

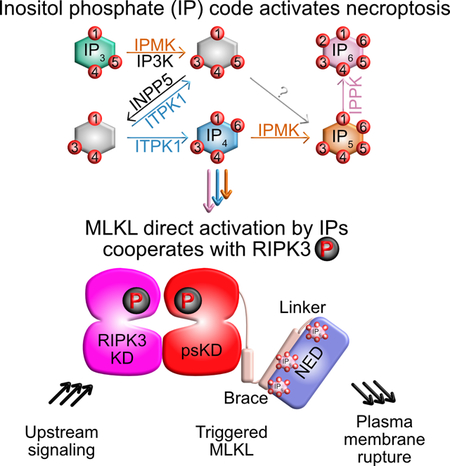
SUMMARY
Necroptosis is an inflammatory form of programmed cell death executed through plasma membrane rupture by the pseudokinase mixed lineage kinase domain-like (MLKL). We previously showed that MLKL activation requires metabolites of the inositol phosphate (IP) pathway. Here we reveal that I(1,3,4,6)P4, I(1,3,4,5,6)P5, and IP6 promote membrane permeabilization by MLKL through directly binding the N-terminal executioner domain (NED) and dissociating its auto-inhibitory region. We show that IP6 and inositol pentakisphosphate 2-kinase (IPPK) are required for necroptosis as IPPK deletion ablated IP6 production and inhibited necroptosis. The NED auto-inhibitory region is more extensive than originally described and single amino acid substitutions along this region induce spontaneous necroptosis by MLKL. Activating IPs bind three sites with affinity of 100–600 μM to destabilize contacts between the auto-inhibitory region and NED, thereby promoting MLKL activation. We therefore uncover MLKL’s activating switch in NED triggered by a select repertoire of IP metabolites.
Keywords: Cell death, membrane permeabilization, MLKL, necroptosis, plasma membrane rupture, inositol phosphate, metabolite
Graphical Abstract
eTOC IN BRIEF
McNamara et al. demonstrate that MLKL is extensively auto-inhibited and that several inositol phosphates and their enzymes, but not the putative lipid ligand phosphatidylinositol-4,5-bisphosphate, are required for MLKL activation in necroptosis. These inositol phosphates cooperate with RIPK3 phosphorylation potently activating MLKL to rupture the plasma membrane in necroptosis.
INTRODUCTION
Necroptosis is a form of programmed cell death executed through plasma membrane rupture by the pseudokinase mixed lineage kinase domain-like (MLKL) (Murphy et al., 2013; Petrie et al., 2019; Sun et al., 2012; Sun and Wang, 2014). Human MLKL is activated by the combined upstream actions of receptor-interacting protein kinase 3 (RIPK3) (Declercq et al., 2009; Peter, 2011), inositol phosphate multi kinase (IPMK), and inositol tetrakisphosphate kinase 1 (ITPK1) (Dovey et al., 2018). The precise role of necroptosis in development and adult homeostasis is still emerging (Grootjans et al., 2017). Its involvement in pathology has been established by genetic and pharmacologic analyses, but the exact mechanism by which necroptosis contributes to different diseases is complex and remains poorly delineated (Newton et al., 2016; Wegner et al., 2017). Necroptosis has been linked to autoimmunity including multiple sclerosis (Alvarez-Diaz et al., 2016; Ofengeim et al., 2015), neurodegeneration including Parkinson’s disease (Caccamo et al., 2017; Iannielli et al., 2018; Ito et al., 2016), ischemic injury (Degterev et al., 2005), infectious diseases (Kaiser et al., 2013; Mocarski et al., 2015; Pearson et al., 2017; Upton and Kaiser, 2017), and cancer (Najafov et al., 2017; Seifert et al., 2016), suggesting that targeting the necroptosis pathway may lead to promising therapies for these diseases.
Necroptosis is activated by death and Toll-like receptors or virus-induced signaling, all of which engage and activate RIPK3 (Grootjans et al., 2017). Death receptor ligation under conditions of caspase-8 inhibition activates RIPK1 (Weinlich et al., 2017), which in turn activates RIPK3 (Li et al., 2012). RIPK3 phosphorylates MLKL to activate it (Rodriguez et al., 2016; Wang et al., 2014b). Currently, MLKL is the most downstream component of the pathway essential for membrane rupture (Dondelinger et al., 2014; Huang et al., 2017; Quarato et al., 2016; Wang et al., 2014b; Xia et al., 2016). Although other downstream components such as ion channels have been proposed as indirect executioners of plasma membrane rupture, they have not been validated (Cai et al., 2014; Chen et al., 2014). The endosomal sorting complexes required for transport (ESCRT) machinery antagonizes necroptosis by repairing membrane damage induced by MLKL (Gong et al., 2017; Yoon et al., 2017).
MLKL has an N-terminal helix bundle domain (NB), herein referred to as the N-terminal executioner domain (NED), connected by a two-helix brace (B) to the C-terminal pseudokinase domain (psKD) (Murphy et al., 2013). NED alone induces membrane rupture upon oligomerization, representing the most minimal domain exhibiting this activity (Dondelinger et al., 2014; Hildebrand et al., 2014; Quarato et al., 2016; Tanzer et al., 2015; Wang et al., 2014b). MLKL activation is unresolved (Cai et al., 2014; Davies et al., 2018; Huang et al., 2017; Petrie et al., 2018; Quarato et al., 2016; Su et al., 2014; Wang et al., 2014b). RIPK3 phosphorylation in psKD triggers an allosteric switch, altering the conformation of the brace to promote oligomerization and recruitment to the plasma membrane (Davies et al., 2018; Murphy et al., 2013; Petrie et al., 2019; Petrie et al., 2018; Quarato et al., 2016; Sun et al., 2012; Wang et al., 2014b). We discovered inositol phosphate (IP) kinases (IPKs) IPMK and ITPK1 as essential regulators of human MLKL activation (Dovey et al., 2018). Genetic deletion of IPMK or ITPK1 blocks MLKL-mediated necroptosis in human cells, through complete depletion of the IP5 and IP6 pools. We showed that IP6 displaces the autoinhibitory region of NED.
Here we perform structure-function and genetic analyses providing mechanistic insights into NED auto-inhibition and activation and showing that a repertoire of the IP code directly triggers NED. We propose that the combined effects of IP4, IP5, and IP6 are essential for necroptotic induction by human MLKL and genetically implicate inositol pentakisphosphate 2-kinase (IPPK), which converts IP5 to IP6, as a significant contributor to MLKL activation through production of IP6.
RESULTS
Linker and Brace Auto-Inhibition Blocks Spontaneous Necroptosis by NED
We determined the NMR structure of auto-inhibited NED, hNBB156 (Table S1, Figure S1A). This structure is similar to the 6-helix bundle determined previously for hNBB2–154 (2.4 Å RMSD [root mean square deviation] between all pairwise ensembles) (Su et al., 2014), exhibiting differences in the loop regions associated with the N-terminal pole and details of the interactions between brace helix α6 and NED (NB121; Figures S1A and S1B). Specifically, loops between helices α2-α3 and α5-α6 (the linker) are more flexible in the latest structure correlating with lower heteronuclear nuclear Overhauser effect (NOE) observed in these regions (Figures S1A and S1C). Mouse (4bft) (Murphy et al., 2013) and human auto-inhibited NED (2msv, 6d74) (Su et al., 2014) structures are significantly different throughout NED and at its interface with the linker and brace (Figures S1B and S1D). Additional residues at the C terminus of α6 (148–156) are found in human but not mouse MLKL (Figures S1E and S1F). The electrostatic network at the interface of the brace helix α6 with NED helices α2 and α5 extends along one third of the length of NED near its C-terminal pole. The linker region delimited by Met122 and Trp133 is found in different conformations in the NMR structures being largely disordered in the X-ray structure (Figure 1A).
Figure 1. The linker and brace auto-inhibit NED. (see legend next page).

A. Surface-cartoon representation of the NBB156 NMR structure highlighting the linker region between NED and the brace helix α6. Met122 and Trp133 residues anchor the flexible linker to NED.
B. Necroptosis assays monitoring SYTOX Green uptake by IncuCyte imaging induced by WT and mutant NBB156 linked to two drug-inducible FKBP dimerization domains (2xF) and a Venus (V) tag (NBB156-2xFV) in ripk3−/− mlkl−/− MEFs. Doxycycline (Dox)-mediated transactivation of linker and brace mutants of NBB156-2xFV induced necroptosis even in the absence of dimerizer (Dim).
C and D. Quantification of SYTOX Green uptake by IncuCyte imaging at 3 h and 24 h (C) and propidium iodide (PI) uptake monitored by FACS at 3 h and 24 h (D) for cells analyzed in (B) and (C).
E. Necroptosis monitored by IncuCyte imaging in ripk3−/− mlkl−/− MEFs induced by WT or M122E full-length (FL) MLKL tagged with Venus expressed with Dim-induced Cerulean-2xFV-RIPK3 under the control of Dox transactivation and necrosulfonamide (NSA) inhibition.
F. PI uptake monitored by FACS at 6 h for cell analyzed in (E). Error bars represent mean + SEM of n=4 independent experiments done in triplicate or quadruplicate. Panels E and F contain n=2 independent experiments done in quadruplicate. **** P<0.0001, *** P<0.005, ** P<0.01, and * P<0.05 using one-way ANOVA with Tukey-Kramer’s method for multiple comparisons (B-D) or unpaired, two-tailed t-test with Welch’s correction for individual comparisons (E-F). See also Figures S1, S2, and Table S1.
Although required for stabilization of NED fold (Su et al., 2014), the linker and brace are dispensable for membrane permeabilization (Dondelinger et al., 2014; Su et al., 2014), and therefore may function together in auto-inhibition. To test this concept, we investigated the effect of destabilizing mutagenesis on necroptosis using one of the most inactivated constructs encompassing NED and brace helix α6, NBB156 (Arnez et al., 2015). We previously described a Tet-On 3G expression system of NBB140 fused with the tandem oligomerization cassette 2x-FK506 binding protein (FKBP) and Venus (NBB140-2xFV), which is transactivated using doxycycline (Dox) and activated by forced oligomerization using dimerizer (Dim) to induce robust necroptosis in mlkl−/− mouse embryonic fibroblasts (MEFs) (McNamara et al., 2018; Quarato et al., 2016). Here we used NBB156-2xFV in necroptosis assays measuring uptake of cell-impermeable dyes SYTOX Green and propidium iodide to show that substitutions of single residues stabilizing the interaction of NED with the linker and brace activate this construct even in the absence of oligomerization. WT NBB156-2xFV required Dox and Dim to induce robust necroptosis but was inactive in the absence of Dim, as previously reported (Arnez et al., 2015) (Figures 1B–D). In contrast, substitutions of the linker-anchoring residues M122E and W133A, and of the brace-stabilizing residue D137A induced spontaneous necroptosis upon transactivation with Dox, and this was accelerated by oligomerization with Dim (Figures 1C and 1D). Dox titration of WT, M122E, W133A, and D137A in the absence and presence of Dim revealed similar potency for these mutants, all of which were hyperactive compared to the WT upon oligomerization with Dim at low doses of Dox (<12.5 ng/mL; Figures S2A and S2B). WT and mutants were expressed at similar protein levels (Figure S2C). The WT construct was inactive in the absence of Dim even at the maximum Dox dose tested (2 μg/mL; Figure S2D). To exclude their spontaneous oligomerization as driver of plasma membrane rupture, we produced these NBB156CH mutants in E. coli and purified them to homogeneity. Size exclusion chromatography (SEC) of WT and NBB156CH mutants indicated that all are monomeric (Figure S2E). We corroborated that M122E and D137A in FL MLKL-Venus were hyperactive in necroptosis compared to WT when co-expressed with Cerulean-2xFVRIPK3 in ripk3−/− mlkl−/− MEFs (Figures 1E, 1F, S2F, and S2G). IncuCyte imaging of SYTOX Green uptake indicated that D137A induced necroptosis with faster kinetics than WT upon Dox− and Dox+Dim-induced expression, whereas M122E was barely more active than WT at 5 and 6 hours only with Dox but not Dox+Dim (Figures 1E, 1F, S2F, and S2G). Immunoblot analysis indicated FL MLKL expression with WT≥M122>>D137A. Resistance of D137A to necrosulfonamide (NSA) inhibition is responsible for its low level (Figure S2G). These results demonstrate that the hydrophobic interactions between the linker and NED contribute to inhibition of necroptosis similarly to the electrostatic interactions between NED and the brace.
