

Using an affinity column retention assay, we showed that the purified Tet38 membrane transporter of Staphylococcus aureus bound specifically to host cell CD36 and to the complex CD36–Toll-like receptor 2 (TLR-2), but not to TLR-2 alone or TLR-2 and S. aureus lipoteichoic acid (LTA). We tested the effect of LTA on the internalization of S. aureus tet38 mutant QT7 versus RN6390 by A549 epithelial cells.
KEYWORDS: CD36, S. aureus, TLR-2, Tet38, Congo red, teichoic acids, tunicamycin
ABSTRACT
Using an affinity column retention assay, we showed that the purified Tet38 membrane transporter of Staphylococcus aureus bound specifically to host cell CD36 and to the complex CD36–Toll-like receptor 2 (TLR-2), but not to TLR-2 alone or TLR-2 and S. aureus lipoteichoic acid (LTA). We tested the effect of LTA on the internalization of S. aureus tet38 mutant QT7 versus RN6390 by A549 epithelial cells. Addition of anti-LTA antibody to the bacteria prior to adding to A549 cells reduced internalization of QT7 2-fold compared to that with nonspecific antibody treatment. QT7 internalized 4- to 6-fold less than RN6390 with or without anti-LTA antibody. These data suggested that Tet38 and LTA were independently involved in the invasion process. The wall teichoic acid (WTA) inhibitor tunicamycin had an 8-fold decrease in activity with overexpression of tet38 and a 2-fold increase in activity in QT7 (tet38). Reserpine (an inhibitor of efflux pumps) reduced the effect of tet38 overexpression on tunicamycin resistance 4-fold. In addition, tet38 affected growth in the presence of LTA inhibitor Congo red, with overexpression increasing growth and deletion of tet38 reducing growth. In conclusion, Tet38 contributes to S. aureus invasion of A549 via direct binding to CD36 of the complex CD36–TLR-2, and LTA independently bound to TLR-2. The reduction of tunicamycin resistance in the presence of reserpine and the survival ability of the tet38 overexpressor in the presence of Congo red suggest that Tet38 can also protect the synthesis of LTA and WTA in S. aureus against their inhibitors, possibly functioning as an efflux pump.
INTRODUCTION
Staphylococcus aureus interacts with the human host in multiple and complex ways. Host cell factors such as fibronectin, integrins, Hsp60, Hsc70, and Toll-like receptor (TLR) heterodimers TLR-2/1 and TLR-2/6 form complexes with staphylococcal components, such as fibronectin-binding proteins (FnbPs) (which complex with fibronectin, integrin, and Hsp60), extracellular adherence protein Eap (which complexes with fibronectin), autolysin Atl (which complexes with Hsc70), IsdB (which complexes with integrin), and lipoteichoic acid (LTA) (which complexes with TLR-2/6, TLR-2/CD36, and TLR-2) (1–6). The host cell scavenger receptor CD36 actively participates in the phagocytosis of S. aureus via bacterial LTA, which leads to the production of cytokines in response to bacterial invasion (7, 8). TLR-2 acts as a signaling receptor that is stimulated by intact Gram-positive bacteria, soluble peptidoglycan, and LTA to activate the host innate immune response (9, 10). TLR-2 plays an important role in host defense against S. aureus by organizing an inhibitory response to S. aureus invasion following its recognition of the pathogen either as whole cells or as extracted LTA (11). TLR-2 and CD36 are located separately from each other on the surface of the host cells and form a complex under certain conditions, such as after contact with staphylococcal LTA or diacylated lipoprotein. CD36 acts as a coreceptor for TLR-2 and increases the ability of the complex CD36/TLR-2 to recognize specific bacterial diacylglycerides (8, 12). There is limited information, however, on other S. aureus components that interact directly with CD36 or the complex CD36–TLR-2.
We recently demonstrated that the Tet38 efflux pump, which extrudes diverse substrates such as tetracycline, fosfomycin, free fatty acids, and glycerol-3-phosphate, is involved in the internalization of S. aureus by A549 epithelial cells, as evidenced by a 5-fold reduction in the recovery of a tet38 mutant after A549 cell invasion (13, 14). Treatment of A549 cells with anti-CD36 antibody reduced binding of wild-type cells 2-fold but had no effect on the tet38 mutant, suggesting that Tet38 interacted with CD36 in host cell invasion (13). In contrast, blocking of the A549 cell monolayer with anti-TLR-2 antibody had similar reductions in binding in the wild-type cells (4-fold) and the tet38 mutant (3.6-fold), suggesting that the involvement of TLR-2 in host cell invasion was not dependent on the presence of Tet38 (13). These data indicated that TLR-2 contributes to host cell invasion with a bacterial component(s) other than Tet38.
To evaluate further the interactions of Tet38 with CD36 and TLR2, we used an affinity column retention assay with purified protein components. We showed that purified Tet38 bound directly to CD36 but not to TLR-2, and purified LTA did not affect binding to the complex of Tet38 and CD36. We also observed an additional 2-fold decrease in the number of internalized tet38 mutant cells by the A549 cell monolayer when the bacteria were covered with anti-LTA antibody, suggesting that Tet38 and LTA participated independently in the cell invasion event.
In addition, we showed that Tet38 provides protection from two inhibitors of S. aureus teichoic acid synthesis, tunicamycin (against wall teichoic acid [WTA]) (15–17) and Congo red (against LTA) (17, 18), possibly functioning as an efflux pump.
RESULTS
Tet38-CD36 interaction.
To demonstrate directly that CD36 and Tet38 interact with each other, we used a column retention assay with histidine-tagged Tet38 bound to an Ni affinity column serving as the anchor. Tet38 (∼48 kDa) is a membrane protein with 14 transmembrane segments (TMS). CD36-His (∼68 kDa) was first treated with enterokinase to remove the His tag portion and then added to the Ni column, which had been previously loaded with Tet38-His. The flowthrough from the Ni column (Ni-His-Tet38-CD36) was collected and then the column was washed with buffer A, followed by an elution with 100 mM imidazole. Proteins separated by SDS-PAGE and stained with Coomassie blue indicated that CD36 was found in the flowthrough fraction (FT), absent in the wash fraction, and present in the elution fraction. Tet38-His was also absent from the wash fraction and found in the eluted fractions (Fig. 1). In parallel, CD36 without a His tag was loaded onto another Ni column and was treated in the same manner as the assay column. CD36 was present in the flowthrough, absent in the wash, and absent in the elution fraction (Fig. 1). These data indicate that CD36 retention on the column is dependent on the presence of Tet38-His bound to the column, consistent with a direct interaction between the two proteins.
