

Abstract
Ample evidence has surfaced documenting the neurotoxic effects of various general anesthetic (GA) agents in the mammalian brain when administered at critical periods of synaptogenesis. However, little is known about how this neurotoxic insult affects persisting neuronal excitability after the initial exposure. Here we investigated synaptic activity and intrinsic excitability of the ventrobasal nucleus (VB) of the thalamus caused by neonatal GA administration. We used patch-clamp recordings from acute thalamic slices in young rats up to two weeks after neurotoxic GA exposure of isoflurane and nitrous oxide for 6 hours at postnatal age of 7 (P7) days. We found that GA exposure at P7 increases evoked excitatory postsynaptic currents (eEPSCs) two fold by means through AMPA mediated mechanisms, while NMDA component was spared. In addition, miniature EPSCs showed a faster decay rate in neurons from GA treated animals when compared to sham controls. Likewise, we discovered that the amplitudes of evoked inhibitory postsynaptic currents (eIPSCs) were increased in VB neurons from GA animals about two-fold. Interestingly, these results were observed in female but not male rats. In contrast, intrinsic excitability and properties of T-type voltage gated calcium currents were minimally affected by GA exposure. Together, these data further the idea that GAs cause lasting alterations in synaptic transmission and neuronal excitability depending upon the placing and connectivity of neurons in the thalamus. Given that function of thalamocortical circuits critically depends on the delicate balance between excitation and inhibition, future development of therapies aimed at addressing consequences of altered excitability in the developing brain by GAs may be an attractive possibility.
Keywords: glutamate, GABA, anesthesia, sedation, sensory processing
Introduction
A growing body of work has begun to document electrophysiological changes in neuronal populations following widespread neuronal injury. Indeed, functional alterations in neuronal electrical activity may serve as a bridge between histological markers of cell death and larger scale functional and behavioral modifications produced by neurotoxicity. For example, various groups have reported alterations in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor functionality due to subunit rearrangement following neurotoxic injury in various models (Kwak & Weiss, 2006; Liu & Zukin, 2007; Rivera-Cervantes et al., 2014). Many groups have also examined alterations in gamma amino-butyric acid (GABA)ergic transmission in post ischemia models (Alia et al., 2016; Mayor & Tymianski, 2018). These findings among other contribute to the idea that surviving neurons after injury can alter their functionality, perhaps as a way to compensate for lost neurons in the network.
Each year millions of people undergo general anesthesia (GA) for surgical purposes. In the past few decades ample evidence has surfaced that neonatal exposure to anesthetic agents produces robust neurotoxic effects in rodent and non-human primate animal models through activation of apoptotic pathways (V. Jevtovic-Todorovic, Beals, Benshoff, & Olney, 2003; Yon, Daniel-Johnson, Carter, & Jevtovic-Todorovic, 2005). This neurotoxic insult, which especially damages the hippocampus and thalamic nuclei when administered at the peak of synaptogenesis at the age of P7, has been shown to cause lasting impairment to cognitive functions, learning and memory (Jevtovic-Todorovic et al., 2003).
While we have seen convincing histological and molecular evidence of apoptotic neurotoxicity, along with clear behavioral deficits in animals treated neonatally with GAs, little is known about how this neurotoxic insult affects neuronal excitability after the initial exposure. Recently, our lab investigated GABAergic neurons in the nucleus reticularis thalami (nRT)(DiGruccio et al., 2015), the main inhibitory structure in the thalamus. We found nRT neurons to be hyperexcitable for up to 2 weeks past initial exposure to GAs. These findings showed a decrease in inhibitory synaptic transmission, an increase in excitatory transmission, and increases in activity modulated by voltage gated T-type calcium channels (T-channels). Furthermore, we found that pharmacologically induced spike-and-wave discharges in ensuing electroencephalographic (EEG) recordings in vivo increased in GA-exposed rats. Importantly, we found that neuronal hyperexcitability after GA exposure was reversed with selective antagonism of T-channels ex vivo and in vivo. Hence, we hypothesized that increased excitability of nRT neurons in GA-treated rats may lead to increased inhibitory synaptic currents in their projection neurons in ventrobasal nucleus (VB) of the thalamus.
As the thalamus is involved in the sensory processing and modulation of a vast amount of neuronal signals, it is important to deepen our understanding of how GA-induced neurotoxicity affects function of this structure. Our current study seeks to further characterize functional changes in neurons in the VB nucleus, which largely consists of excitatory glutamatergic neurons that project to the sensory cortex, and is heavily innervated by the nRT. Both nRT and VB are part of thalamocortical network that plays an important role in consciousness, somatosensation, sleep cycles, and pain perception (Ab Aziz & Ahmad, 2006; Pigeat, Chausson, Dreyfus, Leresche, & Lambert, 2015; Soares Potes, Lourença Neto, & Manuel Castro-Lopes, 2006; Sun et al., 2017). Our results support the idea that early exposure to common GAs may cause alterations in the functionality of the thalamocortical circuit beyond the duration of histological markers of apoptosis, which in turn may contribute to the disorders of sensory processing and cognitive deficits during brain development. These findings show functional differences in thalamic neurons from rodents treated with GAs at an early age, and provide a stronger foundation for future development of therapies aimed at addressing consequences of anesthetic neurotoxicity and altered excitability in thalamocortical circuit of the developing brain.
Materials and Methods
Animal care:
Animals were housed in compliance with IACUC protocols through UC Denver, Anschutz Msdical Campus in Aurora on a 12 hour light- dark cycle. Sprague-Dawley rat mothers were ordered through Envigo (Indianapolis, IN) to arrive with litters of pups at postnatal day 4-6.
Anesthesia:
As vulnerability to anesthesia-induced apoptosis is peak at postnatal day 7 (P7), (Yon et al., 2005), pups were separated from their mother at P7, sexed, and randomly assigned to experimental and control (sham) groups. The experimental group animals were placed in an anesthesia chamber equilibrated to (in atm) .75% isoflurane (Iso) and 75% nitrous oxide in 24-25% oxygen for 6 hours, while controls were allowed to breathe room air as a sham anesthesia in an identical type of chamber for 6 hours. An agent-specific vaporizer delivered isoflurane, and dedicated air tanks of N2O and O2 were mixed to achieve desired volatile anesthetic concentrations. During the exposure, gas levels in anesthetic chambers (CO2, O2, N2O, Iso) were measured constantly with a Datex Capnomac Ultima and both sham and GA pups were warmed with a heating blanket adjusted to 35° C. Pups were regularly checked for hypoxia using a MouseOx Plus pulse oximeter (Starr Life Sciences Corp, Oakmont, PA) at the end of exposures to ensure that results would not be confounded by hypoxia. After exposure, both sham and GA animals were returned to the mother and allowed to recover. It is worth mentioning that in our previous study in nRT we administrated 9 mg/kg of midazolam intraperitoneally (i.p.) prior to the exposure with isoflurane and N2O for 6 hours (DiGruccio et al., 2015). However, in our current study done at relatively high altitude, we discontinued the use of midazolam in order to decrease pup mortality during exposures.
Brain slice preparation:
In this study, we used mostly female rats for all electrophysiological and molecular experiments, as females exhibited more drastic changes in plasticity than males in preliminary experiments and are generally underrepresented in previous studies of the thalamocortical circuit. GA-exposed and sham rats were sacrificed for electrophysiology recordings at a timepoint when histological signs of neurotoxicity have faded: from postnatal day 12 to 21 for all recordings except miniature post synaptic inhibitory currents (mIPSCs), which were taken from animals up to P25. For most recordings, both a sham and GA animal were sacrificed on the same day to provide age matched control and treatment recordings. Each type of recording presented contains animals from at least 3 separate litters to account for variability between litters.
