microscopy.
E. HEK-293T cells were co-transfected with MYC-tagged Ubiquitin, HA-tagged cyclin F, and either FLAG-tagged WT activator E2Fs, or the E2F-RL/AA mutants. WCEs were subjected to IP with an anti-FLAG resin under denaturing conditions and immunoblotted.
F. RPE-1 cells stably expressing FF-SS-tagged WT E2F3A or E2F3A-RL/AA were synchronized in G1/S by a TR for the indicated hours. Cells were collected, lysed, and immunoblotted.
G-H-I. U2OS cells were transduced with lentiviruses expressing an EV or FF-SS-tagged WT E2F1 (G), E2F2 (H), E2F3A (I) or their respective E2F-RL/AA mutants under the control of a doxycycline-inducible promoter. Cells were synchronized in G1/S by a double-TR (DTR) for the indicated hours. Doxycycline was added during the second thymidine block and not re-added after release from the block. Cells were collected, lysed, and immunoblotted. (l.e., long exposure; s.e., short exposure).
J. Quantification of the densitometric intensity of three independent experiments, included that shown in (G-I), represented as percentage change compared to time 0 (mean ± SEM).
K. U2OS cells expressing doxycycline inducible E2F1 or E2F1-RL/AA were synchronized in G1/S by a TR in the presence of nocodazole and doxycycline for the last 16 hours. Mitotic cells (M) were collected by mitotic shake-off. Adherent cells were also collected (G2). A fraction of mitotic cells was released for 2 hours after nocodazole washout (G1). Cells were lysed and immunoblotted.
Next, we analyzed RPE1-hTERT cells stably expressing WT E2F3A or E2F3A-RL/AA under the control of a weak retroviral promoter, which were synchronized using a thymidine block and release. Both endogenous and exogenous WT E2F3A were degraded in late S and G2, whereas E2F3A-RL/AA was not (Fig. 3F), in agreement with this mutant’s inability to bind cyclin F. In a complementary experiment, we synchronized cells expressing either WT E2F1/2/3A or the E2F-RL/AA mutants under the control of a doxycycline inducible promoter, using a double-thymidine block in the presence of doxycycline added during the second thymidine treatment (to induce the expression of E2F proteins) and then washed out (to prevent further accumulation of E2Fs). Acute expression of E2F1/2/3A (either WT or mutants) did not significantly affect cell cycle progression as indicated by the dynamics of the phosphorylation of Histone H3 on Ser10. We observed that WT E2F1/2/3A were degraded in late S and G2, whereas the three E2F-RL/AA mutants were stable during the same time periods (Fig. 3G–J and Fig. S3F–K). Accordingly, fusion of amino acid 67–108 of E2F1 to GST made it unstable in late S and this effect depended on the integrity of the CY motif (Koseoglu et al., 2010).
Finally, we analyzed E2F1 (both WT and mutant) protein levels in U2OS cells synchronized in G2, pro-metaphase, and G1. We observed that the APC/C substrates cyclin B1 and Aurora A were degraded when cells exited mitosis and entered G1, while E2F1-RL/AA protein levels did not decrease (Fig. 3K), confirming what observed with exogenous E2F1/2/3A (Fig. 2H–J).
Altogether, these results support the hypothesis that cyclin F targets E2F1/2/3A for degradation in late S and in G2 by recognizing their conserved N-terminal CY motifs as degrons.
Stable E2F1/2/3A mutants deregulate the expression of E2F target genes
Given that E2F activity peaks at the G1/S transition and in S, we asked how the transcriptional landscape is affected by sustained expression of activator E2Fs in G2. Thus, we performed RNA sequencing (RNA-seq) using synchronized U2OS cells expressing either WT or E2F-RL/AA mutants. To identify acute transcriptional effects, we induced E2F expression with doxycycline during the second thymidine treatment of a double-thymidine block (Fig. 4A–B). Using this strategy, we were able to evaluate the specific effects of maintaining high levels of activator E2Fs for just a few (i.e., 3–6) hours in G2 cells (i.e., without extending their expression for several cell cycles). As a control, we sequenced samples from the same cell lines without treating them with doxycycline (Fig. 4B). Synchronization was confirmed by flow cytometry (Fig. S4A–C). In comparing doxycycline-induction versus no doxycycline control, RNA-seq analyses identified numerous differentially expressed genes for each E2F construct (Fig. 4C and Tables S1–3). We next performed expression analyses for genes differentially induced by E2F-RL/AA mutants versus WT E2Fs for each of the individual E2F proteins. Although many of the same genes were significantly induced by both WT E2Fs and E2F-RL/AA mutants, the magnitude of induction was often greater in cells expressing the matched mutant, particularly for E2F2 and E2F3A (Figure 4D).
Figure 4. Stable E2F1/2/3A mutants deregulate the expression of E2F target genes in G2 and M.

A-B. Experimental workflow: U2OS cells expressing either inducible activator WT E2Fs or E2F-RL/AA mutants were synchronized in G2 phase by a DTR for 9 hours. Doxycycline was added during the second thymidine block and re-added after release from the block. Cells were collected and processed for FACS analysis (Fig. S4A–C), RNA sequencing (C-F), qRT-PCR (G), and Western blot (H). n = 3 biologically independent experiments.
C. Venn diagrams of differentially expressed genes (doxycycline treated versus doxycycline untreated; adjusted p-value < 0.05, |log2(fold change)| > 1) for each E2F construct.
D. Expression heatmap of genes differentially induced in E2F-RL/AA versus WT E2F samples (adjusted p-value < 0.05, |log2(fold change)| > log2(1.25); up to 100 most significant genes per E2F gene analysis plotted). Expression values (variance-stabilized for each E2F analysis and, for E2F3A analysis, adjusted for variation-covariates, details in Methods) were Z-scaled per each experiment. Genes of particular interest are indicated. Triplicate samples appear in grouped columns.
E. Bar plots of gene set enrichment testing (CAMERA) results. Data are shown for the top 10 most significant (by adjusted p-value) gene sets differentially induced by the expression of E2F-RL/AA mutants relative to corresponding WT E2Fs expression in independent E2F1, E2F2, and E2F3A analyses. Comprehensive results are presented in Table S4.
F. Barcode plots depicting differential enrichment of an E2F target gene-set (Ren et al., 2002) (see Table S5) for each E2F analysis (E2F-RL/AA induction versus WT E2F induction). Color bar represents all genes ranked by differential expression statistic (F statistic, directional by fold change) for the indicated contrast. E2F target gene set members are indicated in black lines. Line plot displays sliding average of gene set enrichment. Corresponding CAMERA and pre-ranked GSEA p-values for E2F target gene set enrichment appear on each plot.
G. qRT-PCR of indicated transcripts.
H. Western blot of indicated proteins.
I. RPE-1 cells expressing inducible E2F2-RL/AA were synchronized in G2 by a TR for 9 hours. Doxycycline was added after release from the thymidine block. Cells were processed for RNA FISH and hybridized with RRM2-exon and RRM2-intron specific probe sets. Results are from a representative experiment out of two repeats. The scatter dot plot shows the exon intensity at individual loci (median ± interquartile range; Wilcoxon-Mann-Whitney test).