FIG 1.

SDS-PAGE of the binding assays between Tet38-His and CD36 or TLR-2. Enterokinase-treated CD36 or enterokinase-treated TLR-2 was applied to an Ni column that had been previously loaded with Tet38-His. Flowthrough (FT), wash, and elution fractions were collected and submitted to SDS-PAGE (top middle and right). Reference proteins included Tet38, CD36, and TLR-2 (top left). In the cases of CD36 and TLR-2, enterokinase-treated CD36 or TLR-2 was applied to an Ni column (bottom left and middle). For NorA-His plus CD36, purified NorA-His protein was loaded onto an Ni column and used to retain CD36 as was done with Tet38 (bottom right). Reference proteins included CD36 and NorA (bottom right).
Tet38-TLR-2 interaction.
The same column retention assay with a Tet38-His anchor was performed with enterokinase-treated TLR-2. The commercially purified TLR-2 showed three distinct bands corresponding to 84, 78, and 66 kDa, respectively. The TLR-2 protein is 784 amino acids in length and has a molecular weight of 89.8 kDa. In the affinity assay, TLR-2 was found in the flowthrough fraction and only Tet38-His was eluted with imidazole, suggesting no direct binding between Tet38 and TLR-2 (Fig. 1).
As a control, TLR-2 without a His tag was loaded onto another Ni column and was treated in the same manner as CD36 without His tag, as described above. TLR-2 was present in the flowthrough but absent in the wash and elution fractions (Fig. 1).
As an additional control to demonstrate that the binding of CD36 was specific to Tet38, we carried out the column retention assay using another His-tagged S. aureus efflux pump, NorA-His, as the anchor on the affinity column (19). Enterokinase-treated CD36 was applied to a column previously loaded with NorA-His. CD36 was found only in the flowthrough fraction, and NorA alone was eluted by 100 mM imidazole (Fig. 1).
CD36–TLR-2 interaction.
To assess the ability of our affinity column retention assay to identify the known interactions between CD36 and TLR-2, we bound CD36-His to an Ni column as the anchor and applied enterokinase-treated TLR-2. We found CD36 and TLR-2 in the flowthrough fraction, both absent in the wash, and both again present in the eluted fraction. Conversely, enterokinase-treated CD36 was retained on an Ni column loaded with TLR-2–His as the anchor. Thus, direct binding between CD36 and TLR2 was recapitulated in the column retention assay (Fig. 2).
FIG 2.

SDS-PAGE of binding assays between CD36 and TLR-2. Enterokinase-treated TLR-2 was applied to an Ni column that had been previously loaded with CD36-His. CD36 and TLR-2 proteins were found in the flowthrough fraction, absent in the wash fraction, and then were again eluted together by 100 mM imidazole (middle). Enterokinase-treated CD36 was applied to an Ni column that had been previously loaded with TLR-2-His. CD36 protein was found in the flowthrough fraction, absent in the wash fraction, and eluted together with TLR-2 by 100 mM imidazole (right). Reference proteins included CD36 and TLR-2 (left).
Tet38-His–CD36–TLR-2 interaction.
To assess if CD36 binding to Tet38 affects its ability to bind TLR2, we repeated the retention assay with Tet38-His as the anchor, adding first CD36 and then TLR-2 (both enterokinase treated). We found CD36 and TLR-2 in the flowthrough fraction (FT) and absent in the wash fraction, and they both were eluted together with Tet38-His in the presence of imidazole (Fig. 3). Western blotting was carried out using anti-His tag (for Tet38-His), anti-CD36, and anti-TLR-2 antibodies to verify the presence of each protein in the elution fractions. These findings indicate that Tet38 binding with CD36 did not prevent binding between CD36 and TLR-2, suggesting that there are distinct binding sites on CD36 for these two proteins.
FIG 3.
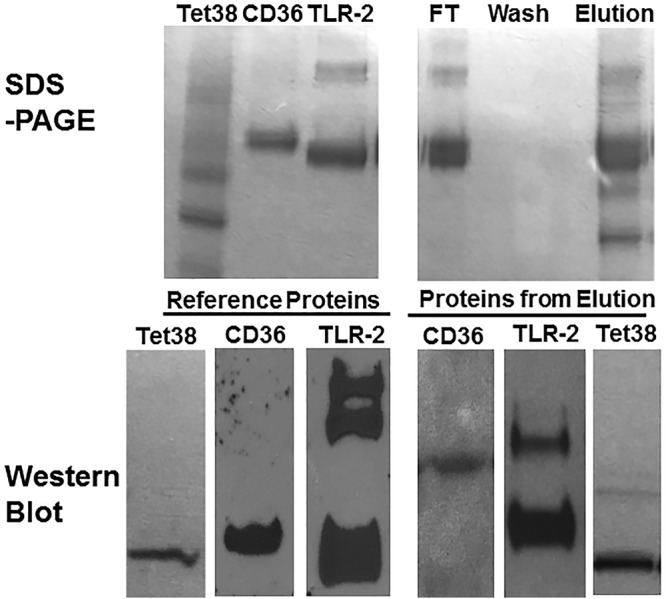
SDS-PAGE and Western blots of binding assays with purified Tet38-His, CD36, and TLR-2. CD36 and then TLR-2 (both enterokinase treated) were applied to an Ni column that had been previously loaded with Tet38-His. CD36 and TLR-2 were found in the flowthrough and elution fractions but not in the wash fraction (top right). Reference proteins included Tet38, CD36, and TLR-2 (top left). Reference proteins (bottom left) and proteins recovered in the elution fractions (bottom right) were submitted to Western blotting using anti-His (for Tet38-His), anti-CD36 (for CD36), and anti-TLR-2 (for TLR-2) antibodies separately to verify their presence in the elution fraction.
All of the bands of TLR-2 were positive on a Western blot with anti-TLR-2 antibody, suggesting that they represented intact TLR-2 and smaller fragments (Fig. 3).
Tet38-His–CD36–TLR-2 complex and LTA.
To verify the affinity of TLR-2 for LTA, we carried out the assay using TLR-2–His as the anchor and added LTA to the Ni column (Fig. 4). LTA was retained on the column by TLR-2, and they were eluted together by imidazole. This assay confirmed the findings by Hashimoto et al. that demonstrated that S. aureus LTA is a ligand of the TLR-2 receptor (10).
FIG 4.

