

Abstract
NRF2, a transcription factor that has been deemed the master regulator of cellular redox homeostasis, declines with age. NRF2 transcriptionally upregulates genes that combat oxidative stress; therefore, loss of NRF2 allows oxidative stress to go unmitigated and drive the aging phenotype. Oxidative stress is a common theme among the key features associated with the aging process, collectively referred to as the “Hallmarks of Aging”, as it disrupts proteostasis, alters genomic stability, and leads to cell death. In this review, we outline the role that oxidative stress and the reduction of NRF2 play in each of the Hallmarks of Aging, including how they contribute to the onset of neurodegenerative disorders, cancer, and other age-related pathologies.
Keywords: NRF2, KEAP1, antioxidant response element (ARE), redox regulation, oxidative stress, age related pathologies, neurodegeneration, cancer, aging
Graphical abstract
Introduction
Reactive oxygen species (ROS) are critical contributors to aging [1]. Under physiological conditions, endogenous ROS (i.e. superoxide, hydroxyl radicals, hydrogen peroxide) can mediate redox signaling, inflammation, and the immune response [2]. Mechanistically, ROS modulate cellular function by modifying proteins, as well as lipids, carbohydrates, and nucleic acids, altering their stability and function. Therefore, when these species are not properly eliminated, their accumulation causes oxidative stress and promotes pathogenesis. This is particularly true in the context of aging, where the accumulation of oxidative stress is considered a key driving force that ultimately increases susceptibility to developing age-related pathologies. This increased susceptibility is due, at least in part, to ROS-induced damage to key intracellular components affecting the overall redox, metabolic, and protein homeostasis of the cell.
Numerous endogenous antioxidant systems are in place to ensure that ROS are properly reduced, including the NRF2-KEAP1 signaling pathway. Nuclear factor (erythroid-derived 2)- like 2 (NRF2) is a member of a family of basic leucine transcription factors that binds to Antioxidant Response Elements (AREs) in the promoter region of genes involved in redox regulation, proteostasis, DNA repair, prevention of apoptosis, iron and heme metabolism, and phase I, II, and III drug/xenobiotic metabolism [3]. Basally, low levels of NRF2 are maintained by an E3 ubiquitin ligase complex containing a substrate adaptor protein that binds to and negatively regulates NRF2: Kelch-like ECH-associated protein 1 (KEAP1) [4]. Two amino acid motifs in NRF2, ETGE and DLG, are recognized and bound by a homodimer of KEAP1, which recruits the Cullin 3-Ring-box protein 1 (CUL3-RBX1) E3 ubiquitin ligase complex, allowing for NRF2 ubiquitylation. Following ubiquitylation, p97 extracts NRF2 from the KEAP1-CUL3- RBX1 E3 ubiquitin ligase complex, delivering it to the 26S proteasome for degradation [5]. Increased oxidative/electrophilic stress induces NRF2 through modifications of key cysteine residues in KEAP1 that cause a conformational change in the NRF2-KEAP1 complex that prevent the ubiquitylation of NRF2 [6]. This allows for the accumulation of newly synthesized NRF2, which can then translocate to the nucleus, heterodimerize with sMAF, bind to AREs, and recruit transcriptional machinery [7]. As mentioned above, NRF2 transcriptional responses are critical not only in maintaining normal homeostasis, but also restoring it following an oxidative insult. Two keys pathways in restoration of redox homeostasis are the glutathione and thioredoxin antioxidant pathways. Glutathione synthesis is controlled by NRF2, as the catalytic and modulatory subunits of the glutathione synthesizing enzyme, glutamate-cysteine ligase (GCLC, GCLM), are NRF2 target genes [8]. Accordingly, it has been shown that NRF2 activity, as well as GCLC and GCLM expression, decrease with age [9], indicating that age- related decline in NRF2 function can significantly decrease the cell’s glutathione pool. Genes associated with the thioredoxin-based antioxidant system (TXN1, TXNRD1) are also regulated by NRF2, indicating that NRF2 regulates the redox state of proteins [10]. As such, it is not surprising that NRF2 dysfunction is a key feature of a wide variety of pathologies, including aging and age-related diseases.
