

Summary
Sickle cell disease (SCD) affects over 2 million people worldwide with high morbidity and mortality in underdeveloped countries. Therapeutic interventions aimed at reactivating fetal haemoglobin (HbF) is an effective approach for improving survival and ameliorating the clinical severity of SCD. A class of agents that inhibit DNA methyltransferase (DNMT) activity show promise as HbF inducers because off‐target effects are not observed at low concentrations. However, these compounds are rapidly degraded by cytidine deaminase when taken by oral administration, creating a critical barrier to clinical development for SCD. We previously demonstrated that microRNA29B (MIR29B) inhibits de novo DNMT synthesis, therefore, the goal of our study was to determine if MIR29 mediates HbF induction. Overexpression of MIR29B in human KU812 cells and primary erythroid progenitors significantly increased the percentage of HbF positive cells, while decreasing the expression of DNMT3A and the HBG repressor MYB. Furthermore, HBG promoter methylation levels decreased significantly following MIR29B overexpression in human erythroid progenitors. We subsequently, observed higher MIR29B expression in SCD patients with higher HbF levels compared to those with low HbF. Our findings provide evidence for the ability of MIR29B to induce HbF and supports further investigation to expand treatment options for SCD.
Keywords: MIR29B, HBG, DNA methylation, fetal haemoglobin, MYB
Sickle cell disease (SCD) is one of the most common genetic disorders affecting over 20 million people worldwide mainly in underdeveloped countries (Weatherall, 2013). Therapeutic intervention aimed at reactivating HBG (γ‐globin) gene transcription and fetal haemoglobin (HbF) expression is an effective strategy for ameliorating the clinical symptoms of SCD and improving long‐term survival (Platt et al, 1994; Poillon et al, 1993). Mechanistically, HbF blocks HbS polymerization and erythrocyte sickling through the formation of hybrid haemoglobin molecules in red blood cells. Based on these findings, hydroxycarbamide (HC) was developed as the only US Food and Drug Administration (FDA)‐approved therapeutic intervention for HbF induction, however it causes bone marrow toxicity if not accurately dosed (Charache et al, 1995). Therefore, alternative approaches to induce HbF through epigenetic mechanisms of HBG βgene silencing targeting DNA hypermethylation of proximal promoter CpG rich regions was investigated in this study.
Therapeutic interventions to decrease HBG gene methylation and reactivate transcription have proven beneficial in clinical studies. For example, SCD patients treated intravenously with the DNA methyltransferase (DNMT) inhibitor decitabine (Dec) demonstrated robust induction of HbF and total haemoglobin levels (Molokie et al, 2017; Akpan et al, 2010; Lavelle et al, 2010; Desimone et al, 2002). However, when administered by mouth, Dec is rapidly inactivated by cytidine deaminase. In a recent Phase I clinical trial, where Dec was combined with tetrahydrouridine to inhibit its metabolism, DNMT1 protein levels and DNA methylation were decreased in peripheral blood mononuclear cells, while HbF levels increased in SCD patients (Molokie et al, 2017). Ideally, treatments for SCD will activate HBG expression without bone marrow toxicity and avoiding off target effects.
An attractive class of molecules under development for therapeutic intervention are microRNA (miRNA) mimics and antagomirs. Due to their capacity to restore control of aberrantly expressed genes, causing a wide array of human diseases, the development of miRNA therapeutics is highly investigated (Bianchi et al, 2009; Walker et al, 2011; Ward et al, 2016; Lulli et al, 2013; Starlard‐Davenport et al, 2013). In previously published work, we demonstrated that MIR29B, inhibits de novo synthesis of the DNMT enzymes DNMT3A and DNMT3B, in breast cancer cells and restores control of aberrantly expressed tumour suppressor genes involved in cell cycle control, including TP73, CDH1, RASSF1, CCNA1, and CDKN1C (Starlard‐Davenport et al, 2013). Recently, we identified MIR144 as a repressor of NFE2L2 and HbF expression in patients with SCD (Li et al, 2018) in a genome‐wide microarray‐based miRNA screen of RNA isolated from reticulocytes. Herein, we demonstrated that MIR29B mediates HBG activation and induces HbF in KU812 cells and human primary erythroid progenitors. To compliment in vitro studies, we observed higher MIR29B levels in the reticulocytes of SCD patients with high HbF compared to those with low HbF levels suggesting a functional role in HBG gene regulation.
Materials and methods
Subject recruitment and blood processing
Blood samples were obtained from individuals (n = 12) with homozygous sickle cell anaemia (HbSS) not on HC therapy followed in the Sickle Cell Program at Augusta University. Medical record review was completed to obtain complete blood counts with differential, reticulocyte count and HbF levels determined by high performance liquid chromatography. Blood samples were processed by ficoll histopaque separation of plasma, red blood cells, and buffy coat. Subsequently, the red blood cells were processed using the magnetic‐activated cell sorting (MACS) column and TFRC + MicroBeads (MACS, Miltenyl Biotec, Auburn, CA, USA) to isolate reticulocytes for total RNA extraction using TRIzol (ThermoFisher, Waltham, MA, USA). All research involving persons with SCD was approved by the Augusta University Institutional Review Board and all clinical investigations were conducted according to the principles expressed in the Declaration of Helsinki.
MicroRNA microarray analysis
Reticulocytes isolated from the peripheral blood were used for miRNA analysis on the miRCURY LNA microRNA Array (Exiqon, Woburn, MA, USA). Raw data were quantile normalized using a model‐based background correction algorithm as recently published by our group (Li et al, 2018). The miRNA genes differentially expressed between the high and low HbF groups were identified (Li et al, 2018) and raw data deposited in the National Center for Biotechnology Information Gene Expression Omnibus database, accession number GSE111356.
Erythroid differentiation of human CD34+ stem cells
Human primary erythroid cells were generated from adult CD34+ stem cells (STEMCELL Technologies, Vancouver, BC, USA) in a two‐phase culture system as previously published (Li et al, 2014). During phase I, stem cells were grown in minimum essential medium‐α (αMEM) containing AB serum, interleukin‐3 (10 ng/ml), stem cell factor (10 ng/ml), and erythropoietin (2 iu/ml). On day 7, cells transitioned to Phase II media where they remained under erythropoietin (2 iu/ml) stimulation. Erythroid progenitors were transfected on day 8 with human mature MIR29B or control scramble (Scr) mimic (Applied Biosystems, Foster City, CA, USA) by nucleofection using the Amaxa® Human CD34+ Cell Nucleofector® Kit. For drug studies, cells were treated with 0·5 μmol/l Dec alone or pretreatment with 100 nmol/l MIR29B on day 8 followed by drug treatment. After 48 h, cells were harvested for reverse transcription‐quantitative PCR (RT‐qPCR), Western blot, and flow cytometry analysis. Giemsa staining was used to monitor cell morphology and cell counts; viability was monitored using 0·4% trypan blue exclusion assay (Gibco, Carlsbad CA, USA).