Pups were deeply anesthetized with isoflurane and decapitated for brain tissue extractions. Their brains were rapidly extracted and placed in an ice cold solution containing in mM, as follows: sucrose 260, D-glucose 10, NaHCO3 26, NaH2PO4 1.25, KCl 3, CaCl2 2, MgCl2 2. Brains were glued ventral side-up to a pedestal and 300 micron slices were cut with a Leica VT 1200S vibratome (Leica Biosystems, Buffalo Grove, IL). Slices were placed in a 37°C solution containing, in mM: NaCl 124, D-glucose 10, NaHCO3 26, NaH2PO4 1.25, KCl 4, CaCl2 2, MgCl2 2. During incubation, slices were constantly perfused with a gas mixture of 95 vol% O2 and 5 vol% CO2. Slices were incubated for 30 minutes and then equilibrated to room temperature, at which electrophysiology experiments were performed.
Whole cell electrophysiology in acute brain slice preparation ex vivo
For miniature excitatory postsynaptic currents (mEPSC) and current clamp recordings, thalamocortical neurons were visualized and using an Olympus BX51WI and signals were amplified with a Multiclamp 200B amplifier (Molecular Devices, Foster City, CA, USA). Recordings of evoked excitatory postsynaptic currents (eEPSCs), evoked inhibitory postsynaptic currents eIPSCS, miniature inhibitory postsynaptic currents (mIPSCs), and spontaneous inhibitory postsynaptic currents (sIPSCs) were amplified with a Multiclamp 700B amplifier. For voltage clamp recordings of T-type calcium currents, VB neurons were visualized under Zeiss optics (Zeiss AXIO Examiner D1, 40x objective) and signals were amplified with a Multiclamp 700B amplifier. All signals were digitized using a Digidata 1440a (Molecular Devices) and filtered at 2 kHz.
Glass microelectrodes (Sutter Instruments, borosilicate glass with filament OD 1.2 mm) were pulled using a Sutter Instruments P-1000 model and fabricated to maintain an initial resistance of 2.5–6 MΩ. Voltage current commands and digitization of the resulting voltages and currents were performed with Clampex 8.3 software (Molecular Devices) running on a PC-compatible computer. Resulting voltage traces were analyzed using Clampfit 10.5 (Molecular Devices).
The external solution for voltage- and current-clamp electrophysiology experiments consisted of the following (in mM), unless otherwise noted: NaCl 125, D-glucose 25, NaHCO3 25, NaH2PO4 1.25, KCl 2.5, MgCl2 1, CaCl2 2. For voltage clamp recordings of synaptic events (IPSCs and EPSCs), cells were held at −70mV. Cells were excluded from analysis if their access resistance rose above 25 MΩ or if their holding current varied by more than 20%. To evoke currents for eEPSCs and eIPSCs we used a bipolar concentric tungsten electrode (Harvard Apparatus, Holliston, MA). Specifics for each type of electrophysiology recording are as follows:
Excitatory Currents
We recorded eEPSCs in the presence of 20 μM picrotoxin and 100 μM CGP35348 in the bath to block inhibitory transmission, while mEPSCs were recorded in the presence of 1 μM tetrodotoxin (TTX) and 20 μM picrotoxin. As mEPSC frequency is fairly low in the VB (typically <1 Hz), we doubled extracellular potassium (raised to 5 mM in bath) in order to increase sample sizeof events. The internal solution used in the pipette was potassium-D-gluconate 130, EGTA 5, NaCl 4, CaCl2 0.5, HEPES 10, Mg-ATP 2, Tris-GTP 0.5, pH 7.2. Recordings of eEPSCs were performed in an external solution containing 0.5 mM MgCl2, as opposed to the 1 mM concentration used for most other recordings. Lidocaine N-ethyl chloride (QX-314) in 5 mM concentration was included in the internal solution to prevent interference from voltage gated sodium channels in the recorded neuron. eEPSC decay time constant (tau) was fitted with a triexponential equation, which fit the curve better than a mono- or bi-exponential fit, as follows,
where τw is weighted tau, An is amplitude, and τη tau.
Inhibitory Currents:
All inhibitory currents were recorded in the presence of 10 μM 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline (NBQX) and 50 μM DL-2-amino-5-phosphonopentanoic acid (AP-V) to block excitatory neurotransmission. We used an internal solution containing (in mM) KCl 130, NaCl 4, CaCl2 0.5, EGTA 5, HEPES 10, Mg-ATP 2, Tris-GTP 0.5, and QX-314 5. QX-314 was omitted for recordings of mIPSCs, as TTX was present in the bath for these recordings to block action potentials. When recording eIPSCs, we raised the concentrations of MgCl2 and CaCl2 each to 3 mM to decrease the frequency of spontaneous events, allowing for more accurate calculation of current kinetics. Time constants of IPSC decay were analyzed with a biexponential fit, using the aforementioned equation with the variables A2 and τ3 excluded.
Current Clamp
For current-clamp recordings cells were excluded from analysis if resting membrane potential (RMP) was more depolarized than −45 mV. Original RMP for each cell was recorded and then cells were injected with small amounts of current (if necessary) to bring them to around −60 mV for excitability studies. A potassium gluconate internal solution equal to that used for mEPSCs was used for current clamp recordings. Liquid junction potential was between 5 and 10 mV and was not corrected in our presentation of this data. As a measure of tonic firing rate we ran an input-output protocol, injecting 100 pA of positive current for 250 ms followed by stepwise increases in current injection by 25 pA for a total of 15 sweeps. Additionally, we ran a hyperpolarization protocol which injected −50 pA of current for 500 ms and the allowed the cell to return to the RMP. This was repeated with each sweep increasing the injected current by −50 pA until either 15 sweeps were recorded or the cell’s potential reached too low to safely continue recording (generally ~−150 mV).
Voltage-clamp recordings
For voltage-clamp recordings of T-type calcium currents, Tetrodotoxin (TTX; 1uM) was added to the extracellular medium as a voltage-dependent sodium current blocker. The internal solution for voltage-clamp experiments consisted of the following (in mM): Cs-methanesulfonate 110, phosphocreatine 14, HEPES 10, EGTA 9, Mg-ATP 5, and Tris-GTP 0.3, pH adjusted to 7.15 to 7.20 with CsOH (standard osmolarity: 300 mOsm) (Stamenic & Todorovic, 2018).
T-channel activation was measured by stepping the membrane potential from an initial holding potential (Vh) of −90 mV to test potentials (Vt) from −80 to −40 mV in 2.5 mV increments over a period of 320 ms. Current-voltage (I-V) curves were generated, and peak current amplitudes and inactivation properties of current waveforms were established. We calculated current densities by measuring average peak current divided by the capacitance of the neuron. Steady-state inactivation curves were generated by using a standard double-pulse protocol with 3.6-s-long prepulses to variable voltages (from −120 to −60 mV in 5 mV increments) and test potentials to −50 mV. The voltage dependencies of activation and steady-state inactivation were described with single Boltzmann distributions of the following forms:
In these forms, Imax is the maximal amplitude of current, Gmax is the maximal conductance (calculated by dividing current amplitude by estimated reversal potential), V50 is the voltage at which half of the current is activated or inactivated, and k represents the voltage dependence (slope) of the distribution.