Traditionally, aging is associated with a gradual decrease in cell signaling, increased protein dysfunction/misfolding/aggregation, and an increased risk of cell death. The gradual reduction of NRF2 with age has been attributed to increased expression of its negative regulators and a general decrease in NRF2 protein expression and pathway responsiveness [11]. A number of other factors associated with decreased NRF2 signaling include epigenetic silencing of its promoter, competition with other transcription factors for overlapping cofactors (i.e. nuclear factor kappa-light chain-enhancer of activated B cells (NF- κB)/CREB-binding protein (CBP)), and increased degradation of NRF2 mRNA and protein [12]. All of these factors can ultimately negate NRF2-mediated transcription and thus expression of its target genes. When NRF2-mediated transcription is reduced, cells are more sensitive to oxidative stress, ER stress, and protein aggregation, which in turn promote the aging phenotype. There are a number of key pathological features associated with aging, collectively termed the “Hallmarks of Aging” that play an important role in driving age-related pathologies. In this review, we highlight recent evidence demonstrating that reduction of NRF2 in aging cells contributes to each of the nine Hallmarks of Aging, with a particular focus on the progression of age-related pathologies, including neurodegeneration and cancer.
The Hallmarks of Aging and NRF2
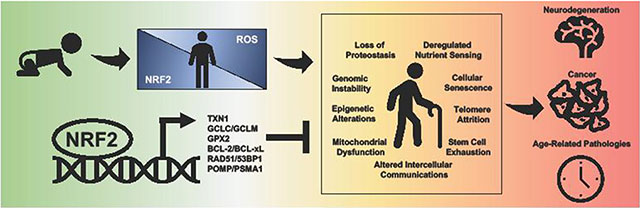
In 2013, Lopez-Otin et al. outlined a series of criteria that encompass the behavior of aging cells deemed the Hallmarks of Aging, which include: 1) Loss of proteostasis, 2) Genomic instability, 3) Telomere attrition, 4) Epigenetic alterations, 5) Mitochondrial dysfunction, 6) Deregulated nutrient sensing, 7) Cellular senescence, 8) Stem cell function, and 9) Altered intercellular communications [13]. Below, we will highlight how oxidative stress and reduction of NRF2 drives each of the Hallmarks of Aging (Figure 1).
Figure 1. Loss of NRF2 and the Hallmarks of Aging.

Loss of NRF2 plays a direct role in each of the Hallmarks of Aging.
1. Loss of Proteostasis
As cells age, an accumulation of unfolded and misfolded proteins can form aggregates that disrupt the normal function of the cell. As misfolded proteins accumulate in the endoplasmic reticulum (ER), the Unfolded Protein Response (UPR) is triggered. The UPR consists of three separate branches responsible for combatting proteotoxic stress: 1) XBP1 mRNA splicing via inositol requiring enzyme 1 (IRE1) to activate transcription of chaperones and endoplasmic reticulum associated protein degradation (ERAD) proteins, 2) PERK phosphorylation of eIF2𝛼 to activate ATF4-mediated transcription of redox enzymes and cell death proteins while simultaneously attenuating translation, and 3) proteolytic cleavage of ATF6 to activate transcription of UPR target genes. When unmitigated, sodium arsenite and paraquat induced ROS blocks the UPR via cysteine sulfenylation of IRE1, causing it to lose its kinase activity, increasing ER stress, and activating NRF2 in a p38-mitogen activated protein kinase (MAPK)- dependent manner [14]. Conversely, UPR activation of PERK causes an induction of NRF2 due to PERK-dependent phosphorylation of NRF2, disrupting NRF2-KEAP1 binding [15]. In both of these contexts, induction of redox regulating proteins reduces ROS in the ER in order to restore normal cellular function. NRF2 activation reduces hydrogen peroxide levels via an upregulation of target genes like glutathione peroxidase 2 (GPX2), which could then prevent subsequent loss of proteostasis [16]. Other NRF2 target genes, like proteasome maturation protein (POMP) and proteasome subunit alpha type-1 (PSMA1) [17, 18], encode proteasome subunits, indicating that a loss of NRF2 function would impair proteasome assembly and result in the accumulation of misfolded proteins. NRF2 also plays a crucial role in autophagy, as it regulates several autophagy-related proteins (ATG5, ATG7) that control autophagosome formation [19]; therefore, should NRF2 be lost, autophagy initiation would be decreased, and misfolded or damaged proteins would accumulate. Thus, NRF2 regulates the UPR, proteasome, and autophagy, inferring that a loss of NRF2 signaling could be a key driving force underlying the proteotoxic phenotype observed in many age-related diseases.