SDS-PAGE of the binding assays between Tet38-His, CD36, and TLR-2, with or without LTA. Enterokinase-treated CD36 and TLR-2 were applied successively to an Ni column that had been previously loaded with Tet38-His. TLR-2 and CD36 were found in the flowthrough fraction, and CD36, TLR-2, and Tet38-His were eluted together by 100 mM imidazole (third image). Enterokinase-treated CD36, TLR-2, and LTA were applied successively to an Ni column that had been previously loaded with Tet38-His. TLR-2, CD36, and LTA were found in the flowthrough. CD36 and Tet38 were eluted together by 100 mM imidazole (fourth image). As a control, LTA was applied to an Ni column that had been previously loaded with TLR-2-His. TLR-2-His and LTA were found in the flowthrough fraction, and they were eluted together by 100 mM imidazole (second image).
To assess the participation of LTA in the binding complex between Tet38-His, CD36, and TLR-2, we first loaded Tet38-His on the Ni column to serve as the anchor and then added successively CD36, TLR-2, and LTA (Fig. 4). We found a sequence of proteins coming off the column. LTA, TLR-2, and CD36 were found in the flowthrough, and no protein was found in the wash. We observed dominantly CD36 and Tet38-His in the eluted fraction, suggesting that LTA may have affected the interaction of TLR-2 with the Tet38-CD36 complex.
To further characterize the effects of LTA on the interactions of CD36 and TLR-2, we carried out the assay with CD36-His as the anchor and added enterokinase-treated TLR-2 and LTA successively to the column (Fig. 5). We found that LTA, CD36, and TLR-2 were present in the flowthrough fraction, absent in the wash fraction, and then were eluted together in the imidazole elution fraction, indicating that LTA binds to the complex of TLR-2 and CD36 (Fig. 5).
FIG 5.

SDS-PAGE of the binding assays between CD36-His and TLR-2 with or without LTA. Enterokinase-treated TLR-2 was applied to an Ni column that had been previously loaded with CD36-His. TLR-2 and CD36-His were found in the flowthrough fraction, and again were eluted together by 100 mM imidazole (middle panel). Enterokinase-treated TLR-2 and LTA were applied successively to an Ni column that had been previously loaded with protein CD36-His. TLR-2, CD36, and LTA were found in the flowthrough fraction and then again were eluted together by 100 mM imidazole (right).
Tet38 and LTA influence the internalization of S. aureus by A549 cells.
We performed S. aureus internalization assays using A549 epithelial cells and S. aureus strains RN6390 and QT7 (tet38). We added to the A549 cells anti-LTA (50 nM) antibody, anti-CD36 (50 nM), or anti-TLR-2 (50 nM) prior to adding S. aureus. The anti-LTA antibody binds to the added S. aureus cells blocking interactions between S. aureus LTA and host cell receptors. A mouse IgG nonspecific isotype control was used at the same concentration (50 nM) as the tested antibodies in the assays and served as a negative control (Table 1). To confirm that the effect of anti-LTA antibody was related to its effect on S. aureus LTA, we also incubated S. aureus cells with anti-LTA (5 μM) prior to adding the bacteria to A549 cells. A mouse IgG nonspecific isotype control was used at the same concentration (5 μM) as the tested antibody in this assay and also served as a negative control.
TABLE 1.
Internalization of S. aureus by A549 cellsa
| S. aureus | Bacterial CFU/well |
|||
|---|---|---|---|---|
| Nonspecific IgG | +LTA antibodyb | +CD36 antibodyb | +TLR-2 antibodyb | |
| RN6390 | 1,300 ± 100 | 400 ± 25 | 400 ± 30 | 450 ± 25 |
| QT7 | 240 ± 15 | 110 ± 15 | 200 ± 10 | 120 ± 30 |
| QT5 | 1,400 ± 150 | 450 ± 50 | 400 ± 50 | 480 ± 30 |
All values represent the means from three independent experiments. Values represent the differences between the CFU of RN6390 and QT7 in the presence of antibodies and are statistically significant based on Student’s t test (P < 0.05), or values represent the differences between the CFU of QT7 in the presence of nonspecific IgG antibody versus the CFU of QT7 in the presence of anti-LTA and anti-TLR-2 antibodies and are statistically significant based on Student’s t test (P < 0.05). QT7 is a tet38 mutant; QT5 is a norB efllux pump mutant.
Concentrations of LTA, CD36, and TLR-2 antibodies are 50 nM per well.
In the presence of nonspecific IgG, QT7 (tet38) internalized 5-fold less efficiently than RN6390, confirming prior findings (20). QT5 (norB), which lacks the NorB efflux pump, was internalized similarly to the RN6390. In the presence of anti-LTA, anti-CD36, and anti-TLR-2 antibodies, RN6390 and QT5 showed approximately 3-fold reductions in internalized CFU. In contrast, QT7 showed minimal reduction in internalization in the presence of anti-CD36 and about 2-fold reductions in the presence of anti-LTA or anti-TLR-2 in comparison to nonspecific IgG (Table 1). The effect of anti-LTA on the ability to internalize S. aureus was similar in the assays in which anti-LTA was added to the A549 cell monolayer and to S. aureus cells.
Tet38 protein was also used to block access of anti-CD36 to the CD36 protein of A549 cells. In the presence of extracellular purified Tet38 and anti-CD36 antibody, internalized RN6390 increased from 450 CFU/well to 700 CFU/well. In contrast, when NorA protein was added prior to addition of anti-CD36 antibody, the CFU of internalized bacteria remained similar in the presence or absence of NorA (450 CFU/well, compared to 440 CFU/well). These data suggested that Tet38 interacts with CD36 and partially blocks the binding of the anti-CD36 antibody to the CD36 of the host cells (Table 2).
TABLE 2.
Effect of purified Tet38 protein on the internalization of S. aureus RN6390 by A549 cells
| Condition |
S. aureus RN6390 (CFU/well)a
|
|
|---|---|---|
| +Nonspecific IgG | +CD36 antibody | |
| No purified proteins | 1,540 ± 100 | 450 ± 25 |
| Purified Tet38 | 1,499 ± 95 | 700 ± 50 |
| Purified NorA | 1,480 ± 80 | 440 ± 30 |
All values represent the means from three independent experiments. Values represent the differences between the CFU of RN6390 in the presence of anti-CD36 antibody, with and without purified Tet38 proteins, and are statistically significant based on Student’s t test (P < 0.05). Concentrations of IgG and CD36 antibodies are 50 nM per well. Proteins (Tet38 or NorA) were used at 5 μg per assay.