To assess percentage of recovery from inactivation we used a paired-pulse protocol in which a 200 ms step to −50 mV was used to inactivate all of the T-currents. After a variable recovery interval (from 2 to 10000 ms) at either −90 or −120 mV, a second step to −50 mV was used to determine the amount of T-current that had recovered from the inactivation during the recovery protocol.
Quantitative real time-PCR
After receiving a sham or GA exposure, P21 rat pups were deeply anesthetized with isoflurane and transcardially perfused with ice cold phosphate buffered saline (PBS). Brains were rapidly extracted and the thalamus was exposed and removed for RNA extraction using dissection forceps. Samples were flash frozen in liquid nitrogen and stored at −80 degrees C. We isolated RNA from the thalamus using an RNeasy kit (Qiagen, Germantown, MD), after which cDNA was synthesized with an iScript reverse transcriptase kit (Bio-Rad, Hercules, CA). RNA and cDNA were measured for yield and purity spectrophotometrically.
PCR plates were run using primers for Gria1, Gria2, Gria3, Gria4 and housekeeping gene Gapdh. Primers were obtained from Bio-Rad and melting curves were checked on each plate to assess specificity. A CFX Connect rt-PCR Detection System (Bio-Rad) was used for quantification of sample cDNA. Each reaction well contained 100 ng of sample cDNA template, 7 μL nuclease-free water, 1 μL 20xPrimePCRAssay (primer), and 10 μL 2x SsoAdvanced universal SYBR Green supermix (Bio-Rad) for a final reaction volume of 20 μL. A PCR protocol was run consisting of the following parameters:
Denaturation/Annealing/Extension:
40 cycles. Each consisting of 5 sec at 95° C followed by 30 seconds at 60°
Melting Curve:
1 cycle of 5 step increments starting at 65°C and increasing temperature by 0.5 °C until 95° was reached
For each cDNA sample, cycle threshold (Cq) was determined for target and housekeeping gene in triplicate reactions, and if outliers were found in each triplicate they were excluded from analysis. ΔCq was determined for each target gene by calculating the difference in Cq between each target gene and Gapdh, which were run on the same plate. ΔCq for each sample (sham and GA) was normalized to the average of the sham Gria1 ΔCq values, and are presented as a ratio.
Analysis
pCLAMP software 10.0 (Molecular Devices) was used to determine current characteristics (amplitude, paired pulse ratio, kinetics), while Minianalysis (Synaptosoft, Fort Lee, NJ) was used for analysis of spontaneous synaptic events (mIPSCs, sIPSCs, mEPSCs). Statistical and graphical analyses were performed using GraphPad Prism 7.0 software (GraphPad Software) or Origin 2018 (OriginLab).
Statistical tests were performed using two-way factorial Analysis of Variance (ANOVA) with repeated measures and a Fisher’s least significant difference (LSD) post hoc test where appropriate. Otherwise, a two-tailed unpaired t-test was used to assess statistical significance. Results are presented as means ± SEM unless stated otherwise.
Results
Excitatory synaptic transmission
Evoked EPSCs
The VB nucleus receives excitatory input from the cortex via the internal capsule (IC) and from the periphery via the medial lemniscus (ML). As we have previously shown increased excitatory input in the nRT by stimulating the IC, we wanted to investigate whether GA-induced alterations in plasticity could occur in other thalamic afferent pathways by stimulating the ML. Using a concentric bipolar tungsten electrode, we stimulated the ML with a 300 ms interval paired-pulse protocol to elicit a large, “all-or-nothing” response with slight paired-pulse depression, consistent with previous characterization of this circuitry (Castro-Alamancos, 2002). Data presented are currents shown at threshold stimulus, as increasing the stimulus past threshold had little-to-no effect on current amplitude. The average threshold stimulus required to evoke an excitatory response by stimulating the ML in our preparation was 18.54 ± 2.17 mA in the sham group, and 23.25 ± 1.84 mA in the GA group (t(44)=1.608, p=.115).
Analysis of eEPSCs showed a significant increase in current amplitude of GA-treated animals of about two-fold when compared to sham controls (Figure 1A,B). Comparison of sham and GA neurons also yielded a significant decrease in decay time constant (tau) in the GA group of about 50% (Figure 1C,D). In contrast, paired pulse ratio (PPR) was unaffected by GA exposure (Figure 1E,F). This led us to speculate that the shown changes in decay and amplitude were due to either presynaptic fiber excitability, and/or postsynaptic mechanisms not involved in short-term plasticity. In contrast, increase in amplitude and decrease in decay time constant was not observed in age- matched male rats (sham group: n=11 neurons in 6 animals, amplitude = 619±114 pA, tau = 18.5±3.5 ms, threshold stimulus=21,1±1.8 mA; GA group n=11 neurons in 5 animals, amplitude = 468±78 pA, tau = 20.9±2.6 ms, threshold stimulus=19.8±2.6 mA; amplitude p=.304, tau p=.576, threshold stimulus p=.681).
Figure 1: Neonatal GA exposure increases eEPSC amplitudes in VB neurons.

A Representative “all or nothing” eEPSC traces held at −70 mV from a GA treated animal and an animal that received a sham exposure, showing a two-fold increase in eEPSC current amplitude in GA-exposed animals, expressed in bar graph B (sham n=13 cells in 6 animals, GA n=12 cells in 5 animals; t(23)=2.186, *p=.039). C Representative traces from A scaled to equal amplitudes to show the decrease in decay time constant (tau), shown in bar graph D (t(23)=2.845, **p=.009). E Though current amplitudes and decay rates were altered, paired pulse ration (PPR) was not significantly different between the sham and GA treated animals, as shown in bar graph F.
To test the hypothesis that the alterations in receptor functionality could underly the observed phenomena in eEPSCs, we investigated the relative contribution of AMPA and NMDA receptors by applying an AMPA antagonist NBQX, and an NMDA antagonist AP-V to some neurons from sham and GA-exposed animals. Original traces from a representative VB neuron exposed to NBQX and AP-V are depicted on Figure 2A. We then subtracted the resulting AP-V-sensitive current from the overall eEPSC to find the amplitude of the NBQX-sensitive portion and compared results between sham and GA animals. Summary of these experiments are depicted on the bar graph of Figure 2B, and traces from representative neurons from GA and sham-treated rats are depicted on Figure 2C. Interestingly, the NBQX-sensitive (AMPA) portion of the current was significantly increased in VB neurons from GA animals about two-fold when compared to sham controls, whereas the NBQX-insensitive (NMDA) component was spared. This led us to conclude that functional alterations in postsynaptic AMPA receptors were likely responsible for increased eEPSC amplitude observed in GA animals.
Figure 2. Neonatal GA exposure alters AMPA/NMDA ratio due to an increase in AMPA-mediated portion of currents.

