

Abstract
Rationale:
Understanding and manipulating the cardiomyocyte cell cycle has been the focus of decades of research, however the ultimate goal of activating mitotic activity in adult mammalian cardiomyocytes remains elusive and controversial. The relentless pursuit of controlling cardiomyocyte mitosis has been complicated and obfuscated by a multitude of indices used as evidence of cardiomyocyte cell cycle activity that lack clear identification of cardiomyocyte “proliferation” versus cell cycle progression, endoreplication, endomitosis, and even DNA damage. Unambiguous appreciation of the complexity of cardiomyocyte replication that avoids oversimplification and misinterpretation is desperately needed.
Objective:
Track cardiomyocyte cell cycle activity and authenticate fidelity of proliferation markers as indicators of de novo cardiomyogenesis in post-mitotic cardiomyocytes.
Methods and Results:
Cardiomyocytes expressing the FUCCI construct driven by the α-myosin heavy chain promoter were readily and uniformly detected through the myocardium of transgenic mice. Cardiomyocyte cell cycle activity peaks at postnatal day 2 and rapidly declines thereafter with almost all cardiomyocytes arrested at the G1/S cell cycle transition. Myocardial infarction injury in adult hearts prompts transient small increases in myocytes progressing through cell cycle without concurrent mitotic activity, indicating lack of cardiomyogenesis. In comparison, cardiomyogenic activity during early postnatal development correlated with coincidence of FUCCI and cKit+ cells that were undetectable in the adult myocardium.
Conclusions:
Cardiomyocyte-specific expression of Fluorescence Ubiquitination-based Cell Cycle Indicators (FUCCI) reveals previously unappreciated aspects of cardiomyocyte cell cycle arrest and biological activity in postnatal development and in response to pathologic damage. Compared to many other methods and model systems, the FUCCI transgenic (FUCCI-Tg) mouse represents a valuable tool to unambiguously track cell cycle and proliferation of the entire cardiomyocyte population in the adult murine heart. FUCCI-Tg provides a desperately needed novel approach in the armamentarium of tools to validate cardiomyocyte proliferative activity that will reveal cell cycle progression, discriminate between cycle progression, DNA replication, and proliferation, and provide important insight for enhancing cardiomyocyte proliferation in the context of adult myocardial tissue.
Keywords: cardiomyocyte, cell-cycle, FUCCI, regeneration, myocardial infarct
1. Introduction
Cardiomyocyte loss is a major contributory factor to heart failure from acute pathologic injury or chronic stress, so generating additional cardiomyocytes to restore structural and functional integrity of the heart is a worthwhile endeavor. Research dedicated to prompting adult mammalian cardiomyocytes to re-enter cell cycle and complete mitosis has been frustratingly difficult to achieve due to inherent biological properties of the adult mammalian myocardium[1–6]. Regardless of the approach, the conclusion from collective efforts is that adult mammalian cardiomyocytes are remarkably refractory to mitotic activity, unlike those found in either early postnatal mice or zebrafish. Moreover, results in the adult mammalian context report widely varying observations of cardiomyocyte “proliferation” using a plethora of markers and metrics to assess de novo cardiomyogenesis[7–9]. Lack of standardization, varied experimental approaches, and underappreciation for distinctive cell cycle regulation of cardiomyocytes has led to claims of translational potential yet to be actualized[10–12]. Unambiguous demarcation of de novo cardiomyocyte formation and the perceived mitotic exit remains difficult to ascertain using current approaches. Successfully identifying reentry into the cell cycle and de novo cardiomyocyte formation requires a clearly identified mitotic exit point that currently remains ill-defined in cardiomyocytes[4,13,14].
The challenge of augmenting adult mammalian cardiomyocyte proliferation can be attributed to myocardial biology approaches taken to understand and overcome them. Major contributory considerations include: structural and functional demands of the adult mammalian heart, distinctly tight control of molecular arrest checkpoints for cardiomyocyte mitosis, the source of de novo cardiomyogenesis from pre-existing cardiomyocytes versus cardiac progenitor cells (CPCs), defining “proliferation” using various markers of mitosis, extrapolation from studies of early stage development or lower vertebrate models, blurring of cardiomyogenesis with related processes of DNA replication without mitosis, and technical approaches to measurement of cardiomyogenesis[10,15–17]. Given the wide range of sources for potential disconnects, accumulating discrepant findings seems inevitable. It stands to reason that cardiomyogenic testing for adult mammalian hearts is best performed in an in vivo adult mammal model to achieve the most dependable and reliable results. A recent consensus statement from the American Heart Association focused upon endogenous cardiomyogenesis concluded: “1) Cardiomyocyte renewal rates may be higher after injury than under normal conditions, and 2) The experimental determination of cardiomyocyte turnover after cardiac injury can be challenging owing to inflammation, proliferation of stromal and vascular cells, and scar formation.”[18] After decades of unrelenting investigation, the consensus is that answers related to cardiomyocyte turnover in the pathological setting remain unresolved. Clearly, new approaches and additional knowledge are required.
A primary issue hampering studies of adult mammalian cardiomyogenesis has been difficulty in determining cardiomyocyte proliferation using markers of cell cycling. The biological responses of adult cardiomyocytes to mitotic stimuli render typical measures of cell division inconclusive. Multiple markers of cell cycle have been developed for investigations of non-myocardial cell biology and co-opted for documenting evidence of cardiomyocyte proliferation (Fig. 1). Each marker has served to document evidence of mitosis, yet none alone are truly definitive indicators of authentic cell division in cardiomyocytes. Specifically, these markers indicate progression through cell cycle or events occurring during progression through mitosis. However, in the context of cardiomyocytes, these markers are present at multiple stages of cell cycle and it is impossible to distinguish cells progressing through mitosis from those arrested at various mitotic checkpoints. Limitations using these markers have been highlighted in previous publications[8,19,20], yet the presentation of these labels as definitive evidence of cardiomyocyte proliferation continues[11,12,17,21,22]. A recent study confirms these observations and offers a way forward using two novel proteins as definitive markers of cardiomyocyte division, but also rests upon confocal analysis of intracellular localization at a critical transient moment in the penultimate steps of mitosis[23]. Contributing to the confusion, biological phenomena of endomitosis, endoreplication, and DNA damage are often unaccounted for in assessments of cardiomyocyte proliferation. Cardiomyocytes can enter mitosis and exit without generating daughter cells by mere duplication of DNA without new nucleation or by adding additional nuclei. Inattention to these normal aspects of cardiomyocyte biology leads to controversial claims of proliferation rates and potentially erroneous claims of regeneration. These collective concerns highlight the critical unmet need for a straightforward model enabling in vivo assessment of cardiomyocyte proliferation.
Fig. 1. Markers of division and cell-cycle status.
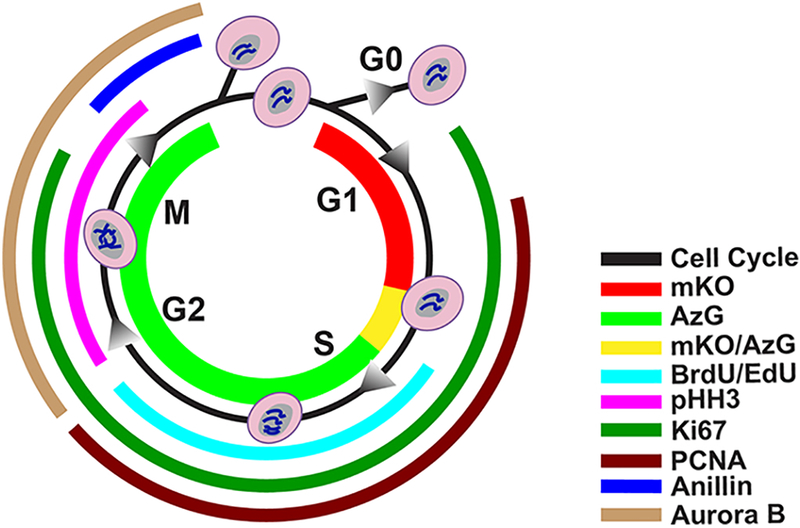
FUCCI fluorescence mKO (red) presents in G1 phase, and AzG (green) presents during S/G2/M phases, where during the G1/S transition both fluorescence (mKO/AzG) present simultaneously and merge into a yellow color. BrdU or Edu, both thymidine analogs incorporate into DNA during synthesis (cyan). Phosphorylated Histone 3 (pHH3) is responsible for chromatin condensation and is thus present during G2 through M phase (magenta). Nuclear antigen Ki67 is present from G1 to M phase (emerald). PCNA is presents between G1 and G2 phase in response to DNA synthesis (burgundy). Anillin plays a role in creating the cleavage furrow formation and begin to accumulate in late G2 through late M phase (blue). Aurora B plays a role in mitosis, present from G2 through M phase (sand).