Glucoside Detergents Destabilize Auto-Inhibited NED
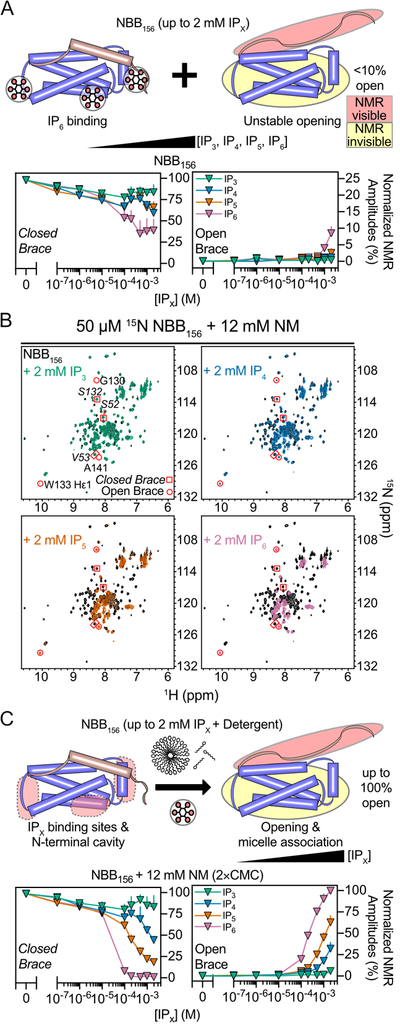
We previously identified dodecylmaltoside detergent (DDM) as inert facilitator of auto-inhibited NED (NBB156) binding to phosphoinositides (PIs) (Quarato et al., 2016). We screened maltoside and glucoside detergents by NMR titrations of 15N-NBB156 and observed that none of the former bound, but all of the latter did as indicated by chemical shift perturbation (CSP) analysis (Figures 2A, S3A, and S3B). We unambiguously assigned resonances of NBB156 in 30 mM deuterated octylglucoside (OG), which did not induce changes in the secondary structure compared to apo-NBB156 (Figure 2B). The CSPs induced by 30 mM OG map to the NBB156 structure primarily at the N-terminal pole defining a buried hydrophobic cavity delimited by NED and the linker (Figures 2C, 2D, and S3B). Importantly, glucoside detergent titrations indicate that each detergent induces significant CSPs in NBB156 below the critical micelle concentration (CMC), suggesting interactions of monomeric detergent molecules at this cavity (Figures 2C, 2D). Glucoside detergents alone were unable to displace the inhibitory region as we showed for the combination of DDM and PI(4,5)P2 (Quarato et al., 2016).
Figure 2. Activation of auto-inhibited NED by glucoside detergents and I(1,4,5)P3.

A. Superimposed 15N-1H TROSY spectra of 15N-NBB156 from titrations of octylglucoside (OG) indicate weak binding by monomeric detergents. Many resonances including the ones labeled (squares) exhibit spectral changes at concentrations below the critical micelle concentration (CMC).
B. Cα chemical shift differences from random coil for free and OG-bound NBB156 suggest similar secondary structure.
C. Backbone amide chemical shift perturbations (CSPs) for OG-bound NBB156 in 30 mM OG are mapped onto the cartoon representation of NBB1–149 structure. Putty thickness represents the magnitude of the CSPs in Figure S3B.
D. Zoomed-in view of cartoon in (C) showing the volume of a cavity found within the OG binding site. This cavity may accommodate detergent acyl chains to promote destabilization of auto-inhibited NED.
E. Glucoside detergents significantly promote large unilamellar vesicle permeabilization (LUVP), quantified by area under curve (AUC) analysis, by WT NBB156CH in a detergent dose-dependent manner. Protein was mixed with detergent at 5×CMC and 1×CMC and the mixture was diluted 20-fold into the LUVP reaction. LUVs mimic the plasma membrane lipid composition and contain His tag-recruiting lipid DGS-NTA(Ni).
F. and G. LUVP AUC analysis of NBB140 (F) or FL MLKL (G) mixed with glucoside detergents at 4×CMC and 1×CMC and diluted 20-fold into reactions with LUVs lacking DGS-NTA(Ni). Error bars represent mean + SEM of 2–3 independent experiments done in triplicate.
H. Superimposed 15N-1H TROSY spectra showing that brace-destabilized E136R NBB156 is prone to partial opening by nonylmaltoside (NM) detergent alone, whereas the WT is inert. Under these conditions, 1 mM I(1,4,5)P3 (IP3) induced full opening in E136R NBB156, whereas phosphatidylinositol (PI) did not. In contrast, in the absence of detergent 1 mM IP3 did not open this mutant. See also Figure S3.
Glucosides but not maltosides activated NBB156 in a concentration-dependent manner in liposome permeabilization assays (Figures 2E, S3C, and S3D). We optimized these assays with C-terminal His6-tagged NBB156 (NBB156CH) and liposomes of lipid composition mimicking mammalian plasma membranes supplemented with His tag-binding lipid DGS-NTA(Ni) to recruit the construct to membranes (Asciolla et al., 2012; van Meer et al., 2008). Without His tag recruitment, the potently autoinhibited NBB156 is inactive in liposome permeabilization. We reasoned that His tag recruitment serves a similar purpose as oligomerization-induced avidity-based membrane translocation of FL MLKL (Quarato et al., 2016). OG, octylthioglucoside (OTG), and nonylglucoside (NG) activated NBB156CH when preincubated in 5×CMC but not 1×CMC prior to 20× dilution into the liposome permeabilization reaction; detergents alone did not permeabilize liposomes (Figures 2E and S3D). Interestingly, the OG analog octyl glucose neopentyl glycol (OGNG), which has 2 branched OG moieties linked at the acyl chain carbon-2 position, does not activate NBB156CH in these assays (Figures 2E and S3D). In contrast, OGNG activates the shorter construct NBB140, which lacks 9 C-terminal residues in the auto-inhibitory brace, albeit slightly less compared to OG (Figures 2F and S3E). Recombinant human FL MLKL is not activated by either detergent suggesting that additional regulation overrides NED activation by detergents (Figures 2G and S3E). Our results indicate a linker-brace dependence on NED activation by glucoside acyl chains through weak destabilizing interactions with the N-terminal pole cavity. Unfortunately, we did not detect nuclear Overhauser effects (NOEs) for binding of OG to NBB156, precluding structure determination and supporting their transient binding.
Although they do not activate it, maltoside detergents enable NED recruitment to their micelles and therefore serve as enabling media to test its activation. We have previously showed that DDM micelles enable NED binding to PIs (McNamara et al., 2018; Quarato et al., 2016). In micellar NM, the hyperactive mutant E136R NBB156 is partially open (~50% brace open and closed conformations; Figure 2H). In the absence of detergent this mutant is 100% found in the apo closed conformation. Interestingly, 125 μM PI in micellar NM did not open this mutant farther compared to the NM detergent alone (Figure 2H). This is consistent with our previous results showing that a small fraction (~15%) of WT NBB156 was opened by 125 μM PI in micellar DDM (Quarato et al., 2016). Remarkably, addition of 1 mM inositol-1,4,5-triphosphate (IP3) in micellar NM resulted in complete opening of E136R NBB156, suggesting that soluble IPs may be activators of auto-inhibited NED. In light of this observation and our recent report on the genetic and biochemical basis of the requirement of IP6 as essential activator of MLKL, we sought to investigate the requirement of the IP code in MLKL activation.
Genetic Demonstration that IP Code Activates MLKL
We previously showed that IPMK and ITPK1 are essential for I(1,3,4,5,6)P5 (IP5) and IP6 production in HT-29 human colorectal adenocarcinoma cells (Dovey et al., 2018). Deletion of either gene blocked MLKL-mediated necroptosis (Dovey et al., 2018). Based on these results, however, we could not conclude that the IP code or individual IPs are essential without formal testing. In addition to IP3 and IP6, which we previously profiled, other IP code metabolites may contribute to MLKL activation (Figure 3A) (Hatch and York, 2010; Otto et al., 2007). Genetically, we could test the contribution of IP6 to MLKL activation by deletion of IPPK, which is exclusively responsible for synthesis of IP6 from IP5 (Figures 3A and S4A). Compared to WT and ΔIPMK HT-29 cells, which respectively, are sensitive and resistant to necroptosis induced by TNF-α, Smac mimetic, and zVAD at 24 h, ΔIPPK exhibited a partial necroptosis phenotype (~50% survival) suggesting that it is not essential but it contributes significantly to necroptosis induction (Figures 3B and S4B) (Dovey et al., 2018). Upstream necroptosis signaling through RIPK1- and RIPK3-mediated phosphorylation was unchanged in ΔIPMK and ΔIPPK compared to WT HT-29 cells, based on their similar levels of phosphorylation at activating serines in RIPK1 (p-S166), RIPK3 (p-S227), and MLKL (p-S358; Figure S4C). This indirectly suggests that TNFR1 shuttling and/or activation are likely unaffected by IP code changes in these cells. Metabolic profiling of IPs in WT and ΔIPPK HT-29 cells indicated complete loss of IP6 and accumulation of IP5 in the mutant, whereas the WT contained abundant levels of IP6 (Figure 3C). This is in contrast to the levels of phosphoinositide phosphate (PIP) species, PI(4)P and PI(4,5)P2, which were unchanged in the 2 cell lines (Figure 3D). Our cellular analyses confirm that robust necroptosis activation through MLKL relies on the combined activation by the IP code rather than activation by IP6 alone.
Figure 3. Inositol pentakisphosphate 2-kinase (IPPK) and IP6 are required but not essential in necroptosis.

A. The inositol phosphate (IP) code as observed in HT-29 cells and based on studies of the mammalian enzymes (Chang and Majerus, 2006; Chang et al., 2002; Frederick et al., 2005; Nalaskowski et al., 2002), highlighting lipid kinases, IP kinases and phosphatases, and phospholipase C (PLC) involved in the synthesis and degradation of phosphoinositide phosphates (PIPs) and IPs. Thus far, we have formally interrogated the role of three IP kinases, including inositol phosphate multikinase (IPMK), inositol tetrakisphosphate kinase 1 (ITPK1), and IPPK, which contribute to the combined synthesis of I(1,3,4,6)P4, IP5, and IP6.
B. HT-29 cell viability measured by cellular ATP assay at 24 h for WT, IPMK-deficient (ΔIPMK), or IPPK-deficient (ΔIPPK) cells. Recombinant human TNF-α (T), Smac mimetic (S), zVAD (Z), and Nec-1 were used at 10 ng/mL, 10 nM, 25 μM, and 30 μM, respectively. Error bars represent mean + SEM of 3 independent experiments done in triplicate.
C. IP levels in WT and ΔIPPK HT-29 cells were assessed by steady-state metabolic profiling of [3H]-inositol-labeled cells, followed by separation and quantification via anion-exchange HPLC. Representative HPLC traces are shown.
D. Phosphoinositide phosphate (PIP) levels in WT and ΔIPPK HT-29 cell were similarly assessed by metabolic profiling of [3H]-inositol-labeled cells, followed by separation and quantitation via anion-exchange HPLC. Representative HPLC traces are shown.