A Representative trace showing the experimental protocol of a recorded eEPSC (GA-treated animal), the EPSC after bath application of NBQX, and the complete blockade of currents from combined bath application of NBQX and APV in the same cell. B Comparison of the NBQX sensitive and NBQX insensitive portions of the current amplitude shows an increase of the amplitude of current sensitive to NBQX in cells from GA treated animals (sham n=5 cells in 3 rats, GA n=7 cells in 5 rats; t(30)=2.934, **p=.006). The NBQX resistant portion of the current is unaltered between the two groups (t(30)=.078, p=.938). C Representative traces from GA treated animals (green color) and sham group (gray color) that display findings in graph B.
mEPSCs
In order to further investigate GA-induced changes in AMPA receptor functionality, we recorded spontaneous synaptic activity in the presence of tetrodotoxin (TTX) and picrotoxin to inhibit voltage-gated sodium currents and GABA-gated currents, respectively. Cells were held at-70 mV where NMDA currents would be electrically inactivated, so any observed events were mostlikely due to AMPA receptor response to spontaneous glutamate vesicle release. Traces from the representative neurons illustrate characteristic mEPSCs on Figures 3A and 3B. Summary graphs from these experiments depicted on Figure 3C-F illustrate that we observed a moderate decrease in mEPSC decay tau of about 25% (Figure 3C-D) in neurons from GA animals, while event frequency (Figure 3E) and amplitude (Figure 3F) were not significantly altered. It is important to mention that mEPSCs represent action potential-independent recordings from all synaptic inputs, while in our eEPSC recordings we stimulated only one of two main efferent pathways, which may provide some explanation for the seeming incongruity of the two recording types.
Figure 3: VB mEPSCs exhibit faster decay in cells from GA-treated animals.

A Representative mEPSC recordings in VB neurons from sham or GA animals showing no difference in event frequency or amplitude, as expressed in graphs E and F. However traces displayed on panel B, which represents the averaged values of all events in the cells used for trace A, illustrate the acceleration in mEPSC decay time caused by GA exposure. C Analysis of cell averages shows a significant decrease in decay tau in neurons from GA-treated animals (sham n= 9 cells from 5 animals, GA n=10 cells in 6 animals; t(17)= 2.975, **p=.008). D A cumulative probability plot of individual event decay tau values reveals an overall leftward shift towards a faster decay rate of mEPSCs in neurons from GA treated animals (sham n=1242 events, GA n=1331 events).
Global thalamic expression of AMPA receptor subunit mRNA
As an increase in AMPA current amplitude was also previously shown in nRT neurons after GA exposure (DiGruccio et al., 2015), we speculated that the neurotoxic insult from GA exposure could cause thalamus-wide alterations in AMPA receptor activity. To provide a possible molecular mechanism for this phenomenon, we assessed expression of AMPA receptor subunit mRNAs in samples taken from the whole thalamus through quantitative RT-PCR. We found that GA exposure did not significantly impact expression of Gria1, Gria2, Gria3 and Gria4 (Figure 4). However, bar graphs on Figure 4 indicate that we were able to confirm that Gria2 and Gria4 were the most abundantly expressed AMPA subunit mRNA in the thalamus of young female rats. This falls in line with previous work investigating distribution of AMPA receptor subunits in the thalamus (Mineff & Weinberg, 2000).
Figure 4: Global thalamic mRNA expression of AMPA receptor subunits is unaffected by GA exposure.
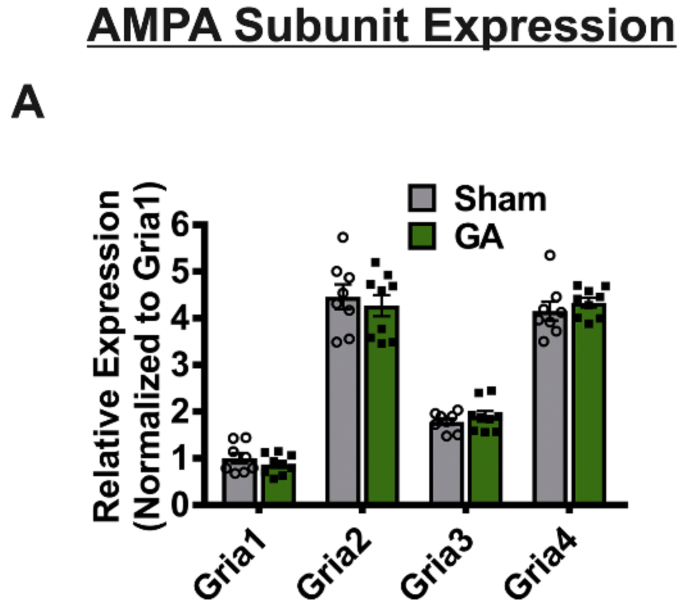
A A graph depicting relative mRNA expression of Gria1, Gria2, Gria 3, and Gria 4 shows that subunit expression in sham animals is not statistically different from GA treated animals (n=8 sham and 9 GA; F (1,60) = 0.002, p=0.962). Overall, relative expression level of each subunit’s mRNA is significant when normalized to Grial’s ΔCq value (F (3, 60) = 233.8, P<0.0001).
Inhibitory neurotransmission
eIPSCs
The nRT provides a main inhibitory tone to the VB thalamus and plasticity of its ionic currents after anesthesia exposure has been well studied in our previous report (DiGruccio et al., 2015). To gain deeper insight into how neurotoxic insult could affect modulatory mechanisms in surviving thalamocortical glutamatergic neurons, we decided to investigate GABAergic transmission between the nRT and VB. In the presence NBQX and AP-V which block excitatory synaptic transmission, an electrode placed in the nRT produced a robust inhibitory current in VB neurons. To assess inhibitory transmission mechanisms, we performed a paired pulse protocol with 250 ms between pulses, starting at very low levels of stimulation. Stimulation was increased until a threshold eIPSC was evoked in the VB neuron, after which neuronal responses were tested at 2 through 5 times the threshold current (Figure 5A,B). The average threshold stimulus for eIPSCs was 708.3 ± 484.5 μA in the sham group and 330.3 ± 113.2 in the GA group (t(27)=1.135, p=.262). Analysis of resulting current amplitudes across the stimulus intensities showed a significant increase in eIPSCs in GA treated animals of about two-fold when compared to the sham control group (Figure 5B). In contrast, average values for PPR depicted on Figure 5C, and decay time constant depicted on Figure 5D showed no significant change between two cohorts. While female rats showed increased amplitude across the I-O protocol, male rats exhibited no significant changes (p=.993) between the sham (n=14 neurons in 5 animals; 1x to 5x threshold currents: 309±107, 535±132, 586±77, 729±188, 784±132 pA) and GA cohorts (n=16 neurons in 7 animals; 1x-5x threshold currents: 162±31, 548±107, 654±147, 752±133, 821±139 pA). Threshold stimulus required to evoke an eIPSC was not statistically different among the male cohorts (408±126 μA for sham, 432±127 μA for GA (p=.941).
Figure 5: VB neurons from GA exposed animals exhibit increased eIPSC amplitude.