2. Genomic Instability
Similar to cancer, genomic instability in aging is marked by a lack of DNA damage repair, resulting in increased frequency of mutations and chromosomal aberrations. Replication errors during DNA duplication occur at a higher frequency with age. Base excision repair machinery is also less functional; therefore, neurodegenerative and cancer-causing mutations are more likely to occur. Target genes of NRF2 play a key role in both Homologous Recombination (HR) and Non-Homologous End Joining (NHEJ), including p53-binding protein (53BP1) and DNA repair protein RAD51 homolog 1 (RAD51). For example, 53BP1 promotes NHEJ by preventing the resection step of HR by binding to the exposed ends of broken DNA strands and blocking the recruitment of repair complexes [20]. This then allows for the Ku dimeric protein complex to bind to the broken ends of the DNA, ultimately leading to the ligation of the strands. Oppositely, RAD51 is an important protein in HR, as it forms a nucleoprotein filament on DNA that aids in the pairing stage of homologous strands [21]. In both cases, these NRF2 target genes facilitate the repair of DNA to prevent aging and disease. Thus, should NRF2 be reduced, DNA repair would be inhibited and an overall increase in mutations would occur.
3. Telomere Attrition
A byproduct of aging is a shortening of telomeres that prevents further duplication of chromosomes during mitosis. Lacking the ability to regenerate and divide yields cells susceptible to accumulating damage that ultimately can lead to loss of function, senescence, or death. Recently, a link was established between NRF2 and telomerase reverse transcriptase (TERT), which indicated that increased NRF2 levels lead to increased TERT expression [22]. TERT is an essential part of the telomerase complex that promotes the extension of telomeres at the ends of DNA, thus allowing for continual cell division, prevention of chromosomal deterioration, and avoidance of apoptosis. This positive loop with NRF2 and TERT was shown to promote cell survival. Thus, a reduction of NRF2-driven TERT expression could play a key role in driving telomere attrition.
4. Epigenetic Alterations
Epigenetic alterations promote and prevent the transcription of DNA via methylation or acetylation of histones or DNA itself. DNA methyltransferases increase methylation on cytosine residues, particularly those found in CpG islands, preventing the activation of transcription. Due to increased inflammation and decreased formation of metabolic intermediates, there is an increase in DNA methylation with age. It has been shown that hypermethylation in the promoter region of NRF2, blocks its transcription and leads to tumorigenesis [23]. Interestingly, microRNAs have also been shown to play a role in the epigenetic regulation of genes, as they control the expression of key DNA methyltransferases and histone deacetylases [24]. Along these lines, NRF2 expression is also decreased by microRNAs (i.e. miRNA-144), leading to increased hydrogen peroxide levels [25]. While many of the links between epigenetic alterations, NRF2, and aging have yet to be established, work thus far has demonstrated that epigenetic control of NRF2 expression is a critical mediator of oxidative stress in cancer, indicating that this could be an important mechanism driving aging as well.