J-K. U2OS cells expressing inducible E2F-RL/AA mutants were synchronized in mitosis by a 16-hour treatment with nocodazole in the presence of doxycycline. Mitotic cells were collected by mitotic shake-off and processed for FACS analysis (Fig. S4G), qRT-PCR (J) and Western blot (K). Random hexamer-primed cDNAs were prepared from the isolated RNA and analyzed by qRT-PCR for the levels of the indicated transcripts, and the significance of differences in E2F targets calculated (mean ± SEM; Multiple t-tests without correction for multiple comparisons). Primary transcripts were detected using intron-exon-directed primers (Fig. S4D). (J) shows the relative expression of primary transcript levels as the ratio Target over GAPDH, corrected for PCR primer efficiency.
To further characterize the gene expression patterns differentially induced by E2F-RL/AA mutants relative to WT E2Fs, we performed gene set enrichment testing for select gene sets [(Hallmark and Canonical Pathways collections from the Molecular Signatures Database (Liberzon et al., 2011)] with the CAMERA tool (Wu and Smyth, 2012). The gene sets most significantly enriched by the induction of E2F-RL/AA mutants (as compared to WT E2Fs) are related to G1/S specific transcription, activation of the pre-replication complex, and the MCM pathway (Fig. 4E and Table S4). Surprisingly, we did not observe significant differential enrichment of apoptosis related gene sets. This is in contrast to the list of apoptotic genes that were reported to be induced by activator E2F expression (Müller et al., 2001). This might be explained by the relatively short time and the specific cell cycle phase (i.e., G2) during which the E2F proteins were induced by doxycycline in our study. We also directly evaluated the enrichment of a defined E2F target gene set [Table S5, as determined by (Ren et al., 2002)] and found that, upon doxycycline treatment, E2F-RL/AA mutants induced the E2F target genes set to a significantly (p-value <0.001) greater level than respective WT E2F controls (Fig. 4F). Similar results were observed in a complementary analysis using the pre-rankedGSEA tool (Subramanian et al., 2005) (Fig 4F).
To validate the RNA-seq results, we performed quantitative PCR (qPCR) and Western blot analyses of the same samples, determining relative gene expression and protein levels of a subset of E2F targets. In agreement with the RNA-seq data, E2F1, E2F2, E2F3A, CCNE1, CDC6, and FBXO5 mRNA levels increased after induction of all three E2F-RL/AA mutants (Fig. 4G). Accordingly, the protein levels of cyclin E, CDC6, CDT1, Geminin, Claspin, and RRM2 increased after induction of the three E2F-RL/AA mutants (Fig. 4H). Finally, to test whether the increased gene and protein expression is caused by increased transcriptional activity, we performed RNA fluorescence in situ hybridization (FISH). A transcriptional event at the RRM2 loci is indicated by the co-localization of exon and intron probes, together with chromatin. RPE1-hTERT cells expressing E2F2-RL/AA were hybridized with intron and exon probe sets for the RRM2 gene. The exon intensity was used as a measure for transcriptional activity at the RRM2 loci. We found that exon intensities increased after induction of E2F2-RL/AA (Fig. 4I), demonstrating increased transcription.
It has long been thought that during mitosis, genes are not transcribed. However, recent studies showed low-level transcription of a subset of genes (such as ATF3) during mitosis (Palozola et al., 2017). We asked whether the degradation of activator E2Fs, might assist in restricting mitotic transcription. So, we examined the transcription of E2F target genes using intron-exon-directed primers to detect nascent transcripts (i.e., before splicing). The primers were confirmed to detect primary transcripts, as treatment with the RNA pol II inhibitor Triptolide decreased their signals (Fig. S4D). Using these primers, we observed that the transcription of E2F1/2/3A target genes, but not ATF3, is reduced in mitotic U2OS cells when compared to asynchronous cells (Fig. S4E–F). We found that expression of all three E2F-RL/AA mutants enhanced the transcription of E2F target genes during mitosis (Fig. 4J and Fig. S4G). The enrichment of mitotic cells (~85%) was verified by staining for the mitotic marker MPM2 and flow cytometry (Fig. S4F–G) and transcriptional events in mitotic cells expressing E2F2-RL/AA were demonstrated by RNA FISH (not shown). Finally, the increased mitotic transcription was reflected at the protein level (Fig. 4K).
Altogether, these results suggest that the degradation of E2F1/2/3A in late S and in G2 contributes to turning off the transcription of E2F target genes.
Stable E2F1/2/3A mutants deregulate cell cycle progression and DNA replication
Because of the nature of the identified deregulated gene-sets and the critical role of the E2F family in controlling cell proliferation, we hypothesized that E2F-RL/AA stable mutants may affect cell cycle progression. To test this, we analyzed cell cycle phase distributions of U2OS cells within the first 24 hours of doxycycline treatment. We observed an increase in the subpopulation of cells in S-phase and a parallel decrease of G1 cells upon the induction of E2F-RL/AA mutants and, to a minor extent, of WT E2Fs (Fig. 5A–C and Fig. S5A–C). These results were confirmed in RPE1-hTERT cells (Fig. S5D). The changes in cell cycle phase distribution depended on total levels of the activator E2Fs, because increasing further the expression of WT E2Fs with a higher concentration of doxycycline caused a cell cycle profile identical to that caused by the induction of E2F-RL/AA mutants (Fig. 5D and S5E–F). These cell cycle alterations could be caused by enhanced DNA synthesis, DNA replication stress (which may cause cells spending more time in S-phase), or both. DNA synthesis requires the assembly of the pre-replication complexes (pre-RCs) at origins of replication during late M and early G1. This process involves the loading of the replicative helicase MCM2–7 onto chromatin by the licensing proteins CDC6 and CDT1. At the G1/S transition, pre-RCs are converted into pre-initiation complexes, wherein the MCM helicase is activated by S-phase kinases (CDKs and DDK), resulting in DNA unwinding, the recruitment of replisome proteins and polymerases, and initiation of DNA synthesis (Fragkos et al., 2015). Interestingly, upon induction of E2Fs, we identified multiple differentially regulated gene-sets related to the pre-RC formation and activation (i.e., MCM pathway, Activation of the pre-RC, Unwinding of DNA, and Association of licensing factors with the pre-RC). Therefore, we examined the levels of MCM3 (as a marker for the MCM2–7 complex) loaded into DNA during G1, as a measure for replication licensing. To this end, we use a flowcytometry assay that involves pre-extraction (to remove soluble MCM), fixation of chromatin-bound proteins, and immunofluorescence with an anti-MCM3 antibody, together with the evaluation of the DNA content (using DAPI) and DNA synthesis (using EdU) to measure cell cycle phases (Matson et al., 2017). U2OS cells were either left untreated or treated with doxycycline for 24 hours (to induce the expression of E2Fs) and stained for DNA, EdU, and DNA-loaded MCM3. Cells expressing E2F-RL/AA mutants showed increased levels of DNA-loaded MCM in G1, as observed by the MCM3 signal in G1 cells (i.e., MCM3 positive, EdU negative, and with a 2n DNA content) that were colored in blue in Fig. 5E and Fig. S5G and quantified in Fig. 5F. The unidirectional nature of MCM loading would predict a broad distribution of DNA-loaded MCM in G1 (i.e., as seen in untreated control cells) (Matson et al., 2017). In contrast, G1 cells expressing stable E2F-RL/AA mutants showed higher levels of DNA-loaded MCM (Fig. 5F). This indicates that the stable E2F-RL/AA mutants, and to a lesser extent WT E2F1 and E2F2, enhance replication licensing, possibly, as a result of a faster MCM loading, in line with the higher levels of the licensing factors CDC6 and CDT1 (Fig. 4H and 4K). We further suggested that stable E2F2-RL/AA enhances replication licensing in G1 using chromatin-fractionation experiments. U2OS cells were synchronized in early G1 phase (releasing them for 1.5 or 3 hours from a mitotic block) and chromatin-bound MCM2 and MCM4 protein levels were evaluated in the chromatin-fraction by western blotting. In agreement with the flowcytometry assay, G1 cells expressing the stable E2F2-RL/AA mutant displayed enhanced replication licensing, as observed by the increased levels of MCM2 and MCM4 in the chromatin-fraction, compared to cells expressing either WT E2F2 or an empty vector (Fig. 5G).