These data suggested that the role of Tet38 in cellular invasion was dominantly dependent on CD36 itself and that additional Tet38-independent effects involved both LTA and TLR-2. These findings further support the demonstrated direct binding of Tet38 to CD36 with indirect binding effects of LTA and TLR-2.
Tet38 affects the action of inhibitors of LTA and WTA synthesis.
We determined the inhibitory activity of tunicamycin, a WTA synthesis inhibitor, and Congo red, an LTA synthesis inhibitor (17, 18), for RN6390, QT7 (tet38), RN6390(pLI50-tet38), and QT7(pLI50-tet38) overexpressing tet38 from a plasmid. To ensure that all strains were treated equally, we compared the MICs of RN6390(pLI50) and QT7(pLI50) to the MICs of the overexpressor RN6390(pLI50-tet38) and the complemented strain QT7(pLI50-tet38). For tunicamycin, MICs increased 8-fold with tet38 plasmid overexpression and decreased 2-fold in the tet38 mutant with plasmid alone. Reserpine, an inhibitor of efflux pumps, including Tet38 (21, 22), had a partial effect, reducing the MIC of RN6390 2-fold and the overexpressor 4-fold, with no effect on QT7 (Table 3). The tunicamycin MIC of the complemented strain QT7(pLI50-tet38) was 4-fold higher than that of QT7 alone. The overexpression of tet38 was verified by real-time PCR, which showed an increase of 15-fold in the transcript level of tet38 of the overexpressor RN6390(pLI50-tet38) (data not shown).
TABLE 3.
Susceptibility of S. aureus to tunicamycin
| S. aureusa | MIC (μg/ml)b
|
|
|---|---|---|
| TUN | TUN + RES | |
| RN6390 | 8 | 4 |
| QT7c | 4 | 4 |
| RN6390(pLI50) | 8 | 4 |
| RN6390(pLI50-tet38) | 64 | 16 |
| QT7(pLI50) | 4 | 4 |
| QT7(pLI50-tet38) | 16 | 4 |
All strains harboring plasmid pLI50 were grown in the presence of chloramphenicol (20 μg/ml) and at 37°C.
TUN, tunicamycin; RES, reserpine at 20 μg/ml.
QT7 is a tet38 mutant.
For Congo red, we used a previously described Congo red growth assay in which growth of serial dilutions of a standardized S. aureus suspension on 0.08% of Congo red agar is measured. All S. aureus strains started at the same number of 106 CFU/μl (107 CFU in 10 μl per Congo red plate) (17). We compared the survival of RN6390(pLI50) and QT7(pLI50) to the survival of the overexpressor RN6390(pLI50-tet38) and the complemented strain QT7(pLI50-tet38). We found that relative to RN6390, growth in Congo red occurred only at lower dilutions with QT7 (tet38) and occurred at higher dilutions with plasmid overexpression of tet38 (Table 4).
TABLE 4.
Growth of S. aureus on LB agar supplemented with 0.08% (wt/vol) Congo red
| S. aureusa | CFUb
of bacteria on LB plate at indicated dilution |
CFUb
of bacteria on LB + Congo red plate at indicated dilution |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 102 | 103 | 104 | 105 | 106 | 10 | 102 | 103 | 104 | 105 | 106 | |
| RN6390 | 25 | 200 | TNTC | TNTC | TNTC | TNTC | 0 | 0 | 0 | 20 | 150 | TNTC |
| QT7 | 20 | 195 | TNTC | TNTC | TNTC | TNTC | 0 | 0 | 0 | 0 | 1 | 9 |
| RN6390(pLI50) | 20 | 220 | TNTC | TNTC | TNTC | TNTC | 0 | 0 | 0 | 12 | 130 | TNTC |
| RN6390(pLI50-tet38) | 22 | 220 | TNTC | TNTC | TNTC | TNTC | 0 | 2 | 13 | 110 | 210 | TNTC |
| QT7(pLI50) | 22 | 190 | TNTC | TNTC | TNTC | TNTC | 0 | 0 | 0 | 0 | 2 | 7 |
| QT7(pLI50-tet38) | 20 | 200 | TNTC | TNTC | TNTC | TNTC | 0 | 1 | 10 | 100 | 195 | TNTC |
All strains harboring plasmid pLI50 were grown in the presence of chloramphenicol (20 μg/ml) and at 37°C. QT7, tet38 mutant.
All values represent the means from three independent experiments. Statistical differences between S. aureus strains on Congo red plates are significant (shown in bold) and were determined by the Student’s t test (P < 0.05). TNTC, too numerous to count. The starting CFU for all strains is 106/μl of bacteria.
DISCUSSION
Tet38 functions as a broad-spectrum membrane transporter in S. aureus, and its substrate spectrum includes chemically unrelated compounds (palmitoleic acid and glycerol-3-phosphate) and antibiotics (tetracycline and fosfomycin). Tet38 also contributes to S. aureus invasion of A549 epithelial cells (14, 20, 21, 23), but there is limited understanding of the mechanism by which it facilitates bacterial internalization into host cells. Using antibodies against host cell receptors CD36 and TLR-2 to block the entrance of S. aureus, we found that a blockage with anti-CD36 antibody was effective only in the presence of Tet38, and anti-TLR-2 antibody had an additional 2-fold effect on internalization of the tet38 mutant QT7. These studies suggested interactions between Tet38 and CD36, independent from TLR-2 (13).
To assess possible direct protein-protein interactions between Tet38, CD36, and TLR-2, we used an Ni affinity column retention assay that used purified Tet38 with a histidine tag (Tet38-His), as well as commercial purified proteins CD36 and TLR-2 treated with enterokinase to cleave off their His tags. As predicted with the internalization assays, CD36 was retained on the Ni column when Tet38 was bound by its His tag but not in its absence. We were unable to detect any Tet38-His column retention of TLR-2. When the assay was carried out with tagged CD36 (CD36-His) as the anchor bound to the Ni column, we found that both Tet38 and TLR-2 from which the His tags had been removed were retained on the column in the presence of CD36-His but not in its absence. This finding is consistent with prior studies (12) supporting the interaction of CD36 and TLR-2. Thus, Tet38 and TLR-2 bind to CD36 independently. In addition, in a three-step column retention assay, CD36 bound to Tet38-His on the Ni column also resulted in retention of TLR-2, indicating that the interaction of Tet38 and CD36 did not interfere with the binding between CD36 and TLR-2. These findings of direct protein-protein interactions support and extend our prior findings of the roles of Tet38, CD36, and TLR-2 in S. aureus invasion A549 cells.
