

Abstract/Summary
Contemporary endovascular stents are the product of an iterative design and development process that leverages evolving concepts in vascular biology and engineering. Indeed, in many respects the stent is the paradigmatic example of a medically motivated and clinically verified device. In this review, we explain how insights into vascular pathophysiology, materials science, and design mechanics drive stent design and explain modes of stent failure.
Stents were themselves part of a continuum in device development that followed on the heels of the success of balloon angioplasty as well as its limitations, principally of arterial dissection and recoil. Early stent experience was fraught with in-stent restenosis, the inadequate flow that arose from luminally-obstructive neointimal hyperplasia. Controlled release of anti-proliferative agents inhibited growth of the neointima and delayed arterial healing, thereby offsetting restenosis but predisposing to late risk of in-stent thrombosis resulting from mechanical injury, alterations in local flow, or direct cellular toxicity (e.g. endothelial cells specifically or loss of neointima generally). Current knowledge of pathological processes such as neoatherosclerosis and recognition of patient-dependent factors are providing a more complete picture of the factors mediating stent failure. Further evolution of endovascular stents includes fully bioresorbable platforms tailored to treat plaques acutely then disappear after lesion pacification. Ongoing refinement of stent technology will continue to require insights from pathology to understand adverse events, refine clinical protocols, and drive innovation.
Keywords: Coronary artery disease, pathology, bare metal stent, drug-eluting stent, in-stent restenosis, in-stent thrombosis, atherosclerosis, neoatherosclerosis
Introduction
Coronary artery disease (CAD) represents the leading cause of death worldwide, attributed to over 17.5 million deaths annually, accounting for approximately 1 of every 3 deaths.1 In the United States, contemporary decreases in CAD-related mortality correlate with the three decades following the Surgeon’s General report on the ills of tobacco, the Framingham Heart Study’s identification of cardiac risk factors with lifestyle modification, and the widespread acceptance and accessibility of evidence-based use of percutaneous coronary intervention (PCI).2 The technologies underpinning PCI have evolved iteratively from balloon angioplasty to increasingly advanced metallic stent platforms with various drug chemistries to self-degrading non-metallic scaffolds. Large-scale clinical trials have validated the safety and efficacy of successive generations of stents. Equally important preclinical3–5 and pathology studies provide complementary insight to reconcile adverse events, refine clinical protocols such as optimal use of anti-thrombotic therapy, and drive innovation for development of next-generation stents.
Native coronary artery disease
The fundamental pathogenesis of native coronary atherosclerosis has been described for decades but only relatively recently have the specific nuances of these processes been characterized, particularly with respect to coronary intervention. As early as the 1970s, the importance of arterial injury in the establishment of atherosclerosis was recognized in the context of vascular smooth muscle cell activation and proliferation.6,7 The injury model shifted towards a more complex understanding underpinned by inflammation, and integration of recognized cardiac risk factors such as hyperlipidemia, hypertension, diabetes, and smoking.8,9 The pathogenesis of atherosclerosis as a chronic inflammatory disease marked by progressive vascular wall injury became further defined and synchronized with clinical events such as plaque rupture and acute thrombosis comprising the acute coronary syndrome.10–13 Establishment and maturation of atherosclerotic plaque is now well-recognized as a progression from deposition and subsequent oxidation of free cholesterol, intimal thickening, and xanthoma (“fatty streak”) development to infiltration and lipid-avid macrophages, formation of a necrotic core, and progression to fibroatheroma predisposed to rupture and thrombosis.14,15 Introduction of routine percutaneous coronary intervention (PCI) transformed the management of coronary artery disease and provided serial clinical data which emphasized the non-linear nature of atherosclerotic plaque development: luminal stenosis alone is not a predictor of future clinical events16 and bore the concept of the “vulnerable plaque.”17 Clearly, not all plaques are created equal and some are more prone to rupture than others. Our contemporary understanding of atherosclerosis now classifies the vulnerable plaque as thin-cap fibroatheroma (TCFA), predisposed to acute plaque rupture, and incorporates other pathologic mechanisms of thrombosis such as healed plaque rupture, surface erosion, and calcified nodules.13 Ante-mortem identification of such rupture-prone plaques has yet to be realized, frustrating current clinical management paradigms and causing some to question the very existence of such lesions.
Pathology of balloon angioplasty
The introduction of PCI with balloon angioplasty marked the first widely adopted technique to directly alter the natural history of atherosclerosis. Angioplasty alone, however, proved to be a temporizing therapy owing to the traumatic and inconsistent nature of plaque modification.18–20 Though initially perceived and perhaps hoped to result in permanent deformation, the end result was far more elastic, with reversible displacement that more often recoiled back to its original dimension. Associated tissue damage was, however, real and injury to endothelium, intima, and media promoted rapid restenosis within weeks to months of therapy in addition to further precipitating the acute complications of arterial dissection and recoil.21,22 The specific effects of balloon angioplasty on the arterial wall have been defined serially: vascular recoil and contraction following balloon dilation; injury to the intima and dissection of the media; inflammatory activation and proliferation of vascular smooth muscle cells, resulting in rapid neointimal hyperplasia, extracellular matrix deposition, and negative vascular remodeling.23–26 To combat the loss of effect and minimize the extent of injury, PCI incorporated the use of permanent metal mesh implants, bare metal stents (BMS), following balloon angioplasty to rigidly support the arterial lumen, controlling arterial dissection and preventing acute arterial recoil to good effect.22,27 Stent placement, nevertheless, necessarily modifies the stenotic lesion and alters arterial architecture, inducing arterial injury not unlike balloon angioplasty.
Pathology of bare metal stents
Bare metal stent deployment typically results in neointimal hyperplasia, which is driven by vascular smooth muscle cell proliferation and associated with macrophage accumulation and neovascularization. This may be either distributed along the length of the stent or focally.24,28–34 During the first two weeks of BMS placement, fibrin, platelets, and acute inflammatory cells are localized to the stent struts, particularly those embedded within the necrotic plaque core or injured arterial wall media (Figure 1 & 2).35,36 In the weeks and months that follow, neointimal hyperplasia and then increasingly, extracellular matrix deposition contribute to neointimal growth.37 In association with a metallic scaffold, the arterial architecture is altered such that homeostatic expansive remodeling occurs (the so-called Glagov’s phenomenon38) and physiologic vasodilation is impaired. Incremental plaque development thus directly impinges on lumen area, rapidly precipitating in-stent restenosis (ISR) at an accelerated rate compared to native disease. Through these mechanisms, ISR accelerates early and appears to peak at around six months and, in its ultimate state, may precipitate recrudesce of clinical symptoms requiring repeat target lesions revascularization (TLR). By the first year following BMS placement, however, the neointima generally stabilizes and luminal diameter may regress.39,40 Our laboratory reported the importance of the geometric configuration of stent struts in addition to surface material in predicting the degree of arterial injury and inflammation (Figure 3).5 This idea not only led to introduction of a range of stent designs but suggested that drugs might best have their effects if delivered in the vicinity of the implanted devices and to this end, drug-eluting stents engineered with advanced materials and anti-proliferative properties were developed to prevent short-term vascular injury, neointimal hyperplasia, and thrombosis.
Figure 1.
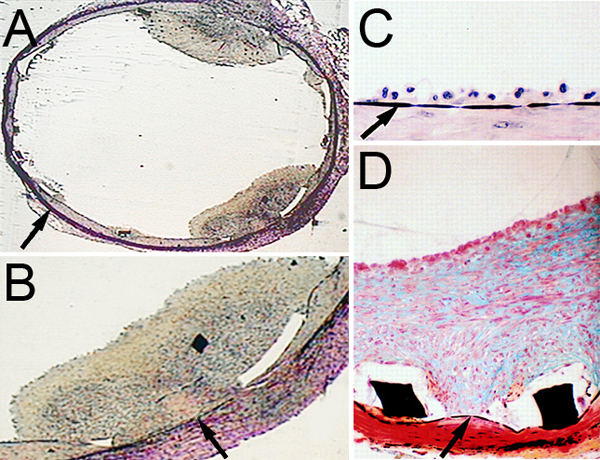
A & B, Acute and subacute thrombosis is observed following endovascular bare metal stent (BMS) placement. C, Three days following endovascular BMS placement (Verhoeff’s elastic tissue stain), adherent mononuclear cells line the internal elastic lamina (arrow). D, Two weeks following BMS placement (Modified Russell-Movat Pentachrome stain), robust neointimal hyperplasia is observed separating the lumen from the internal elastic lamina (arrows). Stent struts (black rectangles, white area is post-processing effect). Modified from Rogers et al.133
Figure 2.