The novel transgenic mouse presented here is based upon well documented and proven Fluorescence Ubiquitination-based Cell Cycle Indicator (FUCCI)[24] technology adapted to in vivo cell cycle monitoring via cardiomyocyte-specific transgenesis (FUCCI-Tg). Briefly, FUCCI system employs two fluorescent probes, monomeric Kusabira Orange (mKO) and monomeric Azami Green (AzG). mKO is fused to chromosome licensing factor hCdt1 that indicates the G1 phase with orange fluorescence. AzG is fused to licensing inhibitor hGeminin, that indicates S/G2/M with green fluorescence. Together, oscillation between mKO-hCdt1 and AzG-hGeminin by ubiquitination during cell cycle progression provides direct visualization of cell cycle phases from G1 to the late stage of mitosis[24]. Although the FUCCI system has previously been studied in the cardiovascular context, prior models did not use cardiomyocyte-specific expression and none were concerned with demonstration of enhanced adult cardiomyogenesis[24–27]. The FUCCI-Tg is particularly valuable as a novel tool to assess cardiomyocyte proliferation because 1) every cardiomyocyte in the heart is visualized for cell cycle status, not just “cycling myocytes”, 2) four distinct stages of cell cycle progression are revealed with inherent fluorophore expression implications for cardiomyocyte mitosis, 3) quantitation of cell cycle status for collective myocyte populations is possible, 4) the system can be used in combination with DNA labeling to correlate cell cycle progression with DNA synthesis versus DNA damage, and most importantly 5) in vivo labeling is assessed in the adult mammalian heart – the only place where induction of proliferation should be tested for authentic preclinically-relevant activity (rather than in vitro or postnatal environments).
Quantitation of cell cycle status of myocyte populations can be used in combination with DNA labeling to correlate cell cycle progression with DNA synthesis versus DNA damage, and most importantly in vivo labeling is assessed in the adult mammalian heart. The findings presented herein demonstrate not only the straightforward simplicity and utility of the FUCCI-Tg for investigation of cardiomyocyte cell cycle activity, but also reveal previously unrecognized aspects of cardiomyocyte biology in the early postnatal and adult myocardium.
2. Methods
Full methods are available at Supplemental Methods.
3. Results
3.1. FUCCI expression is specific to cardiomyocytes
The adult mammalian heart is a post-mitotic organ with highly restricted cardiomyocyte cell cycle activity, yet recent studies point toward pre-existing cardiomyocytes as the primary source of de novo cardiomyogenesis often utilizing common markers of proliferation found in neonatal development to identify new cells[13,16,28,29]. Lack of clarity regarding demonstrable mitotic exit of cardiomyocytes and ambiguous identifiers of de novo cardiomyocyte formation contribute to current disconnects between the universally acknowledged negligible mitotic activity of adult cardiomyocytes with claims of proliferation of adult mammalian cardiomyocytes. Cardiomyocyte-specific resolution of cell cycle dynamics was problematic in prior iterations of FUCCI mouse models because of ubiquitous expression, prompting creation of a cardiomyocyte-specific FUCCI transgenic mouse reporter (FUCCI-Tg) driven by α-Myosin Heavy Chain (αMHC)[22,29,30]. αMHC-mKO2-hCdt1-pA (mKO) and αMHC-Azami Green-hGeminin1-pA (AzG) linearized DNA transgenes were co-injected into 0.5dpc FVB/NJ embryos in 1:1 molar ratio (Fig. 2A). Four potential founders positive for both AzG and mKO resulted from injected embryos transferred into pseudo-pregnant females. Germline transmission was verified by backcrossing to FVB/NJ non-transgenic mice (Fig. 2B) with AzG and mKO protein expression detected exclusively in heart tissue (Fig. 2C) and backcrossed for ten generations into FVB/NJ strain to produce a congenic FUCCI-Tg used in experimental procedures. FUCCI fluorescence AzG and mKO expression was verified by native fluorescence in isolated cardiomyocytes, approximately 65.7% of neonatal (P2) and 90.9% adult (P90) cardiomyocytes are FUCCI positive, expressing either single or double fluorescence AzG (green) or mKO (orange) (Fig. 2D-E). Additionally, cardiomyocyte specific expression of AzG and mKO was verified by co-staining with cardiomyocyte specific marker α-Sarcomeric Actinin (αSA) in both cultured P2 and P90 cardiomyocytes (Fig. 2F-G). This data demonstrates that the FUCCI-Tg reporter mouse faithfully expresses AzG and mKO fluorescence in a cardiomyocytes specific fashion.
Fig. 2. FUCCI-Tg expression is specific to cardiomyocytes.
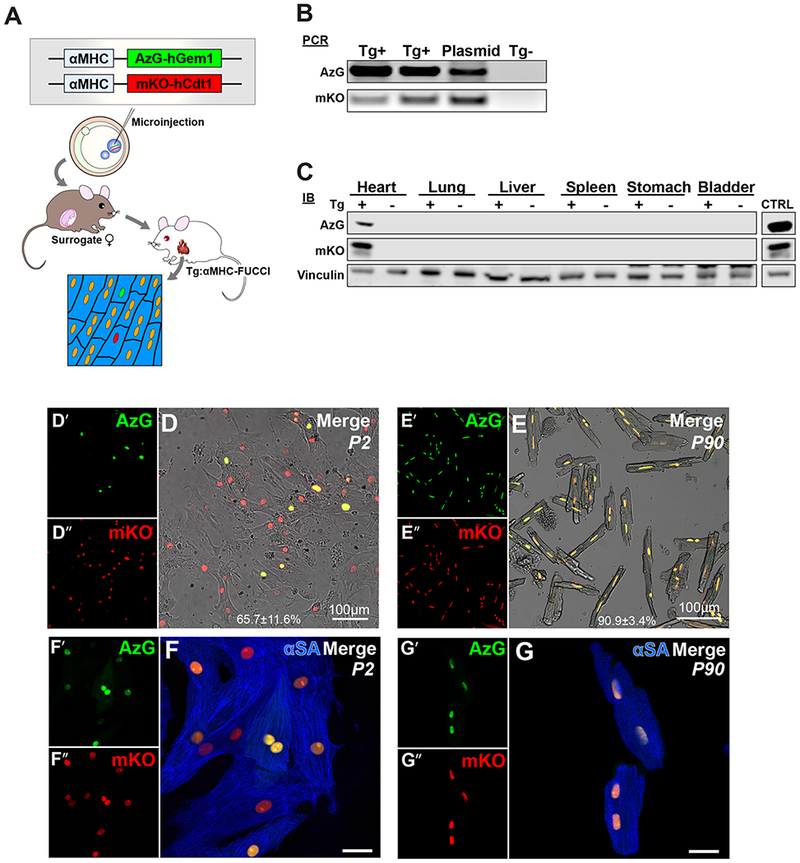
(A) Schematic of transgenic mouse production, n=250 embryos injected, n=28 pups screened for transgene integration, n=1 founder line established. (B) PCR analysis confirm transgene integration in genomic DNA. (C) Immunoblot analysis of founder organs demonstrate cardiac specificity of transgenes. (D-E) Representative images of isolated P2 (D) and P90 (E) mouse cardiomyocytes express FUCCI fluorescence of AzG (green, D’, E’), mKO (red, D”, E”), scale bar 100μm. (F-G) Representative images of nuclear AzG (green) and mKO (red) native fluorescence visualized in α-sarcomeric actinin (αSA, blue) positive P2 (F) and P90 (G) isolated cardiomyocytes, respectively. Scale bar 20μm
To examine if FUCCI expression is exclusive to cardiomyocyte lineage in vivo, frozen tissue sections were co-stained with cardiomyocyte marker Tropomyosin (TPM), smooth muscle cell marker Smooth Muscle 22-α (Sm22α), endothelial cell marker von Willebrand Factor (vWF), and fibroblast marker Vimentin (Vim). AzG and mKO expression were only detected in cardiomyocytes and absent from smooth muscle, endothelial and fibroblast nuclei (Fig. S1A-C). Cardiac progenitor cells (CPCs), a resident cardiac cell type that was previously reported to express low αMHC activity[31], showed very low level of Geminin expression but undetectable level of αMHC driven AzG or mKO fluorescence (Fig. S1D). Furthermore, exclusive expression of AzG and mKO in cardiomyocytes was confirmed by immunoblot analysis, where fused AzG-Geminin and mKO-Cdt1 are only detectable in FUCCI-Tg whole heart lysates but in absent in non-myocytes and CPCs (Fig. S1E). Collectively, FUCCI-Tg allows direct visualization of in vivo cardiomyocyte-specific cell cycle dynamics through direct detection of AzG+ (green) or mKO+ (red) nuclear fluorescence exclusive to the cardiomyocyte population, and not in any other cell lineages in the heart.
3.2. Isolated P2 FUCCI cardlomyocytes In culture allow visualization of cell division and binucleation events
FUCCI-Tg allows for readily resolvable assessment of fixed myocardial tissue sections as well as freshly isolated cells for cardiomyocyte cell cycle activity. As an initial demonstration, P2 neonatal cardiomyocytes were isolated, cultured, and subjected to time-lapse imaging. Neonatal cardiomyocytes underwent limited normal cell cycle division and endoreplication events showed that AzG accumulation in nuclei disappeared, followed by cleavage furrow formation and generation of two separated daughter cells, indicative of completed cardiomyocyte division (Movie S1, Fig. S2A-C). Binucleation events showed nuclear envelope breakdown (NEB) accompanied with endomitosis (acytokinesis, Movie S2, Fig. S2D-G) in culture. Taken together, FUCCI oscillation in cell cycle progression demonstrates the power of FUCCI for faithful in vitro modeling to assess stimulants and suppressors on cardiomyocyte cell cycle re-entry.