5. Mitochondrial Dysfunction
A key source of stress in the cell is the mitochondria, which produce superoxide as a byproduct of the electron transport chain. Mitochondrial dysfunction becomes more prominent with age, and a number of neurodegenerative diseases have been associated with mitochondrial dysfunction [26]. Importantly, NRF2 plays a crucial role in maintaining proper mitochondrial function by mediating the cellular redox balance, fatty acid oxidation, membrane potential, and structural integrity of the mitochondria itself [27]. Thus, an age-related decline in NRF2 function could be a key driving force behind the increased mitochondrial dysfunction associated with age-related diseases. NRF2 also controls the expression of anti-apoptotic mitochondrial proteins like B-cell lymphoma 2 (BCL-2) and B-cell lymphoma-extra large (BCL-xL) [28]. If the expression of these proteins is low, pro-apoptotic cascades can occur, including increased activation of mitochondrial permeability transition pores (MPTP). Increased MPTP disrupts the TCA cycle, lowers ATP generation, enhances superoxide and hydroxyl radical generation, and releases cytochrome c into the cytosol, perpetuating mitochondrial damage and loss of function. Cytochrome c release triggers the formation of the apoptosome and leads to apoptosis. Correspondingly, a reduction of NRF2 is associated with an increased susceptibility to apoptosis, and by inducing NRF2, MPTP can be decreased and mitochondrial function maintained, thus negating age-related mitochondrial dysfunction [29].
6. Deregulated Nutrient Sensing
Deregulation of key nutrient sensing pathways has been associated with extending longevity. AMP-activated protein kinase (AMPK) is a critical nutrient sensing protein that is activated in response to decreased ATP levels; however, it has been shown that AMPK responsiveness decreases with age [30]. AMPK has also been linked to NRF2 activation via phosphorylation of serine 550 that drives its nuclear localization [31]. Therefore, in aged cells, NRF2 signaling is decreased because of a lack of AMPK response. NRF2 is also involved in many metabolic pathways that affect the production and metabolism of nutrients and other macromolecules. NRF2 target genes are important enzymes in a variety of metabolic pathways including the pentose phosphate pathway (glucose-6-phosphaste-dehydrogenase [G6PD], phosphogluconate dehydrogenase [PGD]) and purine biosynthesis (bifunctional methylenetetrahydrofolate dehydrogenase [MTHFD2]) [32, 33]. Therefore, downregulation of NRF2 due to age would significantly alter metabolite flux through key metabolic pathways, promoting the deregulated nutrient metabolism and sensing observed in age-related pathogenesis.
7. Cellular Senescence
While cellular senescence has been proposed to be beneficial in a cancer setting, senescence of healthy cells drives the aging phenotype by stopping proliferation. A link between NRF2 and senescence in fibroblasts has been shown, as NRF2 was downregulated in older compared to younger cells. Thus, older fibroblasts were more sensitive to the effects of general oxidative stress [34]. Studies revealed that young fibroblasts treated with an NRF2 inducer did not become senescent compared to untreated controls due to activation of the proteasome [34]. As mentioned above, NRF2 induction promotes proteasome formation via POMP and PSMA1, which in turn extends survival, enhances cytoprotection, and prevents senescence. In addition, treatment of already senescent cells with an NRF2 inducer reversed the senescent phenotype and restored proliferation [34]. Overall, NRF2 prevents cellular senescence; therefore, when NRF2 is reduced, senescence and the aging phenotype are increased.
8. Stem Cell Exhaustion
Renewal and differentiation of stem cells are a critical part of maintaining tissue homeostasis and repair throughout life. However, it is also known that there is an overall decrease in stem cell functionality in all tissues and organs with advancing age. NRF2 appears to play a dual role in stem cell regeneration during aging. For instance, in the nervous system, studies indicate that while neural stem progenitor cells (NSPCs) progressively decline with advancing age, there is striking reduction in NSPC survival and regeneration (proliferation and neuronal differentiation) during a critical time-period during middle age that is driven by a significant reduction in NRF2 expression [35, 36]. This phenomenon was noted in both the major mammalian NSPC niches, the subventricular zone (SVZ) in the forebrain and the dentate gyrus (DG) of the hippocampus, and had functional consequences in terms of a compromise in NSPC associated olfactory and cognitive behaviors. Moreover, these studies also show that NRF2 upregulation, on the other hand, can improve NSPC survival and function [35,36]. Similarly, it has also been reported that the upregulation of NRF2 promotes human embryonic stem cell differentiation via increased proteasome formation. Nevertheless, a sustained hyperactivation of NRF2 can lead to uncontrolled regeneration and stem cell exhaustion [17]. These data suggest a crucial role NRF2 in stem cells, and highlight a fine balance in NRF2 activity that must be maintained to not only keep stem cells viable and functional but also prevent their depletion. Furthermore, the studies also suggest targeting the NRF2 pathway as a potential approach to optimally modulate stem cell function with age, and potentially age-related disorders.