Hierarchical IP Binding to Auto-Inhibited NED (IP6>IP4≥IP5>>IP3)
We examined IPs produced by IPMK and ITPK1 in MLKL activation (Figure 3A) (Dovey et al., 2018). Since we previously tested the IPMK product I(1,3,4,5)P4, which was not even as potent as IP3 in binding NBB156 (Quarato et al., 2016), we focused on the primary ITPK1 product I(1,3,4,6)P4 (IP4) as a potential contributor to NED activation. We tested IP3, IP4, IP5 and IP6 binding to NBB156 in 2D NMR titrations and performed per residue chemical shift perturbation (CSP) analysis at 2 mM IP concentration (Figures 4A and S5A). We identified 4 categories of resonances: i) resonances exhibiting the strongest binding resulting in line broadening (peak disappearance) are identified as intermediate (int.) exchange; ii) significant (sig.) CSPs are above one standard deviation from the average of sig. and iii) insignificant (insig.) CSPs; iv) resonances excluded due to overlapping peaks (Figure 4B). Based on these considerations, IP6 induced the most CSPs (int. exchange + sig. CSPs) upon binding to NBB156, followed by IP4 and IP5, which induced similar CSPs with slightly more CSPs for the former (Figure 4B), while IP3 induced the fewest CSPs (Figure 4B) (Dovey et al., 2018; Quarato et al., 2016). We traced the sig. CSPs against concentration to establish per residue KD for these resonances (Figures 4B and S5B). Interestingly, for all residues CSP as a function of IP concentration was hyperbolic suggesting that binding is not cooperative, although we do not exclude the possibility that binding of higher affinity sites may allosterically induce CSPs and influence subsequent binding at lower affinity sites (Figure S5B). We mapped the CSPs and KD on the NMR structure of NBB156 to reveal three possible binding sites for IPs, exhibiting binding affinities (KD) of 1–2 mM (site 1 at the N-terminal pole involving N-terminus of helix α1, α2-α3 loop, and C-terminus of α5), 0.2–0.3 mM (site 2 in the middle of the domain involving helices α1 and α2), and 0.1–0.2 mM (site 3 at the C-terminal pole involving C-terminus of helix α3, α4, the N-terminus of α5, and the C-terminus of α6) (Figures 4C, 4D, and S5C). Our binding analysis suggests a hierarchy for IP binding to NED, with IP6>IP4≥IP5>>IP3 (Figure 4D).
Figure 4. Hierarchical multi-pronged binding of IPs to auto-inhibited NED.

A. Superimposed 1H-15N SOFAST-HMQC nuclear magnetic resonance (NMR) spectra of NBB156 in the absence (black) or presence of 2 mM IPs (colored).
B. Backbone amide chemical shift perturbation (CSP) analysis for apo and 2 mM IP-bound NBB156 were calculated for fast exchange resonances (black and orange). Intermediate (int.) exchange resonances were assigned CSP value of 0.06 ppm (red). Overlapping backbone amide resonances (blue) were assigned 0.01 ppm here and in (C). A threshold of the average CSP plus one standard deviation (dashed line) for the remaining residues defined significant (sig.) CSPs (orange). Insignificant (insig.) CSPs are colored black. Plotted above the CSP analysis are per-residue dissociation constants (NMR KD) determined from titrations of IPs in Figure S5B.
C. CSPs and KD were color-coded (as in B) over the putty representation of the NBB156 structure. Putty thickness is proportional to CSPs at 2 mM IP or to inverse NMR KD. Intermediate exchange was not assigned an arbitrary KD and is colored red.
D. Comparison of IP binding to each site summarizes the number of resonances contributing to KD determination or exhibiting intermediate exchange. Error bars represent mean + SD for n = nsig. CSP (KD). See also Figure S5.
Hierarchical Opening of Auto-inhibited NED by IPs (IP6>IP5>IP4>>IP3)
We next sought to test auto-inhibited NED opening by IPs. We examined the apo closed (auto-inhibited) and open (active) NBB156 conformations induced by IPs using 2D NMR titrations (Figures 4A and S5A). The open conformation was weakly induced at 2 mM IPs even in the absence of detergent (~8% IP6; ~2% IP5; ~1% IP4; and 0% IP3; Figures 4A and 5A). In contrast, potent, concentration-dependent conversion of apo closed NBB156 to open conformation is induced by IPs in the presence of enabling NM detergent (12 mM, 2×CMC, Figure 5B). The open brace conformation is detectable under these conditions at 10–100 μM for all IPs but IP3, whose transition started between 1 and 2 mM (Figures 5C and S6). IP6 induced ~100% open conformation at 2 mM, followed by IP5 (~63%), IP4 (~32%), and IP3 (~6%) (Figure 5C). Together, these data indicate that the soluble IP code functions to regulate the transition from auto-inhibited closed to open activated NED, which is likely the form involved in membrane permeabilization.
Figure 5. Hierarchical opening of auto-inhibited NED by IPs.
A. Model of NBB156 interactions with IPs monitored by NMR (top). Normalized amplitudes of resonances from the open (circles) and closed conformation (squares) (Fig. S5A) are plotted against IP dose.
B. Superimposed 1H-15N SOFAST-HMQC NMR spectra of NBB156 with 12 mM NM detergent in the absence (black) or presence of 2 mM IP (colored).
C. Schematic of IP+detergent-induced conformational change in NBB156 monitored by NMR. Normalized amplitudes of resonances from open (circles) and closed conformation (squares) (Fig. S6) are plotted against IP dose. Error bars represent mean + SD for n = 3 NMR amplitudes. See also Figure S6.
IPs Activate MLKL in Membrane Permeabilization (IP6>IP4>IP5>IP3)
We investigated the correlation of binding and inhibitory region displacement by IPs with activation of FL MLKL and NED constructs in membrane permeabilization. First, we tested NED activation with up to 100 μM IP6. NBB156CH was preincubated with up to 2 mM IP6 in 2×CMC NM (12 mM) and the mixture was diluted 20-fold in the assay (Figures S7A and S7B). We observed dose-dependent activation of 2 μM NBB156CH that peaked at 25 μM IP6 (Figures S7A and S7B). At 100 μM IP6 NBB156CH induced less membrane permeabilization than at 25 μM IP6 possibly through inhibitory IP6-induced NBB156CH dimerization observed in analytical ultracentrifugation analysis (Figure S7C). However, we do not exclude the possibility of weak inhibition by alternate mechanisms at higher IP6 doses in the presence of NM. Under similar conditions in the absence of detergent, significant membrane permeabilization was observed at 2 μM NBB156CH even in the absence of IPs (Figures 6A and S7D). Addition of 100 μM IP3, IP4, or IP5 under these conditions induced an insignificant boost in activity, whereas 100 μM IP6 significantly activated NBB156CH by doubling AUC compared to AUC in the absence of IPs. A similar trend was noted at 1 μM and 0.5 μM protein, with IP4 and IP5 achieving significance at the higher dose (Figures 6A and S7D).
Figure 6. Direct activation of NED and FL MLKL by IPs but not by PI(4,5)P2.

A. AUC analysis of LUVP by NBB156CH with liposomes containing DGS-NTA(Ni) reveals activation by 100 μM IPs.
B. AUC analysis of LUVP for liposomes lacking DGS-NTA(Ni) incubated with different doses of NBB140 and 100 μM IPs, reveals IP hierarchy of IP6>IP4>IP5≈IP3 in activating auto-inhibited NED.
C. AUC analysis of LUVP for liposomes lacking DGS-NTA(Ni) incubated with different doses of FL MLKL and 100 μM IPs, reveals MLKL activation hierarchy IP6>IP4>IP5>IP3. D. Comparison of AUC analysis of LUVP for liposomes lacking DGS-NTA(Ni) incubated with different doses of FL MLKL ± 100 μM IP6 ± 100 μM IP3, IP4, or IP5 demonstrates lack of inhibition on IP6 activation by lower IPs.
E, F, and G. AUC analysis of LUVP for liposomes lacking DGS-NTA(Ni) incubated with different doses of NBB140 M122E (E), W133A (F), or D137A (G) and 100 μM IPs reveals decreased sensitivity to IPs caused by spontaneous activation of these mutants exhibiting NED inhibition hierarchy of W133>D137>M122.
H. Comparison of AUC analysis of LUVP for liposomes lacking DGS-NTA(Ni) ± PI(4,5)P2 incubated with different doses of NBB140 and 100 μM IP6.
Error bars represent mean + SEM of 2 independent experiments done in triplicate. See also Figure S7.
We repeated this experiment with tag-less NBB140, which is missing the C-terminus and possibly lacks IP binding site 3 mapped in NBB156 (Figure 4C). By having a shorter brace helix α6, NBB140 is expected to be more prone to activation compared to NBB156. Nonetheless, NBB140 does not exhibit spontaneous activation in liposome permeabilization (Figures S7E and S7F). It instead exhibits significant IP-dependent membrane permeabilization at all protein doses tested. With the exception of IP3, which is the least potent activator, all other IPs significantly activate NBB140 as measured by 2–6× larger AUC values compared with apo NBB140 control (Figures S7E and S7F).
We also tested NBB140 using liposomes without DGS-NTA(Ni) ± PI(4,5)P2 (the putative lipid ligand of NED; Figures 6B and S7G). At 100 μM IPs we observed dose-dependent activation that was significant at 1 μM and 2 μM protein for IP6, and at 1 μM for IP4 (Figures 6B and S7G). In contrast, IP3 or IP5 did not significantly activate NBB140 under these conditions. Apo FL MLKL is alone inactive in this assay but exhibits similar activation hierarchy by IPs as seen with NBB140 and even more pronounced activation by IP6 (Figures 6C and S7H). Moreover, combination of 100 μM IP6 with 100 μM IP3, IP4, or IP5 activated FL MLKL similar to 100 μM IP6 alone in liposome permeabilization assays, suggesting that IP3, IP4, and IP5 do not inhibit MLKL activation by IP6 (Figures 6D and S7I). Expectedly, the hyperactive mutants M122E, W133A, and D137A NBB140 spontaneously permeabilized liposomes in the absence of IPs (Figures 6E–6G, S7J-S7M). Addition of IPs enhanced liposome permeabilization by these mutants with the IP code hierarchy observed for the WT at 1 μM and 0.5 μM protein (Figures 6E–6G, S7J-S7M). In contrast, only IP6 enhanced liposome permeabilization by W133A and D137A at 2 μM protein, which induce more permeabilization in the apo state compared to WT and M122E (Figure S7M). Our data demonstrate that the hyperactive mutants are more readily opened even in the absence of IPs. Remarkably, in liposomes without DGSNTA(Ni) in the presence of increasing amounts of PI(4,5)P2, 100 μM IP6 did not significantly activate NBB140 compared to the levels observed in the absence of PI(4,5)P2 (Figures 6H and S7N). We conclude that highly phosphorylated IPs are activators of NED in membrane permeabilization acting independently of PI(4,5)P2.
DISCUSSION
Two essential events have been discovered for MLKL activation including 1) RIPK3-mediated phosphorylation in the activation loop of the psKD (Murphy et al., 2013; Rodriguez et al., 2016; Sun et al., 2012; Wang et al., 2014b) and 2) binding of essential IP metabolites to the executioner domain (Dovey et al., 2018) (Figure 7). Here we show how MLKL is auto-inhibited and how it is triggered by IP metabolites to permeabilize membranes.
Figure 7. MLKL auto-inhibition and direct activation by the IP code in necroptosis.

MLKL activation involves RIPK3 phosphorylation in the psKD to promote brace-mediated oligomerization. However, these upstream events do not explain how auto-inhibited NED is activated to permeabilize membranes. Here we show that the auto-inhibitory region extends beyond the brace helix α6 involving also the linker between NED and the brace. Single amino acid substitutions along the inhibitory region (hotspots labeled and identified by circles in inactive NED) promoted NED activation and necroptosis. We identified a select repertoire of the IP code, involving I(1,3,4,6)P4, I(1,3,4,5,6)P5, and IP6 as endogenous direct activators of auto-inhibited MLKL, and showed their hierarchical binding to NED and activation of MLKL in membrane permeabilization. We currently do not know the molecular architecture of active NED with the auto-inhibitory region displaced. However, we showed that NED liposome permeabilization is dependent on IPs but independent of PI(4,5)P2, suggesting that the IPs are the potent endogenous activators, whereas PI(4,5)P2 may function downstream.
An unexpected discovery is that MLKL is auto-inhibited not only through the interactions between the brace helix α6 to NED, but also through interactions between NED and the linker. The linker is one of the least conserved regions of MLKL between human and mouse. Specific contacts between hydrophobic residues anchor it to the hydrophobic groove at the N-terminal pole of NED to stabilize auto-inhibited MLKL (Murphy et al., 2013; Su et al., 2014). Accordingly, mutagenesis of the linker resulted in spontaneous activation and necroptosis by NED and FL MLKL. A central mutant in the linker-brace region had the greatest impact on spontaneous activation (Figure 7). WT NED was shown to be incompetent in necroptosis presumably due to potent auto-inhibition in isolation (Arnez et al., 2015), but is readily activated upon oligomerization in the NBB156-2xFV fusion context. Dim-induced oligomerization of this fusion presumably induces a conformational change to destabilize the auto-inhibitory region on the brace side.