The interaction between CD36 and TLR-2 has been demonstrated with S. aureus invasion assays involving various cell lines (7, 8, 12), but there is uncertainty regarding the relative contributions of these two host cell receptors under different conditions. CD36 and TLR-2 form a complex following induction by diacylated lipoproteins, but CD36 was not essential in S. aureus internalization by HEK cells, which lack CD36 receptors. In the absence of CD36, host receptors TLR-2 and TLR-6 form contacts with the S. aureus LTA and the diacylated lipoproteins prior to being internalized by HEK cells (12). These data were in contrast to the findings of Stuart et al. that demonstrated that CD36 was required for the activation of TLR-2/TLR-6 signaling and the internalization of S. aureus and its LTA by macrophages (8).
We investigated further the direct interactions between CD36 and TLR-2 and the effects of Tet38 and LTA on them. We demonstrated that Tet38 does not bind to TLR-2 directly but instead binds directly to CD36 in a manner that does not compete with TLR-2 binding to it, suggesting distinct binding sites on CD36. In addition, it appears that the complex of Tet38 and CD36 affects TLR-2 binding to CD36 in a manner that makes it sensitive to the presence of LTA, a sensitivity not seen with the CD36–TLR-2 interaction in the absence of Tet38. These data suggest that LTA binds to TLR-2 in the Tet38–CD36–TLR-2 complex differently from its binding to TLR-2 in the CD36–TLR-2 complex such that it enables displacement of TLR-2 from the Tet38-CD36 complex.
These direct and indirect binding data for Tet38 are consistent with our invasion assay data showing that CD36 was the most important contributor to Tet38-dependent invasion of A549 epithelial cells but that TLR-2 and LTA had additional, lesser effects independent of Tet38, emphasizing the importance of the involvement of these other components. Several studies have demonstrated that TLR-2 forms a complex with CD36 following stimulation by S. aureus LTA. LTA actively participates in bacterial invasion of host cells via contact with host cell receptor CD36, TLR-2, and TLR-2/6 and TLR-2/1 heterodimers, leading to host immune response, such as pathogen phagocytosis and cytokine production (4, 7, 8, 10, 24, 25).
Tet38 as a membrane protein is surrounded by layers of LTA, wall teichoic acid (WTA), and the peptidoglycan in the same manner as various other membrane proteins as described by Pasquina et al. (26). Since Tet38 and LTA were both involved in bacterial internalization, we tested the possibility that Tet38 could affect the action of inhibitors of teichoic acid synthesis. Susceptibility to tunicamycin, an inhibitor of the WTA synthesis (15, 16), was reduced by increased expression of tet38 and increased in a tet38 mutant. A similar effect was seen for Congo red, an inhibitor of LTA synthesis (17, 18), in the inoculum dilution growth assay. Although the mechanism is uncertain and could involve interaction with the respective drug targets, the prior demonstrated functions of Tet38 as a broad-spectrum efflux pump and the reduction in resistance to tunicamycin by the efflux pump inhibitor reserpine suggest that efflux of these drugs is the most likely mechanism.
In summary, we have demonstrated that Tet38 has a direct interaction with CD36, while LTA has a binding preference for TLR-2. CD36 can form a complex with both Tet38 and TLR-2, and the two binding events were independent from each other. In addition, Tet38 reduces the inhibitory effects of tunicamycin and Congo red on WTA and LTA synthesis, suggesting possible alteration of drug access or binding to their synthesis targets. Further investigation will be needed to define the mechanism of this drug resistance conferred by Tet38, and also to assess potential implication of Tet38 in the biosynthesis of LTA in addition to its role as a protector against an LTA inhibitor. These data further suggest that Tet38 could be a potential target for new therapeutic strategies to interrupt S. aureus cell internalization, which may contribute to the common persistence and recurrence of S. aureus infections even after treatment with active antimicrobials (27).
MATERIALS AND METHODS
Cell lines, bacterial strains, culture media, and other materials.
The bacterial strains, plasmids, primers, and cell lines used in this study are listed in Table 5. Human lung adenocarcinoma A549 cells were purchased from the ATCC (CCL-185) and were cultivated in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) and 4 mM l-glutamine (Fisher Scientific, Waltham, MA). This medium is referred to here as assay medium. A549 cells were grown at 37°C in 5% CO2. Chloramphenicol, ampicillin, gentamicin, Triton-X, tunicamycin, Congo red, n-dodecyl β-d-maltoside (DDM), isopropyl-β-d-thiogalactopyranoside (IPTG), cocktail of protease inhibitors, and lysostaphin were purchased from Sigma-Aldrich (St. Louis, MO). Anti-LTA, anti-CD36, and anti-TLR-2 antibodies, goat anti-mouse and goat anti-rabbit secondary antibodies, and mouse IgG nonspecific isotype control were purchased from Life Technologies. Histidine-tagged proteins CD36 and TLR-2 were purchased from Sino Biological, Inc. (Wayne, PA). LTA was purchased from Sigma-Aldrich. Monoclonal His tag antibody was purchased from EMD Millipore Corp. (Burlington, MA).
TABLE 5.
Bacterial strains, plasmids, cell line, and primers used in this study
| Strain, plasmid, or primer | Genotype or DNA sequence | Reference or source |
|---|---|---|
| S. aureus strains | ||
| RN6390 | Wild type | 30 |
| QT7 | RN6390 tet38 mutant | 21 |
| QT5 | RN6390 norB::cat | 21 |
| RN6390(pLI50) | Cmr | 20 |
| RN6390(pLI50-tet38) | tet38 overexpressor; Cmr | 20 |
| E. coli strains | ||
| DH10B | F− mcrA Δ(mrr-hsdRMS-mcrBC) Φ80dlacZΔM15 | 31 |
| ΔlacX74 endA1 recA1 deoR Δ(ara leu)7697 araD139 | ||
| galU galK nupG rpsL λ− | ||
| BL21 | B F– ompT gal dcm lon hsdSB(rB– mB–) [malB+]K-12(λS) | Invitrogen |
| BL21(pTrcHis 2A-tet38) | Protein purification (Tet38) | 23 |
| BL21(pTrcHis 2A-norA) | Protein purification (NorA) | 19 |
| Plasmids | ||
| pTrcHis2A | Expression vector | Invitrogen |
| pLI50 | Shuttle plasmid E. coli-S. aureus; Cmr | 30 |
| Cell line A549 | Human lung adenocarcinoma (ATCC) | 20 |
| Primers | ||
| Synthesis of tet38 overexpressors | ||
| Forward | TCATTGGTGTAGAAGCTTATGATTATGAATa | |
| Reverse | CGCGAATTTATATCTATGGATCCTTATTTTb | |
| Protein expression, tet38 cloning into pTrcHis2A | ||
| Forward, pTrctet38-BamHI | ATCGGGATCCATGAATGTTGAATATTCTAAAATAAb | |
| Reverse, pTrctet38-HindIII | ATCGAAGCTTTTTTTCAGATTGTGTCCAACGATTTAa |
The HindIII site is underlined.