9. Altered Intercellular Communications
The pathogenic effects associated with altered intercellular communication between aging cells focuses primarily on inflammation. NADPH oxidase-derived superoxide is a critical component of the inflammatory cascade, which in itself is dictated by a number of transcriptional regulators. One prime example is NF-kB, a pro-inflammatory transcription factor that increases interleukin levels and drives the inflammatory phenotype. Intriguingly, NF-kB-driven transcription is negatively correlated with NRF2, as both factors compete for CREB-binding protein (CBP), a cofactor required for both transcriptional responses. This indicates that when the activity of one of these transcription factors is increased, the other cannot bind to CBP, and thus transcription of its target genes is reduced [37]. In the case of inflammation, which is particularly common in many neurodegenerative disorders, NF-kB is upregulated, and thus NRF2 transcription of its targets is decreased, perpetuating the increased pro-inflammatory cytokine production that drive these diseases [38]. Furthermore, α -synuclein is a pro- inflammatory protein that promotes NF-kB activation; however, it has been shown that increased NRF2 can enhance the clearance of α-synuclein [39, 40]. Aggregation of α - synuclein has been linked to several neurodegenerative disorders; therefore, accumulation of α -synuclein as a result of decreased NRF2 could be a key contributor to the onset of neurodegeneration. While there are other examples of altered intercellular communications in aging (i.e. senescent cells triggering senescence in bystander cells [41]), many of the age-related changes in intercellular signaling revolves around superoxide driven inflammation, which goes unmitigated when NRF2 is decreased.
Age-Related Pathologies and Redox Regulation
Unmitigated, ROS increase susceptibility to neurodegeneration, cancer, and other age- related pathologies. This is due to increased damage to proteins, organelles, and DNA that cause cellular dysfunction, mutations, and/or trigger cell death. NRF2 target genes protect against the development of age-related pathologies, both by directly neutralizing free radicals and preventing the damage caused by ROS (Figure 2).
Figure 2. Oxidative stress and age related-pathologies.

Oxidative stress drives neurodegeneration, cancer, and age-related pathologies through organelle dysfunction, DNA damage, and protein aggregation. Target genes of NRF2 (GPX2, TXN1, GCLC, GCLM) facilitate the neutralization of free radicals and prevent damage from occurring. Accumulation of ROS results in mitochondrial dysfunction, DNA damage, and protein aggregates; NRF2 upregulates gene that combat these damages: BCL-2/BCL-xL, 53BP1/RAD51, and POMP/PSMA1 respectively. NRF2 mitigates age-related pathologies.
Neurodegeneration
Neurodegenerative disorders are characterized by neuronal cell damage and protein aggregation that disrupt their function or cause cell death. Since NRF2 plays a key role in maintaining proper proteostasis, a loss of NRF2 drives the proteotoxic phenotype observed in many of these disorders. Based on its anti-proteotoxic and antioxidant roles, NRF2 has been shown to mitigate the effects of Parkinson’s disease (PD), Alzheimer’s disease (AD), and Multiple sclerosis (MS). For example, in PD, triplication of the SNCA gene generates excessive α-synuclein, which promotes the formation of Lewy bodies [42]. As discussed above, NRF2 enhances the clearance of α-synuclein, and thus could be used to prevent Lewy body formation and PD. In AD, hyperphosphorylation of tau leads to its aggregation, which prevents microtubule assembly in neurons [43]. NRF2 was shown to prevent the accumulation of hyperphosphorylated tau by enhancing its autophagic degradation [44]. In the case of MS, oxidative stress leads to demyelination of nerve cells that disrupts the function of the nervous system. To date, the only FDA approved NRF2 inducing drug is Dimethyl Fumarate (DMF). DMF is used in the treatment of early MS in order to reduce oxidative stress in an NRF2-dependent manner [45]. It is now well established that the ability of NRF2 to combat demyelination and oligodendrocyte loss prevents the progression of MS [46]. Overall, the pro-survival role of NRF2 makes it a key factor in understanding neurodegeneration, and gradual loss of NRF2 with aging appears to be a major risk factor for increasing susceptibility to PD, AD, MS, and other neurodegenerative disorders.