KU812 cell culture and MIR29B transfections
Human KU812 cells were grown in Iscove's Modified Dulbecco's medium supplemented with 10% fetal bovine serum, penicillin (100 u/ml), and streptomycin (0·1 mg/ml) in 5% CO2 at 37°C. Cell counts and viability were determined using 0·4% trypan blue exclusion assay. Cells were seeded at a density of 0·5 × 106 viable cells per 100 mm plate for different treatments. During log phase growth, cells were transfected with 25, 50, and 100 nmol/l of pre‐ MIR29B (Applied Biosystems) for 48 h in three independent replicates using Opti‐MEM media (Gibco) and Lipofectamine™ 2000 transfection reagent (Invitrogen Carlsbad, CA, USA) according to the manufacturer's instructions. The cells transfected with Scr oligonucleotide served as control. For drug inductions KU812 cells were treated with Dec (0·5 μmol/l) or HC (75 μmol/l) alone (Lou et al, 2009) or pretreated with 100 nmol/l MIR29B followed by drug treatments for 48 h, then harvested for subsequent analyses.
RT‐qPCR analysis
Total RNA was extracted from KU812 cells using an AllPrep DNA/RNA/Protein Midi Kit (Qiagen, Valencia, CA, USA); TRIzol reagent was used to extract RNA from primary erythroid progenitors. Subsequently, cDNA was generated using the high capacity reverse transcription kit (Applied Biosystems) and qPCR was performed in a QuantStudio 3 Real‐Time System using SYBR Green™ Master Mix (Applied Biosystems). The level of DNMT1, DNMT3A, DNMT3B, MECP2, HBG and HBB gene transcripts were determined using gene specific primers (Table SI). To quantify GYPA (glycophorin A) and TFRC (transferrin receptor) gene expression, we used the RT2‐qPCR Primer system (Qiagen, Germantown, MD, USA). Relative quantification of gene expression was normalized to GAPDH as an internal control. Quantification of MIR29B was performed using the TaqMan miRNA assay (Applied Biosystems) according to the manufacturer's instructions and SNORD48 was used as endogenous control. The 2−ΔΔCt method was used for calculating the relative amount of target mRNA. All RT‐qPCR reactions were performed in triplicate, repeated at least 3 times, and always included a no‐template sample as a negative control. RT‐qPCR results are presented as average fold change of target gene in cells relative to Scr control, which was normalized to one.
Western blot analysis
Total protein was isolated using the AllPrep DNA/RNA/Protein Midi Kit (Qiagen) according to manufacturer's instructions. For Western blot analysis, 20–40 μg of total or nuclear protein was loaded on a 12% acrylamide gel, transferred to a polyvinylidene difluoride membrane, and then blocked in 5% non‐fat milk. Primary antibodies against DNMT3A (sc‐373905), DNMT3B (sc‐376043), MYB (05175 MI), HbF (sc‐21756), and HbA (sc‐21757) purchased from Santa Cruz Biotechnology (Dallas, TX, USA) were diluted in the range of 1:250 to 1:2000, incubated overnight and then followed by treatment with secondary antibody. The immunoblots were developed using SuperSignal® West Pico Chemiluminescent Substrate (Thermo Scientific) and analysed on a LAS‐3000 gel imager (Fujifilm Medical Systems, Stamford, CT, USA). Blots were processed with Restore™ Plus Western blot stripping buffer and probed with β‐actin (also termed ACTB, sc‐53646) antibody as the internal control. The band intensity of different proteins was quantified by densitometry with MultiGauge Software (Fujifilm Medical Systems) and normalized to the band intensity of β‐actin.
Flow cytometry analysis
To measure HbF positive cells (F‐cells), the various experimental samples were harvested, washed in phosphate‐buffered saline and then fixed with 4% paraformaldehyde and permeabilized with an ice‐cold acetone/methanol (4:1) mixture before staining with fluorescein isothiocyanate (FITC) conjugated anti‐HbF antibody (ab19365, Abcam) to quantify F‐cells. Isotype control IgG antibody (MBS524511, MyBioSource, San Diego, CA, USA) was used to detect non‐specific staining. In addition, the false positive attributed to Fc non‐specific binding was subtracted by incubating the unfixed cells with Human TruStain FcX™ (422301, Biolegend, San Diego, CA, USA) prior to antibodies staining. Flow cytometry was conducted on a BD FACSCanto II or LSRII (BD Biosciences, San Jose, CA, USA) machine and percentage of F‐cells determined by FlowJo Version 10.0.7 software (Tree Star, OR) as previously reported (Promsote et al, 2014; Zhu et al, 2017). Briefly, a consistent flow of single events and live cells were sequentially gated using a Time gate, forward scatter (FSC)‐H/FSC‐A plot, FSC/sideways scatter (SSC)‐plot and Live/Dead gates. The resulting population was classified into HbF negative and HbF positive populations.
Epimark site‐specific quantitative analysis
Levels of 5‐methylcytosine and unmodified cytosine at the −54 CCGG site 5′ to the HBG gene cap site were quantitated by Epimark assays as previously described (Ruiz et al, 2015). DNA was purified from cell pellets using QIAGEN AllPrep DNA/RNA/Protein Minikits (Qiagen catalogue number 80234). DNA was analysed by qPCR using primers that target the HBG promoter and spanned the −54 CCGG site as published (Ruiz et al, 2015). The percentage of 5‐methylcytosine and unmodified cytosine was determined using the comparative Ct method according to the manufacturer's instructions (EpiMark 5‐hmC and 5‐mC Analysis Kit).
Statistical analysis
The data are reported as the mean ± standard error of the mean for three to six replicates of independent experiments, each performed in triplicate. All data were analysed by a two‐tailed Student's t‐test and P < 0·05 was considered statistically significant. For the miRNA microarray analysis, two study groups consisting of SCD patients with HbF < 8·6% (low HbF; n = 6) and HbF > 8·6% (high HbF; n = 6) were analysed as previously reported (Li et al, 2018).