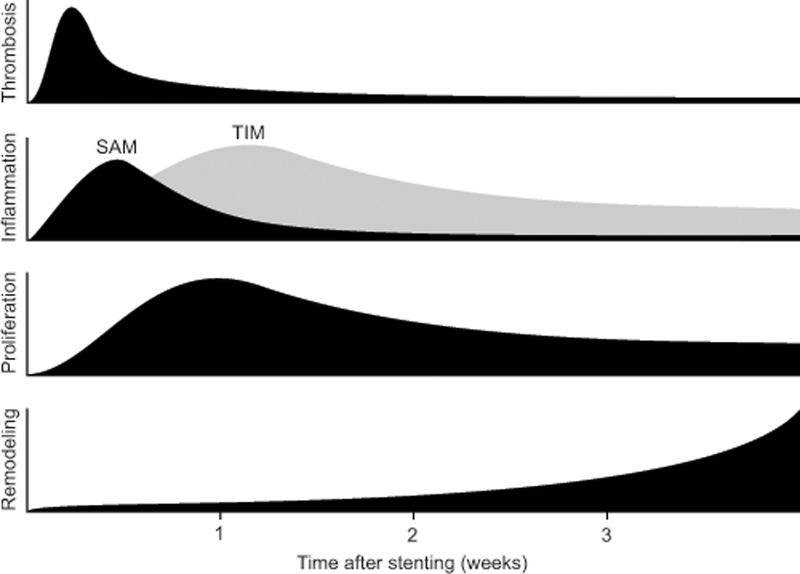
Activity of the four primary components of arterial injury following stent placement. Platelet-rich thrombosis peaks 3–4 days after stent deployment especially over areas of strut injury. Concomitant inflammation is initially mediated by surface-adherent monocytes (SAM) recruited to the injury site that then migrate into the neointima as tissue-infiltrating monocytes (TIM) and accumulate around the stent struts as giant cells. Vascular smooth muscle cell proliferation peaks 7 days after stent deployment coincident with the transition of SAM to TIM and continues for weeks afterward. Extracellular matrix deposition in the adventitia, tunica media, and neointima accelerates at week 3 after stent deployment and underlies arterial remodeling and subsequent luminal narrowing. Adapted from Edelman and Rogers.36
Figure 3.
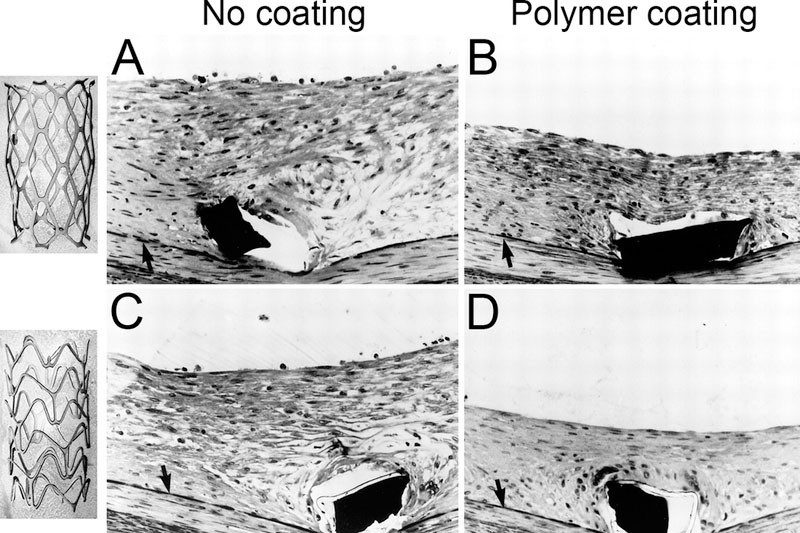
Stent surface and geometry significantly affect vascular injury and neointimal hyperplasia.5 Two bare metal stent (BMS) where fabricated using the same metal, process, and net surface area, on in a slotted tube (upper left panel) and the other corrugated ring (lower left panel) configuration and implanted in rabbit femoral arteries. Histologic examination was performed on methylmethacyrlate-embedded specimens harvested two weeks following placement. A, B uncoated surface; C, D polymer coated surface. Internal elastic lamina (arrow), stent struts (black rectangles), and polymer material (white rim circumscribing stent strut in C & D). Adapted from Edelman and Rogers.5
Pathology of drug-eluting stents: in-stent restenosis
Drug-eluting stents (DES) are defined by an elutable drug delivered to the arterial wall via controlled release from a polymer matrix that uniformly coats the metallic stent. First-generational DES employed either the cytostatic agent sirolimus to suppress smooth muscle cell activation by arresting the G1 phase of the cell cycle or the cytotoxic agent paclitaxel to interfere with microtubular depolymerization.41 Introduction of DES significantly reduced ISR and TLR in several pivotal randomized clinical trials.42–45 It was still the case that lesion complexity drove ISR.46,47 Likely mechanisms to explain ISR with DES were organized into four primary domains: 1) biological factors including vascular injury,33 malapposition,48 non-uniform drug delivery;49–51 2) mechanical factors including stent under-expansion,52,53 hemodynamic stress, and stent fracture;54,55 3) technical factors including barotrauma,43 stent gap and overlap,56 and residual untreated plaque;57 and 4) patient-specific factors such as comorbidities,58 drug resistance,59,60 and hypersensitivity61,62 (Figure 4). Detailed pathological study of stents revealed direct injury to the media or lipid-rich necrotic core of plaques by penetrating stent struts, thus predisposing to ISR.33,63 Unique pathological responses are observed between stent composition, consistent with expected differences in drug biology.64 For example, response to sirolimus-eluting stents is often characterized by malapposition due to robust inflammation and intimal infiltration of eosinophils, lymphocytes, and giant cells whereas response to paclitaxel-eluting stents included malapposition induced by excessive para-strut fibrin deposition.65 These processes ultimately invoke the common pathological response of neointimal hyperplasia and inflammation, leading to extracellular matrix deposition and negative vascular remodeling.
Figure 4.
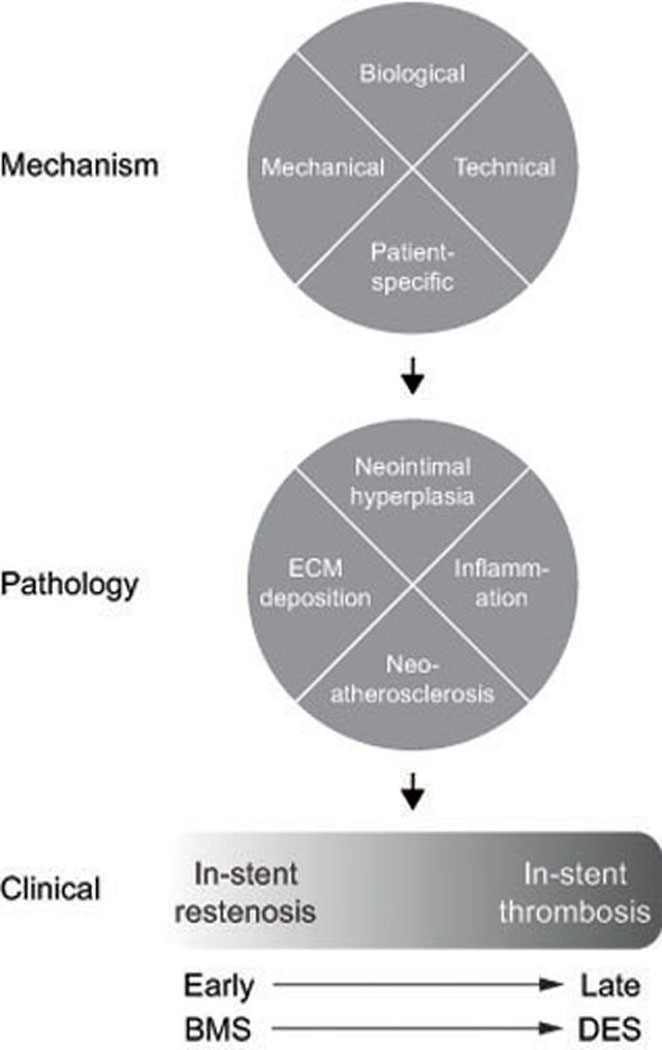
Schematic of the mechanisms and pathology mediating clinical in-stent restenosis and thrombosis, which likely lies on a shared continuum that shifts from early events associated with bare metal stents (BMS) to late events, associated with drug eluting stents (DES). Biological factors include vascular injury, malapposition with fibrin deposition, and non-uniform drug delivery; mechanical factors include stent under-expansion, hemodynamic stress, and stent fracture; technical factors include balloon barotrauma, stent gap and overlap, and residual untreated plaque; and patient-specific factors such as comorbidities, drug resistance, and hypersensitivity. Extracellular matrix = ECM.