3.3. Postnatal cardiomyocyte cell cycle progression transitions toward arrest at G1/S within days after birth
Validation of neonatal cardiomyocyte cell cycle demonstrated by FUCCI (Fig. 2, Fig S1) set the stage for subsequent assessments of cell cycle markers commonly utilized to identify actively cycling cells following neonatal cardiac injury in vivo [15,21,29,30]. Thymidine analog BrdU and phosphorylated histone 3 (pHH3) are typically employed to identify DNA synthesis and mitosis in active cycling cells as well as identification of de novo cardiomyogenesis after injury in both neonatal and adult injury models. Cardiomyocyte cell cycle status in postnatal development from P2 to P90 FUCCI hearts was assessed in combination with BrdU incorporation (150mg/kg). Limiting BrdU incorporation to S phase events was achieved by heart harvesting at two hours after BrdU injection (Fig. 3A). Cardiomyocytes in S/G2/M phase of cell cycle were visualized by direct AzG+ fluorescence and labeled with antibodies to detect cardiac troponin I (cTnI), BrdU, and pHH3 in conjunction with native FUCCI fluorescence in frozen cardiac tissue sections from neonatal P2 to adult P90 (Fig. 3B-F)[24]. Cardiomyocytes in G1 phase of the cell cycle were identified by direct visualization of mKO+ fluorescence together with absence of both BrdU, and pHH3. Active cycling was confirmed in FUCCI cardiomyocytes at P2, P7 and P14 by mKO+ only (G1, Fig. 3B-C; white arrowheads) or AzG+ only (S/G2/M, Fig. 3B-D; yellow arrowheads). At P14, multiple AzG+/pHH3+ cardiomyocytes were identified in several sections, representing cardiomyocytes possibly undergoing binucleation prior to mitotic exit or a cellular division[17,32] (Fig. 3D, yellow arrowhead). At P21-90, Cardiomyocyte cycling activity subsided as confirmed by stark decrease of either AzG+ only or mKO+ only cardiomyocytes, whereas majority of cardiomyocytes express double fluorescence AzG+/mKO+ at this time point (Fig. 3E-F). BrdU incorporation peaked at P2 and rapidly declined with ongoing postnatal maturation by P90 (Fig. 3G), consistent with previous observations[33–35].
Fig. 3. Postnatal cardiomyocyte cell cycle progression transitions toward arrest at G1/S within days after birth.
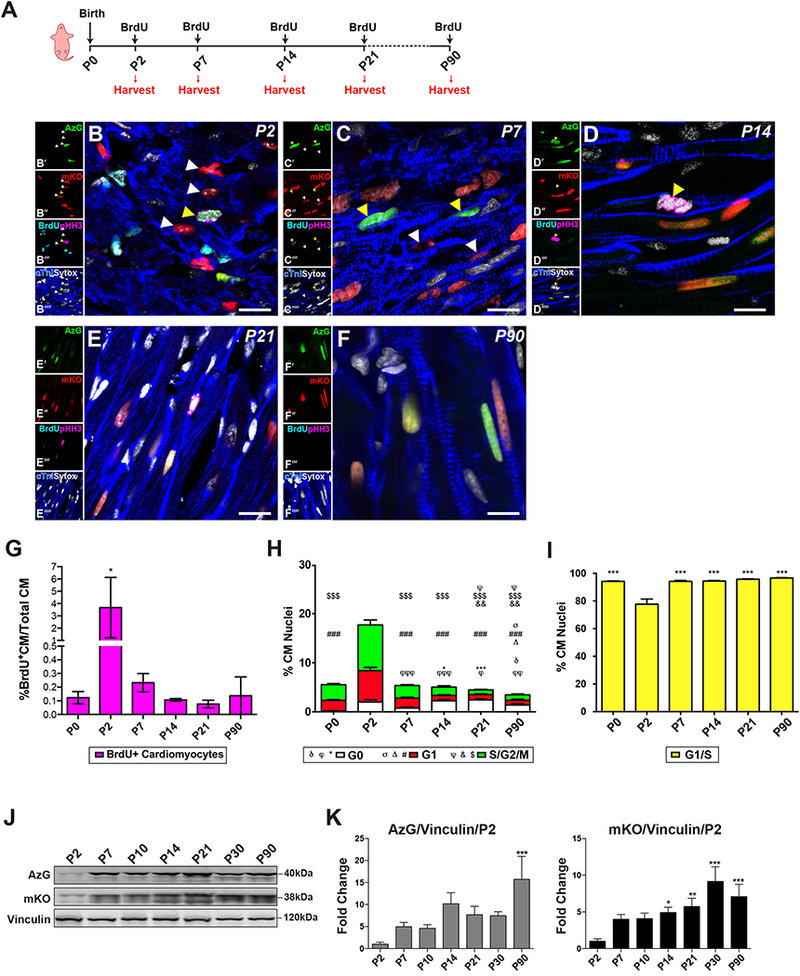
(A) Schematic of developmental time-point isolation, single BrdU injection (150mg/kg) given 2 hours prior to harvest. (B-F) Merged representative confocal images showing AzG and mKO expression in cardiomyocytes during postnatal development visualized by immunostaining and confocal microscopy in cardiac tissue sections at P2 (B), P7 (C), P14 (D), P21 (E), and P90 (F); AzG (green, B’-F’), mKO (red, B”-F”), pHH3 (magenta, B”‘-F”‘), BrdU (cyan, B”‘-F”‘), Sytox (white, B”“-F”“) and cardiac Troponin I (blue, B”“-F”“), Scale bar 10μm, n=3–5 hearts per time point. (G) Quantification of percent of BrdU+ cardiomyocytes in all cardiomyocytes counted peaks at P2 and decreases thereafter, * P <0.05 vs. P0. (H) Quantification of percent of cardiomyocyte nuclei in different cell cycle phases shows G0(mKO−/AzG−), G1(mKO+) and S/G2/M (AzG+). G0: φP<0.05, φφP<0.001, φφφP<0.0001 vs. P0; *P<0.05, ***P<0.001 vs. P7, δP<0.05 vs. P21. G1: σP<0.05 vs. P7, ΔP<0.05 vs. P0. ### P<0.0001 vs. P2. S/G2/M: ψP<0.05 vs. P7, &P<0.001 vs. P0, $$$ P<0.0001 vs. P2. (I) Quantification of percent of cardiomyocyte nuclei in G1/S transition of the cell cycle, ***P<0.0001 vs. P2. (J) Representative immunoblot of whole heart lysates indicate AzG/mKOA/Vinculin protein expression from P2 to P90 show increased protein expression with age. (K) Quantitation of AzG (left) and mKO (right) protein expression relative to loading Vinculin vs P2. *P<0.05, **P<0.001, ***P<0.0001 vs P2. n = 4055 (P0), 1953 (P2), 7536 (P7), 2861 (P14), 4765 (P21), 1873 (P90) CM nuclei from 6 hearts per time point. One-way ANOVA, Tukey’s post hoc test.
Interestingly, persistence of AzG fluorescence observed in P21 and P90 hearts did not correlate with prior FUCCI studies, in which AzG expression was reported to decrease in cardiomyocytes shortly after birth [26,27]. Instead, dual positive AzG+/mKO+ cardiomyocyte nuclei accumulated in FUCCI-Tg mice, indicating cardiomyocytes arrest in G1/S transition phase during postnatal adolescent aging (Fig. 3H-I). Co-expression of nuclear AzG and mKO has been defined as the G1/S transition in previous FUCCI reporter systems[24,25]. Indeed, dual AzG+/mKO+ labeling was evident from P0 to P90 in myocardial sections. Cardiomyocyte cell cycle dynamics in postnatal development observed with the FUCCI-Tg demonstrate peak AzG+/mKO− labeling at P2 (9.3%) indicative of cardiomyocytes in S/G2/M phase, thereafter decreasing significantly to level off at 1 % by P21 (Fig. 3H, green). Similarly, AzG−/mKO+ labeling also peaked at P2 (6.8%) indicative of cardiomyocytes in G1 phase significantly decreased by P14 and also leveled off at 1.1% through P90 (Fig. 3H, red). In contrast, dual labeling of AzG+/mKO+ cardiomyocytes was lowest at P2 (76.5%) but climbs from 94% at P14 to 96% in P90 hearts (Fig. 3I) indicative of cell cycle arrest at the G1/S transition, well known as the restriction point (R-point) of cell cycle withdrawal[36,37]. Dual AzG+/mKO+ fluorescence in postmitotic cardiomyocytes is consistent with R-point arrest at G1/S rather than mitotic exit at G0/G1 or arresting at G2 as previously reported[4,32,33,38–40] R-point cell cycle arrest consistent with dual AzG+/mKO+ fluorescence was validated in vitro using FUCCI-Tg isolated adult cardiomyocytes to demonstrate normal degradation activity of the ubiquitin/proteasome system (UPS), ruling out FUCCI labeling artefacts consequential to impaired protein turnover (Fig. S3).