The N-terminal pole of NBB156 bears a cavity delimited on one side by the auto-inhibitory linker and on the other side by a hydrophobic groove within NED. This cavity serves as weak binding site of monomeric glucoside detergents, which can significantly potentiate NBB156 and NBB140 activity in liposome permeabilization, but not that of FL MLKL, presumably by destabilizing the linker:NED interaction as achieved through linker mutagenesis. We do not know why glucoside detergents do not activate FL MLKL, but presume that they cannot override auto-inhibition imposed by the brace and psKD even when the N-terminal pole of NED is perturbed. Intriguingly, we determined that IP4, IP5, and IP6 bind with affinity in the 600–900 μM KD range to IP-binding site 1 encompassing some (IP4, IP5) or all of the structural elements (IP6) that define the N-terminal pole of NED and linker. This site involves up to 5 residues (IP6) that have been found in inositol phosphate binding sites including K5, Q46, Q48, K50, and R51 (Gosein et al., 2012; Wang et al., 2014a). Its weak affinity for site 1 (KD > 2 mM) suggests that IP3 most likely does not engage this site (Liu et al., 2009).
We demonstrate stabilization of auto-inhibitory region by electrostatic interactions to NED made by another conserved amino acid in the brace helix α6 (D136). This residue contributes similarly to auto-inhibition as previously characterized E136, whose destabilizing substitutions activated the executioner domain NBB2–154 in vitro (Su et al., 2014), and FL-MLKL and NBB182 constructs in cells (Quarato et al., 2016). Our linker and brace mutagenesis results are consistent with the observation that the executioner domain alone, NB125, is able to induce robust necroptosis (Dondelinger et al., 2014) and liposome permeabilization (Su et al., 2014).
The IP-binding sites 2 and 3 (KD 100–300 μM) map adjacent to the brace helix α6 and their engagement by IP4, IP5, and IP6 is expected to destabilize the brace interaction with NED. In the absence of high-resolution structures of complexes between NBB156 and IPs, it is difficult to predict exactly which brace-stabilizing NB side chains are repurposed for binding IPs, but both of these sites contain lysine, arginine, asparagine, or glutamine residues that coordinate IPs in other proteins (Endo-Streeter et al., 2012; Gonzalez et al., 2004; Gosein et al., 2012; Holmes and Jogl, 2006; Miller et al., 2005; Wang et al., 2014a). Based on their higher binding affinity for IPs compared to site 1, we propose that sites 2 and 3 are likely implicated in MLKL activation in cells. Intriguingly, IP6 exhibits the most potent binding to site 3, which in human MLKL contains 9 additional residues compared to mouse MLKL. The extra sequence may confer more stringent auto-inhibition of human MLKL compared to other species. IP binding site 3 may have evolved to overcome potent auto-inhibition in human MLKL. How the IP code contributes to MLKL-mediated necroptosis in other species remains to be formally tested. Our structure-based sequence alignment of NED from different species reveals conservation of sites 1 and 2 in mammals and the divergence of site 3 in primates.
We establish a significant cellular role for IP4 and IP5 in MLKL activation through genetic deletion of IPPK, which eliminated the entire IP6 pool while increasing that of IP5 without disturbing PI(4,5)P2 levels, yet partially blocked necroptosis (50% survival compared to 16% for WT). In contrast, individual deletion of IPMK or ITPK resulted in eradication of both IP5 and IP6, with the latter likely affecting I(1,3,4,6)P4 to fully inhibit necroptosis (Dovey et al., 2018). Our data suggests a combined role of the IP4, IP5, and IP6 repertoire of the IP code in direct activation of auto-inhibited NED independent of PI(4,5)P2. Accordingly, we reveal a selective metabolic requirement for triggering of human MLKL in necroptosis.
Our study suggests that IP4 is a significant contributor to MLKL activation (Figure 7). Reported cellular levels for IP4, IP5, and IP6 range around 5 μM to 100 μM (Irvine, 2005; Irvine and Schell, 2001; Pittet et al., 1989; Stuart et al., 1994; Szwergold et al., 1987) and therefore these IPs are expected to readily bind sites 2 and 3 to activate human MLKL (KD ~100 μM-300 μM). Our functional studies with IP combinations suggest that none of the IPs tested inhibit MLKL activation by IP6, indicating that the IP code indeed triggers MLKL. Moreover, we showed potentiation of IP-induced NED activation by detergents. Endogenously, fatty acids may cooperate with IPs to activate MLKL in necroptosis (Parisi et al., 2017). Nonetheless, despite strong evidence for the role of the IP code in human MLKL activation in necroptosis, we conclude that the IP code alone cannot activate WT NED or FL MLKL in the cells tested. Therefore, additional activation by RIPK3 phosphorylation is essential.
Identifying signaling roles for higher order IPs has been historically difficult as their roles mainly consist of static structural cofactors used in protein folding and assembly (Dick et al., 2018; Liu et al., 2017; Macbeth et al., 2005; Mallery et al., 2018; Montpetit et al., 2011; Scherer et al., 2016; Tan et al., 2007; Watson et al., 2012). IP6 has been shown to affect protein function dynamically (Lupardus et al., 2008) and to contribute to signaling pathways with inositol polyphosphates (Chakraborty et al., 2011). To our knowledge, this is the first report of a direct role for I(1,3,4,6)P4 in cell signaling. Inositol phosphates, such as IP3, have classically been characterized as chemical signaling messengers that are generated upon receptor stimulation to propagate intracellular signaling by transiently altering conformation of their target proteins (Irvine and Schell, 2001). Thus, it could be reasonably hypothesized that other IPs may selectively increase to perform similar cellular functions and enhance signaling specificity. Intriguingly, we have previously shown using steady-state metabolic labeling that under conditions of constitutive G-protein coupled receptor activation and over-expression of IP kinases, the relative pools of IPs can fluctuate (Otto et al., 2007). Thus, we hypothesize that exposing IP pools through activation of pro-necroptotic signaling mechanisms may similarly trigger differential production of IPs and thus downstream engagement of MLKL. Our data suggest that certain IPs remarkably act as transient activators of MLKL required for the execution of necroptosis.
Necroptosis may be targeted in light of MLKL regulation by the IP code. Our studies suggest that human IPMK and ITPK1 inhibitors would be ideal candidates to blunt MLKL activation and necroptosis even when MLKL is phosphorylated by RIPK3, whereas IPPK inhibitors are expected to exhibit marginal effects in similar context. We therefore validated IPMK and ITPK1 as bona fide targets in necroptosis involving essential IP metabolite synthesis as mechanism of action. Previously, NSA was identified as a covalent inhibitor of human MLKL (Sun et al., 2012). More recently, NSA was shown to inhibit gasdermin pores in pyroptosis by covalently binding the pore-forming gasdermin D (Rathkey et al., 2018). IPK inhibitors could serve as selective chemical probes in pathophysiological circumstances driven by excessive necroptosis.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tudor Moldoveanu (tudor.moldoveanu@stjude.org)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
XL10-Gold E. coli
Site-directed mutagenesis and ligation-independent cloning procedures were carried out according to the manufacturer’s or published protocols, respectively, using XL10-Gold competent cells (Agilent Technologies) transformed and grown on LB medium and LB-agar plates at 37 °C overnight (Savitsky et al., 20 10).
BL21 Star (DE3) and T7 Express E. coli
BL21 Star (DE3) (Thermo Fisher Scientific) and T7 Express (New England Biolabs) E. coli containing NBB156 constructs in pNIC28-Bsa4 or pNIC-CH vectors were grown in LB medium (Thermo Fisher Scientific) supplemented with 50 g/mL kanamycin (Gold Biotechnology) to an OD600 of 0.8–1.2 before induction with 0.42 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) (Gold Biotechnology) at 18–22°C overnight. NBB 156-expressing E. coli were grown in MOPS-based minimal media supplemented with 15NH4Cl and unlabeled glucose or 15NH4Cl and 13C-glucose (Neidhardt et al., 1974) for isotope labeling under the same growth and induction conditions.
DH10EMBacY E. coli
DH10EMBacY competent cells (Geneva Biotech) for bacmid DNA generation were grown in LB medium supplemented with 7 μg/mL gentamicin, 50 μg/mL kanamycin, and 10 μg/mL tetracycline.
Sf9 S. frugiperda insect cells
Sf9 cells were maintained in ESF921 media at 27 °C (Kalathur et al., 2016).
Mouse embryonic fibroblasts
SV40 large T antigen immortalized ripk3−/− mlkl−/− primary mouse embryonic fibroblasts (MEFs) were maintained in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL penicillin and streptomycin, 1 mM sodium pyruvate, nonessential amino acids, and 55 μM β-mercaptoethanol (Dillon et al., 2014; Quarato et al., 2016; Rodriguez et al., 2016).
Human cell culture
HT-29 (sex: female; source: William Kaiser laboratory) were cultured at 37 °C and 5% CO2 in DMEM (Hyclone) supplemented with 10% fetal bovine serum and L-glutamine/penicillin/streptomycin (Sigma Aldrich).
METHOD DETAILS
Protein expression and purification
Human MLKL NBB156 constructs were expressed from pNIC28-Bsa4 and pNIC-CH vectors in BL21 Star (DE3) and T7 Express E. coli. TEV protease produced in-house was used to cleave the N-terminal His6-tagged protein expressed from the pNIC28-Bsa4 vector. Protein was purified as previously described and flash-frozen in liquid nitrogen for storage at −80°C (Quarato et al., 2016 ). In brief, harvested cells were resuspended in 50 mM Tris pH 7.6, 250 mM NaCl, and 5 mM imidazole. Cells were lysed by 3 passages at 17K psi using an Emulsiflex C3 (Avestin, Inc.) and clarified at 33,000×g for 30 min. Clarified lysates were bound to nickel agarose resin slurry (Gold Biotechnology) for 1 h with stirring on ice. Gravity immobilized metal affinity chromatography (IMAC) utilized binding, wash, and elution buffers containing 25, 40, and 250 mM imidazole, respectively. N-terminal His-tagged constructs were digested with TEV protease at <10:1 molar ratio for 24–48 hours. Samples were concentrated in 3K or 10K molecular-weight cutoff centrifugal filters (MilliporeSigma). Size-exclusion chromatography (SEC) of the proteolysis sample utilized a HiPrep 16/60 Sephacryl S-100 HR column in 20 mM 2-(N-morpholino)ethanesulfonic acid (MES) pH 6.0 or 20 mM sodium acetate pH 5.5 and 50–150 mM NaCl. Concentrated NBB-containing fractions were resolved with 0 mM to 1 M NaCl gradient cation-exchange chromatography on MonoS 5/50 GL or SP FF 16/60 columns using an AKTA Pure (GE Healthcare). Greater than 15-fold dilution in centrifugal filters was performed three times to buffer exchange NBB156 to 20 mM sodium phosphate pH 6.8 for NMR samples or 20 mM HEPES pH 6.8 and 2 mM dithiothreitol (DTT) for unlabeled samples. Human full-length (FL) MLKL with N-terminal hexahistidine and GST tags with a TEV protease cleavage site was expressed for 72 h in Sf9 insect cells following baculovirus infection. Cells were harvested from 1.5 L media by centrifugation at 1,000×g for 10 min and resuspended in 75 mL lysis buffer supplemented with 10% glycerol (Babon and Murphy, 2013; Murphy et al., 2013). Cells were lysed in 2 passages at 10K psi using an Emulsiflex C3 and clarified at 33,000×g for 45 min. Gravity IMAC at ~0.5 mL/min utilized binding, wash, and elution buffers containing 5, 20, and 250 mM imidazole, respectively. Five eluates of 5 mL from 1 mL beads were mixed directly into 25 mL lysis buffer, then TEV protease was added at 1:10 molar ratio with 0.5 mM EDTA and 2 mM DTT for 72 hours. TEV-digested FL MLKL was further purified by subtractive IMAC. Flowthrough was concentrated and FL MLKL was resolved by SEC on a Superdex 200 Increase 10/300 GL column in 20 mM Tris pH 8.5, 150 mM NaCl, and 5% glycerol. Fractions collected at retention volumes corresponding to human FL MLKL were combined and stored at −80°C (Petrie et al., 2018). Protein concentrations were determined from primary sequence-derived extinction coefficients and absorbance measured at 280 nm. Purified protein masses were verified by intact MS.