Neoatherosclerosis
Contemporary long-term pathological studies have identified a novel mechanism of in-stent atherosclerosis, which is independent of native atherosclerosis and develops rapidly within the neointima. This is so-called neoatherosclerosis, an important mediator of ISR with DES.66–68 Neoatherosclerosis is characterized by accelerated plaque progression in which there is accumulation of peri-strut macrophages and lipid-rich foam cells which organize as fibroatheroma on the luminal surface and deeper within the neointima.67 Neoatherosclerosis predisposes to plaque fissure and rupture that may present symptomatically as acute, often catastrophic, thrombosis or may be clinically silent, forming substrate for ISR and chronic thrombotic occlusion.68 Compared to BMS, DES are associated with neointima with more abundant proteoglycan in the extracellular matrix which is highly avid to lipoproteins.69,70 Local disruption of laminar blood flow by stent struts induces alterations in shear stress that stimulates endothelium to express the intercellular adhesion molecules ICAM-1 and VCAM-1, allowing transmigration of circulating monocytes into the neointima where they activate into macrophages, load with lipid, and form foam cells.71–73 The disrupted fluid dynamics also promote platelet and fibrin deposition,67 and predispose to thrombus formation. Over time, a necrotic core of free cholesterol devoid of extracellular matrix and sometimes exhibiting calcification forms within the neoatherosclerotic plaque from direct apoptosis of foam cells and smooth muscle cells.74–76 Unlike native atherosclerosis, pathologic neointimal thickening does not occur within the stented neoatherosclerotic plaques, and lesions progress rapidly with superficial necrotic cores that are inherently less stable and progress more rapidly into late fibroatheromas.68 Moreover, unlike intimal xanthomas or “fatty steaks” in native atherosclerosis which may remain stable or regress,77,78 neoatherosclerosis lesions invariably progress to necrotic cores though apoptosis.13 Intramural hemorrhage from fissuring of the luminal surface or leaking of the adventitial vaso vasorum further destabilizes the neoatherosclerotic plaque.79 The fibrous cap eventually thins, forming “vulnerable plaque” or thin-cap fibroatheroma (TCFA) at high-risk for plaque rupture67 and a histologic predictor of future coronary event.13
The precise mechanism of neoatherosclerosis remains unclear, but dysfunctional endothelial barrier function due to incompetent or incomplete endothelial coverage of the stent is thought to play a key role.79,80 This is supported by clinical evidence that neoatherosclerosis develops more rapidly with DES than BMS.65 Mechanical injury by stent struts denudes the arterial wall of endothelium. The anti-proliferative effects of DES prevents maturation of the regenerating endothelium, further impairing endothelial integrity. This may explain the more pronounced and rapid development of neoatherosclerosis in DES than in BMS.65,81 Specifically, poorly functional endothelium is characterized by reduced intercellular junctions, antithrombin expression, and nitric oxide production, which are observed more commonly in DES compared to BMS.71,80 The anti-proliferative effects of drug and delayed endothelial healing is further compounded when stent struts violate the necrotic core where drug clearance and repair mechanisms are reduced in the avascular space.67 Classically, circulating monocytes infiltrate the intima through the damaged endothelium and differentiate into macrophages, which load with lipid and undergo apoptosis, leaving xanthomas.67,80 More recently, direct transdifferation of smooth muscle cells into macrophages has been described, using elegant in vitro lineage tracing experiments of native atherosclerosis.82 Whether a similar process of phenotype transition occurs in neoatherosclerosis is unknown. Neoatherosclerosis may result in ISR or more often serves as substrate for plaque instability and eventual rupture and thrombosis.
Pathology of drug-eluting stents: late in-stent thrombosis
A complementary pathological process to ISR is in-stent thrombosis (IST)83,84 (Figure 4). Whereas ISR is a progressive process often leading to stable angina and rarely (~ 10%), myocardial infarction (MI), IST is a catastrophic cause of acute MI and sudden death84 and may present as late stent failure.58,85–87 The biological, mechanical, technical, and patient-specific factors described for ISR also contribute to a lesser extent to IST.65,79,88,89 Notably, neoatherosclerosis has more recently been studied as a significant substrate for LST/VLST in both BMS and DES. Late stent failure has been ascribed to neoatherosclerosis in numerous pathology and intra-coronary imaging studies.66,90,91 While DES successfully forestalls early development of ISR through inhibition of intimal hyperplasia, delayed healing of the stented region also predisposes to the complication of late IST.79,85,88,92–95 Although initial studies reported comparable rates of IST with DES compared with BMS within the first six to twelve months of stenting,96–98 late thrombosis became recognized with longer follow-up.99 The benefit of decreased ISR with DES was thus partially counter-balanced by increased risk of IST. Long-term rates of MI and death beyond one year after stenting were not significantly different between first-generation DES and BMS.99 In addition to acute and subacute IST within one month of stenting, late stent thrombosis (LST) occurring 30 days to one year and very late stent thrombosis (LVST) occurring after one year are well-recognized complications of first-generation DES.92 The primary substrate for LST/VLST in DES is delayed arterial healing and possibly inadequate stent coverage and incorporation into the vessel wall.80,88,93,100 The ratio of uncovered to total stent area as determined by histology is a significant predictor of LST.93 Thrombosis by plaque rupture may be mediated by lesions in the vicinity of thrombus initiation, likely either within the stented region or in the immediate vicinity. Intra-coronary imaging studies suggest that plaque rupture occurs more frequently at the non-stented region immediately adjacent to the stent edge, causing thrombosis of the stented region.101,102 Autopsy studies, however, show neoatherosclerosis originating within and restricted to stented regions, suggesting flow disturbances at the native-stent transition may play a role in stented plaque rupture and thrombosis.67
Second-generation DES
Second-generation DES technology provided improvements in drug, polymer, and metal properties together with lower profile geometries that address many of the biological, mechanical, and technical factors that underlie first-generation DES failure. Nevertheless, second-generation DES remain vulnerable to long-term stent failure. The second-generation cobalt-chromium everolimus-eluting stent (CoCr-EES) has been consistently superior to first-generation DES with reduction in ISR and TRL, myocardial infarction, and cardiac death in high-quality randomized control trials.94,103–107 Second-generation DES technology has ameliorated the risk of ISR with improved arterial healing and reduced the risk of IST,108–110 yet stent failure remains a known complication and cumulative incidence of ISR and TLR increases with all generations of DES platforms over time.86,111–113 It is suggested that in total, all of the design improvements that accompany CoCr-EES enhance healing or reduce initial injury with greater endothelial regeneration114 and less inflammation and fibrin deposition compared to sirolimus- and paclitaxel-eluting first-generation DES.115 Evidence for neoatherosclerosis was noted for CoCr-EES at 270 days, much later than first-generation siroliumus- and paclitaxel-DES in which neoatherosclerosis was observed at 120 and 70 days, respectively.115 The more gradual development of neoatherosclerosis in second- versus first-generation DES may promote more stable lesion development, as high-risk features like TCFA and plaque rupture were not observed with second-generation DES.116 While second-generation DES appears to promote greater endothelial healing and relatively more stable neoatherosclerosis, the overall incidence of neoatherosclerosis, however, is similar across DES generations,110,115,117 and thrombosis risk remains a concern clinically.111 Second-generation DES is increasingly acknowledged to have a catch-up phenomenon of neointimal growth that correlates with delayed arterial healing.111,118
Further improvements in PCI technology and techniques have enabled treatment of more complex disease such as bifurcation lesions posing higher risk for complications. The pathology associated with DES in bifurcation lesions is accentuated at the bifurcation carina, which is a high shear stress area where arterial healing is impaired compared to the lateral wall. This results in greater fibrin deposition, necrotic core accumulation, and plaque thickness at the carina compared to the lower shear stress lateral walls.119 Efforts to improve stent material and architecture as well as polymer coating have been eclipsed by technology to mitigate or eliminate these components from next generation devices.