Immunoblot analysis of whole heart lysates confirmed that both AzG and mKO protein expression increased significantly as development progressed relative to P2 (Fig. 3J-K). Accumulation of AzG and mKO expression in whole heart lysates prompted examination of endogenous Geminin and Cdt1 levels in postnatal cardiomyocyte development further substantiating expression of FUCCI-Tg probes in postnatal heart development (Fig. S4A). Endogenous Geminin (Fig. S4B, left) significantly increased with age as did endogenous Cdt1 (Fig. S4B, right) showing a 2.6 and 2.4-fold change respectively by P30 relative to P2. Endogenous Geminin was further visualized in ACMs isolated from non-transgenic mice at P30 (data not shown) as well as P90 that exhibited nuclear localization (Fig. S4C-E). These results demonstrate FUCCI-Tg probes faithfully identified postnatal cycling cardiomyocytes, corroborated by colocalization of BrdU and pHH3. Nevertheless, AzG and mKO expression persist after the proliferative window of postnatal development consistent with endogenous cognate expression of Geminin and Cdt1, serving as further validation of FUCCI-Tg labeling to accurately represent cardiomyocyte cell cycle dynamics. Collectively, these results validate dual AzG+/mKO+ FUCCI labeling resulting from G1/S boundary arrest as observed in mature cardiomyocytes.
Taken together, these results demonstrate for the first time that mature cardiomyocytes remain poised at the G1/S interface of the cell cycle and do not undergo a full mitotic exit.
3.4. Cell cycle progression but not cardiomyogenesis in border zone cardiomyocytes following infarction damage.
Reactivation of the cardiomyocyte cycle and de novo cardiomyocyte formation has been proposed following myocardial infarction (MI)[28,29]. Adult cardiomyocyte cell cycle re-entry consequential to MI injury as an inductive stimulus was tested in FUCCI-Tg subjected to left anterior descending artery (LAD) ligation in conjunction with daily BrdU injection (50mg/kg) to detect DNA synthesis until harvest (Fig. 4A). However, despite increased FUCCI labeling consistent with cardiomyocyte cell cycle re-entry, BrdU+ labeling indicative of DNA synthesis was restricted solely to infiltrating or interstitial cells as early as 3-10 days post injury (dpi) (Fig. S5A-C), and pHH3 immunolabeling was absent at all tested time points within the cardiomyocyte population (data not shown). Cardiomyocyte cell cycle re-entry consistent with appearance of AzG+ expression within the border zone (BZ) at 14dpi (Fig. 4B-C, arrowhead) rarely correlate with BrdU incorporation, which instead was prevalent throughout the interstitial population. At 21 dpi, BrdU incorporation was detected at very low level in cardiomyocytes (Fig. 4D-F), with either dual fluorescence AzG+/mKO+ (arrowhead) or single fluorescence mKO+ (open arrowhead). BrdU+/AzG−/mKO− (GO; 0.042%), BrdU+/AzG−/mKO+ (G1; 0.179%), and BrdU+/AzG+/mKO+ (G1/S; 1.112%) cardiomyocytes were found sporadically throughout 149 quantified sections of a total 6,705 nuclei at 21dpi (N=4 hearts; Fig. S6A-C). Similar observations were made in the remote zone (RZ) distant from damage in MI hearts (Fig. S7).
Fig. 4. Border zone cardiomyocytes exhibit signs of cell cycle re-entry but fail to show new cardiomyocyte formation.
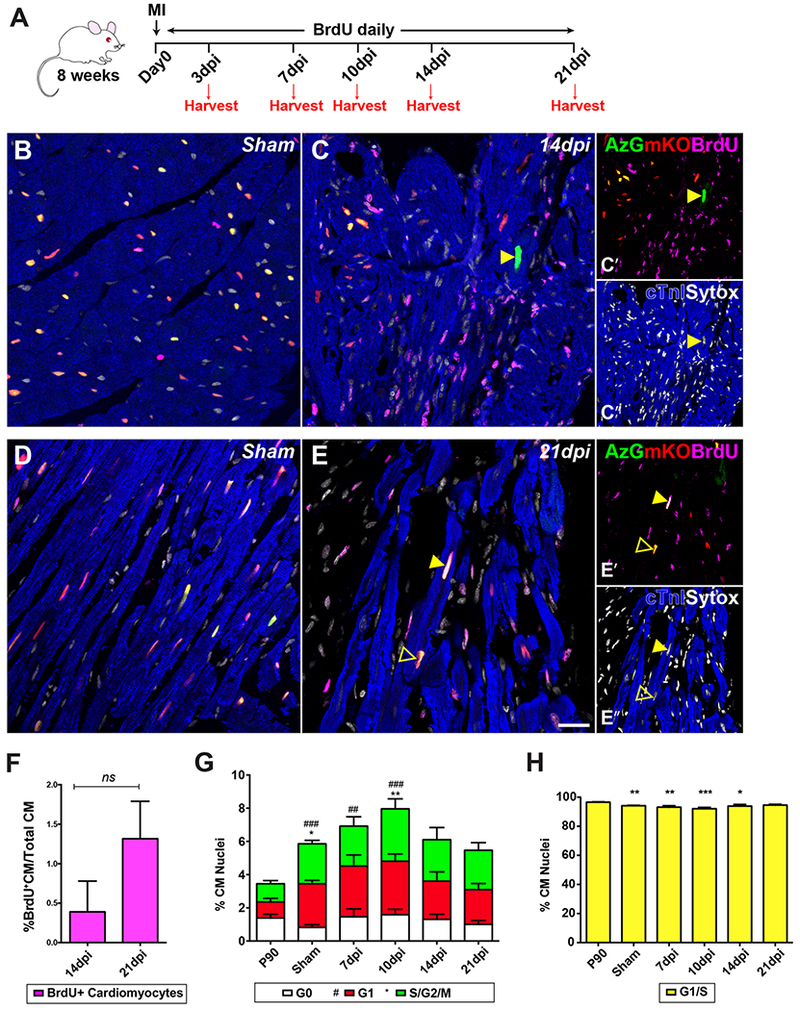
(A) Schematic timeline showing MI and daily BrdU pulse (50mg/kg). (B, D) Representative confocal images of sham tissue sections at 14 and 21 days post operation. (C) Border zone (BZ) cardiomyocytes at 14dpi show AzG expression (yellow arrowhead); BrdU incorporation is restricted to interstitial population. (E) BZ cardiomyocytes at 21dpi show mKO+/AzG+/BrdU+ (yellow arrowhead) and mKO+/BrdU+ (open arrowhead). Native fluorescence AzG (green, C’, E’), mKO (red, C’, E’), and immunolabeled for cardiac Troponin I (blue, C”, E”), BrdU (magenta, C’, E’), and Sytox (white, C”, E”) in (B-E) scale bar 20μm. (F) Quantification of percent BrdU+ cardiomyocytes in all cardiomyocytes counted, n = 4 BrdU+CM in 1,802 CM at 14dpi, n=85 BrdU+CM in 6,705 CM at 21 dpi. (G) Percent CM nuclei at GO, G1, S/G2/M phase in tissue sections at 7, 10, 14 and 21dpi. G0: ##P<0.001, ###P<0.0001 vs. P90. S/G2/M: *P<0.05, **P<0.001 vs. P90. (H) Percent CM nuclei at G1/S interface; *P<0.05, **P<0.001, ***P<0.0001 vs. P90. n=3351 (P90), 2116 (sham), 987 (7dpi), 936 (10dpi), 1299 (14dpi), 1235 (21dpi) CM nuclei counted from 9 sections along IZ/BZ for each heart. One-way ANOVA, Tukey’s post hoc test.
Quantification of FUCCI labeling for cardiomyocytes in the injured myocardium at 7 through 21dpi showed highest induction of single positive AzG+/mKO− or AzG−/mKO+ myocytes occurring at 10dpi (G1; 3.2%, S/G2/M; 3.2%) returning to sham levels by 21 dpi. Double positive AzG+/mKO+ myocyte levels were inversely correlated to changes in single positive AzG+/mKO− or AzG−/mKO+ myocytes (Fig. 4G-H) suggesting that R-point arrested myocytes had been prompted to re-enter cell cycle. Notably, a small but significant elevation of single positive AzG+/mKO− or AzG−/mKO+ myocytes occurs in sham operated hearts throughout the myocardium relative to basal FUCCI labeling in unoperated P90 hearts (Fig. 4G, 2.4% and 2.6% respectively). Thus, relatively modest surgical ex vivo manipulation of the heart prompts low level cardiomyocyte cell cycle re-entry without BrdU incorporation or pHH3 immunolabeling indicative of mitotic chromatin that could be interpreted as cardiomyogenesis (Fig. 4G). Collectively, these findings are consistent with concluding that cardiomyocytes are prompted to re-enter cell cycle following MI injury, however this induction does not lead to significant cardiomyogenesis.