Figure 5. Stable E2F1/2/3A mutants deregulate cell cycle progression.

A-C. U2OS cells expressing inducible activator E2Fs or corresponding E2F-RL/AA mutants were treated with or without doxycycline and, 24 hours later, pulsed with BrdU and processed for Western blot (top) and FACS analysis (bottom). BrdU incorporation and propidium iodide (PI) staining were analyzed using flow cytometry. The bar graph shows the percentage of cells in the different cell cycle phases from five independent experiments (mean ± SEM; Ordinary one-way ANOVA and Holm-Sidak’s multiple comparisons test, on S-phase population).
D. The experiment was performed as in (C) using the indicated amounts of doxycycline.
E. U2OS cells expressing inducible E2F3A or E2F3A-RL/AA were treated with doxycycline and, two days later, pulsed with EdU and processed for chromatin flow cytometry. EdU incorporation, DAPI-, and DNA-loaded MCM3-staining were analyzed using flow cytometry. Cells with DNA-bound MCM3 in G1 are indicated in blue.
F. Histograms of G1 cells that contain DNA-loaded MCM represent the quantification of four independent experiments.
G. U2OS cells expressing EV, inducible E2F2, or inducible E2F2-RL/AA were synchronized as in (3K). Mitotic cells were harvested by mitotic shake-off, released from the block, and replated in fresh medium without doxycycline. At the indicated time points, cells were collected, and processed for chromatin-fractionation and Western blot.
H. U2OS cells expressing EV, inducible E2F3A, or inducible E2F3A-RL/AA were synchronized as in (3K), except that BrdU was added 2 hours after replating. At the indicated time points, cells were collected, and processed for Western blot (Fig. S5I) and FACS analysis to measure BrdU incorporation and PI staining. The graphs show the percentage of cells entering S phase as observed by BrdU positivity from three independent experiments (mean ± SEM).
Next, to examine replication initiation, we determined the length of the G1 phase by measuring the time necessary for cells to go from mitosis to the start of DNA synthesis. We had observed that a decreased degradation of E2F-RL/AA mutants during G2 corresponds to a higher level of activator E2Fs in the next G1 (Fig. 3K). Likely because of this, U2OS cells expressing E2F-RL/AA mutants entered S-phase sooner than cells expressing WT E2Fs or an empty vector (Fig. 5H and S5H–I).
Stable E2F1/2/3A mutants induce DNA damage and apoptosis differentially
Cells that express E2F-RL/AA mutants for just 24 hours display a higher percentage of their populations in S-phase, enhanced replication licensing, and a faster G1/S transition. Likely because of this, two days after expression of E2F-RL/AA mutants, U2OS cells started to display signs of replication stress, such as enhanced phosphorylation of CHK1 and RPA2 (Fig. S6A), as well as DNA damage, as shown by an increased tail moment using a neutral comet assay (Fig. 6A). When we counted the number of U2OS cells for up to six days after induction of E2F proteins, we observed that cells expressing E2F1-RL/AA and E2F2-RL/AA mutants proliferated less than cells expressing WT counterparts or an empty vector (Fig. 6B–D). This decrease in cell number was likely caused by an induction of apoptosis, as indicated by the appearance of the cleaved form of PARP1 (Fig. 6E) and an increase in Annexin V-positive cells (Fig. 6F). U2OS cells expressing the E2F3A-RL/AA mutant did not undergo apoptosis and did not proliferate significantly less than cells expressing WT E2F3A or an empty vector. This could be, at least in part, attributed to the fact that, whereas E2F1-RL/AA and E2F2-RL/AA promoted the expression of the pro-apoptotic genes TP73 and NOXA, E2F3A-RL/AA did not (Fig. 6G–H).
Figure 6. Prolonged expression of stable E2F1/2/3A mutants impairs cell fitness.

A. U2OS cells expressing inducible E2F-RL/AA mutants were treated with doxycycline for two days, collected, and processed for a neutral comet assay. Top panels, representative images of comets. The box plot (bottom) shows the quantification of tailmoment of a representative experiment out of three repeats (median ± interquartile range, and 5–95% whiskers; two-tailed Wilcoxon-Mann-Whitney test).
B-D. U2OS cells expressing EV, inducible activator E2Fs, or E2F-RL/AA mutants were treated with doxycycline for the indicated days, collected, and counted. Proliferation was plotted as the number of cells (mean ± SEM; n = 2 independent experiments with technical triplicates; One-way ANOVA with multiple comparisons to EV and Dunnett’s multiple comparisons test).
E. U2OS cells expressing inducible activator E2Fs, or E2F-RL/AA mutants were treated with doxycycline for three days, collected, and processed for Western blot.
F. U2OS cells expressing inducible activator E2Fs or E2F-RL/AA mutants were treated with doxycycline for four days, collected and processed for Annexin V/PI staining. The graph shows the percentage of apoptotic cells as observed by Annexin V (mean ± SEM; n = 3 independent experiments; Multiple t-tests without correction for multiple comparisons).
G-H. U2OS cells expressing inducible activator E2Fs or E2F-RL/AA mutants were treated with doxycycline for three days, collected and total RNA was isolated. PolyAAA-primed cDNAs were prepared from the RNA and analyzed by qRT-PCR for the levels of TP73 (G) and NOXA (H) transcripts. Relative expression of mRNA levels was indicated as the ratio Target over GAPDH, and corrected for PCR primer efficiency (mean ± SEM; representative out of two repeats; Multiple t-tests without correction for multiple comparisons).
Discussion
Here we reveal a major molecular mechanism required for irreversibly shutting off E2F activity by showing that SCFcyclin F targets all three activator E2Fs for degradation in late S- and in G2-phases of the cell division cycle. We propose a model wherein the cyclin F-mediated degradation of activating E2Fs confines their activity to early/mid S-phase and resets the transcriptional cell cycle program to prevent unscheduled entry into the next S-phase and, ultimately, cell death.
During G2, cyclin F controls genome integrity by mediating the degradation of a variety of substrates (see Introduction). With this study, we add three substrates to this list, which further underscores cyclin F’s role in controlling cell proliferation and fitness, and extends it to controlling transcription. Specifically, failure to degrade E2F1, E2F2, or E2F3A in late S and in G2 maintains E2F activity. This, in turn, results in an imbalanced transcriptional landscape in G2 and M, and in an unscheduled DNA synthesis in the next cell cycle, which is accompanied by DNA replication stress and DNA damage. This is in line with the evidence that DNA replication stress can be caused by overexpression of oncoproteins (Jones et al., 2013; Macheret and Halazonetis, 2018) and that the control of the RB1-E2F pathway is important for safeguarding genomic stability. E2F1 overexpression has previously been shown to promote apoptosis (Bertoli et al., 2013). Previous studies evaluated the effects of E2F1 long-term expression. Conversely, we studied the immediate and short-term defects during G2, M, and the next G1/S upon stabilization of E2F1/2/3A. We propose that, short-term, these alterations cause an accelerated G1/S transition. The premature S-phase entry, combined with enhanced replication licensing, may cause various forms of DNA replication defects, such as conflicts with transcription and disruption of the origin firing schedule. In addition, continued presence of activator E2Fs during mitosis may affect the mitotic ‘bookmarking’ of specific loci and/or change the temporal order of transcriptional waves during mitotic exit. As a long-term effect of not degrading activator E2Fs, cells undergo apoptosis.