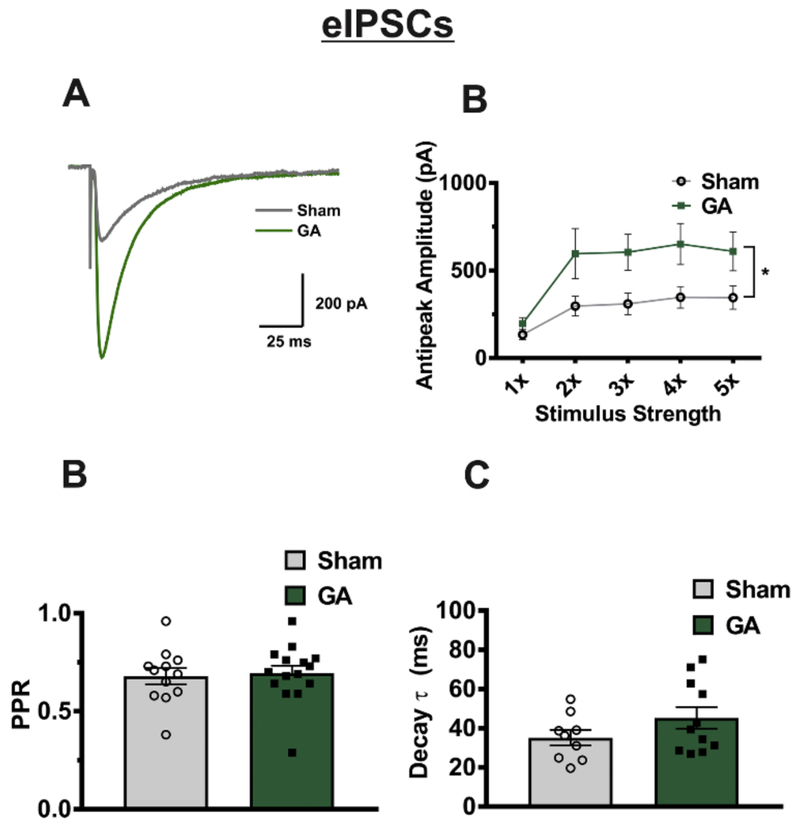
A Representative recordings of eIPSCs at 5x threshold stimulation in neurons from sham (gray trace) and GA (green trace) animals B Cells from GA treated animals show in average a two-fold increase of current amplitudes compared to sham controls (sham n=11 cells in 6 animals, GA n=11 cells in 5 animals; F (1,20)= 5.38, *p=.031) across all stimulus intensities. C While amplitude increased in GA animals, paired pulse ratio (PPR) was not significantly different between the two groups (t(25)= .264, p=.794). D Decay tau was likewise unaffected by GA exposure (t(18)= 1.437, p=.168).
m/sIPSCs
To further investigate mechanisms of GA-induced inhibitory plasticity, we recorded spontaneous inhibitory synaptic events that are summarized on Figure 6. We found that mIPSCs recorded in the presence of TTX showed minimal changes in all parameters tested (amplitude, frequency, and decay tau). As expected, baseline amplitudes of sIPSC recorded without TTX were larger and more frequent than mIPSCS. However, comparison of tested sIPSC parameters showed no significant difference in neurons from sham and GA-treated cohorts. Based on these data, we speculate that changes in eIPSC amplitude could be due to increased excitability of presynaptic fibers, but without alterations in probability of vesicular release.
Figure 6. Miniature and spontaneous IPSCs are unaffected by neonatal GA exposure.

A,B Representative traces from an mIPSC and sIPSC, respectively, recorded from two different VB neurons (sham animal). Averages from multiple VB cells show no significant difference between sham and GA treated animals in event frequency for both mIPSCs (sham n=13 cells from 6 animals, GA n=11 cells from 5 animals; t(22)=.152, p=.880) and sIPSCs (sham n=13 cells from 7 animals, GA n=9 cells from 5 animals; t(20)=.639, p=.53). E GA exposure likewise has no significant effect on amplitude of mIPSCs (t(22)=.541, p=.594) or sIPSCs (t(20)=.348, p=.731) G Decay tau is also unaffected by GA exposure in mIPSCs (t(22)= .063, p=.950) and sIPSCs (t(20)=.987, p=.336) D, F, and H are respective cumulative probability plots of said parameters illustrating the lack of change in mIPSCs (sham n= 1733 events, GA n=1186 events) and sIPSCs (sham n=915 events, GA n=1204 events).
Intrinsic Excitability
Current-clamp studies
As the study had focused primarily on synaptic input to the VB thalamus, we also wanted to explore the possibility that GA exposure could modify the output of these neurons as we previously reported in the nRT (DiGruccio et al., 2015). It is well known that most of the thalamic neurons exhibit depolarization-induced tonic firing pattern and post-inhibitory rebound burst firing mode (Perez Velazquez & Carlen, 1996). Hence, in current-clamp mode, we utilized two recording protocols to assess neuronal excitability. The first injected stepwise positive current pulses to artificially raise membrane potential, allowing us to investigate tonic firing, while the second injected negative current pulses to investigate functionality of T-channel dependent activity. The hyperpolarizing pulse removes inactivation of T-channels, thereby allowing for a “rebound” low threshold calcium spike (LTCS) and subsequent rebound burst firing of one or more action potentials (APs).
As depicted in Figure 7A-D, we found that exposure to GA had no significant effect on the active membrane properties such as tonic firing rate and number of APs fired in rebound burst mode. Furthermore, no significant differences were found in LTCS amplitude (Figure 7E) and latencyto LTCS between sham and GA groups (sham n=9 neurons in 5 rats, GA n=9 neurons in 6 rats; p>.05 for all values). Finally, we did not find any significant alterations in passive membrane properties such as RMP and the membrane input resistance between sham and GA groups (Figures 7G and 7H, respectively).
Figure 7. Intrinsic excitability of VB neurons is not altered by GA exposure.

A Figure showing select steps and representative traces of input-output protocol (sham cohort). B A graph depicting no statistical difference in the average firing rates of VB neurons across all depolarizing current injections between cells from sham and GA animals (sham n=19 neurons in 8 animals, GA n=19 neurons in 8 animals; F(2,41)=. 167, p=.847) C, Figure showing hyperpolarization, rebound burst firing, and LTCS (above) due to hyperpolarizing current injection (below) in the same neuron from a sham group. D In neurons from sham and GA treated animals, analysis of rebound burst action potentials across steps of hyperpolarization shows no difference across current injections (sham n=19 cells from 8 animals, GA n= 17 cells from 8 animals; F(2,38) = .093, p=.911). E Bar graph of −200 pA current injection to illustrate variability and spread of LTCS action potential number F Analysis of latency to rebound action potential likewise showed no difference between sham and GA groups (p>.05 at all current injections). G Resting membrane potential is not altered between experimental (GA) and control (sham) groups (t(26)=0.574, p=.571). H Input resistance is similar between sham and GA groups (p>.05 for all current injections).
Minimal alterations of voltage-dependent T-channel inactivation properties in VB neurons after exposure to GA during brain development
Our previous study demonstrated significant up-regulation of T-currents in nRT neurons after exposure to GA at P7 that was accompanied with a depolarizing shift in voltage-dependent inactivation kinetics (DiGruccio et al., 2015). Hence, we compared kinetic properties and amplitudes of T-currents in VB neurons from control (sham) animals and animals exposed to GA at the age of P7. Representative traces showing T-current recovery from inactivation from GA animal at −90 mV (recovery interval from 20 to 100 ms - top and from 200 to 1000 ms - bottom) are presented at Figure 8A. Figures 8B and 8C show average data points and fitted values for recovery from inactivation at −90 or −120 mV, respectively in the sham and GA exposed animals. The time constants presented in Figures 8B and 8C summarize the effects of sham and GA animals on T-currents with regards to recovery from inactivation at membrane potentials of −120 and −90 mV, respectively. The recovery from inactivation was significantly faster in SHAM animals at −120 mV, while at −90 mV that difference was not present. Figure 8D shows plots for the average steady-state inactivation curves and average V50 values −75.9 ± 0.5 mV (gray curve, sham animals, 5 cells) and −73.0 ± 0.8 mV (green curve, GA animals, 6 cells). The small depolarizing shift of 2.9 mV in GA animals in steady-state inactivation was statistically significant. Inset in Figure 8D shows no significant difference in the T-current densities recorded at a prepulse potential of −120 mV. In contrast to voltage-dependent inactivation, there were no difference in voltage-dependence of steady-state activation of VB neurons between two cohorts: Figure 8E shows average steady-state activation curves and average V50 values −62.2 ± 0.5 mV (gray curve, sham animals, 6 cells) and −62.5 ± 0.6 mV (green curve, GA animals, 5 cells). Inset in Figure E shows no difference in the T-current densities recorded at a test potential of −50 mV between GA and sham groups.