Adrenergic stress has been reported to stimulate cardiac regeneration in experimental mouse models, but these findings remain controversial[41–44]. The FUCCI-Tg was used to interrogate the impact of acute adrenergic stress upon cardiomyocyte cell cycle re-entry and induction of a cardiac progenitor response with a single high dose of isoproterenol (50mg/kg; Fig. S8A). BrdU incorporation and pHH3 immunolabeling were not detected in CMs at any time-point screened, whereas cKit+ cardiac progenitor cells were detected as early as 3-7dpi as previously reported[45,46] (Fig. S8B-C). FUCCI labeling indicative of cell cycle re-entry at 3dpi is modest and lost by 7dpi (Fig. S8D-E). These findings argue against cardiomyocyte proliferation as a contributory factor to myocardial repair.
In summary for the injury model experiments, a very small population of cardiomyocytes re-enter cell cycle as revealed by FUCCI labeling in the adult myocardium as result of myocardial injury. However, cardiomyocyte cell cycle re-entry is disconnected from mitotic activity. Instead, the evidence points toward an alternative conclusion of DNA damage response[47,48] (BrdU+/G1/S). In the context of these injury models, unlike the postnatal myocardium, adult cardiomyocytes are severely limited in their ability to progress through the cell cycle and their contribution to cardiomyogenesis is insignificant[8,49,50].
3.5. Amplifying cardiac progenitors express αMHC-FUCCI AzG in vivo.
Unlike the context of adult myocardium, cardiomyocytes and/or cardiac progenitor cells participate in cardiomyogenesis during development and neonatal response to injury[18,41,51–54]. The FUCCI-Tg provides an opportunity to assess cardiomyocyte cycling in postnatal development and upregulation of αMHC promoter activity indicative of cardiomyogenic commitment. Indeed, a cluster of cKit+ cells within the myocardium at was detected at P2, in which only one unique rare amplifying cardiomyocyte progenitor labeled as AzG+/cKit+/BrdU+/pHH3+ was identified among 1,953 cardiomyocytes screened (Fig. 5A, arrowhead). Additionally, a primarily cKit+/AzG” cluster that may have committed to early cardiogenic potential (cTnI+) was detected in P7 myocardium (Fig. 5B, arrowheads). One unique primitive amplifying AzG+/cTnI+ cardiomyocyte lacking mKO expression and BrdU/pHH3 labeling was observed at P7 in 7,536 cardiomyocyte nuclei analyzed at this time point (Fig. 5C, arrowhead). These extremely rare amplifying cardiomyogenic precursor cells were never detected in adult myocardium, indicating they are lost during early postnatal growth. Nevertheless, amplifying cardiomyocyte progenitors may be an important source for de novo cardiomyocyte formation participating in cardiac growth and proliferation in early myocardial development.
Fig. 5. Amplifying progenitors express AzG in vivo.
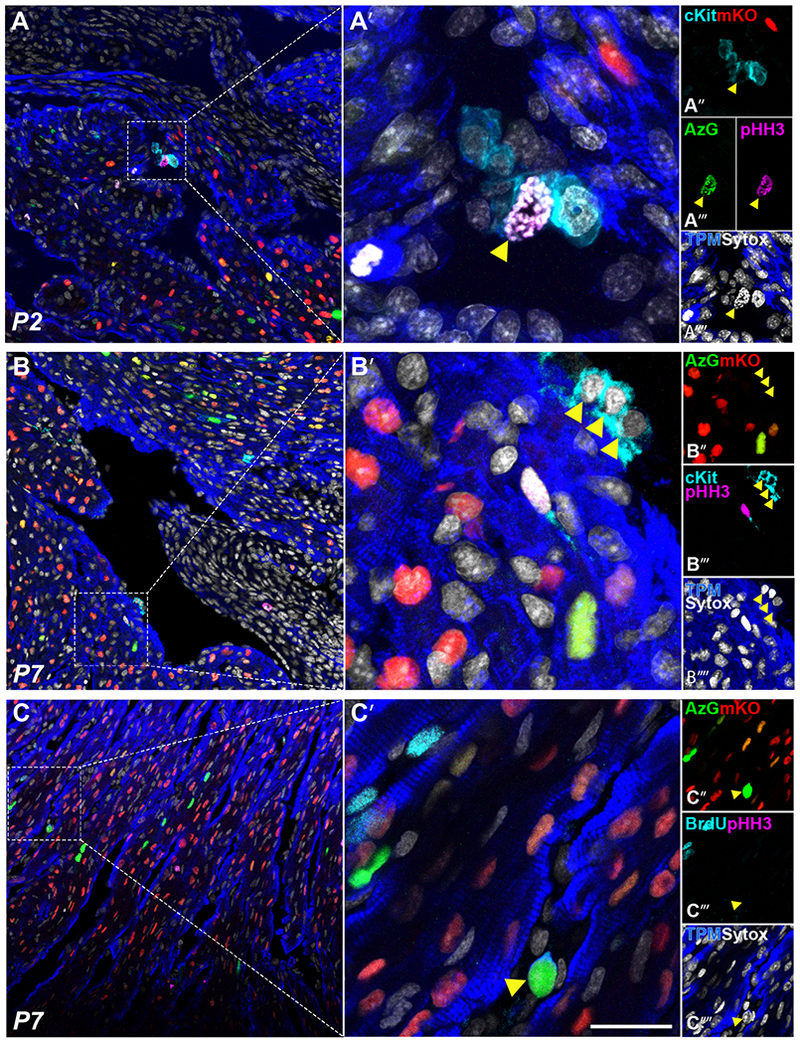
Representative confocal images of early postnatal development tissue sections P2 (A) and P7 (B, C) respectively, showing native fluorescence of AzG (green, A’”, B”), mKO (red, A”-C”), and immunolabeling for cKit (cyan, A”, B’”, C’”), pHH3 (magenta, A”‘-C”‘), sytox (white, A”“-C”“) and tropomyosin (blue, A”“-C”“), scale bar 50μm in A, B, C; 10μm in A’-C’. Yellow arrow shows cKit+/AzG+/pHH3+ amplifying cardiac progenitor in P2 cardiac sections (A), and AzG+/cTnl+ immature cardiomyocyte in P7 section (C).
4. Discussion
Cardiomyocyte proliferation induction in adult mammalian myocardium remains one of the most sought-after yet least successful endeavors of cardiovascular research. Application of rigorous and consistent measures to determine induction of cardiomyocyte proliferation in the adult mammalian myocardium is essential to validate and compare the ever-expanding series of methods and practices developed throughout the world. Inconsistent measures, inappropriately applied measures, and overinterpretation of findings have been and continue to be problematic for achieving resolution in advancing mechanistic understanding of cardiomyocyte cell cycle regulation. Paradoxically, while substantial information has been gathered on unique characteristics of the cardiomyocyte cell cycle relative to other cell types, proliferative measures often fail to fully and faithfully encompass the spectrum of possible outcomes with high rigor and reproducibility.
A primary issue hampering studies of adult mammalian cardiomyogenesis has been difficulty of determining cardiomyocyte proliferation using markers of cell cycling. While such demonstrations are readily reproduced in neonatal mice or zebrafish, the biological responses of adult cardiomyocytes to mitotic stimuli render typical measures of cell division irrelevant. For example, multiple markers of cell cycle have been developed for investigations of non-myocardial cell biology and co-opted for documenting evidence of cardiomyocyte proliferation (Fig. 1). Each of these markers has served to document evidence of mitosis, yet none of them alone are truly definitive indicators of authentic cell division when working with cardiomyocytes. Specifically, these markers indicate progression through cell cycle or events occurring during progression through mitosis. However, in the context of cardiomyocytes, many of these markers are present at multiple stages of cell cycle and it is impossible to distinguish cells that are progressing through mitosis from those that are arrested at various mitotic checkpoints. Indeed, prior studies suggest proliferation markers PCNA and Ki67 are reexpressed during hypertrophic events that do not end in the formation of new myocytes[7,55–57] Limitations of using these markers to document cardiomyocyte proliferation have been highlighted in previous publications[8, 19] but despite these admonitions the presentation of these labels as evidence of cardiomyocyte proliferation continues. This serious problem for the field is indicative of disconnects in recognizing the atypical mitotic resistance of cardiomyocytes relative to other cell types where such labels could be accurate and appropriate. A recent study pointed out these limitations and offered a way forward using two novel proteins (RhoA and IQGAP3) as definitive markers of cardiomyocyte division, but unfortunately use of these two markers also rests upon a tour-de-force confocal analysis of intracellular localization at a critical transient moment in the penultimate steps of mitosis[23]. Demonstration of cardiomyocyte mitosis using RhoA and IQGAP3 will require further development of tools to monitor these proteins in real-time to follow intracellular localization that is beyond capabilities of current typical investigations of cardiomyocyte proliferation. Inability to visualize the final mitotic event, cytokinesis, remains a challenge with almost all markers of cell cycle including the FUCCI-Tg. Loss of AzG+ fluorescence in late mitosis before cleavage furrow formation makes it difficult to unambiguously determine if cardiomyocytes undergo this event in vivo. Crossing FUCCI-Tg with potential RhoA and IQGAP3 reporter models[23] to observe cytokinesis in ACMs may help document cell cycle resolution.