NMR structure determination
NMR spectroscopy experiments for structure determination were performed on 500 μM WT NBB156 in 20 mM phosphate buffer pH 6.8, 10% D2O at 303 K on Bruker Avance 600-MHz and 800-MHz spectrometers equipped with 5-mm triple-resonance cyroprobe and single-axis pulsed-field gradient. Backbone and side-chain assignments were obtained from standard 3D NMR experiments, including HNCACB, CBCA(CO)NH, HNCO, 15N-edited TOCSY-HSQC, 15N-edited NOESY, HCCH-13C-edited TOCSY, HCC(CO)HN, (HB)CB(CGCD)HD, and (HB)CB(CGCDCE)HE spectra. NMR data were processed in TopSpin and analyzed in CARA (Keller, 2004). NOE distance restraints were obtained from 15N-edited NOESY-HSQC and 13C-edited aliphatic and aromatic NOESY-HSQC spectra with mixing times of 120 ms. Backbone dihedral angle restraints were predicted in TALOS+ (Shen et al., 2009). Hydrogen bonds for helical regions were predicted based on missing water-exchange cross-peaks of amides in 15N-edited NOESY spectra. Structures were initially calculated in UNIO (Herrmann et al., 2002), using CYANA for energy minimization, and final calculations were done manually in CYANA with the final 20 lowest energy structures energy minimized in CNS (Brunger, 2007; Brunger et al., 1998; Herrmann et al., 2002). Quality of the final 20 lowest-energy conformers from the 100 calculated structures was verified in MOLMOL for NOE violations and PROCHECK for Ramachandran statistics (Koradi et al., 1996; Laskowski et al., 1993). The structural statistics of the 20 lowest-energy NMR structures are summarized in Table S1. The steady-state heteronuclear NOE experiment was measured in an interleaved manner using standard Bruker pulse program at 298K, with an interscan delay of 3 secs and a saturation time of 3 secs. RMSD between NMR ensembles of NBB156 and NBB2–154 was calculated in Chimera using Ensemble Match with no iterative pruning of matched atoms between alpha-carbons for residues 2–149 (Pettersen et al., 2004). Solvent-accessible cavities in the NBB156 structure were detected with a 1.4 Å probe radius using HOLLOW (Ho and Gruswitz, 2008) and prepared for visualization with all other structural figures using PyMOL.
NMR titrations
Titrations of 40–200 μM WT or Glu136Arg 15N-NBB156 were done with detergents, lipids in micellar detergents, or inositol-1,4,5-trisphosphate (IP3) in 20 mM phosphate buffer in the presence of 0.3–2.0 mM deuterated DTT (dDTT) and 10% D2O at 298 K as previously described (Quarato et al., 2016). Lipids were reconstituted from films comprising chloroform stocks evaporated under nitrogen and stored at −20 °C. Lipids were serially diluted in constant detergent concentrations with 10–30 minutes of water bath sonication and allowed to cool to room temperature before adding protein and dDTT.
Comparative NMR titrations measuring apo, closed-brace and micellar, open-brace NBB156 resonances amplitudes were performed as described previously with 1H-15N SOFAST-HMQC experiments (Dovey et al., 2018; McNamara et al., 2018; Quarato et al., 2016; Schanda and Brutscher, 2006). Three well-dispersed resonances from apo-NBB156 were used to normalize titrations across conditions. Three resonances previously assigned for the disordered linker-brace segment (122–156) were averaged as the normalized fraction of 100% micellar NBB156 in the presence of inositol phosphate species with and without detergents.
Titrations of 50 μM WT 15N-NBB156 were done as previously described with inositol-1,4,5-triphosphate (IP3), inositol-1,3,4,6-tetraphosphate (IP4), inositol-1,3,4,5,6-pentaphosphate (IP5), and inositol hexakisphosphate (IP6) using 150 mM HEPES pH 6.8 buffer supplemented with 2 mM dDTT, 2 mM ethylenediaminetetraacetic acid (EDTA), and 10% D2O in the absence or presence of n-nonyl-b-D-maltopyranoside (NM) at 298 K (Dovey et al., 2018).
Chemical shift perturbations (CSPs) in parts per million (ppm) were calculated as Euclidean distances with 20% weighting on nitrogen chemical shifts from 1H-15N resonance assignments. All-residue CSPs for detergents were used to determine significance cutoffs (σcorr) as previously described for localized interactions (Dou et al., 2012; Schumann et al., 2007). Briefly, CSPs greater than three times the standard deviation (SD) of all-residue CSPs were iteratively filtered until no more than 25% of data were excluded from the calculation of σcorr. Residue CSPs throughout NBB156 for IPs were similarly calculated and categorized based on a threshold of average CSP plus one standard deviation for significance. CSPs were mapped onto NBB156 structure, and cartoon putty radii were colored and scaled relative to global maximum CSP or inverse dissociation constant (1/KD) for graphical representation using PyMOL. The per-residue KD from 0–2 mM titration of IPs was calculated from nonlinear regression of CSP as a function of IP concentration with a quadratic binding model (Pollard, 2010; Teilum et al., 2017). Dissociation constants were compared by averaging across sites derived from similar affinities and shared structural elements.
Cell lines and western blotting
Stable expression of SV40 immortalized ripk3−/− mlkl−/− MEFs with WT and mutant NBB156 linked to two drug-inducible FKBP dimerization domains (2xF) and a Venus (V) tag (NBB156-2xFV) was designed similarly to the previously described NBB140-2xFV construct (McNamara et al., 2018; Quarato et al., 2016). Sf9 insect cells expressing FL MLKL were generated using established baculovirus methods to transfect bacmid DNA and initiate viral infection (Kalathur et al., 2016). Chemiluminescent western blotting using a polyclonal anti-GFP antibody was used to monitor protein expression, and Ponceau S staining and a monoclonal anti-actin antibody were used for loading controls.
Cell death assays
Necroptosis induced by WT or mutant NBB156-2xFV was monitored by fluorescent dye uptake using IncuCyte imaging (SYTOX Green) and FACS (PI) as previously described (McNamara et al., 2018; Quarato et al., 2016). Briefly, protein expression in ripkS/- mlkf−/− MEFs was induced by incubation with up to 200 ng/mL doxycycline (Dox) in the absence or presence of 25 nM B/B homodimerizer (Dim). SYTOX Green uptake was integrated and averaged over 4 fields-of-view per well on 24-well plates or 2 fields-of-view per well on 96-well plates in triplicate or quadruplicate. Counts per well were normalized by confluence measured using IncuCyte FLR and ZOOM software (Essen BioScience Inc.). Cells were trypsinized in wells at the end of 24 hours and stained with propidium iodide. FACS populations were analyzed in FlowJo (FlowJo, LLC).
Generation of CRISPR mutants
A wild-type clonal HT-29 line was derived by single-cell sorting using a BD Influx cell sorter following transduction with a lentivirus carrying the reverse tetracycline transactivator (TA) fusion protein (pLenti CMV rtTA3G Blast (R980-M38–658), gift of Dominic Esposito, Addgene #31797). Although the functionality of the TA system was not employed here, it was included in anticipation of potential downstream applications. Clonal mutant cell lines were generated from this cell line (“HT-29 ta”) as described previously (Dovey et al., 2018). Briefly, specific sgRNA sequences for IPMK (GCCCGGCCACCTGATGCGAG) (Dovey et al., 2018) and IPPK (TGAATGGGGGTACCACGGAG) were designed using the tool at http://crispr.mit.edu/ and cloned into pSpCas9(BB)-2A-GFP (PX458), a gift from Feng Zhang (Addgene #48138). The constructs (IPMK CRISPR construct: pCD623; IPPK CRISPR construct: pJD001) were transfected into HT-29 using Lipofectamine LTX, single-cell sorted using the BD Influx cell sorter, and cultured in conditioned media.
PCR sequencing was performed from genomic DNA as described (Marceau et al., 2016) and confirmation of the desired CRISPR editing was done prior to any downstream phenotypic analyses. Briefly, genomic region of ~500 bp encompassing the CRISPR sgRNA target site was PCR amplified and Sanger sequenced. We have routinely observed two types of data corresponding to clonal CRISPR mutants in HT-29 (Dovey et al., 2018). First, we find homozygous identical indels at a given locus. Second, we find multiple non-identical mutations. As described previously, we selected only clones containing frame-shift mutations at the sgRNA target site that were predicted to result in complete gene disruption. Sanger sequencing data were initially processed with the CRISP-ID software to decode multiple indel sequences (Dehairs et al., 2016). For downstream analyses, mutants were compared to the clonal wild-type control from which they were derived.
Cell viability measurements
Cell viability measurements were performed as described (Dovey et al., 2018). Recombinant human TNF-α, Smac mimetic RMT5265 (also referred to as SMAC007) (Li et al., 2004), and zVAD (TSZ) were used at 10 ng/mL, 10 nM, and 25 μM, respectively. Nec-1 was used at 30 μM. Briefly, HT-29 cells were seeded on the day preceding the experiment at 5,000 cells per well in 96-well tissue culture plates. Following induction with TSZ for 24 h, cell viability was assayed using the CellTiter-Glo assay (Promega) according to the manufacturer’s recommendations. Viability data are presented as a percentage compared to untreated control cells in each independent experiment performed three times with three biological replicates.
Mutagenesis and ligation-independent cloning
Mutagenesis of NBB156 and NBB156-2xFV was performed using the QuikChange II XL kit (Agilent Technologies) according to manufacturer’s instructions. Ligation-independent cloning was performed with hMLKL sequences amplified by PCR and the vectors pNIC28-Bsa4, pNIC-CH, or pFB-HGT-LIC (Strain-Damerell et al., 2014). Sequences were verified by Sanger sequencing.
LUVP assays
Lipid films with defined molar compositions were prepared using chloroform lipid stock solutions (Avanti Polar Lipids). LUVP assays primarily used lipids mimicking the plasma membrane containing 30% phosphatidylcholine (PC), 23% phosphatidylethanolamine, 4% phosphatidylinositol, 10% phosphatidylserine, and 25% sphingomyelin. Ni2+-affinity lipid 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (nickel salt) [DGS-NTA(Ni)] composed 8% of each LUV total lipid replacing corresponding % of PC when noted (Zheng et al., 2018). Phosphatidylinositol-4,5-bisphosphate was added to 1–4% molar composition where indicated. Nitrogen-dried films were further evaporated overnight under vacuum before storage under nitrogen at −20 °C.
Films were suspended in 10 mM HEPES pH 6.8, 200 mM KCl, and 5 mM MgCl2 assay buffer containing as the fluorophore-quencher pair 12.5 mM 8-aminonaphthalene-1,3,6-trisulfonic acid, disodium salt (ANTS) and 45 mM p-xylene-bis-pyridinium bromide (DPX) (Molecular Probes), by water bath sonication and then extruded to a homogenous population of LUVs through a 0.2-μm membrane (Avanti Polar Lipids) at least 30 times. LUVs were purified from free dyes by fast protein liquid chromatography on a packed S500 gel filtration column and stored under nitrogen at 4 °C in the dark. LUVP assays in 96-well format were prepared on ice, and kinetic fluorescence intensity measurements were recorded at 37 °C every 2 min for up to 90 min in a CLARIOstar monochromator microplate reader (BMG LABTECH GmbH).
LUVP data analysis involved normalization of ANTS fluorescence relative to the maximum fluorescence induced by 2.5% (w/v) CHAPS and minimum fluorescence measured relative to buffer control. Numerical integration with Simpson’s rule over even numbers of 2-min intervals approximated AUC for normalized LUVP data measured over defined ranges of time as previously described (Zheng et al., 2018). Data processing and analysis were done in Excel (Microsoft) and statistical analysis and presentation in Prism (GraphPad Software Inc.).