Next-generation stents and scaffolds
Persistence of polymer coating in first-generation DES after drug delivery is problematic as a stimulus of peri-strut inflammation, delayed arterial healing, ISR, and IST. This has led to efforts to minimize polymer or develop polymer-free scaffolds.80,120–122 The next iteration of the DES involves further refinement of drug and polymer matrix, including stents with asymmetric coatings, biodegradable polymer materials or controlled drug delivery not requiring polymer coating. These DES with novel polymer materials or drug delivery strategies are thought be transformed into BMS after the acute and short-term benefit of drug elution is complete. The underlying hypothesis in support of the totally erodible materials is that long-term presence of a rigid metallic stent in the arterial environment inhibits physiologic vascular tone and vasomotion123 and is a nidus for late complications like fracture, neoatherosclerosis, and LST/LVST. To this end, bioresorbable scaffolds (BRS) that degrade after lesion pacification have been in development since the early conceptualization of endovascular stents with the pioneering Igaki-Tamai BRS and succeeded by several BRS currently in clinical trial. Notably, the Absorb everolimus-eluting bioresorbable vascular scaffold (BVS) (Abbott Vascular; Abbott Park, Illinois) and DESolve novolimus-eluting bioresorbable coronary scaffold (Elixir Medical Corporation; Sunnyvale, California) have received Conformité Européenne (CE) mark approval in 2011 and 2013, respectively, and await FDA approval.124 Early experience with the Absorb BVS has been promising,125,126 but must be tempered by reports of increased rates of acute thrombosis.127,128 While BRS appears to address the longer-term complications of delayed arterial healing and neoatherosclerosis by complete degradation, they may be subject to the complications encountered with early stent designs. Specifically, large and bulky strut architecture and propensity for under expansion creates local disruption of laminar flow and increased hemodynamic stress.5,63,129 The inflammatory response to the Absorb BVS appears to peak at one month and largely abates by three months with positive expansile remodeling after 12 months in non-atherosclerotic swine but may persist for longer in the human.130,131 Small cohorts of patients have demonstrated similar increases in lumen diameter in imaging studies.132 The anticipated benefits of BRS technology, notably restoration of vascular tone and vasomotion and reduced risk of LST and LVST, require further clinical study. Human pathology and imaging studies will again provide important insights into the pathophysiology and performance of BRS technology.
Summary/Conclusions
The generalized pathology associated with endovascular stents is characterized by acute arterial injury, neointimal hyperplasia and inflammation, extracellular matrix deposition, and negative vascular remodeling. Numerous biological, mechanical, and technical factors contribute to ISR and IST, and evolution of PCI technologies has led to an amazing array of studies defining this vascular pathobiology in a synergistic manner and addressed these pathologies, seeking to balance the acute, chronic, and long-term requirements for lesion pacification and vascular healing. Commercially available DES are effective but limited by late and very late complications of IST, mediated principally by incomplete endothelial healing as a consequence of the anti-proliferative effects that oppose early ISR. Neoatherosclerosis is another mechanism increasingly recognized as a cause of LST and LVST. Next-generation DES and BRS platforms address the pathology observed with prior stent designs and will likely be complemented by adjunctive devices like drug-eluting balloons and specialized bifurcation designs. Careful attention to the pathophysiological response to these new technologies, with rigorous preclinical, autopsy, and in vivo imaging studies, will inform continued advancements. Successful endovascular stent design will ultimately match the specific attributes of the stent with the expected pathology underlying the exact clinical setting, including integration of lesion- and patient-specific determinants of stent failure.
Key Points.
Improvement in endovascular stent performance has occurred iteratively over decades and highlights the ability to optimize physiologic function and minimize pathologic response through design.
Pathology associated with endovascular stents most commonly manifests clinically as progressive angina due to in-stent restenosis or acute myocardial infarction due to in-stent thrombosis.
- In-stent restenosis is mediated by neointima hyperplasia due to a complex interaction of
- biological factors including vascular injury, malapposition with fibrin deposition, and non-uniform drug delivery;
- mechanical factors including stent under-expansion, hemodynamic stress, and stent fracture;
- technical factors including balloon barotrauma, stent gap and overlap, and residual untreated plaque; and
- patient-specific factors such as comorbidities, drug resistance, and hypersensitivity.
Delayed arterial healing and possibly incomplete stent coverage by endothelium predominantly mediate in-stent thrombosis, leading to plaque fissuring and rupture.
Neoatherosclerosis develops over months to years as opposed to decades with native coronary atherosclerosis and contributes to late and very late in-stent thrombosis.
Acknowledgements
The authors thank Dr. Abraham R. Tzafriri for thoughtful discussion and editorial assistance.
Funding
This manuscript was supported in part by the National Institutes of Health (R01 GM-49039) to Dr. Edelman.
Footnotes
Financial Disclosure: Nothing to disclose for all authors.