Results
Using in silico analysis, we discovered that MIR29B has a consensus sequence complimentary to the 3′‐untranslated region of DNMT3A and DNMT3B (Starlard‐Davenport et al, 2013). Since HBG is silenced via DNA hypermethylation in adult erythroid cells (Lavelle et al, 2006; Ruiz et al, 2015) and MIR29B silences MYB (Martinez et al, 2011), a known HBG repressor protein, we determined whether MIR29B alters DNMT and MYB expression as a mechanism of HbF induction. Using KU812 cells, which express the HBG and HBB genes, we confirmed efficient transfection of MIR29B mimic into KU812 cells after 48 h by RT‐qPCR analysis. In fact, KU812 cells treated with 25 nmol/l MIR29B showed a 200‐fold (P < 0·05) increase in expression levels compared to Scr control (Fig 1A). Furthermore, a dose‐dependent increase in MIR29B expression up to the 100 nmol/l concentration was observed. We next quantified DNMT3A, DNMT3B, and MYB mRNA and protein levels using RT‐qPCR and Western blot, respectively. Transfection of KU812 cells with 100 nmol/l MIR29B significantly decreased DNMT3A and DNMT3B mRNA by ~50% and ~25% (P < 0·05) respectively (Fig 1B, C). Transfection of KU812 cells with 100 nmol/l MIR29B also significantly decreased DNMT3A protein expression; by contrast, the effect of MIR29B on DNMT3B levels was not significant. We quantified expression of MECP2 (methyl CpG binding protein 2) which preferentially binds to DNA containing symmetrically methylated CpG dinucleotides and observed a 90% decrease in MECP2 mRNA levels mediated by MIR29B mimic (Fig 1B). We subsequently determined the effects of MIR29B on its gene target the oncogene MYB, a known HBG gene repressor. MYB protein was decreased by 50% in MIR29B transfected cells compared to Scr control (P < 0·01) (Fig 1C). Thus, based on these findings we postulated that MIR29B activates HBG transcription, partly through DNMT and MYB silencing.
Figure 1.
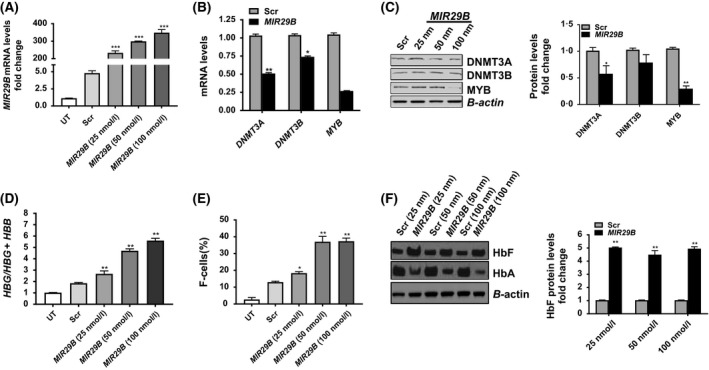
MIR29B regulates HBG gene expression in KU812 cells. (A) Fold change of MIR29B expression in KU812 cells following transfection for 48 h with miR‐29b mimic or Scr control by reverse transcription‐quantitative polymerase chain reaction (RT‐qPCR) analysis. The expression of MIR29B level in Scr control was set to one after normalization to endogenous SNORD48 levels. For all studies, the quantitative data are shown as the mean ± standard error of the mean (SEM) of 3 to 5 independent experiments. *P < 0·05, is considered statistically significant; ***P < 0·001. (B) Fold change of DNMT3A,DNMT3B, and MECP2 expression by RT‐qPCR analysis after MIR29B 100 nmol/l (black bars) or Scr (grey bars) mimic transfection into KU812 cells. (C) Western blot for the proteins shown was completed in KU812 cells transfected with MIR29B (black bars) and Scr control (grey bars); β‐actin was used as the internal control; **P < 0·01. (D) The mRNA levels of HBG and HBB in KU812 cells was quantified by RT‐qPCR and calculated as the ratio: HBG/HBG + HBB for the different conditions. (E) Flow cytometry analysis was completed to determine the % HbF positive cells (F‐cells) after staining with fluorescein isothiocyanate labelled anti‐HbF antibody. Shown in the graph is the quantitative data generated by FlowJo analysis. (F) Western blot of HbF and haemoglobin A (HbA) relative to β‐actin protein levels in KU812 cells transfected with MIR29B mimic or Scr control at the concentrations shown. Scr, scramble; UT, untreated.
To ascertain whether MIR29B mediates HBG activation, we performed RT‐qPCR analysis in KU812 cells. After 48 h of treatment, we observed a 2·3‐fold (P < 0·01) dose‐dependent increase in HBG mRNA mediated by MIR29B compared to Scr control (Fig 1D). By flow cytometry, we demonstrated the ability of MIR29B to increase F‐cells from 15% (Scr control) to 40% (Fig 1E). To further support HBG activation, we observed a 2‐fold increase in HbF protein levels in MIR29B transfected KU812 cells by Western blot, while adult haemoglobin A (HbA) decreased by 50% (P < 0·01) (Fig 1F).
While these data in KU812 cells were supportive of the ability of MIR29B to regulate HBG, we next performed studies under physiological conditions, using primary erythroid progenitors generated from normal CD34+ stem cells. As previously reported by our group erythroid progenitors were generated in a two‐phase culture system (Li et al, 2018). Transfection of erythroid progenitors with 50 and 100 nmol/l MIR29B produced a 7‐ to 8‐fold increase in MIR29B expression (Fig 2A), while combining 100 nmol/l MIR29B with 0·5 μmol/l Dec did not increase levels further. Under the same treatment conditions, we observed a significant decrease in DNMT3A, DNMT3B and MECP2 mRNA levels (Fig 2B). Parallel these effects, MIR29B decreased DNMT3A, DNMT3B, and MYB protein levels by 50% in human primary erythroid progenitors (Fig 2C).
Figure 2.