The BamHI site is underlined.
Drug susceptibility determinations.
The MIC was determined as the lowest concentration of antibiotic in a series of 2-fold dilutions that yielded no visible growth after incubation at 37°C for 24 h. The MICs of antibiotics were determined by the broth microdilution method as previously described (21, 28). Reserpine was added to the medium at a 20 μM final concentration as indicated.
Congo red susceptibility testing was based on the work carried out by Suzuki et al., with minor modifications (17, 18). S. aureus cells starting at 107 CFU in 10 μl (106 CFU/μl), and in a series of 10-fold dilutions in LB media, were spotted onto LB agar plates supplemented with 0.08% (wt/vol) Congo red. A similar series was also carried out in parallel using LB agar plates as a control of bacterial growth. The Congo red and control plates were incubated at 37°C for 24 h in the dark (plates wrapped in aluminum foil) to avoid inactivation of Congo red by light. The Congo red effects on the growth of S. aureus were determined by comparing the colony counts and extent of growth across dilutions among RN6390, QT7, and a tet38 overexpressor on control and Congo red plates. Plasmid pLI50 was introduced into RN6390 and QT7 by electroporation, and the transformants were grown on medium containing chloramphenicol at 20 μg/ml. These transformants were used as controls to be compared with the overexpressor RN6390(pLI50-tet38) and the tet38-complemented strain QT7(pLI50-tet38).
Internalization of S. aureus by A549 epithelial cells.
S. aureus parental strain RN6390 was compared with the tet38 isogenic mutant QT7 (tet38::cat) to assess their ability to survive inside epithelial cells following standard invasion assays, as previously described (13, 20). The A549 cells were cultured in 5 ml of assay medium until 90% confluency in a 25-ml tissue culture flask and then were seeded into 24-well plates (Costar) in assay medium to yield a cell concentration of 104/well. S. aureus RN6390 or QT7 was prepared from overnight cultures, grown to an optical density at 600 nm (OD600) of 0.5, and then adjusted to a concentration of 106 CFU/ml. A549 cells were infected with S. aureus at a multiplicity of infection (MOI) of 100 bacteria per epithelial cell (i.e., 100:1; 106 washed bacteria/104 A549 cells). The bacterium-cell mixtures were incubated at 37°C in 5% CO2 for 2 h. The monolayers were then incubated for 60 min at 37°C in 5% CO2 in assay medium with 200 μg/ml of gentamicin and 20 μg/ml of lysostaphin and lysed with 200 μl of Triton X-100 (0.1%). Bacteria were plated on LB agar plates, and colony counts were performed to determine the number of viable internalized bacteria.
Internalization of S. aureus into A549 cells previously treated with anti-CD36, anti-TLR-2, and anti-LTA antibodies.
The invasion assay was carried out as described above. Prior to adding S. aureus to the monolayers, the host cells were preincubated at 37°C under 5% CO2 for 30 min in assay medium containing 1% bovine serum albumin (BSA) plus anti-CD36, anti-TLR-2, or anti-LTA antibodies (Life Technologies, Grand Island, NY) at a final concentration of 50 nM (13, 20). The CFU/monolayer of intracellular bacteria was enumerated as described above, and each assay was repeated three times. A mouse IgG nonspecific isotype (Life Technologies) was used at the same concentration (50 nM) as the tested antibody in the assays and served as negative control.
For assays using anti-LTA antibodies, we also carried out a second approach consisting of adding anti-LTA antibodies at 5 μM to the bacterial suspension for 30 min at 37°C prior to adding the mixture (S. aureus/anti-LTA) to A549 cells. This concentration of antibody was found to be the most efficient for the assay. The same mouse IgG nonspecific isotype was used at the same concentration (5 μM) as tested antibody in this assay and served as a negative control.
Internalization of S. aureus into A549 cells previously treated with purified Tet38 or purified NorA proteins and anti-CD36 antibody.
The invasion assay was carried out as described above, with some modifications. Prior to adding S. aureus to the monolayers, the host cells were preincubated at 37°C under 5% CO2 for 30 min in assay medium containing 1% BSA plus purified Tet38 or purified NorA, and then anti-CD36 antibody (50 nM) was added for an additional 30 min. The concentration of Tet38 or NorA proteins was 5 μg/well. The CFU/monolayer of intracellular bacteria was enumerated as described above, and each assay was repeated three times. A mouse IgG nonspecific isotype (Life Technologies, Grand Island, NY) was used at the same concentration (50 nM) as the tested antibody in the assays and served as a negative control.
Tet38 and NorA protein purifications.
The histidine-tagged Tet38 (Tet38-His) and histidine-tagged NorA (NorA-His) expressed from Escherichia coli BL21(pTrchis2A-tet38) and E. coli BL21(pTrchis2A-norA) were purified using an Ni affinity column as previously described, with some modifications for the Tet38 purification (23). The tet38 gene amplified from genomic DNA of S. aureus RN6390 was cloned into the expression vector pTrcHis2A and introduced into E. coli BL21 by electroporation.
E. coli BL21 which harbored construct pTrcHis2A-tet38 was cultured in 500 ml of LB medium supplemented with ampicillin (100 μg/ml) under shaking at 37°C until an OD600 of ∼0.6 was reached. IPTG at 1 mM was then added, and the incubation was continued at 30°C for ∼20 h under shaking. The bacteria were harvested by centrifugation at 6,000 rpm for 20 min at 4°C, and then the pellet was resuspended in 10 ml of lysis buffer (1× phosphate-buffered saline [PBS; pH 7.4] with 5% glycerol, 1% DDM, 10 mg/ml of lysozyme, 1 μl of benzonase, and 1 tablet of a protease inhibitor cocktail which inhibits serine, cysteine, aspartic, and metalloproteases [Sigma]) and was left on ice for 45 min. The mixture was vortexed every 15 min and then centrifuged at 8,000 rpm for 45 min at 4°C. The lysate was filtered and applied to a nickel affinity column preequilibrated with 5 column volumes of equilibration buffer (1× PBS [pH 7.4] with 5% glycerol, 0.25% DDM). The sample-loaded nickel column was washed with 5 column volumes of equilibration buffer, followed by 10 mM imidazole in equilibration buffer (5 column volumes). The Tet38 protein was eluted with 150 mM imidazole in equilibration buffer (elution buffer). Purified Tet38 was desalted with a PD-10 column (GE Healthecare) and concentrated with Amicon Ultra 30K (EMD Millipore). The protein concentration was measured in a NanoDrop spectrophotometer (ND1000 spectrophotometer V3.6.0), and the homogeneity of the protein was estimated by SDS-PAGE.