In yeast ~ 1.3% of all genes encode ubiquitin ligases (75 of ~5,300 genes), whereas in human the fraction is ~3% (~600 of 20K genes) (Li et al., 2008b). This indicates that proteolysis plays a more significant role in higher eukaryotes, likely because of its irreversible nature, which ensures a more robust inactivation than that achieved by post-translational modifications or protein-protein interactions. This is particularly important for cell cycle events that need to occur only once per cell cycle and require a high level of coordination. SBF is the Saccharomyces cerevisiae functional equivalent of the activator E2Fs. In late S-phase, CDK-dependent phosphorylation of SBF releases it from the promoters of target genes, and this event, together with the contribution of repressors, leads to the inactivation of transcription. Persistent activation of SBF has been associated to genome instability (Bertoli et al., 2013). However, unicellular organisms tend to adapt/mutate and move on. Our current study shows that inactivation of mammalian activator E2Fs based solely on phosphorylation or other post-transcriptional modification would be a risky business and that inhibition by degradation is a preferable way to prevent cell death and other deleterious consequences.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michele Pagano (Michele.Pagano@nyumc.org)
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell culture and synchronization.
The human embryonic kidney cell line HEK-293T (female origin) and telomerase-expressing non-transformed epithelial retina cell line RPE1-TERT (female origin) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS (HyClone) and antibiotics (Corning), at 37C. The human osteosarcoma cell line U-2OS (female origin) was cultured in McCoy 5A Medium (Gibco) containing 8% FBS and antibiotics, at 37C. Cell lines transduced with pTRIPZ plasmids were cultured with Tet-system approved FBS (Clontech). Cells were plated on 4-well glass-bottom chambered coverglass (Lab-Tek II), or on 10-cm Falcon dishes. Cells were plated 24–48 h before synchronization at G1/S by a double-thymidine protocol (2.5 mM final concentration; Sigma-Aldrich). G2 cells were collected 9 h after release from a double-thymidine block. Cells were synchronized in mitosis by a 24 h thymidine block and released from thymidine in the presence of nocodazole (830 nM final concentration; Sigma-Aldrich). Mitotic cells were collected by mitotic shake-off. G1 cells were obtained by releasing and replating cells arrested in mitosis for 2 h. Other drugs, used as indicated: proteasome inhibitor MG-132 (5 uM final concentration; Peptide International); neddylation inhibitor MLN-4924 (1 uM final concentration; Active Biochem), doxycycline (25 ng/mL for 24 hours, or as indicated, Sigma-Aldrich), RNA pol II inhibitor Triptolide (1 uM final concentration, Sigma-Aldrich).
METHOD DETAILS
Plasmids and siRNA.
F-box proteins were STREP-STREP-tagged and cloned into a modified pcDNA3 (Clontech), as previously described (Busino et al., 2003). Homo sapiens cDNAs were PCR amplified and cloned into a modified pcDNA3 vector containing 2xFLAG-tag and 2xSTREP-tag (pcDNA3-FF-SS), or HA (pcDNA3-HA), or 2xSTREP-tag (pcDNA3-SS), from the following sources: E2F1 (pcDNA3-HA-E2F1), E2F2 (pCMVHA-E2F2; #24226; Addgene), E2F3A (pCMV-Neo-Bam E2F3a; #37970; Addgene), E2F3B (pCMV-Neo-Bam E2F3b; #37975; Addgene), E2F4 (a kind gift from Brian Dynlacht, Perlmutter Cancer Center, NYU), E2F5 (pCMVHA-E2F5; #24213, Addgene), E2F6 (Clone ID 3535798, Dharmacon), E2F7 (Clone ID 9020354, Dharmacon), E2F8 (Clone ID 8992116, Dharmacon), DP1 (pCMV-Neo-Bam DP1; #37968; Addgene), DP2 (pOTB7-TFDP2; TCH1003; transomic). Mus musculus E2F3A and E2F3B cDNA was PCR amplified and cloned into pcDNA3-FF-SS. pCMV-HIS-HA-Ubiquitin and HIS-HA-Ubiquitin(K48R) were a gift from A. Iavarone, Columbia University.
The putative RxL mutants in E2F1, E2F2 and E2F3A were made by site-directed mutagenesis using the pcDNA3-FF-SS WT plasmids as templates. The cyclin-box mutants of cyclin F were made by site-directed mutagenesis using pcDNA3-FF-SS-cyclin F as template, and subcloned into pcDNA3-HA and pcDNA3-SS using gibson cloning (Gibson et al., 2009). The non-targetable version of cyclin F was made by site-directed mutagenesis using pcDNA3-FF-SS-cyclin F as template. The retroviral plasmids pLIB-NLS-Cherry-PCNA-P2A-Venus-E2F1/E2F2/E2F3A were constructed by cloning a linker containing the NLS sequence, a linker containing the P2A sequence, and PCR amplification and cloning of Venus sequence and Cherry-PCNA sequence into pLIB-N1 (a kind gift from Rob Wolthuis, VU Amsterdam, Netherlands). This resulted in the empty backbone pLIB-NLS-Cherry-PCNA-P2A-Venus-MCS. cDNAs of E2F1, E2F2 or E2F3A were PCR amplified and cloned into the empty backbone using gibson cloning (Gibson et al., 2009). The retroviral plasmids pBABE-FF-SS-E2F were constructed by PCR amplification of the cDNA from the corresponding pcDNA3-FF-SS plasmids and cloning into pBABE backbone. The lentiviral plasmids pTRIPZ-FF-SS-E2F were constructed by PCR amplification of the cDNA from the corresponding pcDNA3-FF-SS plasmids and cloning into pTRIPZ backbone (Dharmacon) using AgeI-XhoI restriction sites. For expression of E2F2 or E2F2-R87A,L89A, an NLS was introduced: pTRIPZ-FF-SS-NLS-E2F2. All plasmids were sequence verified. The siRNA oligos to target cyclin F (CCNF) were purchased from Dharmacon and verified. Cyclin F si-RNA #10: UAGCCUACCTCTACAATGA.
Cell transfection.
HEK-293T cells were transfected with expression plasmids using polyethylenimine (Polysciences) or Lipofectamine 3000 (Invitrogen). For retrovirus mediated gene transfer, HEK-293T cells were co-transfected with retroviral vectors (pLIB or pBABE), vesicular stomatitis virus G protein (VSV-G) and pCMV-Gag-Pol. For lentivirus mediated gene transfer, HEK-293T cells were co-transfected with lentiviral vector (pTRIPZ), VSV-G, and pCMV Delta R8.2. Retrovirus- or lentivirus-containing medium was, 48 h after transfection, collected and supplemented with 8 mg/mL polybrene (Sigma-Aldrich). U2OS or RPE-1 cells were infected for 24 h by replacing the culture medium with the virus-containing medium. Selection was carried out using puromycin. For transfection of siRNAs, we used Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions.
Immuno/Affinity-precipitation and Western blots.