Analytical Ultracentrifugation
Sedimentation velocity experiments were conducted in a ProteomeLab XL-I analytical ultracentrifuge (Beckman Coulter) following standard protocols unless mentioned otherwise (Zhao et al., 2013). Samples in 150 mM HEPES pH 6.8, 2 mM DTT, 2 mM EDTA, with or without 2 mM IP6 were loaded into cell assemblies comprised of double sector charcoal-filled centerpieces with 12 mm path length and sapphire windows. Buffer density and viscosity were determined in a DMA 5000 M density meter and an AMVn automated micro-viscometer (Anton Paar), respectively. The partial specific volumes and molecular mass was calculated based on amino acid composition in SEDFIT (Cohn and Edsall, 1943). The cell assemblies, containing identical sample and reference buffer volumes of 390 μL, were placed in a rotor and temperature equilibrated at rest at 20 °C for 2 hours before acceleration from 0 to 50,000 rpm. Rayleigh interference optical data were collected at 1 min intervals for 12 hours. The velocity data were modeled with diffusion-deconvoluted sedimentation coefficient distributions c(s) in SEDFIT, using algebraic noise decomposition and with signal-average frictional ratio and meniscus position refined with non-linear regression (Schuck, 2000). The s-value was corrected for time and finite acceleration of the rotor was accounted for in the evaluation of Lamm equation solutions (Zhao et al., 2015). Maximum entropy regularization was applied at a confidence level of P-0.68. All plots were created in GUSSI (Brautigam, 2015) (kindly provided by Dr. Chad Brautigam).
Mass spectrometry
Protein samples were diluted to 2 mg/mL, and 20 μL samples were desalted in C4 or C18 Zip tips (MilliporeSigma). Desalted samples were loaded into nano-spray emitter tips for static nano-electrospray ionization and time-of-flight MS analysis (Waters LCT Premier XE MS, Waters). Mass spectra were analyzed in MaxEnt (Waters) software for peak mass identification corresponding to purified protein constructs.
Inositol Phosphate and Phosphoinositide Metabolic Labeling
WT and ΔIPPK cells were grown in DMEM media supplemented with 10% FBS and penicillin-streptomycin. Cells were then plated at 25,000 cells/well in inositol-free DMEM with 10% dialyzed FBS for 24 hours. The media was then supplemented with 50 μCi 3H-inositol (Perkin Elmer) and cells were grown for approximately 3 doublings (~4 days). Cells were then washed in PBS and harvested in 0.5 M HCl. Soluble IPs were separated from phosphoinositide lipids by adding 372 μL of CHCl3:methanol (1:2) and vortexed. Next, 125 μL of CHCl3 and 125 μL KCl was added to each sample followed by vortexing. The soluble (upper aqueous) and lipid (lower organic) fractions were separated by centrifugation. Soluble IPs were then run on HPLC as previously described (Otto and York, 2010). Lipids were then deacylated and separated by HPLC as previously described (Otto and York, 2010). Fractions of 1 mL were collected and mixed with 6 mL scintillation fluid. Disintegrations per minute (DPM) were measured by a liquid scintillation analyzer and data were normalized to total counts of radioactivity.
QUANTIFICATION AND STATISTICAL ANALYSIS
Summary data are plotted as averages with error bars of standard error of the mean (SEM) from means of independent experiments with specific details in each corresponding figure legend. Other data plots contributing to summary data are from experiments performed in triplicate or quadruplicate being represented as means with error bars of SD where indicated. Statistical analyses presented in all figures were performed as outlined on sets of mutants or experimental conditions compared by unpaired t-test with Welch’s correction or one-way ANOVA using the Tukey-Kramer method for multiple comparisons with two-tailed or multiplicity adjusted, family-wise 0.05 confidence level in Prism. P-values in all figures are indicated where **** P<0.0001, *** P<0.005, ** P<0.01, and * P<0.05.
DATA AND SOFTWARE AVAILABILITY
Atomic coordinates and assignments have been deposited in the PDB (6D74) and BMRB (30458).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GFP | Santa Cruz | Cat# sc-18334 RRID:AB_641123 |
| Anti-actin | MilliporeSigma | Cat# MAB1501 RRID: AB_2223041 |
| Anti-RIPK1 (pS166) | Cell Signaling Technology |
Cat# 65746 |
| Anti-RIPK3 (pS227) | Abcam | Cat# ab209384 RRID: AB_2714035 |
| Anti-MLKL (pS358) | Abcam | Cat# ab187091 RRID: AB_2619685 |
| Anti-MLKL | Abcam | Cat# ab184718 RRID: AB_2755030 |
| Anti-GAPDH | Santa Cruz | Cat# sc-32233 RRID: AB_627679 |
| Bacterial and Virus Strains | ||
| Escherichia coli strain: XL10-Gold | Agilent Technologies | Cat# C404010 |
| Escherichia coli strain: BL21 Star (DE3) | Thermo Fisher Scientific |
Cat# C601003 |
| Escherichia coli strain: T7 Express | New England Biolabs | Cat# C2566I |
| Escherichia coli strain: DH10EMBacY | Geneva Biotech | Cat#DM10EMBacY |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fetal Bovine Serum - Premium Select | Atlanta Biologicals | Cat# S11550 |
| DMEM, high glucose, no phosphates | Thermo Fisher Scientific |
Cat# 11971025 |
| Gibco L-glutamine | Thermo Fisher Scientific |
Cat# 25030081 CAS# 56-85-9 |
| Gibco Penicillin-Streptomycin | Thermo Fisher Scientific |
Cat# 15140122 CAS# 69-57-8, 57-92-1 |
| Gibco Sodium Pyruvate | Thermo Fisher Scientific |
Cat# 11360070 CAS# 113-24-6 |
| Gibco MEM Non-Essential Amino Acids Solution | Thermo Fisher Scientific |
Cat# 11140050 |
| Gibco 2-Mercaptoethanol | Thermo Fisher Scientific |
Cat# 21985023 CAS# 60-24-2 |
| 0.25% Trypsin, 0.1% EDTA in HBSS
w/o Calcium, Magnesium and Sodium Bicarbonate |
Corning | Cat# 25053CI |
| L-α-phosphatidylcholine (Egg, Chicken) | Avanti Polar Lipids | Cat# 840051C CAS# 97281-44-2 |
| L-α-phosphatidylinositol (Liver,
Bovine) (sodium salt) |
Avanti Polar Lipids | Cat# 840042C CAS# 383907-33-3 |
| L-α-phosphatidylserine (Brain, Porcine)
(sodium salt) |
Avanti Polar Lipids | Cat# 840032C CAS# 383907-32-2 |
| 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1- carboxypentyl)iminodiacetic acid)succinyl] (nickel salt) |
Avanti Polar Lipids | Cat# 790404C CAS# 231615-77-3 |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine | Avanti Polar Lipids | Cat# 850725C CAS# 4004-05-1 |
| L-α-phosphatidylcholine (Egg, Chicken) | Avanti Polar Lipids | Cat# 840051C CAS# 97281-44-2 |
| 18:0 octadecanoyl sphingomyelin
(Braine, Porcine) |
Avanti Polar Lipids | Cat# 860062C CAS# 383907-91-3 |
| L-α-phosphatidylinositol-4,5-bisphosphate
(Brain, Porcine) (ammonium salt) |
Avanti Polar Lipids | Cat# 840046X CAS# 383907-42-4 |
| D-myo-inositol-1,4,5-triphosphate (sodium salt) | Cayman Chemical | Cat# 10008205 CAS# 141611-10-1 |
| D-myo-inositol-1,3,4,6-tetraphosphate (ammonium salt) |
Cayman Chemical | Cat# 10008442 CAS# 142507-74-2 |
| D-myo-inositol-1,3,4,5,6-pentaphosphate
(sodium salt) |
Cayman Chemical | Cat# 10007784 CAS# 20298-95-7 |
| Phytic acid sodium salt | MilliporeSigma | Cat# P8810 CAS# 14306-25-3 |
| n-Hexyl-β-D-Glucopyranoside | Anatrace | Cat# H305 CAS# 59080-45-4 |
| n-Heptyl-β-D-Glucopyranoside | Anatrace | Cat# H300 CAS# 78617-12-6 |
| n-Octyl-β-D-Glucopyranoside | Anatrace | Cat# 0311 CAS# 29836-26-8 |
| Deuterated n-Octyl-β-D-Glucopyranoside (D24) | Cambridge Isotope Laboratories | Cat# DLM-6726-PK CAS# 29836-26-8 |
| n-Octyl-β-D-Thioglucopyranoside | Anatrace | Cat# O314 CAS# 85618-21-9 |
| Octyl Glucose Neopentyl Glycol | Anatrace | Cat# NG311 CAS# 1257853-32-9 |
| n-Nonyl-β-D-Glucopyranoside | Anatrace | Cat# N324 CAS# 69984-73-2 |
| n-Nonyl-β-D-Maltopyranoside | Anatrace | Cat# N330 CAS# 106402-05-5 |
| n-Decyl-β-D-Maltopyranoside | Anatrace | Cat# D322 CAS# 82494-09-5 |
| n-Undecyl-β-D-Maltopyranoside | Anatrace | Cat# U300 CAS# 253678-67-0 |
| n-Dodecyl-β-D-Maltopyranoside | Anatrace | Cat# D310 CAS# 69227-93-6 |
| n-Tridecyl-β-D-Maltopyranoside | Anatrace | Cat# T323 CAS# 93911-12-7 |
| n-Dodecyl-β-D-Thiomaltopyranoside | Anatrace | Cat# D342 CAS# 148565-58-6 |
| Necrosulfonamide | MilliporeSigma | Cat# 480073 CAS# 432531-71-0 |
| ANTS
(8-aminonpaphthalene-1,3,6-trisulfonic acid, disodium salt) |
Molecular Probes | Cat# 1278701 CAS# 5398-34-5 |
| DPX (p-xylene-bis-pyridiunium bromide) | Molecular Probes | Cat# X1525 CAS# 14208-10-7 |
| Propidium Iodide | Invitrogen | Cat# P3566 CAS# 25535-16-4 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat# L3000015 |
| Lipofectamine LTX | Thermo Fisher Scientific | Cat# 15338500 |
| Doxycycline | Clontech | Cat# 631311 CAS# 24390-14-5 |
| SYTOX Green | Thermo Fisher Scientific | Cat# S7020 CAS# 163795-75-3 |
| B/B Homodimerizer AP20187 | Takara | Cat# 635059 CAS# 195514-80-8 |
| Recombinant human TNF-α | Invivogen | Cat# Rhtnf-a |
| Smac mimetic RMT5265 (SMAC007) | (Li et al., 2004) | N/A |
| Z-VAD-FMK | Santa Cruz | Cat# sc-311561 |
| Nec-1 | Caymen Chemical | Cat# 11658 CAS# 4311-88-0 |
| 15N Ammonium Chloride | Cambridge Isotope Laboratories | Cat# NLM-467–10 CAS# 12125-02-9 |
| 13C Glucose | Cambridge Isotope Laboratories | Cat# CLM-1396–1 CAS# 50-99-7 |
| Deuterated DTT | Cambridge Isotope Laboratories | Cat# DLM-2622–1 |
| Deuterium Oxide | Sigma Aldrich | Cat# 617385–1 CAS# 7789-20-0 |
| Ponceau S | G-Biosciences | Cat# 786–576 |
| 3H-inositol | American Radiolabled Chemicals | Cat# ART-116A |
| ESF921 Insect Cell Culture Medium, Protein Free | Expression Systems, LLC | Cat# 96-001-01 |
| Transfection Medium | Expression Systems, LLC | Cat# 95-020-020 |
| Critical Commercial Assays | ||
| Supersignal West Dura | Thermo Fisher Scientific | Cat# 34075 |
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent | Cat# 200517 |
| CellTiter-Glo Assay | Promega | Cat# G7572 |
| Deposited Data | ||
| Human NBB156 structure | This paper | PDB: 6D74 BMRB: 30458 |
| Human NBB2–154 structure | Su et al.,2014 | PDB: 2MSV BMRB: 25135 |
| Mouse full-length MLKL structure | Murphy et al., 2013 | PDB: 4BTF |
| Experimental Models: Cell Lines | ||
| Mouse Embryonic Fibroblasts (MEFs)
ripk3−/− Mlkl−/− |
Dillon et al., 2014 | N/A |
| Human HT-29 | William Kaiser Laboratory | RRID: CVCL_0320 |
| Human HT-29 CRISPR mutants | This study | N/A |
| Spodoptera frugiperda (Sf9) insect cells | Expression Systems, LLC | Cat# 94–001F |
| Recombinant DNA | ||
| pRetroX-TRE3G | Clontech | Cat# 631188 |
| pNIC28-Bsa4 | Savitsky et al., 2010 | Addgene #26103 |
| pNIC-CH | Savitsky et al., 2010 | Addgene #26117 |
| pFB-HGT-LIC | Strain-Damerell et al., 2014 | N/A |
| Human MLKL in pNIC28-Bsa4 | Quarato et al., 2016 | N/A |
| Human MLKL and mutants in pRetroX-TRE3G | Quarato et al., 2016 | N/A |
| Human MLKL in pNIC-CH | This paper | N/A |
| Human MLKL in pFB-HGT-LIC | This paper | N/A |
| Software and Algorithms | ||
| CARA v1.9.1.7 | (Keller, 2004) | http://cara.nmr.ch/ |
| Chimera v1.10.2 (build 40686) | (Pettersen et al., 2004) | http://www.cgl.ucsf.edu/chimera |
| CNS v1.3 | (Brunger,
2007; Brunger et al., 1998) |
http://cns-online.org/v1.3/ |
| CYANA v2.1 | (Herrmann et al., 2002) | http://www.cyana.org |
| FlowJo X v10.0.7r2 | FlowJo, LLC | http://www.flowjo.com |
| GUSSI | (Brautigam, 2015) | http://biophysics.swmed.edu/MBR/software.html |
| IncuCyte 2011A Rev2 v20111.3.4288 | Essen BioScience, Inc. | http://www.essenbioscience.com |
| IncuCyte ZOOM 2016A v20161.1.5932.22771 | Essen BioScience, Inc. | http://www.essenbioscience.com |
| MacPyMOL v1.7.6.3 | Schrödinger, LLC | http://www.pymol.or9 |
| MOLMOL | (Koradi et al., 1996) | |
| Prism v7.0a | GraphPad Software, Inc. | http://www.graphpad.com |
| PROCHECK v3.5.4 | (Laskowski et al., 1993) | http://www.ebi.acuk/thornton-srv/software/PROCHECK |
| SEDFIT | (Schuck, 2000) | https://sedfitsedphat.nibib.nih.gov/software/default.aspx |