Figure 8. Minimal alterations in T-current biophysical properties due to GA exposure: recovery from inactivation, inactivation and activation of isolated T-currents in VB neurons.

A Representative traces showing T-current recovery from inactivation from GA animal at −90 mV (recovery interval from 20 to 100 ms - top and from 200 to 1000 ms - bottom). Graphs B and C show recovery from inactivation at −90 and −120 mV, respectively, in sham and GA exposed animals. The fitted values for time constants presented in Figures B and C summarize the effects of neonatal GA exposure on T-currents with regards to recovery from inactivation at membrane potentials of −120 and −90 mV, respectively. The recovery from inactivation was faster in cells from sham animals at −120 mV (t(10) = 2.47, p = 0.033), while at −90 mV that difference was lost (t(10) = 1.02, p = 0.333). D Plots for the average steady-state inactivation curves and average V50 values −75.9 ± 0.5 mV (gray curve, sham animals, 5 cells) and −73.0 ± 0.8 mV (green curve, GA animals, 6 cells). The depolarizing shift of 2.9 mV in GA animals in steady-state inactivation was statistically significant (t(9) = 2.84, p = 0.019). Inset in Figure D shows no difference in current densities between two groups recorded at a conditioning potential of −120 mV. E Plots show that there was no significant difference in voltage-dependence of steady-state activation of VB neurons between GA and sham cohorts. The average V50 values was −62.2 ± 0.5 mV (sham, grey curve, n= 6 cells) and −62.5 ± 0.6mV (GA, green curve n= 5 cells) (t(9) = 0.30, p = 774). Inset in Figure E shows no significant difference in current densities between two groups recorded at a test potential of −50 mV.
Discussion
The ability of sedative/anesthetic drugs to induce safe and reversible loss of consciousness is of paramount importance. However, recent data from in vivo animal models have suggested that GAs are neurotoxic to the developing mammalian brain and have implicated them in causing cognitive deficits later in life. Every year tens of millions of patients, including very young children, are exposed to GAs, which underscores the importance of studies to address current safe practices of anesthesia. Recent animal data in rodents and non-human primates strongly suggest that common GAs may harm the developing mammalian brain by inducing neuroapoptosis, and cause lasting cognitive deficits (Brambrink et al., 2010, 2012; V. Jevtovic-Todorovic et al., 2003; Zou et al., 2009, 2011). Although human studies addressing the issue of safety of clinical anesthesia in the developing brain still are not conclusive, at least some concerns have been raised (Flick et al., 2009). Further research is warranted to elucidate cellular mechanisms for long-lasting effects of currently available GAs on neuronal function and to develop possible therapeutic strategies that could be used to make clinical anesthesia practice safer.
Many studies have established that most commonly used classes of GAs can cause widespread neuroapoptosis in the developing brain which, depending on the model and anesthetic used, can be detected 3 to 24 hours post exposure (Brambrink et al., 2010, 2012; V. Jevtovic-Todorovic et al., 2003; Zou et al., 2009, 2011). Much less is known about functional disturbances of individual neurons and networks that survived acute exposure to GAs during critical developmental age. Specifically, the possibility that common GAs may cause lasting alterations of ion channels that control neuronal excitability in the thalamocortical loop has not been taken into consideration. However, we addressed this issue in our recent studies using ex vivo and in vivo approaches to demonstrate that rats exposed to GAs (a clinically-relevant combination of midazolam, isoflurane, and nitrous oxide (N2O) at postnatal day 7 (P7) display lasting neuronal hyperexcitability of nRT neurons (DiGruccio et al., 2015; Joksovic, Lunardi, Jevtovic-Todorovic, & Todorovic, 2015). Specifically, we found that a single 6 hour-long exposure to GAs at P7, the critical age for brain development, caused an increase in excitatory synaptic transmission, decrease in inhibitory synaptic transmission and concomitant increase in the amplitude of T- currents in nRT neurons. Collectively, this plasticity of ionic currents leads to increased action potential firing ex vivo and increased strength of pharmacologically-induced spike and wave discharges in vivo as determined by electroencephalographic (EEG) recordings in adolescent rats (DiGruccio et al., 2015). This could offer at least in part, an explanation for our findings that inhibitory currents evoked from the nRT are larger in VB neurons of GA-exposed animals. Furthermore, it is well documented that thalamic dysfunction in humans may underlie disorders of cognition, sleep and wakefulness, tinnitus, neurogenic pain, as well as epilepsy—collectively termed “thalamocortical dysrhythmias” (Walton & Llinás, 2010). Hence, we examined here plasticity of inhibitory and excitatory synaptic transmission in VB thalamic neurons, the main target area for the projections of nRT neurons.
To our knowledge, this represents the first study to investigate the electrophysiology of the VB thalamus of young female rats and provides applications in the fields of anesthesia, neurotoxicity, and thalamocortical circuitry. We decided to focus the study on female subjects as preliminary recordings found the female VB to be more sensitive to GA exposure in terms of synaptic transmission, driving our interest to the female thalamus. From a toxicity standpoint, this is likely the first study to investigate functionality of surviving thalamocortical neurons populations in the VB nucleus in any post-neurotoxicity model. The VB’s role in thalamocortical circuitry has been well studied, as it helps to regulate consciousness (and thus anesthetic endpoints) as well as the transmission of peripheral noxious and non-noxious stimuli to cortical brain regions.
Our finding that neonatal GA neurotoxicity causes lasting changes in AMPA currents is of particular interest, as many other groups have documented similar plasticity in AMPA receptors in post-neonatal neuronal insult models (for a review, see Kwak & Weiss, 2006). Often, an increase in decay kinetics as we saw is linked to an increase in Ca2+ permeability via the removal of the GluA2 subunit, which renders the receptor impermeable to Ca2+ions when present (Geiger et al., 1995). Though we found no significant differences in mRNA levels of Gria2, it is known that the GluA2 subunit undergoes RNA editing (Isaac, Ashby, & McBain, 2007), which may be a post-translational mechanism to investigate in future studies. Additionally, mRNA editing, alterations in AMPA receptor splice variants, local insertion of receptors into the membrane, and interactions with accessory proteins could all be investigated as post-translational mechanisms behind altered excitatory transmission after GA exposure (Salussolia & Wollmuth, 2012). As previous groups have shown, differential distribution of AMPA receptor subunits in various thalamic nuclei may account for the lack of change we observed when investigating global thalamic mRNA expression (Mineff & Weinberg, 2000). Thus, future molecular investigation of GA-induced AMPA alteration should take nucleus specificity into account when assessing mechanisms of AMPA receptors in the thalamus. As we found an increase in amplitudes of eEPSCs but not mEPSCs, there may be an interesting interplay of presynaptic and postsynaptic mechanisms that contribute to altered glutamatergic transmission after GA exposure.