One day after transfection, cells were harvested in PBS using a cell scraper and washed once with PBS by centrifugation. Cell pellets were lysed for 30 minutes on ice in IP-lysisbuffer: 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 2.5 mM MgCl2, 10% glycerol, 0.2% NP-40 substitute, supplemented with 1 mM DTT, protease inhibitor cocktail (Roche), phosphatase inhibitor cocktail (Sigma-Aldrich) and benzonase nuclease (Sigma-Aldrich). Cleared lysate was incubated with either Strep-Tactin Superflow beads (50%, IBA) or M2-FLAG beads (Sigma-Aldrich) for 2 hours while tumbling at 4C. Beads were washed once with IP-lysisbuffer, incubated with benzonase nuclease for 20 minutes, and washed twice with IP-lysisbuffer. For the detection of ubiquitin-conjugated proteins, cells were lysed for 10 minutes on 95C in denaturing lysisbuffer: 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 10% SDS. After cooling down, lysates were diluted 10 times using IP lysisbuffer (see above, and supplemented with NEM (Sigma)), and disrupted by short pulse (5–10s) sonication (Sonic Dismembrator 60, Fisher Scientific). For detection of endogenous protein interactions, cells were lysed in IP lysisbuffer (see above), pulse sonicated (see above), and pre-incubated with 4–5 ug antibody for 1 hour. Then, lysates where incubated for an additional 1 hour with magnetic dynabeads protein G (Invitrogen-Novex). Elution of the immunoprecipitated was carried out using NuPAGE LDS 4× Sample buffer (Fisher Scientific) supplemented with b-mercaptoethanol (Sigma-Aldrich) and boiling for 10 minutes at 95C. Protein was separated on NuPAGE 4–12% Bis-Tris Gels (Novex) and blotted on PVDF membranes (Millipore). Membranes were blocked in 4% milk powder (Bio-Rad). Primary antibody (listed in the Key Resources Table) incubation was performed overnight in 4% BSA (Sigma-Aldrich). For chemiluminescent detection of HRP activity from antibodies, we used SuperSignal Chemiluminescent Substrate (Fisher Scientific). Analysis was done using Image Quant LAS 4000 (GE).
Flow cytometry.
For analysis of DNA replication, cells were pulsed with BrdU (10 uM final concentration, Millipore Sigma) for 30 minutes, ethanol fixed, followed by 30 min HCl/Triton treatment, borate neutralization, and staining with anti-BrdU antibodies. For analysis by flow cytometry, cells were counterstained with propidium iodide (Sigma-Aldrich) diluted in RNase containing PBS. For analysis of DNA replication and DNA-bound MCM3, cells were pulsed with EdU (10 uM final concentration, Click Chemistry Tools) for 30 minutes, pre-extracted with CSK, followed by fixation in 4% PFA, and washing with 1%BSA (Roche) in PBS. EdU was detected with click-chemistry using 2mM CuSO4, 1 uM AF647 Picolyl Azide (Click Chemistry Tools), and 10 mM sodium ascorbate (Sigma). Cells were incubated with primary anti-MCM3 antibody in 0.1% NP40 (Sigma-Aldrich)C1%BSA-PBS. After secondary antibody-staining, cells were kept in 0.1% NP40 (Sigma-Aldrich)C1%BSA-PBS supplemented with DAPI (1 ug/mL, Molecular Probes) and RNAse A (100 ug/mL). For analysis of apoptosis, we used the Alexa Fluor 488 Annexin V/Dead cell Apoptosis Kit (Invitrogen) according to the manufacturer’s instructions. Measurement was done on the FACSCalibur cell analyzer using CellQuest Pro (BD Biosciences) software, or on the LSR-UV/HTS cell analyzer using FACSDiva (BD Biosciences). Annexin V/PI and BrdU positivity was analyzed using FlowJo software.
RNA sequencing.
RNA was isolated using the RNeasy Mini kit and on-column DNA digestion (Qiagen). Libraries were prepared with the TruSeq Stranded mRNA kit (Illumina). High throughput sequencing was performed on an Illumina HiSeq 4000 instrument in 150 nt paired-end configuration. The samples were sequenced at a median depth of 30 million reads per sample.
RNA-Seq read mapping and quantification.
Following quality assessment by fastQC (version 0.11.3), adaptor sequences were trimmed from the reads using cutadapt (version 4.0) (Martin, 2013). Trimmed reads were mapped using STAR (version 2.5.0b) (Dobin et al., 2012) to the human genome reference (version GRCh37) with Ensembl transcript annotation (version 75) and additional plasmid specific annotations. Read counts per gene were quantified with STAR (column 4 of the gene count output table, equivalent to htseq-count option -s reverse (Anders et al, 2015)). Genes were designated “expressed” for each E2F experiment (i.e., E2F1 experiment, E2F2 experiment, E2F3A experiment) if there were at least 5 read counts in at least 3 samples. Genes not satisfying these criteria were designated “not expressed” and removed from further analyses.
RNA-Seq analysis.
Differential gene expression analyses were conducted with EdgeR (version 3.20.9) (Chen et al., 2016; Robinson et al., 2010) in the R statistical framework. Independent analyses were performed for each E2F experiment (i.e., E2F1 experiment, E2F2 experiment, E2F3A experiment). For the E2F3A experiment, exploratory data analysis by principal component analysis revealed substantial variation across biological replicates; variation unrelated to experimental design was modeled with RUVSeq (function RUVs in version 1.12.0) (Davide Risso et al., 2014), the output of which was used to adjust downstream differential expression analysis (E2F3A analysis only). For each E2F experiment, read counts were TMM-normalized (using the calcNormFactors function in edgeR) and differential expression was assessed with edgeR using a linear model including factors for E2F mutation (wild-type, E2F-RL/AA) and doxycycline treatment (untreated, treated) (E2F3A model also included RUVSeq-determined covariates).
To identify genes differentially expressed upon induction of each E2F variant, doxycycline-treated samples were contrasted with corresponding doxycycline-untreated samples (e.g. E2F1 WT [doxycycline-treated] versus E2F1 WT [doxycycline-untreated], E2F2-RL/AA [doxycycline-treated] versus E2F2-RL/AA [doxycycline-untreated], etc.). Significant differential expression was defined as adjusted p-value < 0.05, |log2(fold-change)| > 1 (Fig. 4C). We next tested for differential induction of genes by E2F-RL/AA mutant (doxycycline-treated versus doxycycline-untreated) relative to corresponding WT E2F (doxycycline-treated versus doxycycline-untreated). Significant differential induction was defined as adjusted p-value < 0.05, |log2(fold-change)| > log2(1.25)). Genes identified as significant in any E2F experiment (up to 100 top genes ranked by adjusted p-value) were selected for plotting on heatmap in Figure 4D. Gene expression counts were log-transformed and variance stabilized by regularized log transformation (rlog function in DESeq2 (version 1.18.1) (Love et al., 2014) and z-scaled (per E2F experiment) for heatmap visualization. For E2F3A experiment only, sample variation in variance stabilized counts was corrected with the removeBatchEffect function (Limma version 3.34.9) (Ritchie et al., 2015) using the RUVSeq modeled covariates. Gene set enrichment testing was performed with CAMERA (Wu and Smyth, 2012) and the hallmark (H) and canonical pathway (C2-CP) gene set collections from the Molecular Signatures Database (Liberzon et al., 2011). Analyses were conducted with the same linear model as in differential gene expression analysis (above), testing for enrichment of gene sets differential induced by E2F-RL/AA (doxycycline-treated versus doxycycline-untreated) relative to corresponding WT E2F (doxycycline-treated versus doxycycline-untreated). All significantly enriched gene sets (adjusted p-value<0.01) per E2F contrast are reported in Table S4. To present the most significant differentially enriched gene sets in Figure 4E, we selected the top 10 gene sets (ranked by adjusted p-value) from each E2F experiment. In those cases where more than 50% of genes overlapped with an additional gene set, only the highest ranked (by sum of -log10 adjusted p-values for each E2F experiment) gene set is presented. For gene set enrichment testing of the E2F target gene-set (Ren et al., 2002) (Table S5), CAMERA analysis was performed as above. In addition, set enrichment was analyzed by pre-Ranked Gene Set Enrichment Analysis (preRankedGSEA) (version 6.0.11) (Subramanian et al., 2005) on genes ranked by differential expression statistic (F statistic, directional by fold change) for each E2F experiment. Barcode plots displaying enrichment profiles of the E2F target gene-set (Fig. 4F) were generated by modified barcodeplot function in Limma.