References
- 1.World Health Organization Global Status Report on Noncommunicable Diseases. 2014. http://apps.who.int/iris/bitstream/10665/148114/1/9789241564854_eng.pdf. Accessed 12/20/2015. [DOI] [PubMed]
- 2.Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics−−2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29–322. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz RS, Edelman ER, Carter A, et al. Drug-eluting stents in preclinical studies: recommended evaluation from a consensus group. Circulation. 2002;106(14):1867–1873. [DOI] [PubMed] [Google Scholar]
- 4.Joner M, Byrne RA. The importance of preclinical research in contemporary interventional cardiology. EuroIntervention : journal of EuroPCR in collaboration with the Working Group on Interventional Cardiology of the European Society of Cardiology. 2010;6(1):19–23. [PubMed] [Google Scholar]
- 5.Rogers C, Edelman ER. Endovascular stent design dictates experimental restenosis and thrombosis. Circulation. 1995;91(12):2995–3001. [DOI] [PubMed] [Google Scholar]
- 6.Ross R, Glomset JA. The pathogenesis of atherosclerosis (first of two parts). The New England journal of medicine. 1976;295(7):369–377. [DOI] [PubMed] [Google Scholar]
- 7.Ross R, Glomset JA. The pathogenesis of atherosclerosis (second of two parts). The New England journal of medicine. 1976;295(8):420–425. [DOI] [PubMed] [Google Scholar]
- 8.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. The New England journal of medicine. 2005;352(16):1685–1695. [DOI] [PubMed] [Google Scholar]
- 9.Libby P Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874. [DOI] [PubMed] [Google Scholar]
- 10.Fuster V, Badimon L, Badimon JJ, Chesebro JH. The pathogenesis of coronary artery disease and the acute coronary syndromes (1). The New England journal of medicine. 1992;326(4):242–250. [DOI] [PubMed] [Google Scholar]
- 11.Fuster V, Badimon L, Badimon JJ, Chesebro JH. The pathogenesis of coronary artery disease and the acute coronary syndromes (2). The New England journal of medicine. 1992;326(5):310–318. [DOI] [PubMed] [Google Scholar]
- 12.Stary HC, Chandler AB, Dinsmore RE, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92(5):1355–1374. [DOI] [PubMed] [Google Scholar]
- 13.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arteriosclerosis, thrombosis, and vascular biology. 2000;20(5):1262–1275. [DOI] [PubMed] [Google Scholar]
- 14.Davies MJ, Thomas A. Thrombosis and acute coronary-artery lesions in sudden cardiac ischemic death. The New England journal of medicine. 1984;310(18):1137–1140. [DOI] [PubMed] [Google Scholar]
- 15.Falk E, Nakano M, Bentzon JF, Finn AV, Virmani R. Update on acute coronary syndromes: the pathologists’ view. European heart journal. 2013;34(10):719–728. [DOI] [PubMed] [Google Scholar]
- 16.Glaser R, Selzer F, Faxon DP, et al. Clinical progression of incidental, asymptomatic lesions discovered during culprit vessel coronary intervention. Circulation. 2005;111(2):143–149. [DOI] [PubMed] [Google Scholar]
- 17.Stary HC, Blankenhorn DH, Chandler AB, et al. A definition of the intima of human arteries and of its atherosclerosis-prone regions. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1992;85(1):391–405. [DOI] [PubMed] [Google Scholar]
- 18.Dangas G, Fuster V. Management of restenosis after coronary intervention. American heart journal. 1996;132(2 Pt 1):428–436. [DOI] [PubMed] [Google Scholar]
- 19.Ormiston JA, Stewart FM, Roche AH, Webber BJ, Whitlock RM, Webster MW. Late regression of the dilated site after coronary angioplasty: a 5-year quantitative angiographic study. Circulation. 1997;96(2):468–474. [DOI] [PubMed] [Google Scholar]
- 20.Dangas GD, Claessen BE, Caixeta A, Sanidas EA, Mintz GS, Mehran R. In-stent restenosis in the drug-eluting stent era. Journal of the American College of Cardiology. 2010;56(23):1897–1907. [DOI] [PubMed] [Google Scholar]
- 21.Block PC, Myler RK, Stertzer S, Fallon JT. Morphology after transluminal angioplasty in human beings. The New England journal of medicine. 1981;305(7):382–385. [DOI] [PubMed] [Google Scholar]
- 22.Serruys PW, de Jaegere P, Kiemeneij F, et al. A comparison of balloon-expandable-stent implantation with balloon angioplasty in patients with coronary artery disease. Benestent Study Group. The New England journal of medicine. 1994;331(8):489–495. [DOI] [PubMed] [Google Scholar]
- 23.Libby P, Tanaka H. The molecular bases of restenosis. Progress in cardiovascular diseases. 1997;40(2):97–106. [DOI] [PubMed] [Google Scholar]
- 24.Lowe HC, Oesterle SN, Khachigian LM. Coronary in-stent restenosis: current status and future strategies. Journal of the American College of Cardiology. 2002;39(2):183–193. [DOI] [PubMed] [Google Scholar]
- 25.Danenberg HD, Welt FG, Walker M, 3rd, Seifert P, Toegel GS, Edelman ER. Systemic inflammation induced by lipopolysaccharide increases neointimal formation after balloon and stent injury in rabbits. Circulation. 2002;105(24):2917–2922. [DOI] [PubMed] [Google Scholar]
- 26.Schoenhagen P, Ziada KM, Vince DG, Nissen SE, Tuzcu EM. Arterial remodeling and coronary artery disease: the concept of “dilated” versus “obstructive” coronary atherosclerosis. Journal of the American College of Cardiology. 2001;38(2):297–306. [DOI] [PubMed] [Google Scholar]
- 27.Fischman DL, Leon MB, Baim DS, et al. A randomized comparison of coronary-stent placement and balloon angioplasty in the treatment of coronary artery disease. Stent Restenosis Study Investigators. The New England journal of medicine. 1994;331(8):496–501. [DOI] [PubMed] [Google Scholar]
- 28.Hoffmann R, Mintz GS, Dussaillant GR, et al. Patterns and mechanisms of in-stent restenosis. A serial intravascular ultrasound study. Circulation. 1996;94(6):1247–1254. [DOI] [PubMed] [Google Scholar]
- 29.Kearney M, Pieczek A, Haley L, et al. Histopathology of in-stent restenosis in patients with peripheral artery disease. Circulation. 1997;95(8):1998–2002. [DOI] [PubMed] [Google Scholar]
- 30.Kornowski R, Hong MK, Tio FO, Bramwell O, Wu H, Leon MB. In-stent restenosis: contributions of inflammatory responses and arterial injury to neointimal hyperplasia. Journal of the American College of Cardiology. 1998;31(1):224–230. [DOI] [PubMed] [Google Scholar]
- 31.Rogers C, Welt FG, Karnovsky MJ, Edelman ER. Monocyte recruitment and neointimal hyperplasia in rabbits. Coupled inhibitory effects of heparin. Arteriosclerosis, thrombosis, and vascular biology. 1996;16(10):1312–1318. [DOI] [PubMed] [Google Scholar]
- 32.Komatsu R, Ueda M, Naruko T, Kojima A, Becker AE. Neointimal tissue response at sites of coronary stenting in humans: macroscopic, histological, and immunohistochemical analyses. Circulation. 1998;98(3):224–233. [DOI] [PubMed] [Google Scholar]
- 33.Farb A, Weber DK, Kolodgie FD, Burke AP, Virmani R. Morphological predictors of restenosis after coronary stenting in humans. Circulation. 2002;105(25):2974–2980. [DOI] [PubMed] [Google Scholar]
- 34.Farb A, Kolodgie FD, Hwang JY, et al. Extracellular matrix changes in stented human coronary arteries. Circulation. 2004;110(8):940–947. [DOI] [PubMed] [Google Scholar]
- 35.Farb A, Sangiorgi G, Carter AJ, et al. Pathology of acute and chronic coronary stenting in humans. Circulation. 1999;99(1):44–52. [DOI] [PubMed] [Google Scholar]
- 36.Edelman ER, Rogers C. Pathobiologic responses to stenting. The American journal of cardiology. 1998;81(7A):4E–6E. [DOI] [PubMed] [Google Scholar]
- 37.Chung IM, Gold HK, Schwartz SM, Ikari Y, Reidy MA, Wight TN. Enhanced extracellular matrix accumulation in restenosis of coronary arteries after stent deployment. Journal of the American College of Cardiology. 2002;40(12):2072–2081. [DOI] [PubMed] [Google Scholar]
- 38.Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. The New England journal of medicine. 1987;316(22):1371–1375. [DOI] [PubMed] [Google Scholar]