| TopSpin v3.5 | Bruker BioSpin | http://www.bruker.com |
| UNIO v2.02 | (Herrmann et al., 2002) | http://perso.ens-lyon.fr/torsten.herrmann/Herrmann/Software.html |
| HOLLOW v1.2 | (Ho and Gruswitz, 2008) | http://hollow.sourceforge.net/ |
| Other | ||
| Nickel agarose beads (high density) | Gold Biotechnology | Cat# H-320–500 |
| Amicon Ultra 15-mL 3K MWCO centrifugal filter | MilliporeSigma | Cat# UFC900324 |
| Amicon Ultra 15-mL 10K MWCO centrifugal filter | MilliporeSigma | Cat# UFC800324 |
| HiPrep Sephacryl S-100 HR column | GE Healthcare | Cat# 17116501 |
| Superdex 200 Increase 10/300 GL | GE Healthcare | Cat# 28990944 |
| Superdex 75 Increase 10/300 GL | GE Healthcare | Cat# 29148721 |
| MonoS 5/50 GL column | GE Healthcare | Cat# 17-5168-01 |
| HiPrep SP FF 16/10 | GE Healthcare | Cat# 28-9365-44 |
| HPLC with autosampler | Waters | Cat# WAT078905 |
| Partisphere SAX 4.6mm × 125mm | Whatman | Cat# 46210505 |
| Scintillation counter Tri-Carb | Packard | Cat# 2100TR |
| Scintillation fluid | Perkin Elmer | Cat# 509050584 |
SIGNIFICANCE.
The mechanism of plasma membrane rupture by MLKL is poorly understood. Early studies discovered RIPK3-mediated MLKL phosphorylation as major signaling event essential for MLKL activation. MLKL phosphorylation in psKD is thought to allosterically induce oligomerization into trimers or tetramers involving poorly understood rearrangements in its structure. Additionally, MLKL binds via NED several membrane lipids including phosphoinositides, and therefore lipids were thought to act as additional activators. Moreover, genetic and biochemical studies have identified IP kinases IPMK and ITPK1 as essential genes in necroptosis, through synthesis of IPs required for activation of NED. Even a RIPK3 phospho-mimetic hyperactive MLKL was unable to overcome deficiency in the IP code enzymes and metabolites. Despite these efforts, the activation mechanism remains unsatisfactorily resolved. Our study demonstrates in great detail that triggering of auto-inhibited NED by a selective repertoire of IPs is essential in MLKL activation. The auto-inhibition mechanism was validated in light of the NMR structure of NED to reveal a more extensive auto-inhibitory region than originally thought. The IP kinase IPPK, which converts IP5 to IP6, was directly implicated genetically in necroptosis regulation based on its deletion ablating cellular IP6 and significantly inhibiting necroptosis. We identified I(1,3,4,6)P4, I(1,3,4,5,6)P5, and IP6 as NED ligands. Remarkably, these IPs bind three sites to activate NED and FL MLKL. Importantly, we show that one of the best putative lipid ligands of MLKL, PI(4,5)P2, does not act as activator in the context of liposome permeabilization, suggesting that IP-mediated activation is essential upstream by opening up NED for lipid binding. Our study cements the role of the IP code in MLKL activation revealing innovative opportunities in targeting necroptosis.
HIGHLIGHTS.
MLKL N-terminal executioner domain (NED) is auto-inhibited by the linker-brace
Inositol pentakisphosphate 2-kinase (IPPK) synthesizes IP6 to regulate necroptosis
Selective inositol phosphate (IP) repertoire binds three distinct sites in NED
Hierarchical direct activation of MLKL is induced by three IPs (IP6>IP4≥IP5)
ACKNOWLEDGMENTS
We thank Kanisha Kavdia and Drs. Weixing Zhang and Paolo Rossi for assistance with mass spectrometry and NMR studies, and Prakhar Singal for assistance with preliminary studies as part of the Pediatric Oncology Education program at St. Jude. GSK (Drs. Peter Gough and John Bertin) is acknowledged for providing Smac mimetic RMT5265 (also referred to as SMAC007). This work was supported by the St. Jude Academic Programs Office Special Postdoctoral Fellowship (to D.E.M.), ALSAC, The David and Lucile Packard Foundation (to J.E.C.), and National Institutes of Health grants to D.R.G (R01CA169291, R01CA231620), A.T.H. (F30HL143826), J.D.Y. (R01GM124404), C.M.D. (T32AI007328), J.E.C. (DP2AI104557), and T.M. through the St. Jude Comprehensive Cancer Center Developmental Funds mechanism (P30CA021765).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alvarez-Diaz S, Dillon CP, Lalaoui N, Tanzer MC, Rodriguez DA, Lin A, Lebois M, Hakem R, Josefsson EC, O’Reilly LA, et al. (2016). The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 45, 513–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnez KH, Kindlova M, Bokil NJ, Murphy JM, Sweet MJ, and Guncar G (2015). Analysis of the N-terminal region of human MLKL, as well as two distinct MLKL isoforms, reveals new insights into necroptotic cell death. Biosci Rep 36, e00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asciolla JJ, Renault TT, and Chipuk JE (2012). Examining BCL-2 family function with large unilamellar vesicles. J Vis Exp 68, e4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babon JJ, and Murphy JM (2013). In vitro JAK kinase activity and inhibition assays. Methods Mol Biol 967, 39–55. [DOI] [PubMed] [Google Scholar]
- Brautigam CA (2015). Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data. Methods Enzymol 562, 109–133. [DOI] [PubMed] [Google Scholar]
- Brunger AT (2007). Version 1.2 of the Crystallography and NMR system. Nat Protoc 2, 2728–2733. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. (1998). Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, Readhead B, Dudley JT, Spangenberg EE, Green KN, et al. (2017). Necroptosis activation in Alzheimer’s disease. Nat Neurosci 20, 1236–1246. [DOI] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, and Liu ZG (2014). Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 16, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Kim S, and Snyder SH (2011). Inositol pyrophosphates as mammalian cell signals. Sci Signal 4, re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SC, and Majerus PW (2006). Inositol polyphosphate multikinase regulates inositol 1,4,5,6-tetrakisphosphate. Biochem Biophys Res Commun 339, 209–216. [DOI] [PubMed] [Google Scholar]
- Chang SC, Miller AL, Feng Y, Wente SR, and Majerus PW (2002). The human homolog of the rat inositol phosphate multikinase is an inositol 1,3,4,6-tetrakisphosphate 5-kinase. J Biol Chem 277, 43836–43843. [DOI] [PubMed] [Google Scholar]
- Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, et al. (2014). Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 24, 105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn EJ, and Edsall JT (1943). Density and apparent specific volume of proteins In Proteins, amino acids, and peptides, Cohn EJ, and Edsall JT, eds. (Princeton, NJ: Van Nostrand-Reinhold; ), pp. 370–381. [Google Scholar]
- Davies KA, Tanzer MC, Griffin MDW, Mok YF, Young SN, Qin R, Petrie EJ, Czabotar PE, Silke J, and Murphy JM (2018). The brace helices of MLKL mediate interdomain communication and oligomerisation to regulate cell death by necroptosis. Cell Death Differ 25, 1567–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Declercq W, Vanden Berghe T, and Vandenabeele P (2009). RIP kinases at the crossroads of cell death and survival. Cell 138, 229–232. [DOI] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, and Yuan J (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1, 112–119. [DOI] [PubMed] [Google Scholar]
- Dehairs J, Talebi A, Cherifi Y, and Swinnen JV (2016). CRISP-ID: decoding CRISPR mediated indels by Sanger sequencing. Sci Rep 6, 28973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick RA, Zadrozny KK, Xu C, Schur FKM, Lyddon TD, Ricana CL, Wagner JM, Perilla JR, Ganser-Pornillos BK, Johnson MC, et al. (2018). Inositol phosphates are assembly co-factors for HIV-1. Nature 560, 509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, et al. (2014). RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, et al. (2014). MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 7, 971–981. [DOI] [PubMed] [Google Scholar]
- Dou H, Buetow L, Sibbet GJ, Cameron K, and Huang DT (2012). BIRC7-E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat Struct Mol Biol 19, 876–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovey CM, Diep J, Clarke BP, Hale AT, McNamara DE, Guo H, Brown NW Jr., Cao JY, Grace CR, Gough PJ, et al. (2018). MLKL Requires the Inositol Phosphate Code to Execute Necroptosis. Mol Cell 70, 936–948 e937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo-Streeter S, Tsui MK, Odom AR, Block J, and York JD (2012). Structural studies and protein engineering of inositol phosphate multikinase. J Biol Chem 287, 35360–35369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick JP, Mattiske D, Wofford JA, Megosh LC, Drake LY, Chiou ST, Hogan BL, and York JD (2005). An essential role for an inositol polyphosphate multikinase, Ipk2, in mouse embryogenesis and second messenger production. Proc Natl Acad Sci U S A 102, 8454–8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, and Green DR (2017). ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 169, 286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez B, Schell MJ, Letcher AJ, Veprintsev DB, Irvine RF, and Williams RL (2004). Structure of a human inositol 1,4,5-trisphosphate 3-kinase: substrate binding reveals why it is not a phosphoinositide 3-kinase. Mol Cell 15, 689–701. [DOI] [PubMed] [Google Scholar]
- Gosein V, Leung TF, Krajden O, and Miller GJ (2012). Inositol phosphate-induced stabilization of inositol 1,3,4,5,6-pentakisphosphate 2-kinase and its role in substrate specificity. Protein Sci 21, 737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grootjans S, Vanden Berghe T, and Vandenabeele P (2017). Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ 24, 1184–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch AJ, and York JD (2010). SnapShot: Inositol phosphates. Cell 143, 1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann T, Guntert P, and Wuthrich K (2002). Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol 319, 209–227. [DOI] [PubMed] [Google Scholar]
- Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, Pierotti C, Garnier JM, Dobson RC, Webb AI, et al. (2014). Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A 111, 15072–15077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho BK, and Gruswitz F (2008). HOLLOW: generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct Biol 8, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes W, and Jogl G (2006). Crystal structure of inositol phosphate multikinase 2 and implications for substrate specificity. J Biol Chem 281, 38109–38116. [DOI] [PubMed] [Google Scholar]
- Huang D, Zheng X, Wang ZA, Chen X, He WT, Zhang Y, Xu JG, Zhao H, Shi W, Wang X, et al. (2017). The MLKL Channel in Necroptosis Is an Octamer Formed by Tetramers in a Dyadic Process. Mol Cell Biol 37, e00497–00416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannielli A, Bido S, Folladori L, Segnali A, Cancellieri C, Maresca A, Massimino L, Rubio A, Morabito G, Caporali L, et al. (2018). Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep 22, 2066–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine RF (2005). Inositide evolution - towards turtle domination? J Physiol 566, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine RF, and Schell MJ (2001). Back in the water: the return of the inositol phosphates. Nat Rev Mol Cell Biol 2, 327–338. [DOI] [PubMed] [Google Scholar]
- Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L, et al. (2016). RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, and Mocarski ES (2013). Viral modulation of programmed necrosis. Curr Opin Virol 3, 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalathur RC, Panganiban M, and Bruni R (2016). High-Throughput Baculovirus Expression System for Membrane Protein Production. Methods Mol Biol 1432, 187–202. [DOI] [PubMed] [Google Scholar]