It has been shown that upregulation of the NMDA NR1 subunit occurs 6-24 hours after acute ketamine exposure in primate and murine cortical culture (Liu et al., 2013; Wang et al., 2006). If the same occurs in vivo, then the data presented may be recorded from a time point when fluctuations in NMDA receptor activity and composition have ceased. The present study, while perhaps lacking in its ability to capture the mechanistic component of NMDA alterations directly after exposure, provides interesting evidence that AMPA current functionality is altered in a more long-term manner. We speculate that more transient NMDA changes may play a role in causing the lasting increase in thalamic AMPA currents, which would be an interesting topic to pursue in future studies.
Nucleus and ion channel-specific alterations due to GA exposure comprise another interesting aspect of this study. Our previous exploration of the nRT showed robust changes in evoked synaptic excitation similar to what we see in the VB. However, we observed much more prominent alteration in evoked inhibitory currents in the VB, while we found considerably less changes in the functionality of T-channels in the VB presently than in the nRT. As the VB is rich in a mostly homogenous population of the Cav3.1 isoform of T-channels while the nRT is comprised mostly of Cav3.2 and Cav3.3 isoforms (Talley et al., 1999), it is possible that the different isoforms of T-channels exhibit distinct adaptation to GA-induced neurotoxicity. As output of measured neurons in the VB was minimally affected by GAs while we found the nRT’s output to be robustly increased, it would be of interest to investigate how these relative changes in excitability contribute to the overall functionality of corticothalamic circuitry in vivo and how that could be implicated in neuropathological conditions. For example, it would be expected that stronger eEPSCs in VB would lead to enhanced tonic firing mode in GA animals, while stronger eIPSCs could lead to enhanced burst firing mode in vivo in GA-treated animals since resulting inhibitory postsynaptic potentials would be expected to deinactivate T-channels in VB neurons. Somewhat slower recovery from inactivation of T-currents that we observed in VB from GA animals may represent a compensatory mechanism to constrain synaptic-driven hyperexcitability, but it is unlikely sufficient to offset the effects of large increase in eIPSCs and eEPSCs.
It is also possible that the discrepancies in the plasticity of ionic currents in GA animals between the current VB study and our previous nRT study (DiGruccio et al., 2015) could be explained by differential sensitivities between males and females to each anesthetic, and/or mode of delivery (e.g. use of midazolam). A future study consisting of an electrophysiological survey in various brain regions nuclei with a consistent anesthesia regimen would be able to more accurately assess whether excitability parameters and synaptic transmission in different nuclei/brain regions are preferentially affected by neonatal GA treatment.
Previous studies have identified AMPA receptors as a potential target for the treatment of absence seizures (Citraro et al., 2017; Hu, Cortez, Man, Wang, & Snead, 2001). Interestingly, we found that animals exposed neonatally to anesthesia experience higher severity of absence-like seizures induced by administration of gamma butyrolactone (GBL) (DiGruccio et al., 2015). As dysregulation of network excitability in the corticothalamic circuit is highly implicated in the pathology of absence seizures (Hu et al., 2001), it follows that a therapeutic which acts on multiple components of the circuit could prove effective as treatment. Now that our studies in two separate thalamic nuclei (VB and nRT) now shown increased AMPA receptor functionality after GA exposure, the AMPA receptor antagonists appear to be promising as a pharmacological therapies for treating absence seizures following GA exposure. Further experiments involving in vivo pharmacological and EEG studies would be required to scrutinize this notion.
Conclusion.
Our studies may provide a rationale for new strategies to improve cognitive dysfunctions or other unidentified persisting changes in the function of excitatory and inhibitory synaptic transmission in thalamocortical circuitry after GA exposure. Understanding the effects of GAs on different classes of ion channels that are crucial to controlling the excitability and synaptic transmission in the thalamus could be used to develop novel and potentially safer GAs that could be better tailored to individual needs.
Highlights.
We investigated hypothesis that exposure to general anesthesia at early age may cause lasting plasticity of ion channels that regulate excitability of thalamocortical neurons in ventrobasal thalamic nucleus.
Amplitudes of AMPA-mediated evoked excitatory and GABA-mediated inhibitory synaptic currents were increased in female, but not in male adolescent rats following a single exposure to general anesthesia with isoflurane and nitrous oxide at age of postnatal day 7.
Amplitude of T-type calcium channels, NMDA synaptic currents, properties of tonic and rebound burst firing were not altered in animals exposed to general anesthesia when compared to sham groups.
Pharmacological targeting of altered synaptic transmission may be a novel therapeutic approach for treating cognitive impairment after exposure to general anesthesia during brain development.
Acknowledgements
This study was funded in part by grants from the National Institutes of Health (GRANT# R01GM102525 to S.M.T.). We thank Dr. Steven Mennerick on critical evaluation of our manuscript. We thank Dr. Shelby Chastain and Dr. Vesna Tesic on technical assistance with molecular studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors received no compensation nor do they have any conflicting financial interests in regards to the work described in this manuscript.
References
- Ab Aziz CB, & Ahmad AH (2006). The role of the thalamus in modulating pain. Malaysian Journal of Medical Sciences, 13(2), 11–18. 10.1056/NEJMra1313875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alia C, Spalletti C, Lai S, Panarese A, Micera S, & Caleo M (2016). Reducing GABA A-mediated inhibition improves forelimb motor function after focal cortical stroke in mice. Scientific Reports, 6(November), 1–15. 10.1038/srep37823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambrink AM, Back SA, Riddle A, Gong X, Moravec MD, Dissen GA, … Olney JW (2012). Isoflurane-induced apoptosis of oligodendrocytes in the neonatal primate brain. Annals of Neurology, 72(4), 525–535. 10.1002/ana.23652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambrink AM, Evers AS, Avidan MS, Farber NB, Smith DJ, Zhang X, … Olney JW (2010). Isoflurane-induced Neuroapoptosis in the Neonatal Rhesus Macaque Brain. Anesthesiology, 112(4), 834–841. 10.1097/ALN.0b013e3181d049cd [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA (2002). Properties of primary sensory (lemniscal) synapses in the ventrobasal thalamus and the relay of high-frequency sensory inputs. Journal of Neurophysiology, 10.1013/jphysiol.2001.013283 [DOI] [PubMed] [Google Scholar]
- Citraro R, Leo A, Franco V, Marchiselli R, Perucca E, De Sarro G, & Russo E (2017). Perampanel effects in the WAG/Rij rat model of epileptogenesis, absence epilepsy, and comorbid depressive-like behavior. Epilepsia, 58(2), 231–238. 10.1111/epi.13629 [DOI] [PubMed] [Google Scholar]