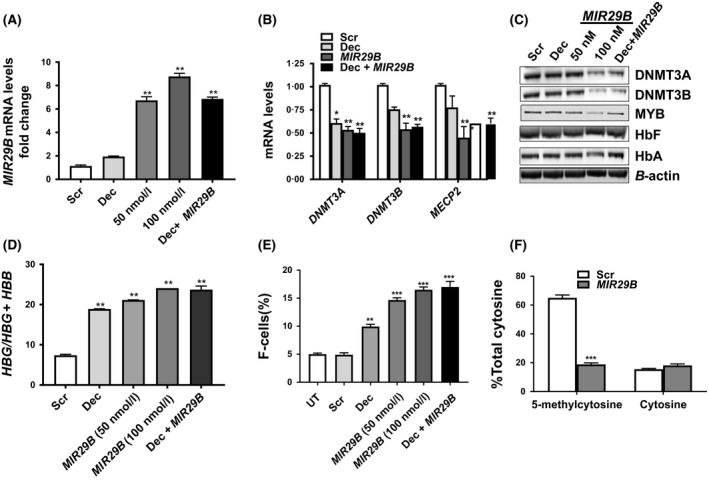
MIR29B regulates HBG gene expression through proximal promoter 5‐hydroxymethylation in human primary erythroid progenitors. (A) Shown is the fold change of MIR29B expression by reverse transcription‐quantitative polymerase chain reaction (RT‐qPCR) in erythroid progenitors generated from CD34+ stem cells (see Material and Methods), treated with Scr (100 nmol/l), MIR29B (50 or 100 nmol/l) or 0·5 μmol/l Dec alone or combined treatment with 0·5 μmol/l Dec and MIR29B 100 nmol/l. The expression of MIR29B in Scr control was set to one after normalization to the SNORD48 endogenous control. The data are shown as the mean ± SEM. (B) Shown is the fold change of DNMT3A,DNMT3B, and MECP2 mRNA levels in erythroid progenitors under the various conditions normalized to the internal control β‐actin by RT‐qPCR analysis. (C) Western blot analysis of the various proteins shown in erythroid progenitors for the different treatment conditions. (D) The mRNA levels of HBG and HBB in erythroid progenitors was calculated as the ratio: HBG/HBG + HBB for the different conditions by RT‐qPCR. (E) Flow cytometry analysis was completed to determine F‐cell levels in erythroid progenitors stained with fluorescein isothiocyanate‐labelledanti‐HbF antibody under the different treatment conditions. (F) Levels of 5‐methylcytosine and unmodified cytosine (cytosine) at the HBG promoter −54 CCGG site for Scr control and MIR29B transfected erythroid progenitor cells. Scr, scramble; UT, untreated. *P < 0·05, **P < 0·01, ***P < 0·005
In our next set of experiments, we sought to determine whether combined treatment with MIR29B (silencing DNMT gene family expression) and Dec (incorporation into DNA to inhibit the action of DNMT), would produce an additive effect on HBG activation. Therefore, erythroid progenitors were treated with 100 nmol/l MIR29B alone or combined with Dec (0·5 μmol/l) as shown in Fig 2D, E. Similar to findings in KU812 cells, the HBG/HBG + HBB mRNA ratio was significantly increased 2·5‐fold (P < 0·01) in erythroid progenitors treated with MIR29B; however, combination treatment with Dec did not increase HBG further (Fig 2D). Similarly, F‐cell levels changed from 5·6% (Scr) to 9% and 16% with Dec and MIR29B (100 nmol/l) alone, respectively (Fig 2E, Figure S1). When both agents were combined, F‐cells remained at levels observed for single agent treatments.
Lastly, we determined the effects of MIR29B on HBG promoter methylation levels in primary erythroid progenitors using the Epimark assay as previously described (Walker et al, 2011). Levels of 5‐methylcytosine and unmodified cytosine at the CCGG MspI restriction site located at nucleotide −54 relative to the HBG gene cap site were determined. We observed an 85% decrease in 5‐methylcytosine levels in erythroid progenitors transfected with MIR29B alone compared to Scr control, whereas levels of unmodified cytosine remained unchanged (Fig 2F). These findings support the ability of MIR29B to mediate demethylation of the HBG promoter and HbF induction in normal human erythroid progenitors.
Given that Dec and MIR29B combined did not produce additive HbF induction, we performed studies with HC, which activated HBG expression by different mechanisms involving cytotoxicity and rapid erythroid regeneration, (Dover et al, 1986; Rodgers, 1992; Maier‐Redelsperger et al, 1998) and activation of cyclic guanosine monophosphate signalling (Ikuta et al, 2011; Lou et al, 2009). Primary erythroid progenitors were treated with 75 μmol/l HC or MIR29B (50 and 100 nmol/l) alone or in combination (Fig 3A, B). Treatment with MIR29B 100 nmol/l and HC alone increased F‐cells to 8% and 9% respectively, compared to 2% for Scr control (Fig 3A, B). For combined studies with MIR29B (100 nmol/l), F‐cell levels remained at 9%, which was similar to single agent treatments.
Figure 3.
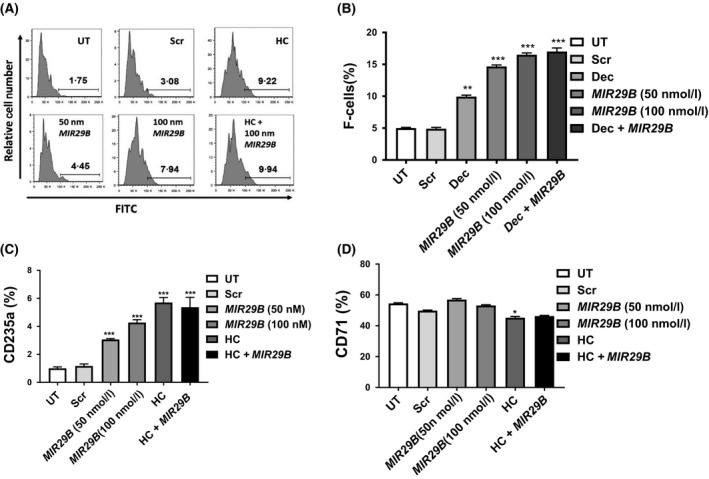
MIR29B increases expression of the late erythroid marker GYPA during erythropoiesis. (A) Shown is representative histograms generated by flow cytometry analysis of erythroid progenitors under untreated (UT), scrambled (Scr), hydroxycarbamide (HC) or MIR29B treatment conditions. (B) F‐cell levels were determined by flow cytometry. Shown in the bar graph is the quantitative data generated by FlowJO analysis. (C and D) Shown in the graphs are data obtained by reverse transcription‐quantitative polymerase chain reaction analysis of RNA isolated from erythroid progenitors under the various treatment conditions using gene‐specific primers for erythroid differentiation markers GYPA (glycophorin A) and TFRC (transferrin receptor). FITC, fluorescein isothiocyanate; HC, hydroxycarbamide; Scr, scramble; UT, untreated. *P < 0·05, **P < 0·01, ***P < 0·005
To determine if MIR29B has an effect on differentiation, we performed Giemsa stain for cell morphology to quantify erythroid cells at different stages of maturation by light microscopy. Both MIR29B (100 nmol/l) and HC (75 μmol/l) produced an increase in early stage mononuclear cells by 1·9 and 2·5‐fold; respectively, with a concomitant decrease in polychromatophilic and orthochromatophilic cells. Of note, MIR29B and HC also increased late stage reticulocytes and red blood cells by 4·3 and 5·3‐fold respectively (Table 1); combined treatment produced similar changes in erythroid maturation kinetics. To compliment cell morphology, we also measured expression of the erythroid differentiation markers GYPA and TFRC (Fig 3C, D). The expression of GYPA significantly increased in erythroid progenitors overexpressing MIR29B or treated with HC alone and combination treatment (Fig 3C); by contrast, only HC produced a 19% decrease (P < 0·05) in TFRC expression (Fig 3D). These data suggest that MIR29B expanded early erythroid progenitors and accelerated differentiation similar to HC.