The FUCCI-Tg model assesses validity of commonly used markers of proliferation in measuring de novo cardiomyogenesis by allowing direct visualization of cell cycle dynamics in G1 and S/G2/M phase cardiomyocytes. Ubiquitous expression of FUCCI reporters in the original model obscured distinction of cardiomyocytes from other cardiac cell types also expressing FUCCI[24,26,27]. Previous attempts to demonstrate cardiomyocyte cell cycle activity using CMV chicken beta-actin beta-globin synthetic promoter-driven-FUCCI (CAG-FUCCI) used whole and thin sliced heart tissue ex-vivo culture methods[26]. Whole and sliced cardiac tissue from CAG-FUCCI in culture exhibited increased mKO+ levels over time indicating quiescent (G0) or a G1 arrested state[26,27]. Endothelial, smooth muscle, fibroblast and cardiomyocytes within the whole and sliced tissue culture ubiquitously expressed FUCCI that, together with uncontrollable consequences of tissue culture, obfuscate analyses. The FUCCI-Tg mouse model circumvents the problem of nonspecific cardiomyocyte labeling and allows unambiguous visualization of cardiomyocyte specific cell cycle activity. FUCCI-Tg further highlights the nominal progression of cardiomyocytes through the cell cycle following two different myocardial injury stimuli. Recent reports postulate surviving cardiomyocytes as the only contributors to de novo cardiomyogenesis in postnatal development and following injury[10,58,59]. Flowever, BrdU+ cardiomyocytes spanning the border zone comprised less than 100 cells in all sections analyzed, suggesting that cardiomyocytes re-entering the cell cycle are a limited source of cardiomyogenesis in response to injury.
The FUCCI-Tg has identified cardiomyocyte “mitotic” cell cycle arrest at the G1/S restriction point (Fig. 3). Over 95% of CMs express mKO and AzG by postnatal day 21 indicative of seized cycling. The G1/S interface Restriction point (R-point) in yeast and mammals has been documented as the stage at which mitogenic stimuli no longer exert effects on cell cycle progression and is also the point of maximal ATP production[37,60]. Given the role of cardiomyocytes and the energy requirements necessary to execute these mechanical demands, arrest at R-point seems logical. Together with previously reported observations of mitochondrial organization in adult CMs and the biological demand for energy production, arrest at R-point is intuitively appealing for optimal cardiomyocyte function.
Lack of cell cycle progression observed in previous models reporting a switch from cardiomyocyte proliferation to hypertrophy between one and two weeks after birth[6,22,32,39,61,62] correlate with results obtained with the FUCCI-Tg. AzG+ nuclei indicative of S/G2/M phase incorporate BrdU and show pHH3 through P14 (Fig. 3) as previously reported in postnatal development, decreasing to undetectable levels by P90[17,25,35]. Proliferation markers BrdU and pHH3 do not appear to identify actively cycling FUCCI-Tg cardiomyocytes in response to injury and instead may be representative of myocytes undergoing DNA repair or endoreplication events as a consequence of injury[7,8]. Cardiac trauma in both border and remote zones of the injured myocardium induced a small population of cardiomyocyte to re-enter cell cycle (Fig. 4, Fig. S5–7), similar to a diffuse injury model using adrenergic stress (Fig. S8) without evidence of de novo cardiomyogenesis. Similarly, lack of profound cell cycle reentry and failure to progress through cell cycle has been shown in studies involving LAD ligation[50,63]. Additionally, absence of pHH3 immunolabeling coupled with extremely low identification of post-MI border zone BrdU+ cardiomyocytes (Fig. S6) at 21dpi contrasts with observations claiming proliferation[34] and may instead represent cells undergoing DNA repair or binucleation in response to injury. Interestingly, a cKit+/AzG+/pHH3+ young amplifying progenitor cell was identified in P2 hearts (Fig. 5), suggesting that progenitors can contribute to de novo cardiomyogenesis in postnatal development. While a comparable progenitor cell was not observed in the adult myocardium, but such a cell could perhaps be coaxed out of the adult myocardium by highly selective in vitro amplification as argued in prior publications[64,65].
This report establishes the utility and functionality of the FUCCI-Tg in response to postnatal development and infarction injury. Developing a shared platform of the FUCCI-Tg with the research community will help dispel misinterpretations of cardiomyocyte cell cycle status and cardiomyogenesis in the context of adult mammalian myocardium by providing a single, unified, straightforward technical approach and model readily interpreted and disseminated to the research community. Finally, it is now possible to identify ACMs in G0, G1, S/G2/M and G1/S without immunolabeling by identifying mKO+ and AzG+ expression patterns indicative of distinct cell cycle phases within the myocardium. Isolated ACMs can be sorted and compared to each other to identify novel chromatin remodeling and/or unique RNA signatures that potentially hold the key to forcing cardiomyocyte cell cycle reentry and completion. Availability of the FUCCI-Tg represents a novel and valuable tool to perform assessments of preclinical testing for interventional strategies intended to boost de novo cardiomyogenesis in adult myocardial tissue in vivo.
Supplementary Material
Acknowledgments
R. Alvarez and M. Sussman designed the overall experiments. R. Alvarez, B. Wang, T. Ho, M. Shaitrit, N. Navarrette, and P. Quijada performed the experiments and analyzed the data. R. Alvarez, B. Wang and M. Sussman wrote the article. D. Avitabile, M. Moshref, S. Siddiqui, N. Gude, F. Firouzi, D. Ebeid, M. Chavarria, F. Firouzi, D. Ebeid, M. Monsanto, K. Broughton, B. Bailey, N. Gude, assisted with experimental execution and/or data analysis. All authors read and approved the final article.
We gratefully acknowledge the San Diego State University Mouse Genomics Core for the generation of the transgenic mouse model created through their facility.
Sources of Funding
M.A. Sussman is supported by NIH grants: R01HL067245, R37HL091102, R01HL105759, R01HL113647, R01HL117163, P01HL085577, and R01HL122525, as well as an award from the Foundation Leducq.
Non-standard Abbreviations and Acronyms
- αMHC
alpha myosin heavy chain
- αSA
alpha sarcomeric actinin
- ACM
adult cardiomyocyte
- AzG
monomeric Azami Green
- BrdU
5’-bromo-deoxy-uridine (bromodeoxyuridine)
- BZ
border zone
- cKit
tyrosine-protein kinase Kit or CD117
- CM
cardiomyocyte
- CPC
cardiac progenitor cell
- cTnI
cardiac troponin I
- DAPI
4’,6-diamidino-2-phenylindole, nuclei stain
- dpc
days post coitum
- dpi
days post injury
- FUCCI
Fluorescent Ubiquitination-based Cell Cycle Indicators
- FBS
fetal bovine serum
- FVB/NJ
Friend leukemia virus B Strain
- G1
Gap 1 (G1-phase)
- G1/S
G1 to S phase transition
- G2
Gap 2 (G2-phase)
- HRP
horse radish peroxidase
- HS
horse serum
- Hsp60
Heat shock protein 60
- hCdt1
human Cdt1
- hGem
human Geminin
- ISO
isoproterenol
- IZ
infarct zone
- M
Mitosis (M-phase)
- MI
myocardial infarction
- mKO
monomeric Kusabira Orange 2
- PCR
polymerase chain reaction
- pHH3
phosphorylated Histone H3
- P
postnatal day
- RZ
remote zone
- S
Synthesis phase (S-phase)
- SA-X
streptavidin (X= LS490, 700, etc.)
- Sm22α
smooth muscle 22 alpha
- Sytox
Sytox blue, nuclei stain
- TPM
Tropomyosin
- Tyr
tyramide
- Vim
Vimentin
- vWF
von Willebrand Factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
M.A. Sussman is a founding member of CardioCreate, Inc.