- 39.Kimura T, Yokoi H, Nakagawa Y, et al. Three-year follow-up after implantation of metallic coronary-artery stents. The New England journal of medicine. 1996;334(9):561–566. [DOI] [PubMed] [Google Scholar]
- 40.Kuroda N, Kobayashi Y, Nameki M, et al. Intimal hyperplasia regression from 6 to 12 months after stenting. The American journal of cardiology. 2002;89(7):869–872. [DOI] [PubMed] [Google Scholar]
- 41.Costa MA, Simon DI. Molecular basis of restenosis and drug-eluting stents. Circulation. 2005;111(17):2257–2273. [DOI] [PubMed] [Google Scholar]
- 42.Stone GW, Ellis SG, Cox DA, et al. A polymer-based, paclitaxel-eluting stent in patients with coronary artery disease. The New England journal of medicine. 2004;350(3):221–231. [DOI] [PubMed] [Google Scholar]
- 43.Moses JW, Leon MB, Popma JJ, et al. Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery. The New England journal of medicine. 2003;349(14):1315–1323. [DOI] [PubMed] [Google Scholar]
- 44.Stettler C, Wandel S, Allemann S, et al. Outcomes associated with drug-eluting and bare-metal stents: a collaborative network meta-analysis. Lancet. 2007;370(9591):937–948. [DOI] [PubMed] [Google Scholar]
- 45.Morice MC, Serruys PW, Sousa JE, et al. A randomized comparison of a sirolimus-eluting stent with a standard stent for coronary revascularization. The New England journal of medicine. 2002;346(23):1773–1780. [DOI] [PubMed] [Google Scholar]
- 46.Windecker S, Serruys PW, Wandel S, et al. Biolimus-eluting stent with biodegradable polymer versus sirolimus-eluting stent with durable polymer for coronary revascularisation (LEADERS): a randomised non-inferiority trial. Lancet. 2008;372(9644):1163–1173. [DOI] [PubMed] [Google Scholar]
- 47.Morice MC, Colombo A, Meier B, et al. Sirolimus- vs paclitaxel-eluting stents in de novo coronary artery lesions: the REALITY trial: a randomized controlled trial. Jama. 2006;295(8):895–904. [DOI] [PubMed] [Google Scholar]
- 48.Cook S, Wenaweser P, Togni M, et al. Incomplete stent apposition and very late stent thrombosis after drug-eluting stent implantation. Circulation. 2007;115(18):2426–2434. [DOI] [PubMed] [Google Scholar]
- 49.Balakrishnan B, Tzafriri AR, Seifert P, Groothuis A, Rogers C, Edelman ER. Strut position, blood flow, and drug deposition: implications for single and overlapping drug-eluting stents. Circulation. 2005;111(22):2958–2965. [DOI] [PubMed] [Google Scholar]
- 50.Hwang CW, Levin AD, Jonas M, Li PH, Edelman ER. Thrombosis modulates arterial drug distribution for drug-eluting stents. Circulation. 2005;111(13):1619–1626. [DOI] [PubMed] [Google Scholar]
- 51.Hwang CW, Wu D, Edelman ER. Physiological transport forces govern drug distribution for stent-based delivery. Circulation. 2001;104(5):600–605. [DOI] [PubMed] [Google Scholar]
- 52.de Jaegere P, Mudra H, Figulla H, et al. Intravascular ultrasound-guided optimized stent deployment. Immediate and 6 months clinical and angiographic results from the Multicenter Ultrasound Stenting in Coronaries Study (MUSIC Study). European heart journal. 1998;19(8):1214–1223. [DOI] [PubMed] [Google Scholar]
- 53.Mintz GS. Features and parameters of drug-eluting stent deployment discoverable by intravascular ultrasound. The American journal of cardiology. 2007;100(8B):26M–35M. [DOI] [PubMed] [Google Scholar]
- 54.Doi H, Maehara A, Mintz GS, et al. Classification and potential mechanisms of intravascular ultrasound patterns of stent fracture. The American journal of cardiology. 2009;103(6):818–823. [DOI] [PubMed] [Google Scholar]
- 55.Scheinert D, Scheinert S, Sax J, et al. Prevalence and clinical impact of stent fractures after femoropopliteal stenting. Journal of the American College of Cardiology. 2005;45(2):312–315. [DOI] [PubMed] [Google Scholar]
- 56.Kereiakes DJ, Wang H, Popma JJ, et al. Periprocedural and late consequences of overlapping Cypher sirolimus-eluting stents: pooled analysis of five clinical trials. Journal of the American College of Cardiology. 2006;48(1):21–31. [DOI] [PubMed] [Google Scholar]
- 57.Costa MA, Angiolillo DJ, Tannenbaum M, et al. Impact of stent deployment procedural factors on long-term effectiveness and safety of sirolimus-eluting stents (final results of the multicenter prospective STLLR trial). The American journal of cardiology. 2008;101(12):1704–1711. [DOI] [PubMed] [Google Scholar]
- 58.Doyle B, Rihal CS, O’Sullivan CJ, et al. Outcomes of stent thrombosis and restenosis during extended follow-up of patients treated with bare-metal coronary stents. Circulation. 2007;116(21):2391–2398. [DOI] [PubMed] [Google Scholar]
- 59.Yusuf RZ, Duan Z, Lamendola DE, Penson RT, Seiden MV. Paclitaxel resistance: molecular mechanisms and pharmacologic manipulation. Current cancer drug targets. 2003;3(1):1–19. [DOI] [PubMed] [Google Scholar]
- 60.Huang S, Houghton PJ. Mechanisms of resistance to rapamycins. Drug resistance updates : reviews and commentaries in antimicrobial and anticancer chemotherapy. 2001;4(6):378–391. [DOI] [PubMed] [Google Scholar]
- 61.Koster R, Vieluf D, Kiehn M, et al. Nickel and molybdenum contact allergies in patients with coronary in-stent restenosis. Lancet. 2000;356(9245):1895–1897. [DOI] [PubMed] [Google Scholar]
- 62.Nebeker JR, Virmani R, Bennett CL, et al. Hypersensitivity cases associated with drug-eluting coronary stents: a review of available cases from the Research on Adverse Drug Events and Reports (RADAR) project. Journal of the American College of Cardiology. 2006;47(1):175–181. [DOI] [PubMed] [Google Scholar]
- 63.Kolandaivelu K, Swaminathan R, Gibson WJ, et al. Stent thrombogenicity early in high-risk interventional settings is driven by stent design and deployment and protected by polymer-drug coatings. Circulation. 2011;123(13):1400–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Levin AD, Vukmirovic N, Hwang CW, Edelman ER. Specific binding to intracellular proteins determines arterial transport properties for rapamycin and paclitaxel. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(25):9463–9467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakazawa G, Finn AV, Vorpahl M, Ladich ER, Kolodgie FD, Virmani R. Coronary responses and differential mechanisms of late stent thrombosis attributed to first-generation sirolimus- and paclitaxel-eluting stents. Journal of the American College of Cardiology. 2011;57(4):390–398. [DOI] [PubMed] [Google Scholar]
- 66.Kang SJ, Mintz GS, Akasaka T, et al. Optical coherence tomographic analysis of in-stent neoatherosclerosis after drug-eluting stent implantation. Circulation. 2011;123(25):2954–2963. [DOI] [PubMed] [Google Scholar]
- 67.Otsuka F, Byrne RA, Yahagi K, et al. Neoatherosclerosis: overview of histopathologic findings and implications for intravascular imaging assessment. European heart journal. 2015;36(32):2147–2159. [DOI] [PubMed] [Google Scholar]
- 68.Otsuka F, Joner M, Prati F, Virmani R, Narula J. Clinical classification of plaque morphology in coronary disease. Nature reviews. Cardiology. 2014;11(7):379–389. [DOI] [PubMed] [Google Scholar]
- 69.Nakano M, Otsuka F, Yahagi K, et al. Human autopsy study of drug-eluting stents restenosis: histomorphological predictors and neointimal characteristics. European heart journal. 2013;34(42):3304–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arteriosclerosis, thrombosis, and vascular biology. 1995;15(5):551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Otsuka F, Finn AV, Yazdani SK, Nakano M, Kolodgie FD, Virmani R. The importance of the endothelium in atherothrombosis and coronary stenting. Nature reviews. Cardiology. 2012;9(8):439–453. [DOI] [PubMed] [Google Scholar]
- 72.Jimenez JM, Davies PF. Hemodynamically driven stent strut design. Annals of biomedical engineering. 2009;37(8):1483–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nature clinical practice. Cardiovascular medicine. 2009;6(1):16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arteriosclerosis, thrombosis, and vascular biology. 2005;25(11):2255–2264. [DOI] [PubMed] [Google Scholar]