Table 1.
Summary of cell morphology from Giemsa stain
| Mononuclear cells (%) | Basophilic erythroblast (%) | Polychromatophilic erythroblast (%) | Orthochromatophilic erythroblast (%) | Reticulocytes/red blood cells (%) | |
|---|---|---|---|---|---|
| UT | 8 | 32 | 11 | 47 | 3 |
| Scr 50 nmol/l | 9 | 41 | 13 | 28 | 5 |
| MIR29B 50 nmol/l | 7 | 44 | 4 | 33 | 2 |
| Scr 100 nmol/l | 15 | 42 | 6 | 33 | 3 |
| MIR29B 100 nmol/l | 16 | 52 | 2 | 16 | 13 |
| HC 75 μmol/l | 20 | 43 | 6 | 16 | 16 |
| MIR29B 100 nmol/l/HC 75 μmol/l | 19 | 45 | 5 | 18 | 13 |
HC, hydroxycarbamide; Scr, scramble; UT, untreated.
While in vitro studies provide evidence for the ability of MIR29B to induce HbF, to gain evidence for its physiological relevance in SCD, we performed genome‐wide miRNA expression profiling as recently reported (Li et al, 2018). After obtaining informed consent, blood samples were collected from individuals with SCD (n = 12) not on HC therapy, ranging from 4 to 20 years old. Male and female participants were equally distributed in the high HbF and low HbF groups. The clinical phenotype data collected included HbF levels, complete blood counts with differential and reticulocyte counts (Table 2). There was no significant difference in haematological parameters between the two groups. The individuals with low HbF (average HbF: 3·4%) and high HbF (average HbF: 23·4%) used for miRNA microarray analysis which generated 180 differentially expressed miRNA genes (Li et al, 2018), are shown in Fig 4A. Of those identified, MIR29B was the only gene with known function as a DNMT inhibitor that was significantly up‐regulated among SCD patients with high HbF compared to those with low HbF levels (Fig 4B). We extracted miRNA gene expression raw data for two MIR29 isoforms; MIR29B levels were 2·5‐fold higher (P = 0·015) among SCD patients with high HbF levels compared to the low HbF group (Fig 4B). However, as shown in Fig 4C, there was no significant difference in expression of miR‐29c between the two groups (P = 0·796).
Table 2.
Summary of clinical phenotype data for SCD patients used in miRNA microarray analysis
| Subject group | Hb (g/l) | Hct (%) | Platelets (×109/l) | WBC (×109/l) | Neut (×109/l) | Lymph (×109/l) | Retic count (%) |
|---|---|---|---|---|---|---|---|
| High HbF (n = 6) | 86·3 ± 4·9 | 26·11 ± 1·42 | 404·67 ± 60·17 | 11·7 ± 2·00 | 5·2 ± 0·72 | 4·917 ± 1·25 | 8·467 ± 1·15 |
| Low HbF (n = 6) | 78·0 ± 13·0 | 24·13 ± 1·56 | 521 ± 54·35 | 12·95 ± 0·94 | 6·35 ± 0·73 | 4·750 ± 0·59 | 9·783 ± 1·42 |
| P‐values | 0·1766 | 0·3698 | 0·1802 | 0·5943 | 0·2876 | 0·9067 | 0·4353 |
Hb, haemoglobin; Hct, haematocrit; Lymph, lymphocytes; Neut, neutrophils; Retic, reticulocytes; WBC, white blood cells.
P < 0·05 is considered significant.
Figure 4.
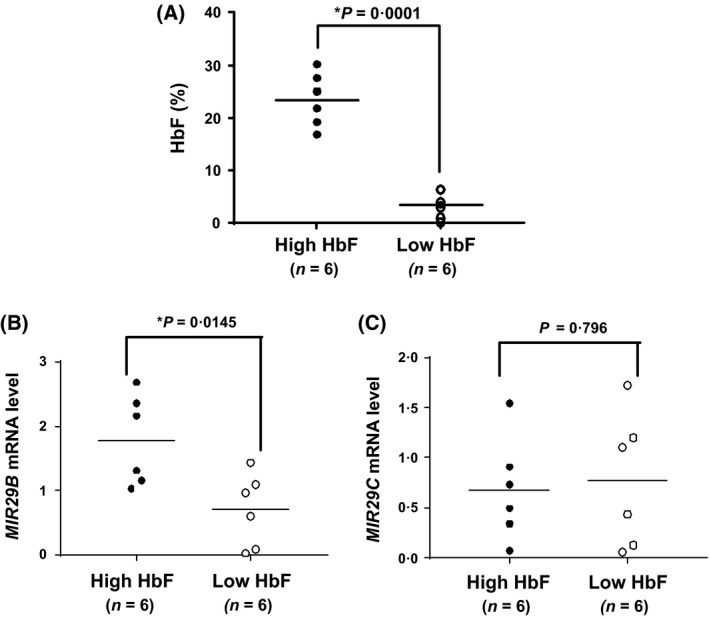
MIR29B correlates with higher peripheral red blood cell HbF levels in SCD patients. (A) Blood samples were obtained after informed consent from subjects with sickle cell disease (SCD)receiving medical care in the Sickle Cell Program at Augusta University. Shown is the distribution of HbF levels determined by clinical high‐performance liquid chromatography for individuals with SCD in the high HbF group (black circles) and low HbF group (white circles); the vertical line represents the mean HbF level. (B) The raw data from miRNA microarray was analysis for MIR29B and (C) miR‐29c, and expression levels correlated with the high HbF (black circles) and low HbF (white circles) SCD groups.