Reference
- [1].Yuan X, Braun T, Multimodal Regulation of Cardiac Myocyte Proliferation, Circ. Res 121 (2017) 293–309. doi: 10.1161/CIRCRESAHA.117.308428. [DOI] [PubMed] [Google Scholar]
- [2].He L, Zhou B, Cardiomyocyte proliferation: remove brakes and push accelerators., Cell Res. 27 (2017) 959–960. doi: 10.1038/cr.2017.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lázár E, Sadek HA, Bergmann O, Cardiomyocyte renewal in the human heart: Insights fromthe fall-out, Eur. Heart J 38 (2017) 2333–2339. doi: 10.1093/eurheartj/ehx343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Siddiqi S, Sussman MA, The Heart: Mostly Postmitotic or Mostly Premitotic? Myocyte Cell Cycle, Senescence, and Quiescence, Can. J. Cardiol 30 (2014) 1270–1278. doi: 10.1016/j.cjca.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yutzey KE, Cardiomyocyte proliferation, Circ. Res 120 (2017) 627–629. doi: 10.1161/CIRCRESAHA.116.310058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pasumarthi KBS, Field LJ, Cardiomyocyte cell cycle regulation., Circ. Res 90 (2002) 1044–54. doi: 10.1161/01.RES.0000020201.44772.67. [DOI] [PubMed] [Google Scholar]
- [7].Zebrowski DC, Engel FB, The cardiomyocyte cell cycle in hypertrophy, tissue homeostasis, and regeneration., Rev. Physiol. Biochem. Pharmacol 165 (2013) 67–96. doi: 10.1007/112_2013_12. [DOI] [PubMed] [Google Scholar]
- [8].Zebrowski DC, Becker R, Engel FB, Towards regenerating the mammalian heart: challenges in evaluating experimentally induced adult mammalian cardiomyocyte proliferation., Am. J. Physiol. Heart Circ. Physiol 310 (2016) H1045–54. doi: 10.1152/ajpheart.00697.2015. [DOI] [PubMed] [Google Scholar]
- [9].Walsh S, Pontén A, Fleischmann BK, Jovinge S, Cardiomyocyte cell cycle control and growth estimation in vivo--an analysis based on cardiomyocyte nuclei., Cardiovasc. Res 86 (2010) 365–73. doi: 10.1093/cvr/cvq005. [DOI] [PubMed] [Google Scholar]
- [10].Vujic A, Lerchenmüller C, Di Wu T, Guillermier C, Rabolli CP, Gonzalez E, Senyo SE, Liu X, Guerquin-Kern JL, Steinhauser ML, Lee RT, Rosenzweig A, Exercise induces new cardiomyocyte generation in the adult mammalian heart, Nat. Commun 9 (2018) 1–9. doi: 10.1038/s41467-018-04083-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mohamed TMA, Ang YS, Radzinsky E, Zhou P, Huang Y, Elfenbein A, Foley A, Magnitsky S, Srivastava D, Regulation of Cell Cycle to Stimulate Adult Cardiomyocyte Proliferation and Cardiac Regeneration, Cell. 173 (2018) 104–116.e12. doi: 10.1016/j.cell.2018.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lesizza P, Prosdocimo G, Martinelli V, Sinagra G, Zacchigna S, Giacca M, Single-Dose Intracardiac Injection of Pro-Regenerative MicroRNAs Improves Cardiac Function after Myocardial Infarction, Circ. Res 120 (2017) 1298–1304. doi: 10.1161/CIRCRESAHA.116.309589. [DOI] [PubMed] [Google Scholar]
- [13].Paradis AN, Gay MS, Zhang L, Binucleation of cardiomyocytes: the transition from a proliferative to a terminally differentiated state., Drug Discov. Today 19 (2014) 602–9. doi: 10.1016/j.drudis.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ahuja P, Sdek P, MacLellan WR, Cardiac Myocyte Cell Cycle Control in Development, Disease, and Regeneration, Physiol. Rev 87 (2007) 521–544. doi: 10.1152/physrev.00032.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Polizzotti BD, Ganapathy B, Haubner BJ, Penninger JM, Kühn B, A cryoinjury model in neonatal mice for cardiac translational and regeneration research, Nat. Protoc 11 (2016) 542–552. doi: 10.1038/nprot.2016.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Darehzereshki A, Rubin N, Gamba L, Kim J, Fraser J, Huang Y, Billings J, Mohammadzadeh R, Wood J, Warburton D, Kaartinen V, Lien CL, Differential regenerative capacity of neonatal mouse hearts after cryoinjury, Dev. Biol 399 (2015) 91–99. doi: 10.1016/j.ydbio.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Naqvi N, Li M, Calvert JW, Tejada T, Lambert JP, Wu J, Kesteven SH, Holman SR, Matsuda T, Lovelock JD, Howard WW, Iismaa SE, Chan AY, Crawford BH, Wagner MB, Martin DIK, Lefer DJ, Graham RM, Husain A, A proliferative burst during preadolescence establishes the final cardiomyocyte number., Cell. 157 (2014) 795–807. doi: 10.1016/j.cell.2014.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Eschenhagen T, Bolli R, Braun T, Field LJ, Fleischmann BK, Frisén J, Giacca M, Hare JM, Houser S, Lee RT, Marbán E, Martin JF, Molkentin JD, Murry CE, Riley PR, Ruiz-Lozano P, Sadek HA, Sussman MA, Hill JA, Cardiomyocyte regeneration: A consensus statement, Circulation. 136 (2017) 680–686. doi: 10.1161/CIRCULATIONAHA.117.029343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Raulf A, Horder H, Tarnawski L, Geisen C, Ottersbach A, Röll W, Jovinge S, Fleischmann BK, Hesse M, Transgenic systems for unequivocal identification of cardiac myocyte nuclei and analysis of cardiomyocyte cell cycle status., Basic Res. Cardiol 110 (2015) 33. doi: 10.1007/s00395-015-0489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Leone M, Magadum A, Engel FB, Cardiomyocyte proliferation in cardiac development and regeneration: a guide to methodologies and interpretations, Am. J. Physiol. - Hear. Circ. Physiol (2015). doi: 10.1152/ajpheart.00559.2015. [DOI] [PubMed] [Google Scholar]
- [21].Mahmoud AI, Porrello ER, Kimura W, Olson EN, Sadek HA, Surgical models for cardiac regeneration in neonatal mice, Nat. Protoc 9 (2014) 305–311. doi: 10.1038/nprot.2014.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Aroumougame A, Shah AM, Szweda LI, Sadek HA, The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response, Cell. 157 (2014) 565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Leone M, Musa G, Engel FB, Cardiomyocyte binucleation is associated with aberrant mitotic microtubule distribution, mislocalization of RhoA and IQGAP3, as well as defective actomyosin ring anchorage and cleavage furrow ingression, Cardiovasc. Res (2018) 1–17. doi: 10.1093/cvr/cvy056. [DOI] [PubMed] [Google Scholar]
- [24].Sakaue-Sawano A, Miyawaki A, Visualizing spatiotemporal dynamics of multicellular cell-cycle progressions with fucci technology, Cold Spring Harb. Protoc 2014 (2014) 525–531. doi: 10.1101/pdb.prot080408. [DOI] [PubMed] [Google Scholar]
- [25].Abe T, Sakaue-Sawano A, Kiyonari H, Shioi G, Inoue K.-i., Horiuchi T, Nakao K, Miyawaki A, Aizawa S, Fujimori T, Visualization of cell cycle in mouse embryos with Fucci2 reporter directed by Rosa26 promoter, Development. 140 (2013) 237–246. doi: 10.1242/dev.084111. [DOI] [PubMed] [Google Scholar]
- [26].Hashimoto H, Yuasa S, Tabata H, Tohyama S, Hayashiji N, Hattori F, Muraoka N, Egashira T, Okata S, Yae K, Seki T, Nishiyama T, Nakajima K, Sakaue-Sawano A, Miyawaki A, Fukuda K, Time-lapse imaging of cell cycle dynamics during development in living cardiomyocyte, JMCC. 72 (2014) 241–249. doi: 10.1016/j.yjmcc.2014.03.020. [DOI] [PubMed] [Google Scholar]
- [27].Hashimoto H, Yuasa S, Tabata H, Tohyama S, Seki T, Egashira T, Hayashiji N, Hattori F, Kusumoto D, Kunitomi A, Takei M, Kashimura S, Yozu G, Shimojima M, Motoda C, Muraoka N, Nakajima K, Sakaue-Sawano A, Miyawaki A, Fukuda K, Analysis of cardiomyocyte movement in the developing murine heart, Biochem. Biophys. Res. Commun 464 (2015) 1000–1007. doi: 10.1016/j.bbrc.2015.07.036. [DOI] [PubMed] [Google Scholar]
- [28].Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN, Sadek HA, Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family, PNAS. 110 (2013) 187–192. doi: 10.1073/pnas.1208863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Porrello ER, Olson EN, A neonatal blueprint for cardiac regeneration, Stem Cell Res. 13 (2014) 556–570. doi: 10.1016/j.scr.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA, Transient regenerative potential of the neonatal mouse heart, Science (80-. ). 331 (2011) 1078–80. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bailey B, Izarra A, Alvarez R, Fischer KM, Cottage CT, Quijada P, Dez-Juan A, Sussman MA, Diez-Juan A, Sussman MA, Dez-Juan A, Sussman MA, Cardiac stem cell genetic engineering using the αmHC promoter, Regen. Med 4 (2009) 823–833. doi: 10.2217/rme.09.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li F, Wang X, Bunger PC, Gerdes AM, Formation of binucleated cardiac myocytes in rat heart: I. Role of actin-myosin contractile ring, JMCC. 29 (1997) 1541–1551. doi: 10.1006/jmcc.1997.0381. [DOI] [PubMed] [Google Scholar]
- [33].Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ, Cardiomyocyte DNA synthesis and binucleation during murine development., Am. J. Physiol 271 (1996) H2183–9. doi: 10.1152/ajpheart.1996.271.5.H2183. [DOI] [PubMed] [Google Scholar]