Affinity column retention assay.
The histidine tags were cleaved from His-CD36 and His–TLR-2 proteins using 1 U of enterokinase in a reaction mixture of 10 μg of protein in 25 μl incubated at 25°C for 16 h. Enterokinase was purchased from New England BioLabs (Beverly, MA). We used ∼5 μg of each protein for the column retention assays. Tet38-His was first loaded to an Ni column, and then enterokinase-treated CD36 was loaded onto the same column. The column was washed with 5 column volumes of buffer A (10 mM Tris-HCl [pH 7.6], 500 mM NaCl, 10 mM imidazole), and then the proteins were eluted with 100 mM imidazole in buffer A. The enterokinase-treated CD36 protein was loaded onto a separate Ni column, washed with buffer A, and then eluted with 100 mM imidazole to verify the absence of nonspecific binding of CD36 to the column. This step was used as a control for the specificity of CD36 binding to the histidine-tagged protein Tet38. Eluted proteins were analyzed by SDS-PAGE. The same procedure was carried out with enterokinase-treated TLR-2. To study the effect of a combination of two receptors on Tet38, CD36 and TLR-2 were applied successively to the Tet38-His-Ni column. Washing and elution steps were performed in the same manner as described above. LTA (25 μg per assay) was added as indicated.
Western blotting.
Tet38-His, CD36, and TLR-2 (∼1 μg) and 50 μl of each elution fraction were subjected to SDS-PAGE, followed by protein transfer onto a nitrocellulose membrane using the iBlot gel transfer system, as recommended by the manufacturer (Life Technologies, Grand Island, NY). The Western blot procedure was carried out as previously described (29). The nitrocellulose membrane was incubated with the specified antibody diluted in TBS-Tween 20 blocking buffer at 4°C overnight, and then the membrane was washed with TBS-Tween/Triton buffer before incubation again with specified secondary antibody. Monoclonal anti-His-tag antibody was used for Tet38-His (primary antibody, 1/2,000, and secondary antibody, goat anti-mouse at 1/5,000). Polyclonal anti-CD36 antibody was used for CD36 (primary antibody, 1/500, and secondary antibody, goat anti-rabbit at 1/1,000). Polyclonal anti-TLR-2 antibody was used for TLR-2 (primary antibody, 1/500, and secondary antibody, goat anti-rabbit at 1/1,000). Membranes were incubated with secondary antibodies for 1 h at room temperature. The chemiluminescence detection reaction was performed, and the membranes were exposed to X-ray film accordingly to the manufacturer’s recommendations.
ACKNOWLEDGMENTS
This work was supported by U.S. Public Health Service grants P01-AI083214 (M. Gilmore, principal investigator; subproject to D.C.H.) and R37-AI23988 (to D.C.H.) from the National Institutes of Health.
REFERENCES
- 1.Palma M, Haggar A, Flock JI. 1999. Adherence of Staphylococcus aureus is enhanced by an endogenous secreted protein with broad binding activity. J Bacteriol 181:2840–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zapotoczna M, Jevnikar Z, Miajlovic H, Kos J, Foster TJ. 2013. Iron-regulated surface determinant B (IsdB) promotes Staphylococcus aureus adherence to and internalization by non-phagocytic human cells. Cell Microbiol 15:1026–1041. doi: 10.1111/cmi.12097. [DOI] [PubMed] [Google Scholar]
- 3.Hirschhausen N, Schlesier T, Schmidt MA, Gotz F, Peters G, Heilmann C. 2010. A novel staphylococcal internalization mechanism involves the major autolysin Atl and heat shock cognate protein Hsc70 as host cell receptor. Cell Microbiol 12:1746–1764. doi: 10.1111/j.1462-5822.2010.01506.x. [DOI] [PubMed] [Google Scholar]
- 4.Nilsen NJ, Deininger S, Nonstad U, Skjeldal F, Husebye H, Rodionov D, von AS, Hartung T, Lien E, Bakke O, Espevik T. 2008. Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling: role of CD14 and CD36. J Leukoc Biol 84:280–291. doi: 10.1189/jlb.0907656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ning R, Zhang X, Guo X, Li Q. 2010. Attachment of Staphylococcus aureus is required for activation of nuclear factor kappa B in human osteoblasts. Acta Biochim Biophys Sin (Shanghai) 42:883–892. doi: 10.1093/abbs/gmq096. [DOI] [PubMed] [Google Scholar]
- 6.Dziewanowska K, Carson AR, Patti JM, Deobald CF, Bayles KW, Bohach GA. 2000. Staphylococcal fibronectin binding protein interacts with heat shock protein 60 and integrins: role in internalization by epithelial cells. Infect Immun 68:6321–6328. doi: 10.1128/IAI.68.11.6321-6328.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baranova IN, Kurlander R, Bocharov AV, Vishnyakova TG, Chen Z, Remaley AT, Csako G, Patterson AP, Eggerman TL. 2008. Role of human CD36 in bacterial recognition, phagocytosis, and pathogen-induced JNK-mediated signaling. J Immunol 181:7147–7156. doi: 10.4049/jimmunol.181.10.7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stuart LM, Deng J, Silver JM, Takahashi K, Tseng AA, Hennessy EJ, Ezekowitz RA, Moore KJ. 2005. Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J Cell Biol 170:477–485. doi: 10.1083/jcb.200501113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. 1999. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by Toll-like receptor 2. J Biol Chem 274:17406–17409. doi: 10.1074/jbc.274.25.17406. [DOI] [PubMed] [Google Scholar]
- 10.Hashimoto M, Tawaratsumida K, Kariya H, Aoyama K, Tamura T, Suda Y. 2006. Lipoprotein is a predominant Toll-like receptor 2 ligand in Staphylococcus aureus cell wall components. Int Immunol 18:355–362. doi: 10.1093/intimm/dxh374. [DOI] [PubMed] [Google Scholar]
- 11.Gillrie MR, Zbytnuik L, McAvoy E, Kapadia R, Lee K, Waterhouse CC, Davis SP, Muruve DA, Kubes P, Ho M. 2010. Divergent roles of Toll-like receptor 2 in response to lipoteichoic acid and Staphylococcus aureus in vivo. Eur J Immunol 40:1639–1650. doi: 10.1002/eji.200939929. [DOI] [PubMed] [Google Scholar]