Discussion
Several research efforts have focused on identifying miRNA genes that regulate HBG expression during haemoglobin switching (Gasparello et al, 2017; Hojjati et al, 2017; Lai et al, 2017; Sun et al, 2017; Obeidi et al, 2016; Saki et al, 2016; Tayebi et al, 2017; Guda et al, 2015; De Vasconcellos et al, 2014; Bianchi et al, 2009). Bianchi et al (2009) identified the upregulation of MIR210 by microarray analysis of erythroid precursors from a thalassaemia patient with high HbF levels; they also demonstrated that mithramycin targeted MIR210 as a mechanism of HBG activation in K562 cells (Bianchi et al, 2009). In a subset of patients with SCD treated with HC, expression of MIR26B and MIR151‐3P was associated with HbF levels at a maximum tolerated dose (Walker et al, 2011). Previous studies by our group demonstrated that MIR34A mediated HbF induction in K562 cells by repression of STAT3 expression, a known repressor of HBG (Ward et al, 2016). Previously, Lee et al (2013) confirmed overexpression of LIN28B decreased MIRLET7 miRNA family expression and increased HbF levels in primary erythroid cells. As further evidence of the therapeutic potential of miRNA, miR‐486‐3p induces HbF in adult erythroid progenitors by inhibiting BCL11A expression (Lulli et al, 2013). In this study, we demonstrated that MIR29B functions as an HbF inducer in human KU812 cells and normal human erythroid progenitors. To our knowledge, there are no other studies generating experimental evidence of a miRNA that targets the DNA methylation machinery as a mechanism of HbF induction.
DNA methylation is an epigenetic event that can be reversed by targeting key genes, such as DNMTs and MECP2, which are involved in mechanisms of gene silencing (Wu & Zhang, 2014). Elevated levels of 5‐methylcytosine at the −54 methylation site in the HBG promoter during differentiation of erythroid progenitors is associated with HBG methylation and transcriptional silencing (Ruiz et al, 2015). In our current study, we observed significantly lower HBG promoter methylation following MIR210 overexpression in normal human erythroid progenitors mediated by decreased 5‐methylcytosine and MECP2 expression. Previous studies also demonstrated that methyl‐CpG binding domain proteins mediate embryonic HBE1 and fetal HBG gene silencing in human adult erythroid cells and their gene silencing produced reactivation of HBG transcription (Gnanapragasam et al, 2011). Furthermore, strategies to inhibit the DNA methylation machinery have proven to be an effective therapeutic approach of HbF induction in SCD patients via HBG promoter hypomethylation (Molokie et al, 2017). We did not observe additive HbF induction with combined HC and MIR29B treatment in erythroid progenitors generated from adult CD34+ stem cells. However, whether additive HbF induction would result for combination treatments under oxidative stress conditions, which exist in SCD, requires future investigations.
Experimental studies demonstrated that MIR29 targets the tumour suppressor MYB (Martinez et al, 2011; Hu et al, 2014). In recent genome‐wide association studies consisting of individuals diagnosed with SCD or β‐thalassaemia, several loci, including the HBS1L‐MYB intergenic interval, were associated with ~40% of the inherited HbF variance. The transcriptional activator MYB is essential for definitive haematopoiesis (Sheiness & Gardinier, 1984) and is highly expressed in immature haematopoietic cells and down‐regulated during erythropoiesis (Bianchi et al, 2009). Overexpression of MYB inhibits HBG expression in K562 cells (Jiang et al, 2006); and its knockdown in primary human erythroid progenitors induces HbF expression (Sankaran et al, 2011). Moreover, MYB has been shown to regulate HbF expression in quantitative trait locus studies and functional assays (Jiang et al, 2006; Sankaran et al, 2011; Thein et al, 2007). Considering the essential role of MYB as a HBG repressor protein, we hypothesized that overexpression of MIR29B to silence MYB would mediate HbF induction. We observed downregulation of MYB by MIR29B and HbF induction in KU812 cells. To support a physiological role, Sankaran et al (2011) showed that overexpression of MIR15A/MIR16‐1 in patients with trisomy 13, down‐regulated MYB protein as a mechanism of persistent HbF expression during adulthood. These findings support the role of MYB as a negative regulator of HBG gene transcription in vivo directly targeted by MIR15A/MIR16‐1 in erythroid progenitors.
In summary, our study shows for the first time that MIR29B, a DNMT inhibitor, reactivates HBG gene transcription and HbF synthesis in human primary erythroid progenitors. Moreover, increased expression of MIR29B in reticulocytes isolated from individuals with SCD and high HbF levels provides additional evidence for the clinical relevance of this molecule. Future studies to evaluate ability of MIR29B to mediate epigenetic effects to activate HBG transcription and HbF expression in SCD progenitors or preclinical mouse models, are desirable to develop treatments for SCD and other β‐haemoglobinopathies.
Conflict of Interest
The authors declare no competing financial interests and no conflict of interests to disclose.
Supporting information
Table SI. Summary of primer sequences.
Figure S1. Shown is representative histograms generated by flow cytometry analysis of erythroid progenitors under untreated (UT), scrambled (Scr), decitabine (Dec) or miR‐29b treatment conditions.
Acknowledgements
AS‐D, AS, LV and BL performed the research; AS‐D and BSP designed the research study; AS‐D, LV and BSP contributed essential reagents and tools; AS‐D, LV and BL analysed the data; AS‐D and BSP wrote the paper. This work was funded through an NIH grant R01HL069234 (B.S.P.) and funding support from the UTHSC Center for Integrative Genomics, Methodist Mission, and Collaborative Research Network (CORNET) Clinical award (AS‐D).