Unless otherwise noted, p values were corrected for multiple testing by the method of Benjamini and Hochberg (Benjamini and Hochberg, 1995).
Python and R scripts used for analysis are available upon request.
qRT-PCR.
PolyAAA-primed or random hexamer-primed cDNAs were prepared from the isolated RNA using RNA to cDNA EcoDry Premix (Clontech/Takara) according manufacturer’s instructions. Relative expression of transcripts, using primer sets as listed in Table S6, was quantified using qRT-PCR with absolute blue qPCR SYBR Green mix (Thermo Scientific) in a LightCycler 480 (Roche), or with PowerUp SYBR Green Master Mix (Applied Biosystems) in a QuantStudio 3 (Applied Biosystems).
Neutral comet assay.
The neutral comet assay was performed as indicated by the manufacturer’s instructions (ENZO Comet SCGE assay kit, #ADI-900–166). Briefly, U2OS cells were collected in PBS at ± 3*105 cell/mL concentration. Cells were diluted at 1:10 ratio in pre-warmed Comet LMAgarose and immediately applied on a pre-warmed comet slide (±75 uL/well), and kept on 4C for 20 minutes. Slides were incubated in Pre-chilled Lysis Solution supplemented with 1:10 DMSO, for 1 hour at 4C. Then, slides were incubated in pre-chilled 1× neutral electrophoresis buffer (1M Tris-Base, 3M Sodium acetate, pH=9.0), for 1 hour at 4C. Electrophoresis was performed using a Comet Assay ES tank (TREVIGEN), at 21 Volts for 1 hour at 4C. Comet DNA precipitation was performed by immersing the slides in DNA precipitation solution (1M NH4Ac in 95% ethanol) for 30 minutes at RT, and 70% ethanol for 30 minutes at RT, and drying at 42C. DNA was stained using SYBR Gold (Invitrogen, # S11494). Acquisition of fluorescence images was done on a DeltaVision Elite microscope system (Applied Precision). The tailmoment of at least 45 cells per condition was quantified using the OpenComet plugin (Gyori et al., 2014) in ImageJ (http://rsbweb.nih.gov/ij/).
Fluorescence time-lapse microscopy.
U2OS cells co-expressing Cherry-PCNA and Venus-E2F1 or Venus-E2F2 or E2F3A, and transfected with si-RNA were followed by fluorescence time-lapse microscopy as described (Clijsters and Wolthuis, 2014). Acquisition of fluorescence images started 24 h after transfection on a DeltaVision Elite microscope system (Applied Precision) in a heated culture chamber (5% CO2 at 37C).
Immunofluorescence.
RPE-1 cells expressing FLAG-FLAG-STREP-STREP-tagged E2F proteins (wild type or mutant) under a doxycycline inducible promoter were plated on 8-chamber glass-bottom culture slides (Falcon), and 24 h later treated with 50 ng/mL doxycycline for 24 h as indicated. Cells were immunostained with FLAG-antibodies and mounted in Prolong Gold Antifade Mountant with DAPI (Molecular Probes, Life Technology). Acquisition of fluorescence images was done on a DeltaVision Elite microscope system (Applied Precision).
Viability assay.
U2OS cells expressing FLAG-FLAG-STREP-STREP-tagged E2F proteins (inducible EV, WT E2F, or E2F-RL/AA mutant) under a doxycycline inducible promoter were counted, and 50.000 cells/well were plated in 12-well plates. Doxycycline (25 ng/mL) was added 24 h after plating. Cells were harvested and viable cells counted at the indicated time-points using Vi-Cell counter (Beckman Coulter).
RNA FISH.
RNA fluorescence in situ hybridization (FISH) was performed as described (Chou et al., 2013). The probe sets (RRM2_intron and RRM2_exon) contained up to 48 single-stranded DNA oligos (20 nucleotides) each labeled with one fluorophore (Quasar670 or Quasar570) (Stellaris, Biosearch Technologies). RPE-1 cells expressing FLAG-FLAG-STREP-STREP-tagged E2F2-RL/AA were plated on 12-mm circular microscope cover glass (Fisher Scientific). Cells were, 8 h after doxycycline induction, fixed in 4% paraformaldehyde in PBS for 10 min at room temperature (RT). The cells were then permeabilized with 0.5% TritonCX-100 in PBS for 10 min at RT. Cells were washed in 10% formamide in 2XSSC for 10 min before hybridization. To detect RNAs, 125 nM of labeled probes in 40 uL hybridization buffer (10% dextran sulfate, 10% formamide, 2 mg/mL BSA, 2XSSC, E.coli tRNA, and salmon sperm ssDNA) was used for each sample. Hybridization was carried out in humidified chambers for 16 hours at 37C. The samples were washed two times with 10% formamide in 2XSSC for 20 min, and one time with PBS for 20 min. DNA was stained with DAPI (1 ug/mL final concentration) by incubation for 5 min. Cells were washed with PBS and mounted in Prolong Gold Antifade Mountant (Molecular Probes, Life Technology). Acquisition of fluorescence images was done on a DeltaVision Elite microscope system (Applied Precision). For quantitative analysis of exon intensity at active transcription sites, CellProfiler software was used.
Image acquisition.
The DeltaVision Elite microscope system (Applied Precision) is equipped with a 40×/1.3 Oil Plan (fluorescence time-lapse) or 60×/1.3 Silicone Plan APO (immunofluorescence, RNA FISH) oil-immersion objective and the appropriate filter blocks. Images were taken using SoftWoRx imaging software with a CoolSnap HQ2 camera at 100-ms exposure times. For RNA FISH acquisition, serial optical sections were obtained 200-nm apart along the z-axis across a range of 2.4 um, processed using the SoftWoRx deconvolution algorithm, and projected into one picture using SoftWoRx software. Images from each data set were acquired on the same day using the same exposure times.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image analysis.
ImageJ (http://rsbweb.nih.gov/ij/) was used to measure the degradation of activator E2Fs by fluorescence time-lapse microscopy. Signal intensities were measured in a defined area with the cell, and normalized against the intensity at time-point t = 0, that is the end of S-phase, which is defined as the moment in which discrete nuclear Cherry-PCNA foci (which represent DNA replication factories) disappear (Clijsters and Wolthuis, 2014), or nuclear envelope breakdown (NEB), as indicated. The intensity of fluorescence was plotted against the time from the disappearance of PCNA dots, or the start of NEB. Background intensity was subtracted from each measurement. Exon intensity at active transcription sites was measured using a custom, automated CellProfiler pipeline (cellprofiler.org, version 3.0). Briefly, nuclei, introns, and exons were segmented using an intensity threshold of a maximum intensity projection of respectively the DAPI, CY5, and TRITC image stacks. Introns in the nucleus were defined as transcription sites. The integrated intensity of exons was measured at locations of transcription sites. Captured images were processed using Photoshop and Illustrator software (Adobe).