Cancer
It has also been well established that cancer is an age-related disease. This is due to a variety of factors, including higher frequency of DNA damage/replication errors in older cells, chronic, low-dose exposure to carcinogens, or changes in the levels of certain pro- or anti-tumorigenic proteins (i.e. p53) [47]. As discussed above, target genes of NRF2 play a critical role in both the HR and NHEJ facets of DNA repair, which are critical in preventing genomic instability, a key driver of cancer initiation. NRF2 target genes are also critical mediators of xenobiotic/drug metabolism, which plays an important role in processing harmful xenobiotics into their less toxic forms, preventing carcinogen accumulation and cancer initiation. Therefore, in a cancer context, the loss of NRF2 as a result of aging can lead to tumorigenesis, mainly as a result of increased DNA damage/mutations and decreased metabolism of pro-tumorigenic xenobiotics, inferring that induction of NRF2 in aging populations could be important therapeutically for treating age-related cancers.
Hutchinson-Gilford Progeria Syndrome (HGPS)
HGPS is a rare disorder that causes premature aging in children. Mechanistically, it has been shown that a spliced form of Lamin A caused by a single nucleotide polymorphism, results in a truncated form of the protein known as progerin [48]. Similar to dysfunctional NRF2, progerin promotes HGPS via a number of the Hallmarks of Aging, including senescence, telomere alterations, and chronic DNA damage [49]. Progerin has also been shown to directly affect NRF2, as it does not maintain the normal scaffolding/structural support role of Lamin A, but instead sequesters NRF2 to the edge of the nuclear envelope [50]. This mislocalization prevents NRF2 from activating transcription; therefore, cells are more susceptible to DNA damage and death. Once again, a lack of redox homeostasis maintained by target genes of NRF2 drives the aging process by increasing susceptibility to stressors.
Conclusion
Loss of NRF2, as a result of increased expression of its negative regulators or epigenetic repression, drives aging. In turn, this allows for superoxide, hydroxyl radicals, and hydrogen peroxide to accumulate in cells shifting them to a pathogenic state. Specifically, a reduction of NRF2 and increased oxidative stress contributes to each of the Hallmarks of Aging: loss of proteostasis, genomic instability, telomere attrition, epigenetic alterations, mitochondrial dysfunction, deregulated nutrient sensing, cellular senescence, stem cell exhaustion, and altered intercellular communications. Thus, a decline in NRF2 function is a critical component of the aging process. In order to effectively combat the effects of NRF2 loss in aging, more therapeutics are needed. Currently, DMF is the only NRF2-based therapeutic to make it through clinical trials. While DMF is efficacious in the treatment of early MS, whether or not it could be used in the treatment of other age-related pathologies, including PD, AD, or age related cancers, is less clear. Thus, a critical gap in targeting NRF2 in age-related pathologies is the existence of proven, targeted therapies that lessen the severity of the Hallmarks of Aging to help mitigate the onset of disease. As such, the development of NRF2-based therapeutics to mitigate aging and its associated diseases remains a much-needed area of study. Contrastingly, NRF2-mediated must be careful studied as hyperactivation of NRF2 can lead to therapeutic resistance and disease. While we cannot stop aging, we can mitigate its effects by restoring the NRF2 transcriptional response to maintain a proper redox balance and combat age-induced pathologies.
Highlights.