References
- Akpan, I. , Banzon, V. , Ibanez, V. , Vaitkus, K. , Desimone, J. & Lavelle, D. (2010) Decitabine increases fetal hemoglobin in Papio anubis by increasing gamma‐globin gene transcription. Experimental Hematology, 38, 989–993 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi, N. , Zuccato, C. , Lampronti, I. , Borgatti, M. & Gambari, R. (2009) Expression of miR‐210 during erythroid differentiation and induction of gamma‐globin gene expression. Biochemistry and Molecular Biology Reports, 42, 493–499. [DOI] [PubMed] [Google Scholar]
- Charache, S. , Terrin, M.L. , Moore, R.D. , Dover, G.J. , Barton, F.B. , Eckert, S.V. , Mcmahon, R.P. & Bonds, D.R. (1995) Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. New England Journal of Medicine, 332, 1317–1322. [DOI] [PubMed] [Google Scholar]
- De Vasconcellos, J.F. , Fasano, R.M. , Lee, Y.T. , Kaushal, M. , Byrnes, C. , Meier, E.R. , Anderson, M. , Rabel, A. , Braylan, R. , Stroncek, D.F. & Miller, J.L. (2014) LIN28A expression reduces sickling of cultured human erythrocytes. PLoS One, 9, e106924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desimone, J. , Koshy, M. , Dorn, L. , Lavelle, D. , Bressler, L. , Molokie, R. & Talischy, N. (2002) Maintenance of elevated fetal hemoglobin levels by decitabine during dose interval treatment of sickle cell anemia. Blood, 99, 3905–3908. [DOI] [PubMed] [Google Scholar]
- Dover, G.J. , Humphries, R.K. , Moore, J.G. , Ley, T.J. , Young, N.S. , Charache, S. & Nienhuis, A.W. (1986) Hydroxyurea induction of hemoglobin F production in sickle cell disease: relationship between cytotoxicity and F cell production. Blood, 67, 735–738. [PubMed] [Google Scholar]
- Gasparello, J. , Fabbri, E. , Bianchi, N. , Breveglieri, G. , Zuccato, C. , Borgatti, M. , Gambari, R. & Finotti, A. (2017) BCL11A mRNA targeting by miR‐210: a possible network regulating gamma‐globin gene expression. International Journal of Molecular Sciences, 18, E2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnanapragasam, M.N. , Scarsdale, J.N. , Amaya, M.L. , Webb, H.D. , Desai, M.A. , Walavalkar, N.M. , Wang, S.Z. , Zu Zhu, S. , Ginder, G.D. & Williams, D.C. Jr (2011) p66Alpha‐MBD2 coiled‐coil interaction and recruitment of Mi‐2 are critical for globin gene silencing by the MBD2‐NuRD complex. Proceedings of the National Academy of Sciences of the United States of America, 108, 7487–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guda, S. , Brendel, C. , Renella, R. , Du, P. , Bauer, D.E. , Canver, M.C. , Grenier, J.K. , Grimson, A.W. , Kamran, S.C. , Thornton, J. , De Boer, H. , Root, D.E. , Milsom, M.D. , Orkin, S.H. , Gregory, R.I. & Williams, D.A. (2015) miRNA‐embedded shRNAs for lineage‐specific BCL11A knockdown and hemoglobin F induction. Molecular Therapy, 23, 1465–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojjati, M.T. , Azarkeivan, A. , Pourfathollah, A.A. & Amirizadeh, N. (2017) Comparison of MicroRNAs mediated in reactivation of the gamma‐globin in beta‐thalassemia patients, responders and non‐responders to hydroxyurea. Hemoglobin, 41, 110–115. [DOI] [PubMed] [Google Scholar]
- Hu, Z. , Klein, J.D. , Mitch, W.E. , Zhang, L. , Martinez, I. & Wang, X.H. (2014) MicroRNA‐29 induces cellular senescence in aging muscle through multiple signaling pathways. Aging (Albany NY), 6, 160–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikuta, T. , Adekile, A.D. , Gutsaeva, D.R. , Parkerson, J.B. , Yerigenahally, S.D. , Clair, B. , Kutlar, A. , Odo, N. & Head, C.A. (2011) The proinflammatory cytokine GM‐CSF downregulates fetal hemoglobin expression by attenuating the cAMP‐dependent pathway in sickle cell disease. Blood Cells, Molecules and Diseases, 47, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, J. , Best, S. , Menzel, S. , Silver, N. , Lai, M.I. , Surdulescu, G.L. , Spector, T.D. & Thein, S.L. (2006) cMYB is involved in the regulation of fetal hemoglobin production in adults. Blood, 108, 1077–1083. [DOI] [PubMed] [Google Scholar]
- Lai, K. , Jia, S. , Yu, S. , Luo, J. & He, Y. (2017) Genome‐wide analysis of aberrantly expressed lncRNAs and miRNAs with associated co‐expression and ceRNA networks in beta‐thalassemia and hereditary persistence of fetal hemoglobin. Oncotarget, 8, 49931–49943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavelle, D. , Vaitkus, K. , Hankewych, M. , Singh, M. & Desimone, J. (2006) Developmental changes in DNA methylation and covalent histone modifications of chromatin associated with the epsilon‐, gamma‐, and beta‐globin gene promoters in Papio anubis. Blood Cells, Molecules and Diseases, 36, 269–278. [DOI] [PubMed] [Google Scholar]
- Lavelle, D. , Saunthararajah, Y. , Vaitkus, K. , Singh, M. , Banzon, V. , Phiasivongsva, P. , Redkar, S. , Kanekal, S. , Bearss, D. , Shi, C. , Inloes, R. & Desimone, J. (2010) S110, a novel decitabine dinucleotide, increases fetal hemoglobin levels in baboons (P. anubis). Journal of Translational Medicine, 8, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y.T. , De Vasconcellos, J.F. , Yuan, J. , Byrnes, C. , Noh, S.J. , Meier, E.R. , Kim, K.S. , Rabel, A. , Kaushal, M. , Muljo, S.A. & Miller, J.L. (2013) LIN28B‐mediated expression of fetal hemoglobin and production of fetal‐like erythrocytes from adult human erythroblasts ex vivo. Blood, 122, 1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B. , Ding, L. , Yang, C. , Kang, B. , Liu, L. , Story, M.D. & Pace, B.S. (2014) Characterization of transcription factor networks involved in umbilical cord blood CD34 + stem cells‐derived erythropoiesis. PLoS One, 9, e107133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B. , Zhu, X. , Ward, C.M. , Starlard‐Davenport, A. , Takezaki, M. , Berry, A. , Ward, A. , Wilder, C. , Neunert, C. , Kutlar, A. & Pace, B.S. (2018) MIR‐144‐mediated NRF2 gene silencing inhibits fetal hemoglobin expression in sickle cell disease. Experimental Hematology, 70, 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou, T.F. , Singh, M. , Mackie, A. , Li, W. & Pace, B.S. (2009) Hydroxyurea generates nitric oxide in human erythroid cells: mechanisms for gamma‐globin gene activation. Experimental Biology and Medicine (Maywood), 234, 1374–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lulli, V. , Romania, P. , Morsilli, O. , Cianciulli, P. , Gabbianelli, M. , Testa, U. , Giuliani, A. & Marziali, G. (2013) MicroRNA‐486‐3p regulates gamma‐globin expression in human erythroid cells by directly modulating BCL11A. PLoS One, 8, e60436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier‐Redelsperger, M. , De Montalembert, M. , Flahault, A. , Neonato, M.G. , Ducrocq, R. , Masson, M.P. , Girot, R. & Elion, J. (1998) Fetal hemoglobin and F‐cell responses to long‐term hydroxyurea treatment in young sickle cell patients. The French Study Group on Sickle Cell Disease. Blood, 91, 4472–4479. [PubMed] [Google Scholar]