Statistics.
All immunoblot images are representative of at least two independent experiments, except where specified in the figure legends. Statistical details of experiments and the statistical tests that were used can be additionally found in the figure legends. P values were derived from unpaired two-tailed Wilcoxon-Mann-Whitney, unpaired two-tailed t-tests, or ordinary one-way ANOVA, with multiple comparisons as indicated, using Prism software (GraphPad). When unpaired, two-tailed t-test or ordinary one-way ANOVA was applied, homogeneity of variance was tested using Bartlett’s test, and normality was tested using Kolmogorov-Smirnov. In addition, when ordinary one-way ANOVA was applied, the mean of each dataset was compared to every other dataset, or otherwise, as indicated. Multiple comparisons were followed up using Holm-Sidak’s or Dunnett’s multiple comparisons test, or otherwise, as indicated. Summarized p-values indicate: ****, p < 0.0001; ***, p < 0.001; **, p < 0.01; *, p < 0.05; n.s., non-significant.
DATA AND SOFTWARE AVAILABILITY
RNA sequencing data has been deposited in the SRA database with SRA accession PRJNA499110. Raw, uncropped images of western blots have been deposited in the Mendeley database, http://dx.doi.org/10.17632/29y4rwj2hp.1.
Supplementary Material
Genes that were differentially induced in mutant versus WT E2F1 upon doxycyline treatment.
Genes that were differentially induced in mutant versus WT E2F2 upon doxycyline treatment.
Genes that were differentially induced in mutant versus WT E2F3A upon doxycyline treatment.
Gene sets variations for each E2F analysis, amongst the genes that were differentially induced in mutant versus WT E2F upon doxycycline treatment, as listed in Table S1–3.
As listed in Ren et al.
Highlights.
Cyclin F targets activating E2Fs for proteasomal degradation during late S and in G2
Cyclin F controls one of the main transcriptional engines of the cell cycle
Degradation of activator E2Fs resets the system and prevents premature S-phase entry
Long-term stabilization of activator E2Fs induces DNA damage and impairs cell fitness
Acknowledgements
We are grateful to R. Wolthuis, B. Dynlacht, B. King, T. Lionnet, and W.F. Marzluff for reagents and advice. We thank J. DeCaprio, B. Dynlacht, and N. Dyson for critically reading the manuscript. We thank NYU Langone’s scientific core facilities, the Genome Technology Center for performing library preparation and RNA sequencing, the Cytometry and Cell Sorting Laboratory for flowcytometry assistance (both are supported by grant P30CA016087 from the National Institutes of Health/National Cancer Institute), and the Microscopy Laboratory for assistance. This work was funded by grants from the National Institutes of Health to MP, and a Rubicon grant from the Netherlands Organisation for Scientific Research (NWO) to LC. MP is an Investigator with the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
M.P. is a member of the scientific advisory boards of CullGen Inc. and Kymera Therapeutics, and a consultant for BeyondSpring Pharmaceutical.
References
- Anders S, Pyl P, and Huber W (2015). HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi S, Weinmann R, and Raychaudhuri P (1991). The retinoblastoma protein copurifies with E2F-I, an E1A-regulated inhibitor of the transcription factor E2F. Cell 65, 1063–1072. [DOI] [PubMed] [Google Scholar]
- Bai C, Richman R, and Elledge SJ (1994). Human cyclin F. EMBO J. 13, 6087–6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandara LR, Buck VM, Zamanian M, Johnston LH, and La TN (1993). Functional synergy between DP-1 and E2F-1 in the cell cycle-regulating transcription factor DRTF1/E2F. Embo J 12, 4317–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A practical and powerful approach to multiple testing. J Royal Stat Soc 57, 289–300. [Google Scholar]
- Bertoli C, Skotheim JM, and De Bruin RAM (2013). Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 14, 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, Hershko A, Pagano M, and Draetta GF (2003). Degradation of Cdc25A by β-TrCP during S phase and in response to DNA damage. Nature 426, 87–91. [DOI] [PubMed] [Google Scholar]
- Chen Y, Lun ATL, and Smyth GK (2016). From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Research 5, 1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou Y. ying, Heaton NS, Gao Q, Palese P, Singer R, and Lionnet T (2013). Colocalization of Different Influenza Viral RNA Segments in the Cytoplasm before Viral Budding as Shown by Single-molecule Sensitivity FISH Analysis. PLoS Pathog 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury R, Bonacci T, Wang X, Truong A, Arceci A, Zhang Y, Mills CA, Kernan JL, Liu P, and Emanuele MJ (2017). The E3 Ubiquitin Ligase SCF(Cyclin F) Transmits AKT Signaling to the Cell-Cycle Machinery. Cell Rep. 20, 3212–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clijsters L, and Wolthuis R (2014). PIP-box-mediated degradation prohibits re-accumulation of Cdc6 during S phase. J. Cell Sci. 127, 1336–1345. [DOI] [PubMed] [Google Scholar]
- D’Angiolella V, Donato V, Vijayakumar S, Saraf A, Florens L, Washburn MP, Dynlacht B, and Pagano M (2010). SCF(CyclinF) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 466, 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, Saraf A, Florens L, Washburn MP, and Pagano M (2012). Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 149, 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angiolella V, Esencay M, and Pagano M (2013). A cyclin without cyclin-dependent kinases: Cyclin F controls genome stability through ubiquitin-mediated proteolysis. Trends Cell Biol. 23, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankert JF, Rona G, Clijsters L, Geter P, Skaar JR, Bermudez-Hernandez K, Sassani E, Fenyö D, Ueberheide B, Schneider R, et al. (2016). Cyclin F-Mediated Degradation of SLBP Limits H2A.X Accumulation and Apoptosis upon Genotoxic Stress in G2. Mol. Cell 64, 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso Davide, Ngai John, Speed Terence P, and Dudoit Sandrine (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol 32, 896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick FA, and Rubin SM (2013). Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 14, 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Zaleski C, Schlesinger F, Drenkow J, Chaisson M, Batut P, Jha S, and Gingeras TR (2012). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dynlacht BD, Lees JA, and Harlow E (1994). Differential regulation of E2F transactivation by cyclin/cdk2 complexes. Genes Dev. 130, 1772–1786. [DOI] [PubMed] [Google Scholar]
- Elia AEH, Boardman AP, Wang DC, Huttlin EL, Everley RA, Dephoure N, Zhou C, Koren I, Gygi SP, and Elledge SJ (2015). Quantitative Proteomic Atlas of Ubiquitination and Acetylation in the DNA Damage Response. Mol. Cell 59, 867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuele MJ, Elia AEH, Xu Q, Thoma CR, Izhar L, Leng Y, Guo A, Chen Y-N, Rush J, Hsu PW-C, et al. (2011). Global identification of modular cullin-RING ligase substrates. Cell 147, 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemington EK, Speck SH, and Kaelin WG (1993). E2F-1-mediated transactivation is inhibited by complex formation with the retinoblastoma susceptibility gene product. Proc. Natl. Acad. Sci 90, 6914–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkos M, Ganier O, Coulombe P, and Méchali M (2015). DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 16, 360–374. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Huntchison CA III, Smith HO, Hutchison CA III, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343. [DOI] [PubMed] [Google Scholar]