- DiGruccio MR, Joksimovic S, Joksovic PM, Lunardi N, Salajegheh R, Jevtovic-Todorovic V, … Todorovic SM (2015). Hyperexcitability of Rat Thalamocortical Networks after Exposure to General Anesthesia during Brain Development. Journal of Neuroscience, 35(4), 1481–1492. 10.1523/JNEUROSCI.4883-13.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flick RP, Wilder RT, Sprung J, Katusic SK, Voigt R, Colligan R, … Warner DO (2009). Anesthesia and cognitive performance in children: no evidence for a causal relationship. Are the conclusions justified by the data? Response to Bartels et al., 2009. Twin Research and Human Genetics : The Official Journal of the International Society for Twin Studies, 12(6), 611–2; discussion 613-4. 10.1375/twin.12.6.611 [DOI] [PubMed] [Google Scholar]
- Geiger JRP, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, & Monyer H (1995). Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron, 15(1), 193–204. 10.1016/0896-6273(95)90076-4 [DOI] [PubMed] [Google Scholar]
- Hu RQ, Cortez MA, Man HY, Wang YT, & Snead OC (2001). Alteration of GLUR2 expression in the rat brain following absence seizures induced by gamma-hydroxybutyric acid. Epilepsy Research, 44(1), 41–51. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11255072 [DOI] [PubMed] [Google Scholar]
- Isaac JTR, Ashby M, & McBain CJ (2007). The Role of the GluR2 Subunit in AMPA Receptor Function and Synaptic Plasticity. Neuron, 54(6), 859–871. 10.1016/j.neuron.2007.06.001 [DOI] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, Beals J, Benshoff N, & Olney JW (2003). Prolonged exposure to inhalational anesthetic nitrous oxide kills neurons in adult rat brain. Neuroscience, 122(3), 609–616. 10.1016/j.neuroscience.2003.07.012 [DOI] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zbrumski CF, … Wozniak DF (2003). Early Exposure to Common Anesthetic Agents Causes Widespread Neurodegeneration in the Developing Rat Brain and Persistent Learning Deficits. The Journal of Neuroscience, 23(3), 876–882. 10.1523/JNEUROSCI.23-03-00876.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joksovic PM, Lunardi N, Jevtovic-Todorovic V, & Todorovic SM (2015). Early Exposure to General Anesthesia with Isoflurane Downregulates Inhibitory Synaptic Neurotransmission in the Rat Thalamus. Molecular Neurobiology, 52(2), 952–958. 10.1007/S12035-015-9247-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak S, & Weiss JH (2006). Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Current Opinion in Neurobiology, 16(3), 281–287. 10.1016/j.conb.2006.05.004 [DOI] [PubMed] [Google Scholar]
- Liu F, Patterson TA, Sadovova N, Zhang X, Liu S, Zou X, … Wang C (2013). Ketamine-induced neuronal damage and altered N-methyl-D-aspartate receptor function in rat primary forebrain culture. Toxicological Sciences: An Official Journal of the Society of Toxicology, 131(2), 548–557. 10.1093/toxsci/kfs296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, & Zukin RS (2007). Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends in Neurosciences, 30(3), 126–134. 10.1016/j.tins.2007.01.006 [DOI] [PubMed] [Google Scholar]
- Mayor D, & Tymianski M (2018). Neurotransmitters in the mediation of cerebral ischemic injury. Neuropharmacology, 134, 178–188. 10.1016/j.neuropharm.2017.11.050 [DOI] [PubMed] [Google Scholar]
- Mineff EM, & Weinberg RJ (2000). Differential synaptic distribution of AMPA receptor subunits in the ventral posterior and reticular thalamic nuclei of the rat. Neuroscience, 101(4), 969–982. 10.1016/S0306-4522(00)00421-8 [DOI] [PubMed] [Google Scholar]
- Perez Velazquez JL, & Carlen PL (1996). Development of firing patterns and electrical properties in neurons of the rat ventrobasal thalamus. Developmental Brain Research, 91(2), 164–170. 10.1016/0165-3806(95)00171-9 [DOI] [PubMed] [Google Scholar]
- Pigeat R, Chausson P, Dreyfus FM, Leresche N, & Lambert RC (2015). Sleep Slow Wave-Related Homo and Heterosynaptic LTD of Intrathalamic GABAAergic Synapses: Involvement of T-TypeCa2+ Channels and Metabotropic Glutamate Receptors. Journal of Neuroscience, 35(1), 64–73. 10.1523/JNEURQSCI.2748-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Cervantes MC, Castañeda-Arellano R, Castro-Torres RD, Gudiño-Cabrera G, y Velasco AIF, Camins A, & Beas-Zárate C (2014). P38 MAPK Inhibition Protects Against Glutamate Neurotoxicity and Modifies NMDA and AMPA Receptor Subunit Expression. Journal of Molecular Neuroscience. 10.1007/s12031-014-0398-0 [DOI] [PubMed] [Google Scholar]
- Salussolia CL, & Wollmuth LP (2012). Flip-flopping to the membrane. Neuron, 76(3), 463–465. 10.1016/j.neuron.2012.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikandar S, West SJ, McMahon SB, Bennett DL, & Dickenson AH (2017). Sensory processing of deep tissue nociception in the rat spinal cord and thalamic ventrobasal complex. Physiological Reports, 5(14), e13323 10.14814/phy2.13323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares Potes C, Lourença Neto F, & Manuel Castro-Lopes J (2006). Inhibition of pain behavior by GABABreceptors in the thalamic ventrobasal complex: Effect on normal rats subjected to the formalin test of nociception. Brain Research, 1115(1), 37–47. 10.1016/j.brainres.2006.07.089 [DOI] [PubMed] [Google Scholar]
- Stamenic TT, & Todorovic SM (2018). Cytosolic ATP Relieves Voltage-Dependent Inactivation of T-Type Calcium Channels and Facilitates Excitability of Neurons in the Rat Central Medial Thalamus. Eneuro, 5(1), ENEURO.0016-18.2018. 10.1523/ENEURO.0016-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LN, Li XL, Wang F, Zhang J, Wang DD, Yuan L, … Qi JS (2017). High-intensity treadmill running impairs cognitive behavior and hippocampal synaptic plasticity of rats via activation of inflammatory response. Journal of Neuroscience Research, 95(8), 1611–1620. 10.1002/jnr.23996 [DOI] [PubMed] [Google Scholar]
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, & Bayliss DA (1999). Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 19(6), 1895–1911. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10066243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton KD, & Llinás RR (2010). Central Pain as a Thalamocortical Dysrhythmia: A Thalamic Efference Disconnection? Translational Pain Research: From Mouse to Man. CRC Press/Taylor & Francis; Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/21882456 [PubMed] [Google Scholar]
- Wang C, Sadovova N, Hotchkiss C, Fu X, Scallet AC, Patterson TA, … Slikker W (2006). Blockade of N-methyl-D-aspartate receptors by ketamine produces loss of postnatal day 3 monkey frontal cortical neurons in culture. Toxicological Sciences, 91(1), 192–201. 10.1093/toxsci/kfj144 [DOI] [PubMed] [Google Scholar]
- Yon JH, Daniel-Johnson J, Carter LB, & Jevtovic-Todorovic V (2005). Anesthesia induces neuronal cell death in the developing rat brain via the intrisic and extrinsic apoptotic pathways. Neuroscience, 135(3), 815–827. 10.1016/j.neuroscience.2005.03.064 [DOI] [PubMed] [Google Scholar]
- Zou X, Liu F, Zhang X, Patterson TA, Callicott R, Liu S, … Wang C (2011). Inhalation anesthetic-induced neuronal damage in the developing rhesus monkey. Neurotoxicology and Teratology, 33(5), 592–597. 10.1016/j.ntt.2011.06.003 [DOI] [PubMed] [Google Scholar]
- Zou X, Patterson TA, Divine RL, Sadovova N, Zhang X, Hanig JP, … Wang C (2009). Prolonged exposure to ketamine increases neurodegeneration in the developing monkey brain. International Journal of Developmental Neuroscience, 27(7), 727–731. 10.1016/J.IJDEVNEU.2009.06.010 [DOI] [PubMed] [Google Scholar]