- 12.Triantafilou M, Gamper FG, Haston RM, Mouratis MA, Morath S, Hartung T, Triantafilou K. 2006. Membrane sorting of Toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J Biol Chem 281:31002–31011. doi: 10.1074/jbc.M602794200. [DOI] [PubMed] [Google Scholar]
- 13.Truong-Bolduc QC, Khan N, Vyas JM, Hooper DC. 2016. Tet38 efflux pump affects Staphylococcus aureus internalization by epithelial cells through interaction with CD36 and contributes to bacterial escape from acidic and non-acidic phagolysosomes. Infect Immun 85:e00862-16. doi: 10.1128/IAI.00862-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Truong-Bolduc QC, Wang Y, Hooper DC. 2018. Tet38 efflux pump contributes to fosfomycin resistance in Staphylococcus aureus. Antimicrob Agents Chemother 62:e00927-18. doi: 10.1128/AAC.00927-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu X, Liu D, Singh AK, Drolia R, Bai X, Tenguria S, Bhunia AK. 2018. Tunicamycin mediated inhibition of wall teichoic acid affects Staphylococcus aureus and Listeria monocytogenes cell morphology, biofilm formation and virulence. Front Microbiol 9:1352. doi: 10.3389/fmicb.2018.01352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mann PA, Muller A, Wolff KA, Fischmann T, Wang H, Reed P, Hou Y, Li W, Muller CE, Xiao J, Murgolo N, Sher X, Mayhood T, Sheth PR, Mirza A, Labroli M, Xiao L, McCoy M, Gill CJ, Pinho MG, Schneider T, Roemer T. 2016. Chemical genetic analysis and functional characterization of staphylococcal wall teichoic acid 2-epimerases reveals unconventional antibiotic drug targets. PLoS Pathog 12:e1005585. doi: 10.1371/journal.ppat.1005585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki T, Campbell J, Kim Y, Swoboda JG, Mylonakis E, Walker S, Gilmore MS. 2012. Wall teichoic acid protects Staphylococcus aureus from inhibition by Congo red and other dyes. J Antimicrob Chemother 67:2143–2151. doi: 10.1093/jac/dks184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vickery CR, Wood BM, Morris HG, Losick R, Walker S. 2018. Reconstitution of Staphylococcus aureus lipoteichoic acid synthase activity identifies Congo red as a selective inhibitor. J Am Chem Soc 140:876–879. doi: 10.1021/jacs.7b11704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu JL, Grinius L, Hooper DC. 2002. NorA functions as a multidrug efflux protein in both cytoplasmic membrane vesicles and reconstituted proteoliposomes. J Bacteriol 184:1370–1377. doi: 10.1128/JB.184.5.1370-1377.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Truong-Bolduc QC, Bolduc GR, Medeiros H, Vyas JM, Wang Y, Hooper DC. 2015. Role of the Tet38 efflux pump in Staphylococcus aureus internalization and survival in epithelial cells. Infect Immun 83:4362–4372. doi: 10.1128/IAI.00723-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Truong-Bolduc QC, Dunman PM, Strahilevitz J, Projan SJ, Hooper DC. 2005. MgrA is a multiple regulator of two new efflux pumps in Staphylococcus aureus. J Bacteriol 187:2395–2405. doi: 10.1128/JB.187.7.2395-2405.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwak YG, Truong-Bolduc QC, Bin KH, Song KH, Kim ES, Hooper DC. 2013. Association of norB overexpression and fluoroquinolone resistance in clinical isolates of Staphylococcus aureus from Korea. J Antimicrob Chemother 68:2766–2772. doi: 10.1093/jac/dkt286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Truong-Bolduc QC, Villet RA, Estabrooks ZA, Hooper DC. 2014. Native efflux pumps contribute resistance to antimicrobials of skin and the ability of Staphylococcus aureus to colonize skin. J Infect Dis 209:1485–1493. doi: 10.1093/infdis/jit660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hattar K, Reinert CP, Sibelius U, Gokyildirim MY, Subtil FSB, Wilhelm J, Eul B, Dahlem G, Grimminger F, Seeger W, Grandel U. 2017. Lipoteichoic acids from Staphylococcus aureus stimulate proliferation of human non-small-cell lung cancer cells in vitro. Cancer Immunol Immunother 66:799–809. doi: 10.1007/s00262-017-1980-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niebuhr M, Langnickel J, Sigel S, Werfel T. 2010. Dysregulation of CD36 upon TLR-2 stimulation in monocytes from patients with atopic dermatitis and the TLR2 R753Q polymorphism. Exp Dermatol 19:e296–e298. doi: 10.1111/j.1600-0625.2009.00989.x. [DOI] [PubMed] [Google Scholar]
- 26.Pasquina L, Santa Maria JP Jr, McKay Wood B, Moussa SH, Matano LM, Santiago M, Martin SE, Lee W, Meredith TC, Walker S. 2016. A synthetic lethal approach for compound and target identification in Staphylococcus aureus. Nat Chem Biol 12:40–45. doi: 10.1038/nchembio.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr. 2015. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28:603–661. doi: 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Truong-Bolduc QC, Wang Y, Chen C, Hooper DC. 2017. Transcriptional regulator TetR21 controls the expression of Staphylococcus aureus LmrS efflux pump. Antimicrob Agents Chemother 61:e00649-17. doi: 10.1128/AAC.00649-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Truong-Bolduc QC, Hooper DC. 2010. Phosphorylation of MgrA and its efect on expression of the NorA and NorB efflux pumps of Staphylococcus aureus. J Bacteriol 192:2525–2534. doi: 10.1128/JB.00018-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Truong-Bolduc QC, Hsing LC, Villet R, Bolduc GR, Estabrooks Z, Taguezem GF, Hooper DC. 2012. Reduced aeration affects the expression of the NorB efflux pump of Staphylococcus aureus by posttranslational modification of MgrA. J Bacteriol 194:1823–1834. doi: 10.1128/JB.06503-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monk IR, Shah IM, Xu M, Tan M-W, Foster TJ. 2012. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3:e00277-11. doi: 10.1128/mBio.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]