- Guardavaccaro D, and Pagano M (2006). Stabilizers and Destabilizers Controlling Cell Cycle Oscillators. Mol. Cell 22, 1–4. [DOI] [PubMed] [Google Scholar]
- Gyori BM, Venkatachalam G, THiagarajan PS, Hsu D, and Clement MV (2014). “OpenComet: An automated tool for comet assay image analysis. Redox Biology 2, 457–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helin K, Harlow E, and Fattaey A (1993). Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol. Cell. Biol 13, 6501–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Heuvel S, and Dyson NJ (2008). Conserved functions of the pRB and E2F families. Nat. Rev. Mol. Cell Biol. 9, 713–724. [DOI] [PubMed] [Google Scholar]
- Hiebert SW, Chellappan SP, Horowitz JM, and Nevins JR (1992). The interaction of pRb with E2F inhibits the transcriptional activity of E2F. Genes Dev. 6, 177–185. [DOI] [PubMed] [Google Scholar]
- Jones RM, Mortusewicz O, Afzal I, Lorvellec M, García P, Helleday T, and Petermann E (2013). Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 32, 3744–3753. [DOI] [PubMed] [Google Scholar]
- Klein DK, Hoffmann S, Ahlskog JK, O’Hanlon K, Quaas M, Larsen BD, Rolland B, Rösner HI, Walter D, Kousholt AN, et al. (2015). Cyclin F suppresses B-Myb activity to promote cell cycle checkpoint control. Nat. Commun 6, 5800. [DOI] [PubMed] [Google Scholar]
- Krek W, Ewen ME, Shirodkar S, Arany Z, Kaelin WG, and Livingston DM (1994). Negative regulation of the growth-promoting transcription factor E2F-1 by a stably bound cyclin A-dependent protein kinase. Cell 78, 161–172. [DOI] [PubMed] [Google Scholar]
- Koseoglu MM, Dong J, and Marzluff WF (2010). Coordinate regulation of histone mRNA metabolism and DNA replication. Cell Cycle 9, 3857–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti A, Wirbelauer C, Scheffner M, and Krek W (1999). Interaction between ubiquitin-protein ligase SCF(SKP2) and E2F-1 underlies the regulation of E2F-1 degradation. Nat. Cell Biol. 1, 14–19. [DOI] [PubMed] [Google Scholar]
- Li J, Ran C, Li E, Gordon F, Comstock G, Siddiqui H, Cleghorn W, Chen HZ, Kornacker K, Liu CG, et al. (2008a). Synergistic Function of E2F7 and E2F8 Is Essential for Cell Survival and Embryonic Development. Dev. Cell 14, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, Chanda SK, Batalov S, and Joazeiro CAP (2008b). Genome-Wide and Functional Annotation of Human E3 Ubiquitin Ligases Identifies MULAN, a Mitochondrial E3 that Regulates the Organelle’s Dynamics and Signaling. PLoS One 3, 612–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M, and Halazonetis TD (2018). Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 555, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2013). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.Journal 17, 10–12. [Google Scholar]
- Matson JP, Dumitru R, Coryell P, Baxley RM, Chen W, Twaroski K, Webber BR, Tolar J, Bielinsky AK, Purvis JE, et al. (2017). Rapid DNA replication origin licensing protects stem cell pluripotency. Elife 6, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrommati I, Faedda R, Galasso G, Li J, Burdova K, Fischer R, Kessler BM, Carrero ZI, Guardavaccaro D, Pagano M, et al. (2018). β-TrCP- and Casein Kinase II-Mediated Degradation of Cyclin F Controls Timely Mitotic Progression. Cell Rep. 24, 3404–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudryj M, Devoto SH, Hiebert SW, Hunter T, Pines J, and Nevins JR (1991). Cell cycle regulation of the E2F transcription factor involves an interaction with cyclin A. Cell 65, 1243–1253. [DOI] [PubMed] [Google Scholar]
- Müller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, Prosperini E, Vigo E, Oliner JD, and Helin K (2001). E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 2, 267–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano M, Draetta G, and Jansen-Dürr P (1992). Association of cdk2 kinase with the transcription factor E2F during S phase. Science (80-.). 255, 1144–1147. [DOI] [PubMed] [Google Scholar]
- Palozola KC, Donahue G, Liu H, Grant GR, Becker JS, Cote A, Yu H, Raj A, and Zaret KS (2017). Mitotic transcription and waves of gene reactivation during mitotic exit. Science (80-.). 358, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peart MJ, Poyurovsky MV, Kass EM, Urist M, Verschuren EW, Summers MK, Jackson PK, and Prives C (2010). APC/C(Cdc20) targets E2F1 for degradation in prometaphase. Cell Cycle 9, 3956–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping Z, Lim R, Bashir T, Pagano M, and Guardavaccaro D (2012). APC/C(Cdh1) controls the proteasome-mediated degradation of E2F3 during cell cycle exit. Cell Cycle 11, 1999–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, and Dynlacht BD (2002). E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes Dev. 16, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Smyth GK, Phipson B, Wu D, Hu Y, Shi W, and Law CW (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ and Smyth GK (2010). edgeR: a Bioconductior package for differential expression analysis of digital gene epxression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SS, Ma L, Bagui TK, Forinash KD, and Pledger WJ (2012). A p27Kip1mutant that does not inhibit CDK activity promotes centrosome amplification and micronucleation. Oncogene 31, 3989–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, and Dagnino L (2017). CDH1 regulates E2F1 degradation in response to differentiation signals in keratinocytes. Oncotarget 8, 4977–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, and Pagano M (2013). Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol. 14, 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Rayman JB, and Dynlacht BD (2000). Analysis of promoter binding by the E2F and pRB families in vivo: Distinct E2F proteins mediate activation and repression. Genes Dev. 14, 804–816. [PMC free article] [PubMed] [Google Scholar]
- Walter D, Hoffmann S, Komseli ES, Rappsilber J, Gorgoulis V, and Sørensen CS (2016). SCF(Cyclin F)-dependent degradation of CDC6 suppresses DNA re-replication. Nat. Commun 7, 10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorp B, Mokry M, Groot Koerkamp MJA, Holstege FCP, Cuppen E, and De Bruin A (2012). E2F7 represses a network of oscillating cell cycle genes to control S-phase progression. Nucleic Acids Res. 40, 3511–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, and Smyth GK (2012). Camera: A competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 40, e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Wang Z, and Wei W (2016). Ubiquitination-mediated degradation of cell cycle-related proteins by F-box proteins. Int. J. Biochem. Cell Biol. 73, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genes that were differentially induced in mutant versus WT E2F1 upon doxycyline treatment.
Genes that were differentially induced in mutant versus WT E2F2 upon doxycyline treatment.
Genes that were differentially induced in mutant versus WT E2F3A upon doxycyline treatment.
Gene sets variations for each E2F analysis, amongst the genes that were differentially induced in mutant versus WT E2F upon doxycycline treatment, as listed in Table S1–3.
As listed in Ren et al.
Data Availability Statement
RNA sequencing data has been deposited in the SRA database with SRA accession PRJNA499110. Raw, uncropped images of western blots have been deposited in the Mendeley database, http://dx.doi.org/10.17632/29y4rwj2hp.1.