- 75.Tulenko TN, Chen M, Mason PE, Mason RP. Physical effects of cholesterol on arterial smooth muscle membranes: evidence of immiscible cholesterol domains and alterations in bilayer width during atherogenesis. Journal of lipid research. 1998;39(5):947–956. [PubMed] [Google Scholar]
- 76.Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. Journal of leukocyte biology. 2009;86(5):1089–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Aikawa M, Rabkin E, Okada Y, et al. Lipid lowering by diet reduces matrix metalloproteinase activity and increases collagen content of rabbit atheroma: a potential mechanism of lesion stabilization. Circulation. 1998;97(24):2433–2444. [DOI] [PubMed] [Google Scholar]
- 78.McGill HC Jr., McMahan CA, Herderick EE, et al. Effects of coronary heart disease risk factors on atherosclerosis of selected regions of the aorta and right coronary artery. PDAY Research Group. Pathobiological Determinants of Atherosclerosis in Youth. Arteriosclerosis, thrombosis, and vascular biology. 2000;20(3):836–845. [DOI] [PubMed] [Google Scholar]
- 79.Nakazawa G, Otsuka F, Nakano M, et al. The pathology of neoatherosclerosis in human coronary implants bare-metal and drug-eluting stents. Journal of the American College of Cardiology. 2011;57(11):1314–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Joner M, Nakazawa G, Finn AV, et al. Endothelial cell recovery between comparator polymer-based drug-eluting stents. Journal of the American College of Cardiology. 2008;52(5):333–342. [DOI] [PubMed] [Google Scholar]
- 81.Nakazawa G, Vorpahl M, Finn AV, Narula J, Virmani R. One step forward and two steps back with drug-eluting-stents: from preventing restenosis to causing late thrombosis and nouveau atherosclerosis. JACC. Cardiovascular imaging. 2009;2(5):625–628. [DOI] [PubMed] [Google Scholar]
- 82.Shankman LS, Gomez D, Cherepanova OA, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nature medicine. 2015;21(6):628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Buja LM. Vascular responses to percutaneous coronary intervention with bare-metal stents and drug-eluting stents: a perspective based on insights from pathological and clinical studies. Journal of the American College of Cardiology. 2011;57(11):1323–1326. [DOI] [PubMed] [Google Scholar]
- 84.Stone GW, Ellis SG, Colombo A, et al. Offsetting impact of thrombosis and restenosis on the occurrence of death and myocardial infarction after paclitaxel-eluting and bare metal stent implantation. Circulation. 2007;115(22):2842–2847. [DOI] [PubMed] [Google Scholar]
- 85.Wenaweser P, Daemen J, Zwahlen M, et al. Incidence and correlates of drug-eluting stent thrombosis in routine clinical practice. 4-year results from a large 2-institutional cohort study. Journal of the American College of Cardiology. 2008;52(14):1134–1140. [DOI] [PubMed] [Google Scholar]
- 86.Natsuaki M, Morimoto T, Furukawa Y, et al. Late adverse events after implantation of sirolimus-eluting stent and bare-metal stent: long-term (5–7 years) follow-up of the Coronary Revascularization Demonstrating Outcome study-Kyoto registry Cohort-2. Circulation. Cardiovascular interventions. 2014;7(2):168–179. [DOI] [PubMed] [Google Scholar]
- 87.Yamaji K, Kimura T, Morimoto T, et al. Very long-term (15 to 20 years) clinical and angiographic outcome after coronary bare metal stent implantation. Circulation. Cardiovascular interventions. 2010;3(5):468–475. [DOI] [PubMed] [Google Scholar]
- 88.Joner M, Finn AV, Farb A, et al. Pathology of drug-eluting stents in humans: delayed healing and late thrombotic risk. Journal of the American College of Cardiology. 2006;48(1):193–202. [DOI] [PubMed] [Google Scholar]
- 89.Nakazawa G, Finn AV, Vorpahl M, et al. Incidence and predictors of drug-eluting stent fracture in human coronary artery a pathologic analysis. Journal of the American College of Cardiology. 2009;54(21):1924–1931. [DOI] [PubMed] [Google Scholar]
- 90.Taniwaki M, Windecker S, Raber L. Neoatherosclerosis as reason for stent failures beyond 5 years after drug-eluting stent implantation. European heart journal. 2014;35(29):1980. [DOI] [PubMed] [Google Scholar]
- 91.Takano M, Yamamoto M, Inami S, et al. Appearance of lipid-laden intima and neovascularization after implantation of bare-metal stents extended late-phase observation by intracoronary optical coherence tomography. Journal of the American College of Cardiology. 2009;55(1):26–32. [DOI] [PubMed] [Google Scholar]
- 92.Holmes DR Jr., Kereiakes DJ, Garg S, et al. Stent thrombosis. Journal of the American College of Cardiology. 2010;56(17):1357–1365. [DOI] [PubMed] [Google Scholar]
- 93.Finn AV, Joner M, Nakazawa G, et al. Pathological correlates of late drug-eluting stent thrombosis: strut coverage as a marker of endothelialization. Circulation. 2007;115(18):2435–2441. [DOI] [PubMed] [Google Scholar]
- 94.Raber L, Magro M, Stefanini GG, et al. Very late coronary stent thrombosis of a newer-generation everolimus-eluting stent compared with early-generation drug-eluting stents: a prospective cohort study. Circulation. 2012;125(9):1110–1121. [DOI] [PubMed] [Google Scholar]
- 95.Daemen J, Wenaweser P, Tsuchida K, et al. Early and late coronary stent thrombosis of sirolimus-eluting and paclitaxel-eluting stents in routine clinical practice: data from a large two-institutional cohort study. Lancet. 2007;369(9562):667–678. [DOI] [PubMed] [Google Scholar]
- 96.Babapulle MN, Joseph L, Belisle P, Brophy JM, Eisenberg MJ. A hierarchical Bayesian meta-analysis of randomised clinical trials of drug-eluting stents. Lancet. 2004;364(9434):583–591. [DOI] [PubMed] [Google Scholar]
- 97.Moreno R, Fernandez C, Hernandez R, et al. Drug-eluting stent thrombosis: results from a pooled analysis including 10 randomized studies. Journal of the American College of Cardiology. 2005;45(6):954–959. [DOI] [PubMed] [Google Scholar]
- 98.Bavry AA, Kumbhani DJ, Helton TJ, Bhatt DL. What is the risk of stent thrombosis associated with the use of paclitaxel-eluting stents for percutaneous coronary intervention?: a meta-analysis. Journal of the American College of Cardiology. 2005;45(6):941–946. [DOI] [PubMed] [Google Scholar]
- 99.Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. The New England journal of medicine. 2007;356(10):998–1008. [DOI] [PubMed] [Google Scholar]
- 100.Awata M, Kotani J, Uematsu M, et al. Serial angioscopic evidence of incomplete neointimal coverage after sirolimus-eluting stent implantation: comparison with bare-metal stents. Circulation. 2007;116(8):910–916. [DOI] [PubMed] [Google Scholar]
- 101.Wakabayashi K, Mintz GS, Weissman NJ, et al. Impact of drug-eluting stents on distal vessels. Circulation. Cardiovascular interventions. 2012;5(2):211–219. [DOI] [PubMed] [Google Scholar]
- 102.Wakabayashi K, Waksman R, Weissman NJ. Edge effect from drug-eluting stents as assessed with serial intravascular ultrasound: a systematic review. Circulation. Cardiovascular interventions. 2012;5(2):305–311. [DOI] [PubMed] [Google Scholar]
- 103.Kedhi E, Joesoef KS, McFadden E, et al. Second-generation everolimus-eluting and paclitaxel-eluting stents in real-life practice (COMPARE): a randomised trial. Lancet. 2010;375(9710):201–209. [DOI] [PubMed] [Google Scholar]
- 104.Smits PC, Kedhi E, Royaards KJ, et al. 2-year follow-up of a randomized controlled trial of everolimus- and paclitaxel-eluting stents for coronary revascularization in daily practice. COMPARE (Comparison of the everolimus eluting XIENCE-V stent with the paclitaxel eluting TAXUS LIBERTE stent in all-comers: a randomized open label trial). Journal of the American College of Cardiology. 2011;58(1):11–18. [DOI] [PubMed] [Google Scholar]