NRF2 plays a role in each of the Hallmarks of Aging
Decreased NRF2 expression contributes to the aging process
Neurodegeneration, cancer, and other age-related pathologies are a result of lower NRF2 levels and increased oxidative stress
Acknowledgements
The authors are funded by the following grants from the National Institutes of Health: ES026845 (D.D.Z.), DK109555 (D.D.Z.), and ES004940 (D.D.Z.).
Disclosure Statement
The authors are unaware of any biases, affiliations, memberships, or funding that would skew the objectivity of this review.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davalli P, et al. , ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid Med Cell Longev, 2016. 2016: p. 3565127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schieber M and Chandel NS, ROS function in redox signaling and oxidative stress. Curr Biol, 2014. 24(10): p. R453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayes JD and Dinkova-Kostova AT, The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci, 2014. 39(4): p. 199–218. [DOI] [PubMed] [Google Scholar]
- 4.Itoh K, et al. , Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev, 1999. 13(1): p. 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tao S, et al. , p97 Negatively Regulates NRF2 by Extracting Ubiquitylated NRF2 from the KEAP1-CUL3 E3 Complex. Mol Cell Biol, 2017. 37(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang DD and Hannink M, Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol, 2003. 23(22): p. 8137–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Itoh K, et al. , An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun, 1997. 236(2): p. 313–22. [DOI] [PubMed] [Google Scholar]
- 8.Wild AC, Moinova HR, and Mulcahy RT, Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem, 1999. 274(47): p. 33627–36. [DOI] [PubMed] [Google Scholar]
- 9.Suh JH, et al. , Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A, 2004. 101(10): p. 3381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reisman SA, et al. , Increased Nrf2 activation in livers from Keap1-knockdown mice increases expression of cytoprotective genes that detoxify electrophiles more than those that detoxify reactive oxygen species. Toxicol Sci, 2009. 108(1): p. 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H, Davies KJ, and Forman HJ, Oxidative stress response and Nrf2 signaling in aging. Free Radic Biol Med, 2015. 88(Pt B): p. 314–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silva-Palacios A, et al. , Nrf2: Molecular and epigenetic regulation during aging. Ageing Res Rev, 2018. 47: p. 31–40. [DOI] [PubMed] [Google Scholar]
- 13.Lopez-Otin C, et al. , The hallmarks of aging. Cell, 2013. 153(6): p. 1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hourihan JM, et al. , Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol Cell, 2016. 63(4): p. 553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cullinan SB, et al. , Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol, 2003. 23(20): p. 7198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh A, et al. , Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am J Respir Cell Mol Biol, 2006. 35(6): p. 639–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jang J, et al. , Nrf2, a regulator of the proteasome, controls self-renewal and pluripotency in human embryonic stem cells. Stem Cells, 2014. 32(10): p. 2616–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwak MK, et al. , Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol Cell Biol, 2003. 23(23): p. 8786–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pajares M, et al. , Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy, 2016. 12(10): p. 1902–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim SB, et al. , Targeting of Nrf2 induces DNA damage signaling and protects colonic epithelial cells from ionizing radiation. Proc Natl Acad Sci U S A, 2012. 109(43): p. E2949–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jayakumar S, Pal D, and Sandur SK, Nrf2 facilitates repair of radiation induced DNA damage through homologous recombination repair pathway in a ROS independent manner in cancer cells. Mutat Res, 2015. 779: p. 33–45. [DOI] [PubMed] [Google Scholar]
- 22.Ahmad F, et al. , Nrf2-driven TERT regulates pentose phosphate pathway in glioblastoma. Cell Death Dis, 2016. 7: p. e2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khor TO, et al. , Epigenetic DNA methylation of antioxidative stress regulator NRF2 in human prostate cancer. Cancer Prev Res (Phila), 2014. 7(12): p. 1186–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sato F, et al. , MicroRNAs and epigenetics. FEBS J, 2011. 278(10): p. 1598–609. [DOI] [PubMed] [Google Scholar]