- Keller R (2004). The computer aided resonance assignment tutorial. . (Cantina-Verlag; ). [Google Scholar]
- Koradi R, Billeter M, and Wuthrich K (1996). MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14, 51–55, 29–32. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Macarthur MW, Moss DS, and Thornton JM (1993). Procheck - a Program to Check the Stereochemical Quality of Protein Structures. J Appl Crystallogr 26, 283–291. [Google Scholar]
- Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et al. (2012). The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, and Harran PG (2004). A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science 305, 1471–1474. [DOI] [PubMed] [Google Scholar]
- Liu S, Li X, Zhang L, Jiang J, Hill RC, Cui Y, Hansen KC, Zhou ZH, and Zhao R (2017). Structure of the yeast spliceosomal postcatalytic P complex. Science 358, 1278–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Villalta PW, and Sturla SJ (2009). Simultaneous determination of inositol and inositol phosphates in complex biological matrices: quantitative ion-exchange chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom 23, 705–712. [DOI] [PubMed] [Google Scholar]
- Lupardus PJ, Shen A, Bogyo M, and Garcia KC (2008). Small molecule-induced allosteric activation of the Vibrio cholerae RTX cysteine protease domain. Science 322, 265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, and Bass BL (2005). Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science 309, 1534–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallery DL, Marquez CL, McEwan WA, Dickson CF, Jacques DA, Anandapadamanaban M, Bichel K, Towers GJ, Saiardi A, Bocking T, et al. (2018). IP6 is an HIV pocket factor that prevents capsid collapse and promotes DNA synthesis. Elife 7, e35335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, Swaminathan K, Mata MA, Elias JE, Sarnow P, et al. (2016). Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 535, 159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara DE, Quarato G, Guy CS, Green DR, and Moldoveanu T (2018). Characterization of MLKL-mediated Plasma Membrane Rupture in Necroptosis. J Vis Exp 138, e58088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GJ, Wilson MP, Majerus PW, and Hurley JH (2005). Specificity determinants in inositol polyphosphate synthesis: crystal structure of inositol 1,3,4-trisphosphate 5/6-kinase. Mol Cell 18, 201–212. [DOI] [PubMed] [Google Scholar]
- Mocarski ES, Guo H, and Kaiser WJ (2015). Necroptosis: The Trojan horse in cell autonomous antiviral host defense. Virology 479–480c, 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montpetit B, Thomsen ND, Helmke KJ, Seeliger MA, Berger JM, and Weis K (2011). A conserved mechanism of DEAD-box ATPase activation by nucleoporins and InsP6 in mRNA export. Nature 472, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453. [DOI] [PubMed] [Google Scholar]
- Najafov A, Chen H, and Yuan J (2017). Necroptosis and Cancer. Trends Cancer 3, 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalaskowski MM, Deschermeier C, Fanick W, and Mayr GW (2002). The human homologue of yeast ArgRIII protein is an inositol phosphate multikinase with predominantly nuclear localization. Biochem J 366, 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidhardt FC, Bloch PL, and Smith DF (1974). Culture medium for enterobacteria. J Bacteriol 119, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, Carano RA, Cao TC, van Bruggen N, Bernstein L, et al. (2016). RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ 23, 1565–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, Ye J, Zhang X, Chang A, Vakifahmetoglu-Norberg H, et al. (2015). Activation of necroptosis in multiple sclerosis. Cell Rep 10, 1836–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto JC, Kelly P, Chiou ST, and York JD (2007). Alterations in an inositol phosphate code through synergistic activation of a G protein and inositol phosphate kinases. Proc Natl Acad Sci U S A 104, 15653–15658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto JC, and York JD (2010). Molecular manipulation and analysis of inositol phosphate and pyrophosphate levels in Mammalian cells. Methods Mol Biol 645, 47–60. [DOI] [PubMed] [Google Scholar]
- Parisi LR, Li N, and Atilla-Gokcumen GE (2017). Very Long Chain Fatty Acids Are Functionally Involved in Necroptosis. Cell Chem Biol 24, 1445–1454. [DOI] [PubMed] [Google Scholar]
- Pearson JS, Giogha C, Muhlen S, Nachbur U, Pham CL, Zhang Y, Hildebrand JM, Oates CV, Lung TW, Ingle D, et al. (2017). EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat Microbiol 2, 16258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter ME (2011). Programmed cell death: Apoptosis meets necrosis. Nature 471, 310–312. [DOI] [PubMed] [Google Scholar]
- Petrie EJ, Czabotar PE, and Murphy JM (2019). The Structural Basis of Necroptotic Cell Death Signaling. Trends Biochem Sci 44, 53–63. [DOI] [PubMed] [Google Scholar]
- Petrie EJ, Sandow JJ, Jacobsen AV, Smith BJ, Griffin MDW, Lucet IS, Dai W, Young SN, Tanzer MC, Wardak A, et al. (2018). Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat Commun 9, 2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pittet D, Schlegel W, Lew DP, Monod A, and Mayr GW (1989). Mass changes in inositol tetrakis- and pentakisphosphate isomers induced by chemotactic peptide stimulation in HL-60 cells. J Biol Chem 264, 18489–18493. [PubMed] [Google Scholar]
- Pollard TD (2010). A guide to simple and informative binding assays. Mol Biol Cell 21, 4061–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, Wakefield R, Frase S, Moldoveanu T, and Green DR (2016). Sequential Engagement of Distinct MLKL Phosphatidylinositol-Binding Sites Executes Necroptosis. Mol Cell 61, 589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, Benson BL, Chirieleison SM, Huang AY, Dubyak GR, et al. (2018). Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol 3, eaat2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, Oberst A, Quarato G, Low J, Cripps JG, et al. (2016). Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ 23, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitsky P, Bray J, Cooper CD, Marsden BD, Mahajan P, Burgess-Brown NA, and Gileadi O (2010). High-throughput production of human proteins for crystallization: the SGC experience. Journal of structural biology 172, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanda P, and Brutscher B (2006). Hadamard frequency-encoded SOFAST-HMQC for ultrafast two-dimensional protein NMR. J Magn Reson 178, 334–339. [DOI] [PubMed] [Google Scholar]
- Scherer PC, Ding Y, Liu Z, Xu J, Mao H, Barrow JC, Wei N, Zheng N, Snyder SH, and Rao F (2016). Inositol hexakisphosphate (IP6) generated by IP5K mediates cullin-COP9 signalosome interactions and CRL function. Proc Natl Acad Sci U S A 113, 3503–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P (2000). Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78, 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann FH, Riepl H, Maurer T, Gronwald W, Neidig KP, and Kalbitzer HR (2007). Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J Biomol NMR 39, 275–289. [DOI] [PubMed] [Google Scholar]
- Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, Avanzi A, Barilla R, Daley D, Greco SH, et al. (2016). The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 532, 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, and Bax A (2009). TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strain-Damerell C, Mahajan P, Gileadi O, and Burgess-Brown NA (2014). Medium-throughput production of recombinant human proteins: ligation-independent cloning. Methods Mol Biol 1091, 55–72. [DOI] [PubMed] [Google Scholar]
- Stuart JA, Anderson KL, French PJ, Kirk CJ, and Michell RH (1994). The intracellular distribution of inositol polyphosphates in HL60 promyeloid cells. Biochem J 303 (Pt 2), 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L, Quade B, Wang H, Sun L, Wang X, and Rizo J (2014). A plug release mechanism for membrane permeation by MLKL. Structure 22, 1489–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. [DOI] [PubMed] [Google Scholar]
- Sun L, and Wang X (2014). A new kind of cell suicide: mechanisms and functions of programmed necrosis. Trends Biochem Sci 39, 587–593. [DOI] [PubMed] [Google Scholar]
- Szwergold BS, Graham RA, and Brown TR (1987). Observation of inositol pentakis- and hexakis-phosphates in mammalian tissues by 31P NMR. Biochem Biophys Res Commun 149, 874–881. [DOI] [PubMed] [Google Scholar]
- Tan X, Calderon-Villalobos LI, Sharon M, Zheng C, Robinson CV, Estelle M, and Zheng N (2007). Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature 446, 640–645. [DOI] [PubMed] [Google Scholar]
- Tanzer MC, Tripaydonis A, Webb AI, Young SN, Varghese LN, Hall C, Alexander WS, Hildebrand JM, Silke J, and Murphy JM (2015). Necroptosis signalling is tuned by phosphorylation of MLKL residues outside the pseudokinase domain activation loop. Biochem J 471, 255–265. [DOI] [PubMed] [Google Scholar]
- Teilum K, Kunze MB, Erlendsson S, and Kragelund BB (2017). (S)Pinning down protein interactions by NMR. Protein Sci 26, 436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, and Kaiser WJ (2017). DAI Another Way: Necroptotic Control of Viral Infection. Cell Host Microbe 21, 290–293. [DOI] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, and Feigenson GW (2008). Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, DeRose EF, London RE, and Shears SB (2014a). IP6K structure and the molecular determinants of catalytic specificity in an inositol phosphate kinase family. Nat Commun 5, 4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, and Wang X (2014b). Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54, 133–146. [DOI] [PubMed] [Google Scholar]
- Watson PJ, Fairall L, Santos GM, and Schwabe JW (2012). Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 481, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegner KW, Saleh D, and Degterev A (2017). Complex Pathologic Roles of RIPK1 and RIPK3: Moving Beyond Necroptosis. Trends Pharmacol Sci 38, 202–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinlich R, Oberst A, Beere HM, and Green DR (2017). Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 18, 127–136. [DOI] [PubMed] [Google Scholar]
- Xia B, Fang S, Chen X, Hu H, Chen P, Wang H, and Gao Z (2016). MLKL forms cation channels. Cell Res 26, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Kovalenko A, Bogdanov K, and Wallach D (2017). MLKL, the Protein that Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 47, 51–65. [DOI] [PubMed] [Google Scholar]
- Zhao H, Brautigam CA, Ghirlando R, and Schuck P (2013). Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr Protoc Protein Sci Chapter 20, Unit20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Ghirlando R, Alfonso C, Arisaka F, Attali I, Bain DL, Bakhtina MM, Becker DF, Bedwell GJ, Bekdemir A, et al. (2015). A multilaboratory comparison of calibration accuracy and the performance of external references in analytical ultracentrifugation. PLoS One 10, e0126420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JH, Grace CR, Guibao CD, McNamara DE, Llambi F, Wang YM, Chen T, and Moldoveanu T (2018). Intrinsic Instability of BOK Enables Membrane Permeabilization in Apoptosis. Cell Rep 23, 2083–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and assignments have been deposited in the PDB (6D74) and BMRB (30458).