- 105.Stone GW, Rizvi A, Sudhir K, et al. Randomized comparison of everolimus- and paclitaxel-eluting stents. 2-year follow-up from the SPIRIT (Clinical Evaluation of the XIENCE V Everolimus Eluting Coronary Stent System) IV trial. Journal of the American College of Cardiology. 2011;58(1):19–25. [DOI] [PubMed] [Google Scholar]
- 106.Palmerini T, Biondi-Zoccai G, Della Riva D, et al. Stent thrombosis with drug-eluting and bare-metal stents: evidence from a comprehensive network meta-analysis. Lancet. 2012;379(9824):1393–1402. [DOI] [PubMed] [Google Scholar]
- 107.Bangalore S, Kumar S, Fusaro M, et al. Short- and long-term outcomes with drug-eluting and bare-metal coronary stents: a mixed-treatment comparison analysis of 117 762 patient-years of follow-up from randomized trials. Circulation. 2012;125(23):2873–2891. [DOI] [PubMed] [Google Scholar]
- 108.Tada T, Byrne RA, Simunovic I, et al. Risk of stent thrombosis among bare-metal stents, first-generation drug-eluting stents, and second-generation drug-eluting stents: results from a registry of 18,334 patients. JACC. Cardiovascular interventions. 2013;6(12):1267–1274. [DOI] [PubMed] [Google Scholar]
- 109.Cassese S, Byrne RA, Tada T, et al. Incidence and predictors of restenosis after coronary stenting in 10 004 patients with surveillance angiography. Heart. 2014;100(2):153–159. [DOI] [PubMed] [Google Scholar]
- 110.Lee SY, Hur SH, Lee SG, et al. Optical coherence tomographic observation of in-stent neoatherosclerosis in lesions with more than 50% neointimal area stenosis after second-generation drug-eluting stent implantation. Circulation. Cardiovascular interventions. 2015;8(2):e001878. [DOI] [PubMed] [Google Scholar]
- 111.Brener SJ, Kereiakes DJ, Simonton CA, et al. Everolimus-eluting stents in patients undergoing percutaneous coronary intervention: final 3-year results of the Clinical Evaluation of the XIENCE V Everolimus Eluting Coronary Stent System in the Treatment of Subjects With de Novo Native Coronary Artery Lesions trial. American heart journal. 2013;166(6):1035–1042. [DOI] [PubMed] [Google Scholar]
- 112.Lee JM, Park KW, Han JK, et al. Three-year patient-related and stent-related outcomes of second-generation everolimus-eluting Xience V stents versus zotarolimus-eluting resolute stents in real-world practice (from the Multicenter Prospective EXCELLENT and RESOLUTE-Korea Registries). The American journal of cardiology. 2014;114(9):1329–1338. [DOI] [PubMed] [Google Scholar]
- 113.Camenzind E, Wijns W, Mauri L, et al. Stent thrombosis and major clinical events at 3 years after zotarolimus-eluting or sirolimus-eluting coronary stent implantation: a randomised, multicentre, open-label, controlled trial. Lancet. 2012;380(9851):1396–1405. [DOI] [PubMed] [Google Scholar]
- 114.Inoue T, Shite J, Yoon J, et al. Optical coherence evaluation of everolimus-eluting stents 8 months after implantation. Heart. 2011;97(17):1379–1384. [DOI] [PubMed] [Google Scholar]
- 115.Otsuka F, Vorpahl M, Nakano M, et al. Pathology of second-generation everolimus-eluting stents versus first-generation sirolimus- and paclitaxel-eluting stents in humans. Circulation. 2014;129(2):211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yonetsu T, Kim JS, Kato K, et al. Comparison of incidence and time course of neoatherosclerosis between bare metal stents and drug-eluting stents using optical coherence tomography. The American journal of cardiology. 2012;110(7):933–939. [DOI] [PubMed] [Google Scholar]
- 117.Taniwaki M, Windecker S, Zaugg S, et al. The association between in-stent neoatherosclerosis and native coronary artery disease progression: a long-term angiographic and optical coherence tomography cohort study. European heart journal. 2015;36(32):2167–2176. [DOI] [PubMed] [Google Scholar]
- 118.Iijima R, Araki T, Nagashima Y, et al. Incidence and predictors of the late catch-up phenomenon after drug-eluting stent implantation. International journal of cardiology. 2013;168(3):2588–2592. [DOI] [PubMed] [Google Scholar]
- 119.Nakazawa G, Yazdani SK, Finn AV, Vorpahl M, Kolodgie FD, Virmani R. Pathological findings at bifurcation lesions: the impact of flow distribution on atherosclerosis and arterial healing after stent implantation. Journal of the American College of Cardiology. 2010;55(16):1679–1687. [DOI] [PubMed] [Google Scholar]
- 120.Wilson GJ, Nakazawa G, Schwartz RS, et al. Comparison of inflammatory response after implantation of sirolimus- and paclitaxel-eluting stents in porcine coronary arteries. Circulation. 2009;120(2):141–149, 141–142. [DOI] [PubMed] [Google Scholar]
- 121.Finn AV, Kolodgie FD, Harnek J, et al. Differential response of delayed healing and persistent inflammation at sites of overlapping sirolimus- or paclitaxel-eluting stents. Circulation. 2005;112(2):270–278. [DOI] [PubMed] [Google Scholar]
- 122.Finn AV, Nakazawa G, Joner M, et al. Vascular responses to drug eluting stents: importance of delayed healing. Arteriosclerosis, thrombosis, and vascular biology. 2007;27(7):1500–1510. [DOI] [PubMed] [Google Scholar]
- 123.Byrne RA. Bioresorbable Vascular Scaffolds--Will Promise Become Reality? The New England journal of medicine. 2015;373(20):1969–1971. [DOI] [PubMed] [Google Scholar]
- 124.Verheye S, Ormiston JA, Stewart J, et al. A next-generation bioresorbable coronary scaffold system: from bench to first clinical evaluation: 6- and 12-month clinical and multimodality imaging results. JACC. Cardiovascular interventions. 2014;7(1):89–99. [DOI] [PubMed] [Google Scholar]
- 125.Serruys PW, Ormiston JA, Onuma Y, et al. A bioabsorbable everolimus-eluting coronary stent system (ABSORB): 2-year outcomes and results from multiple imaging methods. Lancet. 2009;373(9667):897–910. [DOI] [PubMed] [Google Scholar]
- 126.Ellis SG, Kereiakes DJ, Metzger DC, et al. Everolimus-Eluting Bioresorbable Scaffolds for Coronary Artery Disease. The New England journal of medicine. 2015;373(20):1905–1915. [DOI] [PubMed] [Google Scholar]
- 127.Miyazaki T, Panoulas VF, Sato K, Naganuma T, Latib A, Colombo A. Acute stent thrombosis of a bioresorbable vascular scaffold implanted for ST-segment elevation myocardial infarction. International journal of cardiology. 2014;174(2):e72–74. [DOI] [PubMed] [Google Scholar]
- 128.Karanasos A, van Geuns RJ, Zijlstra F, Regar E. Very late bioresorbable scaffold thrombosis after discontinuation of dual antiplatelet therapy. European heart journal. 2014;35(27):1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sanchez OD, Yahagi K, Byrne RA, et al. Pathological aspects of bioresorbable stent implantation. EuroIntervention : journal of EuroPCR in collaboration with the Working Group on Interventional Cardiology of the European Society of Cardiology. 2015;11 Suppl V:V159–165. [DOI] [PubMed] [Google Scholar]
- 130.Otsuka F, Pacheco E, Perkins LE, et al. Long-term safety of an everolimus-eluting bioresorbable vascular scaffold and the cobalt-chromium XIENCE V stent in a porcine coronary artery model. Circulation. Cardiovascular interventions. 2014;7(3):330–342. [DOI] [PubMed] [Google Scholar]
- 131.Danson E, Bhindi R, Hansen P. Follow-up evaluation of unapposed bioresorbable vascular scaffold at a coronary bifurcation using optical coherence tomography. International journal of cardiology. 2014;177(2):e84–86. [DOI] [PubMed] [Google Scholar]
- 132.Simsek C, Karanasos A, Magro M, et al. Long-term invasive follow-up of the everolimus-eluting bioresorbable vascular scaffold: five-year results of multiple invasive imaging modalities. EuroIntervention : journal of EuroPCR in collaboration with the Working Group on Interventional Cardiology of the European Society of Cardiology. 2014. [DOI] [PubMed] [Google Scholar]
- 133.Rogers C, Parikh S, Seifert P, Edelman ER. Endogenous cell seeding. Remnant endothelium after stenting enhances vascular repair. Circulation. 1996;94(11):2909–2914. [DOI] [PubMed] [Google Scholar]