- [34].Soonpaa MH, Field LJ, Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts., Am. J. Physiol 272 (1997) H220–6. doi: 10.1152/ajpheart.1997.272.1.H220. [DOI] [PubMed] [Google Scholar]
- [35].Soonpaa MH, Rubart M, Field LJ, Challenges measuring cardiomyocyte renewal., Biochim. Biophys. Acta 1833 (2013) 799–803. doi: 10.1016/j.bbamcr.2012.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pardee AB, A restriction point for control of normal animal cell proliferation., PNAS. 71 (1974) 1286–90. doi: 10.1073/pnas.71.4.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Blagosklonny MV, Pardee AB, The restriction point of the cell cycle., Cell Cycle. 1 (2002) 103–110. doi: 10.4161/cc.1.2.108. [DOI] [PubMed] [Google Scholar]
- [38].Crescenzi M, Soddu S, Tatò F, Mitotic cycle reactivation in terminally differentiated cells by adenovirus infection., J. Cell. Physiol 162 (1995) 26–35. doi: 10.1002/jcp.1041620105. [DOI] [PubMed] [Google Scholar]
- [39].Li F, Wang X, Capasso JM, Gerdes AM, Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development, JMCC. 28 (1996) 1737–1746. doi: 10.1006/jmcc.1996.0163. [DOI] [PubMed] [Google Scholar]
- [40].Latella L, Sacco A, Pajalunga D, Tiainen M, Macera D, D’Angelo M, Felici A, Sacchi A, Crescenzi M, Reconstitution of Cyclin D1-Associated Kinase Activity Drives Terminally Differentiated Cells into the Cell Cycle, Mol. Cell. Biol 21 (2001) 5631–5643. doi: 10.1128/MCB.21.16.5631-5643.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Angert D, Berretta RM, Kubo H, Zhang H, Chen X, Wang W, Ogorek B, Barbe M, Houser SR, Repair of the injured adult heart involves new myocytes potentially derived from resident cardiac stem cells, Circ. Res 108 (2011) 1226–1237. doi: 10.1161/CIRCRESAHA.110.239046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wallner M, Duran JM, Mohsin S, Troupes CD, Vanhoutte D, Borghetti G, Vagnozzi RJ, Gross P, Yu D, Trappanese DM, Kubo H, Toib A, Sharp TE, Harper SC, Volkert MA, Starosta T, Feldsott EA, Berretta RM, Wang T, Barbe MF, Molkentin JD, Houser SR, Acute Catecholamine Exposure Causes Reversible Myocyte Injury Without Cardiac Regeneration, Circ. Res 119 (2016) 865–879. doi: 10.1161/CIRCRESAHA.116.308687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nadal-Ginard B, Ellison GM, Torella D, The cardiac stem cell compartment is indispensable for myocardial cell homeostasis, repair and regeneration in the adult., Stem Cell Res. 13 (2014) 615–30. doi: 10.1016/j.scr.2014.04.008. [DOI] [PubMed] [Google Scholar]
- [44].Ellison GM, Vicinanza C, Smith AJ, Aquila I, Leone A, Waring CD, Henning BJ, Stirparo GG, Papait R, Scarfò M, Agosti V, Viglietto G, Condorelli G, Indolfi C, Ottolenghi S, Torella D, Nadal-Ginard B, Adult c-kitposcardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair, Cell. 154 (2013) 827–842. doi: 10.1016/j.cell.2013.07.039. [DOI] [PubMed] [Google Scholar]
- [45].Saroff J, Wexler BC, Isoproterenol-induced myocardial infarction in rats. Distribution of corticosterone., Circ. Res 27 (1970) 1101–9. doi: 10.1161/01.RES.27.6.1101. [DOI] [PubMed] [Google Scholar]
- [46].Lobo Filho HG, Ferreira NL, de Sousa RB, de Carvalho ER, Lobo PLD, Lobo Filho JG, Experimental model of myocardial infarction induced by isoproterenol in rats., Rev. Bras. Cir. Cardiovasc 26 (2011) 469–76. http://www.ncbi.nlm.nih.gov/pubmed/22086586 (accessed November 18, 2018). [DOI] [PubMed] [Google Scholar]
- [47].Hesse M, Raulf A, Pilz G-AA, Haberlandt C, Klein AM, Jabs R, Zaehres H, Fügemann CJ, Zimmermann K, Trebicka J, Welz A, Pfeifer A, Röll W, Kotlikoff MI, Steinhäuser C, Götz M, Schöler HR, Fleischmann BK, Direct visualization of cell division using high-resolution imaging of M-phase of the cell cycle, Nat. Commun 3 (2012) 1076. doi: 10.1038/ncomms2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Muskhelishvili L, Latendresse JR, Kodell RL, Henderson EB, Evaluation of Cell Proliferation in Rat Tissues with BrdU, PCNA, Ki-67(MIB-5) Immunohistochemistry and in Situ Hybridization for Histone mRNA, J. Histochem. Cytochem 51 (2003) 1681–1688. doi: 10.1177/002215540305101212. [DOI] [PubMed] [Google Scholar]
- [49].Quaife-Ryan GA, Sim CB, Ziemann M, Kaspi A, Rafehi H, Ramialison M, El-Osta A, Hudson JE, Porrello ER, Multicellular transcriptional analysis of mammalian heart regeneration, Circulation. 136 (2017) 1123–1139. doi: 10.1161/CIRCULATIONAHA.117.028252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zebrowski DC, Vergarajauregui S, Wu CC, Piatkowski T, Becker R, Leone M, Hirth S, Ricciardi F, Falk N, Giessl A, Just S, Braun T, Weidinger G, Engel FB, Developmental alterations in centrosome integrity contribute to the post-mitotic state of mammalian cardiomyocytes, Elife. 4 (2015) 1–16. doi: 10.7554/eLife.05563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chong JJH, Murry CE, Cardiac regeneration using pluripotent stem cells-Progression to large animal models, Stem Cell Res. 13 (2014) 654–665. doi: 10.1016/j.scr.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sadek HA, Martin CM, Latif SS, Garry MG, Garry DJ, Bone-marrow-derived side population cells for myocardial regeneration., J. Cardiovasc. Transl. Res 2 (2009) 173–81. doi: 10.1007/s12265-009-9090-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Urbanek K, Quaini F, Tasca G, Torella D, Castaldo C, Nadal-Ginard B, Leri A, Kajstura J, Quaini E, Anversa P, Intense myocyte formation from cardiac stem cells in human cardiac hypertrophy, PNAS. 100 (2003) 10440–10445. doi: 10.1073/pnas.1832855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhang Y, Mignone J, MacLellan WR, Cardiac Regeneration and Stem Cells, Physiol. Rev (2015). doi: 10.1152/physrev.00021.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Matturri L, Biondo B, Colombo B, Lavezzi AM, Rossi L, Significance of the DNA synthesis in hypertrophic cardiomyopathies, Basic Res. Cardiol 92 (1997) 85–89. doi: 10.1007/BF00805568. [DOI] [PubMed] [Google Scholar]
- [56].Matturri L, Milei J, Grana DR, Lavezzi AM, Characterization of myocardial hypertrophy by DNA content, PCNA expression and apoptotic index, Int. J. Cardiol 82 (2002) 33–39. doi: 10.1016/S0167-5273(01)00578-2. [DOI] [PubMed] [Google Scholar]
- [57].Drenckhahn JD, Strasen J, Heinecke K, Langner P, Yin KV, Skole F, Hennig M, Spallek B, Fischer R, Blaschke F, Heuser A, Cox TC, Black MJ, Thierfelder L, Impaired myocardial development resulting in neonatal cardiac hypoplasia alters postnatal growth and stress response in the heart, Cardiovasc. Res 106 (2015) 43–54. doi: 10.1093/cvr/cvv028. [DOI] [PubMed] [Google Scholar]
- [58].Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Di Wu T, Guerquin-Kern J-LL, Lechene CP, Lee RT, Guerquin-Kern J-LL, Lechene CP, Lee RT, Mammalian heart renewal by pre-existing cardiomyocytes, Nature. 493 (2013) 433–436. doi: 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Senyo SE, Lee RT, Kühn B, Cardiac regeneration based on mechanisms of cardiomyocyte proliferation and differentiation, Stem Cell Res. 13 (2014) 532–541. doi: 10.1016/j.scr.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Finkel T, Hwang PM, The Krebs cycle meets the cell cycle: Mitochondria and the G1-S transition, PNAS. 106 (2009) 11825–11826. doi: 10.1073/pnas.0906430106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Rumyantsev PP, Interrelations of the Proliferation and Differentiation Processes during Cardiac Myogenesis and Regeneration, Int. Rev. Cytol 51 (1977) 187–273. doi: 10.1016/S0074-7696(08)60228-4. [DOI] [PubMed] [Google Scholar]
- [62].Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER, Sadek HA, Meis1 regulates postnatal cardiomyocyte cell cycle arrest., Nature. 497 (2013) 249–253. doi: 10.1038/nature12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Reuter S, Soonpaa MH, Firulli AB, Chang AN, Field LJ, Recombinant neuregulin 1 does not activate cardiomyocyte DNA synthesis in normal or infarcted adult mice., PLoS One. 9 (2014) e115871. doi: 10.1371/journal.pone.0115871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Vicinanza C, Aquila I, Scalise M, Cristiano F, Marino F, Cianflone E, Mancuso T, Marotta P, Sacco W, Lewis FC, Couch L, Shone V, Gritti G, Torella A, Smith AJ, Terracciano CM, Britti D, Veltri P, Indolfi C, Nadal-Ginard B, Ellison-Hughes GM, Torella D, Adult cardiac stem cells are multipotent and robustly myogenic: c-kit expression is necessary but not sufficient for their identification, Cell Death Differ. 24 (2017) 2101–2116. doi: 10.1038/cdd.2017.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Nadal-Ginard B, Ellison GM, Torella D, Absence of Evidence Is Not Evidence of Absence, Circ. Res 115 (2014) 415–418. doi: 10.1161/CIRCRESAHA.114.304676. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.