- Martinez, I. , Cazalla, D. , Almstead, L.L. , Steitz, J.A. & Dimaio, D. (2011) miR‐29 and miR‐30 regulate B‐Myb expression during cellular senescence. Proceedings of the National Academy of Sciences of the United States of America, 108, 522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molokie, R. , Lavelle, D. , Gowhari, M. , Pacini, M. , Krauz, L. , Hassan, J. , Ibanez, V. , Ruiz, M.A. , Ng, K.P. , Woost, P. , Radivoyevitch, T. , Pacelli, D. , Fada, S. , Rump, M. , Hsieh, M. , Tisdale, J.F. , Jacobberger, J. , Phelps, M. , Engel, J.D. , Saraf, S. , Hsu, L.L. , Gordeuk, V. , Desimone, J. & Saunthararajah, Y. (2017) Oral tetrahydrouridine and decitabine for non‐cytotoxic epigenetic gene regulation in sickle cell disease: a randomized phase 1 study. PLoS Medicine, 14, e1002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeidi, N. , Pourfathollah, A.A. , Soleimani, M. , Nikougoftar Zarif, M. & Kouhkan, F. (2016) The effect of Mir‐451 upregulation on erythroid lineage differentiation of murine embryonic stem cells. Cell Journal, 18, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt, O.S. , Brambilla, D.J. , Rosse, W.F. , Milner, P.F. , Castro, O. , Steinberg, M.H. & Klug, P.P. (1994) Mortality in sickle cell disease. Life expectancy and risk factors for early death. New England Journal of Medicine, 330, 1639–1644. [DOI] [PubMed] [Google Scholar]
- Poillon, W.N. , Kim, B.C. , Rodgers, G.P. , Noguchi, C.T. & Schechter, A.N. (1993) Sparing effect of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S at physiologic ligand saturations. Proceedings of the National Academy of Sciences of the United States of America, 90, 5039–5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promsote, W. , Makala, L. , Li, B. , Smith, S.B. , Singh, N. , Ganapathy, V. , Pace, B.S. & Martin, P.M. (2014) Monomethylfumarate induces gamma‐globin expression and fetal hemoglobin production in cultured human retinal pigment epithelial (RPE) and erythroid cells, and in intact retina. Invest Ophthalmology and Visual Science, 55, 5382–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers, G.P. (1992) Spectrum of fetal hemoglobin responses in sickle cell patients treated with hydroxyurea: the National Institutes of Health experience. Seminars in Oncology, 19, 67–73. [PubMed] [Google Scholar]
- Ruiz, M.A. , Rivers, A. , Ibanez, V. , Vaitkus, K. , Mahmud, N. , Desimone, J. & Lavelle, D. (2015) Hydroxymethylcytosine and demethylation of the gamma‐globin gene promoter during erythroid differentiation. Epigenetics, 10, 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saki, N. , Abroun, S. , Soleimani, M. , Kavianpour, M. , Shahjahani, M. , Mohammadi‐Asl, J. & Hajizamani, S. (2016) MicroRNA expression in beta‐thalassemia and sickle cell disease: a role in the induction of fetal hemoglobin. Cell Journal, 17, 583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran, V.G. , Menne, T.F. , Scepanovic, D. , Vergilio, J.A. , Ji, P. , Kim, J. , Thiru, P. , Orkin, S.H. , Lander, E.S. & Lodish, H.F. (2011) MicroRNA‐15a and ‐16‐1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proceedings of the National Academy of Sciences of the United States of America, 108, 1519–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheiness, D. & Gardinier, M. (1984) Expression of a proto‐oncogene (proto‐myb) in hemopoietic tissues of mice. Molecular and Cellular Biology, 4, 1206–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starlard‐Davenport, A. , Kutanzi, K. , Tryndyak, V. , Word, B. & Lyn‐Cook, B. (2013) Restoration of the methylation status of hypermethylated gene promoters by microRNA‐29b in human breast cancer: a novel epigenetic therapeutic approach. Journal of Carcinogenesis, 12, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, K.T. , Huang, Y.N. , Palanisamy, K. , Chang, S.S. , Wang, I.K. , Wu, K.H. , Chen, P. , Peng, C.T. & Li, C.Y. (2017) Reciprocal regulation of gamma‐globin expression by exo‐miRNAs: relevance to gamma‐globin silencing in beta‐thalassemia major. Scientific Reports, 7, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayebi, B. , Abrishami, F. , Alizadeh, S. , Minayi, N. , Mohammadian, M. , Soleimani, M. , Dehghanifard, A. , Atwan, H. , Ajami, M. & Ajami, M. (2017) Modulation of microRNAs expression in hematopoietic stem cells treated with sodium butyrate in inducing fetal hemoglobin expression. Artificial Cells, Nanomedicine, and Biotechnology, 45, 146–156. [DOI] [PubMed] [Google Scholar]
- Thein, S.L. , Menzel, S. , Peng, X. , Best, S. , Jiang, J. , Close, J. , Silver, N. , Gerovasilli, A. , Ping, C. , Yamaguchi, M. , Wahlberg, K. , Ulug, P. , Spector, T.D. , Garner, C. , Matsuda, F. , Farrall, M. & Lathrop, M. (2007) Intergenic variants of HBS1L‐MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proceedings of the National Academy of Sciences of the United States of America, 104, 11346–11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, A.L. , Steward, S. , Howard, T.A. , Mortier, N. , Smeltzer, M. , Wang, Y.D. & Ware, R.E. (2011) Epigenetic and molecular profiles of erythroid cells after hydroxyurea treatment in sickle cell anemia. Blood, 118, 5664–5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, C.M. , Li, B. & Pace, B.S. (2016) Original research: stable expression of miR‐34a mediates fetal hemoglobin induction in K562 cells. Experimental Biology and Medicine (Maywood), 241, 719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherall, D.J. (2013) The role of the inherited disorders of hemoglobin, the first “molecular diseases”, in the future of human genetics. Annual Review of Genomics and Human Genetics, 14, 1–24. [DOI] [PubMed] [Google Scholar]
- Wu, H. & Zhang, Y. (2014) Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell, 156, 45–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X. , Li, B. & Pace, B.S. (2017) NRF2 mediates gamma‐globin gene regulation and fetal hemoglobin induction in human erythroid progenitors. Haematologica, 102, e285–e288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table SI. Summary of primer sequences.
Figure S1. Shown is representative histograms generated by flow cytometry analysis of erythroid progenitors under untreated (UT), scrambled (Scr), decitabine (Dec) or miR‐29b treatment conditions.