- 25.Zhou C, et al. , MicroRNA-144 modulates oxidative stress tolerance in SH-SY5Y cells by regulating nuclear factor erythroid 2-related factor 2-glutathione axis. Neurosci Lett, 2017. 655: p. 21–27. [DOI] [PubMed] [Google Scholar]
- 26.Lin MT and Beal MF, Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 2006. 443(7113): p. 787–95. [DOI] [PubMed] [Google Scholar]
- 27.Dinkova-Kostova AT and Abramov AY, The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dodson M, et al. , Modulating NRF2 in Disease: Timing Is Everything. Annu Rev Pharmacol Toxicol, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greco T, Shafer J, and Fiskum G, Sulforaphane inhibits mitochondrial permeability transition and oxidative stress. Free Radic Biol Med, 2011. 51(12): p. 2164–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salminen A and Kaarniranta K, AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev, 2012. 11(2): p. 230–41. [DOI] [PubMed] [Google Scholar]
- 31.Joo MS, et al. , AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol Cell Biol, 2016. 36(14): p. 1931–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh A, et al. , Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest, 2013. 123(7): p. 2921–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitsuishi Y, et al. , Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell, 2012. 22(1): p. 66–79. [DOI] [PubMed] [Google Scholar]
- 34.Kapeta S, Chondrogianni N, and Gonos ES, Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J Biol Chem, 2010. 285(11): p. 8171–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corenblum MJ, et al. , Reduced Nrf2 expression mediates the decline in neural stem cell function during a critical middle-age period. Aging Cell, 2016. 15(4): p. 725–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ray S, et al. , A Role for Nrf2 Expression in Defining the Aging of Hippocampal Neural Stem Cells. Cell Transplant, 2018. 27(4): p. 589–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu GH, Qu J, and Shen X, NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta, 2008. 1783(5): p. 713–27. [DOI] [PubMed] [Google Scholar]
- 38.Wardyn JD, Ponsford AH, and Sanderson CM, Dissecting molecular cross-talk between Nrf2 and NF-kappaB response pathways. Biochem Soc Trans, 2015. 43(4): p. 621–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aoki R and Li YR, alpha-synuclein promotes neuroprotection through NF-kappaB- mediated transcriptional regulation of protein kinase Cdelta. Sci Signal, 2011. 4(195): p. jc6. [DOI] [PubMed] [Google Scholar]
- 40.Skibinski G, et al. , Nrf2 mitigates LRRK2- and alpha-synuclein-induced neurodegeneration by modulating proteostasis. Proc Natl Acad Sci U S A, 2017. 114(5): p. 1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nelson G, et al. , A senescent cell bystander effect: senescence-induced senescence. Aging Cell, 2012. 11(2): p. 345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heman-Ackah SM, et al. , Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum Mol Genet, 2017. 26(22): p. 4441–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gong CX and Iqbal K, Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem, 2008. 15(23): p. 2321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jo C, et al. , Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun, 2014. 5: p. 3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gopal S, et al. , Evidence of activation of the Nrf2 pathway in multiple sclerosis patients treated with delayed-release dimethyl fumarate in the Phase 3 DEFINE and CONFIRM studies. Mult Scler, 2017. 23(14): p. 1875–1883. [DOI] [PubMed] [Google Scholar]
- 46.Draheim T, et al. , Activation of the astrocytic Nrf2/ARE system ameliorates the formation of demyelinating lesions in a multiple sclerosis animal model. Glia, 2016. 64(12): p. 2219–2230. [DOI] [PubMed] [Google Scholar]
- 47.Serrano M and Blasco MA, Cancer and ageing: convergent and divergent mechanisms. Nat Rev Mol Cell Biol, 2007. 8(9): p. 715–22. [DOI] [PubMed] [Google Scholar]
- 48.McClintock D, et al. , The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS One, 2007. 2(12): p. e1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benson EK, Lee SW, and Aaronson SA, Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J Cell Sci, 2010. 123(Pt 15): p. 2605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kubben N, et al. , Repression of the Antioxidant NRF2 Pathway in Premature Aging. Cell, 2016. 165(6): p. 1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]